

Abstract
The 2.15-Å resolution cocrystal structure of EcoRV endonuclease mutant T93A complexed with DNA and Ca2+ ions reveals two divalent metals bound in one of the active sites. One of these metals is ligated through an inner-sphere water molecule to the phosphate group located 3′ to the scissile phosphate. A second inner-sphere water on this metal is positioned approximately in-line for attack on the scissile phosphate. This structure corroborates the observation that the pro-SP phosphoryl oxygen on the adjacent 3′ phosphate cannot be modified without severe loss of catalytic efficiency. The structural equivalence of key groups, conserved in the active sites of EcoRV, EcoRI, PvuII, and BamHI endonucleases, suggests that ligation of a catalytic divalent metal ion to this phosphate may occur in many type II restriction enzymes. Together with previous cocrystal structures, these data allow construction of a detailed model for the pretransition state configuration in EcoRV. This model features three divalent metal ions per active site and invokes assistance in the bond-making step by a conserved lysine, which stabilizes the attacking hydroxide ion nucleophile.
Recently determined crystal structures of type II restriction endonucleases have produced a wealth of information on the basis for target site sequence selectivity (1–6). However, these structures do not resolve the detailed structural mechanism of catalysis. Although acidic and basic groups in the active sites can be identified, and in some cases divalent-metal binding sites delineated, a convincing picture clarifying the way in which the attacking hydroxide ion is generated, and the leaving group stabilized, has not been elucidated for any of the enzymes.
EcoRV endonuclease is a homodimer of 244 aa per monomer. It cleaves the duplex sequence 5′-GATATC at the central TA step in a blunt-ended fashion (7). As in all type II enzymes, phosphoryl transfer proceeds via attack of a hydroxide ion nucleophile on the scissile phosphorus, generating products containing a 5′ phosphate group. This reaction occurs by in-line displacement through a pentacovalent transition state, with inversion of stereochemistry at phosphorus and an absolute requirement for divalent metal cations (8, 9). High-resolution crystallographic analyses of EcoRV show that the scissile phosphates of the DNA are located adjacent to the carboxylates of Asp-90 and Asp-74, and that a divalent metal ion bridges the enzyme and DNA at this position (2, 3). Although this metal could stabilize the additional negative charge that develops in the transition state, it is not correctly positioned to generate the attacking hydroxide ion. Thus, speculative mechanisms in which the nucleophile arises from dissociation of a metal-ligated water have invoked significant rearrangements of the DNA from its observed conformation in the crystal structures (2, 10).
An alternative mechanism involving substrate-assisted catalysis by the adjacent 3′-phosphate of the DNA also has been proposed (11, 12). In support of this mechanism, it has been found that replacement of the pro-SP oxygen of this phosphate with sulfur reduces catalysis by at least 200-fold (13). Interestingly, superposition of the cocrystal structures of EcoRV, EcoRI, BamHI, and PvuII shows that the adjacent 3′-phosphate of the DNA is in a very similar position relative to the scissile phosphate in each case (14). Moreover, the orientation of this phosphate is approximately in-line with respect to the required direction of attack by the hydroxide nucleophile. Modification experiments provide evidence that this group is crucial to catalysis in EcoRI and in a number of additional type II enzymes of unknown structure (15, 16). Thus, an important role for the 3′-phosphate may be general to the entire enzyme family.
It has been suggested that the 3′-phosphate group operates as a general base by directly deprotonating a water molecule (11–13), but the elevation of the pKa required for this function appears implausible (2, 14). Here, we present the cocrystal structure of the T93A mutant of EcoRV complexed with DNA and Ca2+ ions (Ca2+ promotes specific binding but reduces catalytic rates by at least 105-fold; ref. 17). This structure reveals that one Ca2+ ion both ligates the 3′-phosphate and directs an inner-sphere water toward the scissile phosphate in approximately the required orientation. A second Ca2+ ion in the active site plays a structural role in positioning the essential catalytic residue Asp-74 (18), which ligates both metals. This crystal structure thus provides a plausible structural basis for the role of the 3′-phosphate in a substrate-assisted mechanism. A model of a pretransition state configuration is presented in which each active site binds three divalent metal ions.
MATERIALS AND METHODS
Purification and Crystallization.
EcoRV endonuclease was purified, stored, and prepared for crystallization as described (3). The DNA substrate 5′-CAAGATATCTT was synthesized for cocrystallization trials by standard methods and purified on a Rainin PureDNA HPLC column developed in a gradient of tri-ethyl amine acetate/acetonitrile. Detritylation was performed on the column (19). The DNA peak was collected manually, lyophilized, and stored as dry pellets at −20°C. Cocrystals of the T93A EcoRV-DNA-Ca2+ complex were grown by hanging-drop vapor diffusion from solutions containing 2–8% polyethylene glycol 4000, 10 mM CaCl2, 50 mM Hepes (pH 7.5), and 100 mM NaCl. The final protein concentration was 13 mg/ml, and the protein/DNA stoichiometry was 1:1. Crystals were mounted in capillaries for data collection directly from the drop.
X-Ray Structure Determination.
X-ray diffraction amplitudes were measured on an R-AXIS IIC area detector mounted on a Rigaku RU-200 rotating anode generator. Data were obtained at ambient temperature from one crystal. Determination of the orientation matrix, integration, scaling, and merging of data were performed with the r-axis suite of programs. The crystal structure of EcoRV complexed to 5′-CAAGATATCTT was phased directly by using the isomorphous cocrystal of the wild-type enzyme complexed with 5′-AAAGATATCTT (3) as the starting model. The Thr-93 side chain of each subunit, the 5′-nucleotide of each DNA strand, and 57 water molecules at the enzyme-DNA interface were removed from the model. The starting crystallographic R factor was 34.5%, and it was reduced to 19.2% (Rfree = 29.0% with 10% of the data removed throughout refinement) by several rounds of model building iterated with rigid-body, positional, B-factor, and simulated annealing refinement in xplor (20) (Table 1). Criteria for the inclusion of water molecules were the appearance of peaks at 1.0 σ in (2Fo − Fc) maps, 3.0 σ in (Fo − Fc) maps, and at least one hydrogen-bonding interaction with protein or DNA. All solvent molecules and side chains possessing B-factors above 60.0 Å2 were examined carefully before inclusion in the final model. Model building used the program chain (21). Parameters for the DNA used in xplor refinement were those recently described (22). Least-squares superpositions were performed with insight ii (23) and gem (24). Definitions of the core domains for superpositions were carried out with the aid of difference-distance calculations (3).
Table 1.
Crystallographic data collection and refinement statistics: EcoRV T93A/DNA/Ca2+
| Resolution, Å | 2.15 |
| Space group | P1 |
| Cell dimensions, Å | a = 50.0, b = 64.4, c = 49.2 |
| α = 109.2°, β = 108.7°, γ = 95.8° | |
| % Data coveragea | 75% |
| Rmerge | 0.074 |
| Bover (Å2)b | 40.7 |
| Rcrystc | 0.185 |
| Rfree | 0.288 |
| rms bonds, Å | 0.011 |
| rms angles | 1.94° |
| Number of waters | 168 |
Includes all data in the intensity range I/σ(I) > 0.0. Data coverage in the highest 0.1 Å shell is 73%.
Overall B-factor is determined from a Wilson plot of the structure factor data by using a low-resolution cutoff of 4.6 Å.
Refinement was carried out by using a low-resolution cutoff of 6.0 Å.
RESULTS
Overall Structure of the Mutant Complex.
The crystal structure of the EcoRV T93A-DNA-Ca2+ complex shows that the mutation causes significant structural changes. The two DNA-binding/catalytic domains of the mutant are rotated apart by 1.8° relative to their positions in the wild-type enzyme. Several of the linker segments bridging the dimerization and DNA-binding domains, as well as the minor-groove binding Q-loops, are altered in conformation. The mutation at Thr-93 also shifts the position of the core β-strand at and adjacent to this residue (Fig. 1). This β-strand runs along the DNA backbone and contains the key catalytic amino acids Asp-90 and Lys-92. The DNA backbone is shifted away from this polypeptide segment by 1.0–1.5 Å in the mutant complex, apparently as a result of rearrangements in the interior of the protein arising from the void introduced by the Thr → Ala substitution. The rate of the chemical step for the cleavage of DNA by T93A is reduced by 102-fold compared with the wild-type enzyme, in the presence of either Mg2+ or Mn2+ as the catalytic metal cofactor (M. Sam and J.J.P., unpublished work).
Figure 1.
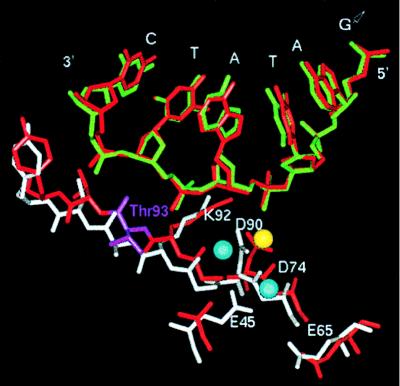
Superposition of the structures of wild-type EcoRV (red) and the T93A mutant (white, green) based on sugar-phosphate atoms of the DNA at the center base pairs of the target site. Thr-93 in the wild-type enzyme is shown in magenta, and the active-site side chains are indicated. The blue spheres indicate the Ca2+ binding sites observed in this mutant structure; the yellow sphere shows the position of the Ca2+ binding site previously found in the wild-type structure.
Structure of the Active Sites.
The structures of the two active sites are not substantially different from those of the wild-type enzyme. Superposition of subunit I of T93A upon that of the wild-type structure cocrystallized with DNA and Ca2+, based on sugar-phosphate backbone atoms at the center four bases of GATATC, gives an rms deviation of only 0.27 Å (Fig. 1). Thus, the conformations of key portions of the DNA in these two complexes are identical within coordinate error. However, because of the shift in the polypeptide backbone at residue 93, the side chains of the adjacent active site residues Lys-92 and Asp-90 are displaced by roughly 1 Å relative to the DNA (Fig. 1). Other active-site amino acids, including Asp-74, Glu-45, and Glu-65, also undergo small reorientations (the Glu-45 side chain is disordered beyond Cβ in the wild-type complex). Superposition based on polypeptide backbone atoms of these five residues gives an rms deviation of 0.29 Å. Therefore, the relative positions of the DNA and the enzyme active site are slightly altered in the mutant, but the internal structure of each is preserved.
The two active sites of the T93A dimer are not identical. In particular, the conformation of the deoxyribose sugar, which bridges the scissile and the adjacent 3′-phosphates, is altered in subunit II of T93A, such that the sugar ring is displaced farther from the active-site residues. The positions of active-site backbone and side-chain atoms also deviate by 0.5–1.0 Å between the two subunits. An apparent consequence of these differences is that Ca2+ binding sites can be identified only in subunit I of this structure. This observation is reminiscent of that made when Mg2+ was soaked into crystals of the wild-type EcoRV–DNA complex in this crystal lattice (2). In this case Mg2+ binding was similarly visualized in only one active site, and the DNA was not cleaved in either strand. It is evident that crystal packing constraints in this P1 lattice give rise to asymmetry in the subunits and also prevent the reactive groups of the enzyme and DNA from achieving correct juxtaposition. However, crystal structures of the EcoRV–DNA complex in four additional lattices (ref. 1; N.C.H. and J.J.P., unpublished work), including one in which activity is observed (1), show similar active-site and DNA conformations. Analysis of these structures suggests that the activity of crystalline EcoRV depends on its quaternary structure, and that inactive conformers of the flexible dimer with active-site separations varying by several Å are readily trapped (3). The detailed structure observed for each individual active site may not, however, deviate markedly from the enzyme in solution.
Metal Binding Sites.
Two Ca2+ binding sites are present in subunit I of the T93A-DNA-Ca2+ structure (Fig. 1). The sites are readily identifiable as 7.5-σ peaks in difference electron density maps calculated with coefficients (Fo − Fc), by using phasing models in which all active-site solvent molecules are removed. Strong density distinguishing the Ca2+ ions from water molecules also is observed in simulated annealing omit maps calculated with coefficients (2Fo − Fc) (Fig. 2). No significant unattributed density remains in the subunit I active site upon completion of refinement. The B-factors of the two calcium ions, CL1 and CL2, (Figs. 2 and 3) refine to 46.3 Å2 and 40.2 Å2, respectively, similar to the overall B-factor of 40.7 Å2.
Figure 2.
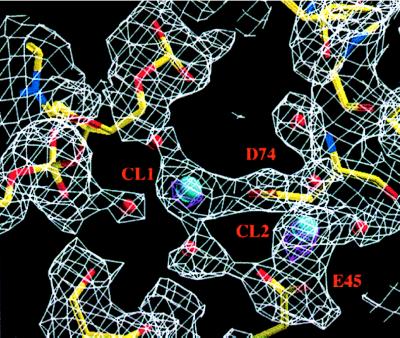
Simulated annealing omit electron density maps in the active site of subunit I of the EcoRV T93A-DNA-Ca2+ complex. Side chains of Asp-90, Asp-74, Glu-45, Lys-92, and Glu-65 and all solvent molecules, were removed and the resulting model subjected to a simulated annealing refinement protocol in xplor. Electron density maps calculated with coefficients (2Fo − Fc) (white) and (Fo − Fc) (magenta) are shown superimposed on the final model. Phases for these maps were derived from the model with these atoms deleted. Each map was computed in the resolution range from 2.15 Å to 20 Å. The density is contoured at 1.0 σ for the (2Fo − Fc) map and 6.5 σ for the (Fo − Fc) map. Blue spheres represent Ca2+ ions and red spheres represent waters.
Figure 3.

Detailed structure of the Ca2+ binding sites in subunit I of the EcoRV T93A-DNA-Ca2+ complex. Metal-ligand distances and distances between the electronegative atoms for selected hydrogen bonds are indicated in Å.
One of the two Ca2+ ions (CL1; Fig. 3) localized in this structure appears in a novel position relative to the divalent metal sites observed in all previous EcoRV–DNA cocrystals (1–3). This metal is coordinated by the side chain of Asp-74, by the main-chain carbonyl oxygen of Ile-91, and by three water molecules. A possible sixth ligand, a carboxylate oxygen of Asp-90, is at a distance of 3.1 Å. One of the waters bridges CL1 with the pro-SP oxygen of the phosphate group located immediately 3′ of the scissile phosphate. A second metal-ligated water molecule is located 4.1 Å from the scissile phosphorus and is oriented approximately in-line with respect to the required direction of nucleophilic attack (Fig. 3). This water also accepts a hydrogen bond from the side-chain amine group of Lys-92, an amino acid crucial to high catalytic activity (18). A previously observed divalent metal binding site also is located between the carboxylates of Asp-74 and Asp-90, but this metal ligates to the scissile phosphate rather than the 3′-phosphate. Divalent metal bound at this alternate position is not detectable in the T93A-DNA-Ca2+ cocrystal, perhaps explaining the decreased catalytic activity of the mutant (25, 26). Superposition of the wild-type and mutant structures, based on the DNA sugar-phosphate backbone at and adjacent to the center TA step, shows that these two metal sites are located 3.2 Å apart. The two sites do not overlap, so that simultaneous occupancy of the active site by metals at both positions, although not observed in any experimental structure, appears possible.
The superpositions of the wild-type and T93A structures provide a rationale for why the two metal sites are not occupied together in either case. Hydrated Ca2+ can bind to site I in T93A because the protein and DNA are shifted slightly apart (Fig. 1). Thus, steric exclusion prevents binding of hydrated Ca2+ at site I in the wild-type enzyme. The mutation-induced structural shift also produces a motion of the scissile phosphate of magnitude 0.8 Å away from Asp-74 and Asp-90 at site III. This shift destabilizes site III Ca2+ binding in T93A, explaining why the metal is not observed at this position. A clear explanation for the inability of Ca2+ to support catalytic activity also is provided by these data: this larger ion cannot bind to site I. Both the radius and the minimum metal-ligand distance in Mg2+ are significantly smaller than in Ca2+ (27), so that Mg2+ may be able to occupy site I in the wild-type enzyme without the structural change caused by the T93A mutation. A further rationale for the inability of Ca2+ to support activity is likely to be its decreased acidity relative to Mg2+. Thus, the direct site III Ca2+ interaction with the scissile phosphate results in a more weakly polarized P-O bond.
The second Ca2+ ion (CL2, Fig. 3) visualized in the T93A active site is not bound to the DNA. It appears instead to play a structural role by ligating to a carboxylate oxygen of Asp-74, stabilizing the conformation of this side chain for its interaction with CL1 (Fig. 3). The other ligands of CL2 are the two carboxylate oxygens of Glu-45 and two water molecules. One of these waters bridges to the adjacent side-chain carboxylate of Glu-65 and to the main-chain carbonyl group of Asp-74, and the other to the Asp-74 side chain. This binding site is similar to one of the two binding sites for Mg2+ observed when crystals of the wild-type EcoRV–DNA complex are soaked in solutions containing this metal (2). In that structure the metal at this site (74/45 site) also ligates both Asp-74 and Glu-45, but the conformation of Glu-45 differs such that only one carboxylate oxygen is directly bound. The two Ca2+ ions CL1 and CL2 are located 5.4 Å apart.
DISCUSSION
Role of the Adjacent 3′-Phosphate in Substrate-Assisted Catalysis.
The crystal structure of the EcoRV T93A-DNA-Ca2+ complex supports the proposal that the adjacent 3′-phosphate plays a key role in the mechanism (11, 12). It has been shown that substitution of the pro-SP oxygen of this phosphate with sulfur reduces Vmax/Km by at least 200-fold, whereas substitution of the pro-RP oxygen has little effect (13). It was proposed, however, that it is the pro-RP oxygen that functions as the catalytic base by abstracting a proton from a water molecule. By this mechanism, the RP-phosphorothioate carrying a negatively charged sulfur also can abstract the proton, whereas the SP-phosphorothioate results in an uncharged double-bonded oxygen in the RP position. This uncharged oxygen cannot function as a base, explaining the inactivity of the SP substitution (13). An unresolved difficulty with this proposal is that the pKa of a nonesterified phosphate oxygen is far too low to efficiently deprotonate water. It seems unlikely that this pKa could be raised sufficiently in the structural environment of the active site, as there are no adjacent negatively charged groups not ligated to metals.
The cocrystal structure reported here now shows that a Ca2+ ion ligates through water to the pro-SP oxygen of the adjacent 3′-phosphate (Fig. 3). Thus, we suggest that the 3′-phosphate functions by helping to assemble the required active-site structure, with the attacking nucleophile instead generated from an inner-sphere water ligated to the divalent metal. Such waters possess greatly lowered pKas in the range of 10–12 (28), so that hydroxide formation is chemically plausible at neutral pH.
Role of Lys-92.
Replacement of Lys-92 with alanine or glutamine reduces cleavage activity by 104-fold or greater (18). It has been suggested that the role of this lysine is to provide electrostatic stabilization at the scissile phosphate in the transition state (8, 18). However, in all structures Lys-92 is positioned on the same face of this phosphate as must be approached by the attacking hydroxide ion. Thus, the nonesterified oxygens move away from this lysine as the geometry at phosphorus changes from tetrahedral in the ground state to trigonal bipyramidal in the transition state. It thus appears very unlikely, on structural grounds, that Lys-92 preferentially stabilizes the incipient additional negative charge on the scissile phosphate.
The crystal structure of EcoRV T93A bound to DNA and Ca2+ shows that Lys-92 donates a hydrogen bond to the metal-ligated water that is best-positioned to attack the scissile phosphate (Fig. 3). Therefore, we suggest that this lysine instead functions to stabilize and orient the attacking hydroxide ion. It is of interest that arginine apparently cannot substitute for lysine in the conserved (E/D) Xn (E/D)ZK motif found in the active sites of some type II restriction endonucleases (ref. 29; A. Pingoud and I. Schildkraut, personal communications). Replacement would be expected in some cases if the function of the lysine were only to provide electrostatic stabilization, particularly given the known large differences in enzyme tertiary structures. Effective stabilization of the attacking hydroxide may require a concentrated localized positive charge, which can be provided only by lysine (30). It is known that water molecules solvating lysine form a tighter, more discrete shell and are more difficult to displace than waters solvating arginine (30, 31). This property of lysine may be exploited to orient the attacking nucleophile in EcoRV and other type II restriction endonucleases.
Lys-92 also donates a second hydrogen bond to the pro-RP oxygen of the adjacent 3′-phosphate (Fig. 3). This interaction may elevate the pKa of the amine group, rendering it a less likely candidate for the catalytic base. In turn, the positively charged amine may well cause a further decrease in the pKa of the metal-bound water, so that hydroxide generation at neutral pH becomes even more facile. An analogous role for an active-site lysine in metal-dependent phosphoryl transfer also was proposed in human myo-inositol monophosphatase (32). In each case, the role of the lysine may be both structural in orienting the water and chemical in lowering its pKa. This role for Lys-92 in EcoRV is also consistent with the phosphorothioate-substitution data: the RP phosphorothioate at the 3′-adjacent phosphate is cleaved well because a negatively charged sulfur at this position still can help elevate the pKa of the lysine. In contrast, the SP phosphorothioate likely slows catalysis because of structural perturbations arising from the bulky sulfur atom, which could directly destabilize the divalent metal binding site. Crystal structures of phosphorothioate-substituted single-stranded DNAs bound to the 3′-5′ exonucleolytic active site of DNA polymerase I show that the sulfur substitution can produce significant structural perturbations, supporting this interpretation (33).
A Three-Metal Ion Mechanism.
No steric hindrance prevents simultaneous occupancy of the two distinct metal binding sites on the scissile and adjacent 3′-phosphates, as observed in the wild-type and T93A mutant cocrystal structures, respectively. Thus, we considered whether a pretransition state configuration including both these metals, together with the metal at the 74/45 site, could be modeled into the active site. One issue that immediately arises is that the proposed nucleophile in the T93A structure is located a distant 4.1 Å from the scissile phosphorus (Fig. 3). Further, the O-P-O angle made by the water oxygen, the phosphorus, and the leaving 3′-oxygen is 132°, compared with the optimal 180° expected in the attack. Because the enzyme in this crystal lattice is catalytically inactive, we superimposed a model of the bound DNA derived from a different 3.0-Å EcoRV–DNA cocrystal structure (1), determined in an alternate lattice in which activity is preserved. This superposition, carried out between the active wild-type structure and the T93A mutant, and using backbone atoms of the active-site residues Asp-90, Asp-74, Glu-45, Glu-65, and Lys-92 (rms deviation = 0.34 Å), shows that in an active lattice the DNA binds more deeply in the active site (Fig. 4). In this configuration, the attacking water-phosphorus distance is shortened to 3.2 Å, and the O-P-O angle improves to 148°.
Figure 4.
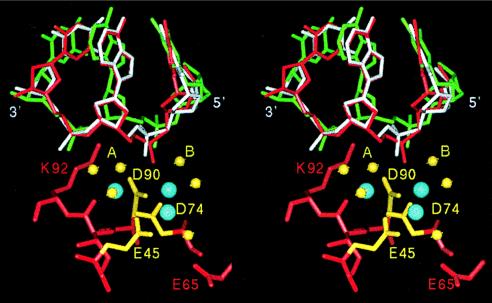
Stereo drawing of a model of the pretransition state configuration for phosphoryl transfer by EcoRV. Three proposed divalent metals bound in the active site are shown in blue. The key carboxylates of Asp-90, Asp-74, and Glu-45, which ligate the three metals, are shown in yellow. The DNA and protein in the energy-minimized model are shown in red. The DNA conformation shown in green is adopted in the T93A mutant structure, and that shown in white is found in a 3.0-Å wild-type cocrystal structure in a crystal lattice where the enzyme is active (refs. 1 and 2; superpositions are described in the text). Yellow spheres represent experimentally observed waters. Water A represents the attacking nucleophile. Water B hydrogen-bonds to the 3′-oxygen leaving group.
Further adjustments were performed by using a model consisting of the DNA in the conformation observed in the active lattice, together with the T93A mutant active site and with the addition of the third metal ion observed bound to the scissile phosphate in the wild-type structure. Waters ligated to this metal were included (3). Torsional angles along the DNA backbone then were systematically altered, while allowing small adjustments in the positions of active-site side chains and the two divalent metals bound to the DNA. The resulting model after energy minimization is shown in Fig. 4. The attacking water is now poised 2.85 Å from the scissile phosphorus with an O-P-O angle of 178°, indicating a nearly direct in-line orientation. All three metals maintain identical ligation states as determined in the experimental structures. The DNA at the scissile phosphate is moved a further 0.7 Å more deeply into the active site, and small rearrangements on the order of 0.2–0.5 Å occur in the positions of some of the active-site side chains. This model represents a plausible pretransition state configuration for phosphoryl transfer by EcoRV.
The chemical mechanism suggested by this model is as follows. A metal ion at site I (Fig. 5) ligates through water to the 3′-phosphate. A second inner-sphere water molecule on this metal dissociates to provide the attacking hydroxide ion, and this dissociation is aided by the immediately adjacent Lys-92 residue. The metal at site III provides stabilization of the incipient negative charge as the transition state develops. An inner-sphere water on this metal, observed in the wild-type structure, is located within hydrogen-bonding distance of the leaving 3′-oxygen (3). Thus, we suggest that the site III metal is also operative in lowering the pKa of this water, so that it may dissociate to immediately protonate the leaving anion. The site II metal is purely structural and serves primarily to correctly orient the crucial Asp-74 carboxylate. The model accounts for facilitation of both the bond-making and bond-breaking steps, as required for the 1015-fold rate enhancement provided by EcoRV relative to the uncatalyzed hydrolytic cleavage of DNA phosphate diesters (34).
Figure 5.

Proposed transition state for the metal ion-mediated DNA cleavage reaction by EcoRV. The metal ion in site I generates the attacking nucleophile, which is stabilized and oriented by Lys-92. The metal in site III facilitates departure of the 3′-anion and compensates the developing negative charge in the transition state. The role of the metal in site II is structural. Metal-ligand inner-sphere contacts are shown as hatched lines. Dotted lines indicate hydrogen bonds.
Several distinctions may be drawn between this catalytic model and the well-described two-metal ion phosphoryl transfer mechanism operative in the 3′-5′ exonuclease active sites of Escherichia coli DNA polymerase I and T4 DNA polymerase, and in E. coli alkaline phosphatase (35–37). In both models the catalytic hydroxide is generated through the agency of a divalent metal ion, which lowers the pKa of an associated inner-sphere water rendering hydroxide generation more facile. Both models also invoke direct metal coordination to the reactive phosphate group as a means of stabilizing the incipient negative charge in the transition state. However, in EcoRV the bond-making step is aided by a lysine residue that orients and further reduces the pKa of the attacking nucleophile, whereas no such assistance is present in the two-metal mechanism. Further, stabilization of the leaving oxygen anion is accomplished by direct metal ligation in the two-metal mechanism, but indirectly through a metal-ligated inner-sphere water molecule in EcoRV. Direct metal ligation may better stabilize the developing negative charge on the leaving group. Therefore, a comparison of the two models suggests catalytic emphasis on the bond-making step in EcoRV, and on the bond-breaking step in the two-metal mechanism.
The proposed three-metal mechanism assigns a central and crucial role to Asp-74. This role is in accord with mutational data, which show that the D74A and D74N mutants are reduced in activity by more than 104-fold (18). Although the mutants D90A and D90N are similarly inactive, it is of interest that the D90E enzyme retains full activity whereas D74E is reduced by roughly 100-fold. This finding suggests a greater importance for the precise positioning of an acidic carboxylate at residue 74, as might be expected for its proposed role in ligating all three of the active-site metal ions. Further, mutants of Glu-45, which remove the negative charge, retain somewhat more activity than similar substitutions of Asp-90 or Asp-74, in accord with the proposed structural role of this residue (18, 38). This three-metal ion mechanism is also consistent with kinetic data. Strong evidence implicating two or more divalent metals per active site has been obtained from stopped-flow fluorescence measurements and from metal-reconstitution experiments (10, 39). Although the most straightforward interpretation of these data has been in terms of two metal binding sites per subunit, the existence of three sites is also consistent with the experimental observations.
Although this proposed mechanism is chemically plausible and consistent with all biochemical data, several ambiguities remain to be resolved. Most importantly, a more detailed rationale for the lack of Mg2+ binding to site I in this P1 lattice, leading to inactivity of the enzyme in the crystalline state, is still needed. Additional kinetic work to more clearly elucidate the role of Lys-92 also is warranted, because the exclusion of this amino acid as the catalytic base presently is based only on structural data.
Crystal structures of EcoRV, EcoRI, and PvuII endonucleases each bound to DNA show that the relative positions of the scissile and adjacent 3′-phosphates are conserved (14). Therefore, although differences in the details of the metal ligations are to be expected, two metal ions bound in site I and site III may have similar functions in each of these enzymes. The structural alignments of Glu-45 are more ambiguous so that the extent to which the site II metal may be conserved is uncertain (29). In BamHI, Glu-113 substitutes for the conserved lysine, and it is possible that this enzyme functions instead by the two-metal mechanism (5, 36). However, an essential metal-ligating role for the adjacent 3′-phosphate may be a conserved feature of many type II restriction endonucleases.
Acknowledgments
We thank Dr. Tom Bruice for helpful discussions and Dr. Stephen Halford for providing the expression vector encoding the EcoRV T93A mutant. This work was supported by National Institutes of Health Grant GM53763 and American Chemical Society-Petroleum Research Fund Grant 30427-G4 (to J.J.P.), and by American Cancer Society Postdoctoral Fellowship PF-98–015-01-GMC (to N.C.H.).
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, Biology Department, Brookhaven National Laboratory, Upton, NY 11973 (PDB ID 1BSS).
References
- 1.Winkler F K, Banner D W, Oefner C, Tsernoglou D, Brown R S, Heathman S P, Bryan R K, Martin P D, Petratos K, Wilson K S. EMBO J. 1993;12:1781–1795. doi: 10.2210/pdb4rve/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kostrewa D, Winkler F K. Biochemistry. 1995;34:683–696. doi: 10.1021/bi00002a036. [DOI] [PubMed] [Google Scholar]
- 3.Perona J J, Martin A M. J Mol Biol. 1997;273:207–225. doi: 10.1006/jmbi.1997.1315. [DOI] [PubMed] [Google Scholar]
- 4.Cheng X, Balendiran K, Schildkraut I, Anderson J E. EMBO J. 1994;17:3927–3935. doi: 10.1002/j.1460-2075.1994.tb06708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newman M, Strzelecka T, Dorner L F, Schildkraut I, Aggarwal A K. Science. 1995;269:656–663. doi: 10.1126/science.7624794. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg J. Curr Opin Struct Biol. 1991;1:104–113. [Google Scholar]
- 7.Schildkraut I, Banner C D B, Rhodes C S, Parekh S. Gene. 1984;27:327–329. doi: 10.1016/0378-1119(84)90078-7. [DOI] [PubMed] [Google Scholar]
- 8.Roberts R J, Halford S E. In: Nucleases. 2nd Ed. Linn S M, Lloyd R S, Roberts R J, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1993. pp. 35–88. [Google Scholar]
- 9.Grasby J A, Connolly B A. Biochemistry. 1992;32:7855–7861. doi: 10.1021/bi00149a016. [DOI] [PubMed] [Google Scholar]
- 10.Vipond I B, Baldwin G S, Halford S E. Biochemistry. 1995;34:697–704. doi: 10.1021/bi00002a037. [DOI] [PubMed] [Google Scholar]
- 11.Jeltsch A, Alves A, Wolfes H, Maass G, Pingoud A. Proc Natl Acad Sci USA. 1993;90:8499–8503. doi: 10.1073/pnas.90.18.8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeltsch A, Alves J, Maass G, Pingoud A. FEBS Lett. 1992;304:4–8. doi: 10.1016/0014-5793(92)80576-3. [DOI] [PubMed] [Google Scholar]
- 13.Thorogood H, Grasby J A, Connolly B A. J Biol Chem. 1996;271:8855–8862. doi: 10.1074/jbc.271.15.8855. [DOI] [PubMed] [Google Scholar]
- 14.Pingoud A, Jeltsch A. Eur J Biochem. 1997;246:1–22. doi: 10.1111/j.1432-1033.1997.t01-6-00001.x. [DOI] [PubMed] [Google Scholar]
- 15.Koziolkiewicz M, Stec W J. Biochemistry. 1992;31:9460–9466. doi: 10.1021/bi00154a019. [DOI] [PubMed] [Google Scholar]
- 16.Jeltsch A, Pleckaityte M, Selent U, Wolfes S, Siksnys V, Pingoud A. Gene. 1995;157:157–162. doi: 10.1016/0378-1119(94)00617-2. [DOI] [PubMed] [Google Scholar]
- 17.Vipond I B, Halford S E. Biochemistry. 1995;34:1113–1119. doi: 10.1021/bi00004a002. [DOI] [PubMed] [Google Scholar]
- 18.Selent U, Ruter T, Kohler E, Liedtke M, Thielking V, Alves J, Oelgeschager T, Wolfes H, Peters F, Pingoud A. Biochemistry. 1992;31:4808–4815. doi: 10.1021/bi00135a010. [DOI] [PubMed] [Google Scholar]
- 19.Aggarwal A K. Methods Companion Methods Enzymol. 1990;1:83–90. [Google Scholar]
- 20.Brunger A T, Kuriyan J, Karplus M. Science. 1987;235:458–460. doi: 10.1126/science.235.4787.458. [DOI] [PubMed] [Google Scholar]
- 21.Sack J S. J Mol Graphics. 1988;6:224–225. [Google Scholar]
- 22.Parkinson G, Vojtechovsky J, Clowney L, Brunger A T, Berman H M. Acta Crystallogr D. 1996;52:57–64. doi: 10.1107/S0907444995011115. [DOI] [PubMed] [Google Scholar]
- 23.Dayringer H, Tramontano A, Sprang S, Fletterick R J. J Mol Graphics. 1986;4:82–87. [Google Scholar]
- 24.Perry K M, Fauman E B, Finer-Moore J S, Montfort W R, Maley G F, Maley F, Stroud R M. Proteins. 1990;8:315–333. doi: 10.1002/prot.340080406. [DOI] [PubMed] [Google Scholar]
- 25.Vipond I B, Halford S E. Biochemistry. 1996;35:1701–1711. doi: 10.1021/bi952391d. [DOI] [PubMed] [Google Scholar]
- 26.Wenz C, Jeltsch A, Pingoud A. J Biol Chem. 1996;271:5565–5573. doi: 10.1074/jbc.271.10.5565. [DOI] [PubMed] [Google Scholar]
- 27.Glusker J P. Adv Protein Chem. 1991;42:1–76. doi: 10.1016/s0065-3233(08)60534-3. [DOI] [PubMed] [Google Scholar]
- 28.Dahm S C, Derrick W B, Uhlenbeck O C. Biochemistry. 1993;32:13040–13045. doi: 10.1021/bi00211a013. [DOI] [PubMed] [Google Scholar]
- 29.Aggarwal A K. Curr Opin Struct Biol. 1995;5:11–19. doi: 10.1016/0959-440x(95)80004-k. [DOI] [PubMed] [Google Scholar]
- 30.Bash P A, Singh U C, Langridge R, Kollman P A. Science. 1987;236:564–568. doi: 10.1126/science.3576184. [DOI] [PubMed] [Google Scholar]
- 31.Perona J J, Tsu C A, McGrath M E, Craik C S, Fletterick R J. J Mol Biol. 1993;230:934–949. doi: 10.1006/jmbi.1993.1211. [DOI] [PubMed] [Google Scholar]
- 32.Ganzhorn A J, Lepage P, Pelton P D, Strasser F, Vincendon P, Rondeau J-M. Biochemistry. 1996;35:10957–10966. doi: 10.1021/bi9603837. [DOI] [PubMed] [Google Scholar]
- 33.Brautigam C, Steitz T A. J Mol Biol. 1998;277:363–377. doi: 10.1006/jmbi.1997.1586. [DOI] [PubMed] [Google Scholar]
- 34.Chin J, Banaszczyk M, Jubian V, Zou X J. J Am Chem Soc. 1989;111:186–190. [Google Scholar]
- 35.Kim E E, Wyckoff H W. J Mol Biol. 1991;218:449–464. doi: 10.1016/0022-2836(91)90724-k. [DOI] [PubMed] [Google Scholar]
- 36.Beese L J, Steitz T A. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J, Yu P, Lin T C, Konigsberg W H, Steitz T A. Biochemistry. 1996;35:8110–8119. doi: 10.1021/bi960178r. [DOI] [PubMed] [Google Scholar]
- 38.Groll D H, Jeltsch A, Pingoud A. Biochemistry. 1997;36:11389–11401. doi: 10.1021/bi9705826. [DOI] [PubMed] [Google Scholar]
- 39.Baldwin G S, Vipond I B, Halford S E. Biochemistry. 1995;34:705–714. doi: 10.1021/bi00002a038. [DOI] [PubMed] [Google Scholar]