

Abstract
AIMS
To evaluate the safety and tolerability of a new oral solution formulation of tolevamer potassium sodium, a nonantibiotic polymer that binds Clostridium difficile toxins A and B.
METHODS
This phase 1 randomized, double-blind, placebo-controlled study evaluated four doses of tolevamer potassium sodium in 40 healthy volunteers using a sequential dose escalation paradigm and doses of 6, 9, 12 and 15 g day−1 for 9 days. Within each 10 patient cohort, eight patients received active treatment and two matching placebo. Placebo subjects were pooled to provide eight per arm. All subjects received three times daily dosing on days 2–8 as well as a loading dose (a single dose equal to the total daily dose) either on day 1 or day 9.
RESULTS
All 40 subjects completed the study per protocol. Treatment-emergent adverse events (TEAEs) were generally mild, transient, and resolved without sequelae. There were no serious AEs or deaths. There was no relationship detected between dose and the incidence of TEAEs, whether drug-related (all gastrointestinal disorders) or not. No clinically significant changes in laboratory parameters, including serum and urinary potassium concentrations, vital signs, and results of physical examination, were observed. A small but statistically significant reduction in 24 h urine potassium excretion was seen in the 15 g day−1 dose group, and on day 10 in the 6 g day−1 group.
CONCLUSIONS
Tolevamer oral solution administered for 9 days at total daily doses up to 15 g, with loading doses of up to 15 g, was generally safe and well-tolerated in healthy volunteers.
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Clostridium difficile-associated diarrhoea (CDAD), which affects about 1% of hospitalized patients whose intestinal flora have been altered by the administration of antimicrobial agents such as the cephalosporins, accounts for 10% to 20% of all cases of antibiotic-associated diarrhoea.
The incidence and severity of CDAD is rising in a number of countries, including the United States, United Kingdom, and Canada, where recent outbreaks of CDAD have been associated with increased mortality.
Since both antibiotics that are currently used for treating CDAD (vancomycin, the only treatment approved by the US Food and Drug Administration for this indication, and metronidazole, the most widely recommended first-line therapy) have serious limitations, including recurrence rates of about 20%, systemic adverse events, and the emergence of drug resistance, there is a large unmet need for the development of novel therapeutic modalities with unique mechanisms of action for the prevention of the disease and/or management of patients with CDAD.
WHAT THIS STUDY ADDS
The results of the phase I study demonstrate that a new phase III formulation of tolevamer, a novel, high-molecular weight, anionic polymer with a unique mechanism of action based on noncovalent binding to C. difficile toxins A and B, may eliminate the increased risk of hypokalaemia observed with the phase II formulation and allow further clinical development of an alternative therapeutic approach to antibiotics for this antibiotic-induced disease.
The results demonstrate that a potassium sodium oral liquid formulation of tolevamer given over a 9 day treatment period is generally safe and well-tolerated.
The information contained herein provides a basis for selection of the appropriate dosing and scheduling of this formulation of tolevamer in future trials.
Keywords: Clostridium difficile, hypokalaemia, nosocomial diarrhoea, safety and efficacy, tolevamer
Introduction
Clostridium difficile-associated diarrhoea (CDAD) typically affects patients whose intestinal flora are altered by the use of antimicrobial agents such as the cephalosporins [1]. In this microbial disorder, C. difficile is able to grow in an intestinal environment ablated of normal, competing intestinal flora. CDAD is associated with a profuse watery or mucoid diarrhoea, abdominal discomfort, fever, leukocytosis, nausea, anorexia, malaise, and haematochezia [2, 3].
An estimated 1% of hospitalized patients are afflicted with CDAD, which accounts for 10% to 20% of all cases of antibiotic-associated diarrhoea [1, 4]. The incidence and severity of CDAD is rising in a number of countries, including the United States, United Kingdom, and Canada [5–7]. In Montreal and other regions of Quebec, recent outbreaks of CDAD have been associated with increased mortality [5, 8, 9]. New cases of CDAD in Quebec have been estimated to be more than five times the national average observed in 1997 [10]. This outbreak is related principally to the emergence of a new hypertoxigenic strain with a possible role for fluoroquinolone use driving its emergence [11–14].
The symptoms of C. difficile infection are mediated by two major virulence factors: toxin A, a potent enterotoxin/cytotoxin, and toxin B, a potent cytotoxin [2, 15]. Upon release from the vegetative form of pathogenic strains of C. difficile, these toxins bind to enterocyte cell-surface receptors, followed by toxin internalization and subsequent disruption of the actin cytoskeleton and widening of tight junctions. This process is accompanied by a robust inflammatory response and ultimately leads to mucosal damage and fluid loss into the intestinal lumen [3, 16, 17].
Antibiotic therapy is currently the most common method of treating patients with CDAD. Vancomycin is the only treatment approved by the US Food and Drug Administration (FDA) for this indication, but metronidazole is the most widely recommended first-line therapy. Both antibiotics are associated with CDAD recurrence rates of about 20% due to the disruption of normal intestinal flora that provide colonization resistance against C. difficile[2, 18–20]. Additionally, metronidazole is associated with systemic adverse effects [2], and vancomycin use is restricted over concerns about the emergence of vancomycin resistance in other bacteria, such as enterococci [20]. Two recent observational studies have raised serious questions about the efficacy of metronidazole for managing CDAD [21–23]. Development of a novel nonantibiotic agent with a unique mechanism of action that does not contribute to adverse microbial ecological selection pressure may represent a significant advance for prevention of CDAD and/or treatment of patients with CDAD.
Tolevamer is a novel, orally administered, soluble, high-molecular weight (> 400 kDa), nonantimicrobial, anionic polymer. Tolevamer has a unique mechanism of action in that, rather than acting directly on the bacteria, it binds noncovalently with high affinity to C. difficile toxins A and B, thus effectively neutralizing these causative toxins [24]. Thus, unlike traditional antibiotics, tolevamer does not interact directly with normal gut bacteria and therefore should not disrupt the protective microflora that provide colonization resistance [25]. The original tolevamer compound (tolevamer sodium, GT160-246) dramatically attenuated CDAD severity and recurrence in preclinical studies and provided clinical benefit in patients with mild to moderate CDAD [25–28]. Tolevamer sodium was well-tolerated and demonstrated similar efficacy for resolution of C. difficile diarrhoea compared with standard therapy with vancomycin, but it was associated with an increased rate of hypokalaemia [27].
To minimize the risk of hypokalaemia, a new mixed potassium sodium salt of tolevamer was developed as an oral solution. In this new formulation [poly(potassium/sodium 4-styrenesulphonate), tolevamer potassium sodium, GT267-004] partial replacement of sodium with potassium is expected to reduce the potential of the polymer, as a result of its ability to bind cations, to contribute to hypokalaemia in patients at risk of potassium loss due to secretory diarrhoea. Based on in vitro experiments with simulated intestinal fluid, it was estimated that 1.8 mEq potassium g−1 of anionic polymer is equivalent to the amount of potassium potentially bound in the colon. Thus, a formulation with this concentration of potassium is expected to be excreted in the faeces carrying approximately the same amount of potassium with which it was administered, thereby being ‘potassium neutral’ and effectively minimizing the risk for hypokalaemia. The active component of tolevamer, the anionic polymer that binds C. difficile toxins A and B, is identical in the two salt formulations. Thus, tolevamer potassium sodium is expected to provide equivalent efficacy as that seen with tolevamer sodium, but without significant hypokalaemia.
We report here the results of a double-blind, randomized, placebo-controlled, dose escalation phase I safety and tolerability study of this new formulation of tolevamer in healthy male volunteers.
Methods
Objectives
The objectives of this study were (i) to examine the safety and tolerability of a new potassium sodium salt formulation of tolevamer, (ii) to investigate the effect of this new tolevamer formulation on urinary potassium excretion and (iii) to provide data to inform decisions regarding acceptable doses of tolevamer potassium sodium in further clinical studies.
Study participants and study design
Males 18–75 years of age who were willing to eat three standardized meals and three snacks per day were eligible. Subjects were required to have no clinically significant abnormal findings on physical examination, medical history, and laboratory testing at screening, and negative urine screens for alcohol and drugs of abuse at screening and on day −1. A level of understanding and a willingness and ability to co-operate with all study procedures and restrictions was required. All subjects gave voluntary written informed consent. The study protocol and informed consent were approved by an Independent Ethics Committee (Strichting Boordeling Ethiek Bio-Medish Onderzoek, Assen, the Netherlands).
Individuals were screened during a 3-week period. Eligible subjects (n = 40, with an additional reserve of nine subjects) were admitted to the study unit on day −1. Subjects were assigned sequentially to one of four treatment cohorts (6 g day−1, 9 g day−1, 12 g day−1, 15 g day−1 total daily dose) starting with the lowest dose (10 subjects per group). Within each cohort, subjects were randomized to receive either active treatment (n = 8) or placebo (n = 2). Subjects were further randomized such that five subjects (four receiving active medication and one receiving placebo) were given a loading dose of tolevamer or placebo on the evening of day 1. The other five subjects received this loading dose of tolevamer or placebo as their last dose on the morning of day 9. Subjects who received a loading dose on the evening of day 1 received a regular single dose on the morning of day 9, while subjects who received a loading dose on the morning of day 9 received a regular single dose on the evening of day 1. The volume of tolevamer potassium sodium solution varied with dose, ranging from 87 ml to 216 ml. Doses were administered with placebo to maintain equivalent volumes. The placebo preparation was matched to the active preparation in taste and consistency. Individual subject medication was prepared by the site pharmacist. The study co-ordinator, investigator, and subjects were blinded to treatment assignment. Study medication was given three times daily, at 8 h intervals, 30 min prior to a scheduled meal or snack.
From the time of admission until the time of discharge the subjects continued on a potassium-controlled diet designed to provide a mean dietary potassium intake of 6600 mg day−1 (40 mg ≈ 1 mEq). A fasting period of 4 h was required before screening and admission procedures were initiated. Subjects remained in the study unit for the 9 day treatment period and were discharged the morning of day 10. A follow-up medical examination was performed within 4–8 days after discharge from the centre.
Dose selection rationale
This study was designed to examine the safety and tolerability of tolevamer potassium sodium dosed at total daily doses of 6 g (2 g three times daily), 9 g (3 g three times daily), 12 g (4 g three times daily) and 15 g (5 g three times daily), as well as to examine the safety and tolerability of a single loading dose at each of these total daily doses. The doses examined were chosen based on prior clinical experience with tolevamer sodium and to provide additional evidence in support of doses planned for phase III studies. Phase II data with tolevamer sodium indicate a dose–response relationship between 3 g day−1 and 6 g day−1, with 6 g day−1 being a clinically effective dose that was statistically noninferior to standard vancomycin therapy (500 mg day−1) [27]. Demonstrating the safety and tolerability of higher tolevamer doses (9 g day−1 and 15 g day−1), with commensurate increases in toxin-binding capacity, may be of clinical importance given the recent epidemic emergence of the hypertoxigenic ribotype 027, toxinotype III C. difficile strain in North America and Europe, which produces in vitro 16- and 23-fold higher concentrations of toxins A and B, respectively, than a reference toxinotype 0 strain [11]. Additionally, the 15 g day−1 dose is equivalent to the total daily dose achieved with a 9 g loading dose plus two additional 3 g doses in a 9 g day−1, 3 g three times daily regimen, a likely regimen for phase III pivotal trials of tolevamer for treatment of CDAD. Optimal treatment of CDAD is likely to include a loading dose of tolevamer as the first dose of treatment to neutralize rapidly and maximally C. difficile toxin-mediated colon damage. In this phase I study, the study population was divided to receive the tolevamer loading dose at either the start or at the conclusion of the treatment phase because it was unknown at the beginning of the study whether high doses of tolevamer would be tolerated. Administering the loading dose to a subset of patients at the conclusion of the study was done to reduce the drop-out rate should high doses be poorly tolerated, and because it allowed examination of whether tolerance to high doses would improve after initial exposure to lower tolevamer doses.
Prior and concomitant medications
Subjects were not permitted to take any prescription or nonprescription medication with the exception of acetaminophen and some topical medications within 7 days prior to the first dose and for the duration of the study. The use of methylxanthine-containing beverages or food, grapefruit juice, and alcohol was not allowed from 48 h (2 days) prior to entrance into the clinical research centre and during the study.
Study assessments
Subjects were queried with nonleading questions to determine the occurrence of adverse events (AEs), which, on dosing days, was done just before each drug administration (three times per day). In addition, all AEs reported spontaneously during the course of the study were recorded. The severity of an AE was rated as mild, moderate, or severe, and it was classified by the investigator as not related, remotely related, possibly related, or probably related to tolevamer administration. Physical examinations, including measurement of vital signs, were performed at screening, on days −1 and 10, and upon follow-up. Fasting serum potassium was measured on study days 1 through 10 prior to administration of the breakfast dose of tolevamer. Twenty-four hour urine samples were collected from the morning of day 1 until discharge on day 10 for analysis of urinary potassium, creatinine, calcium, and magnesium. No 24 h urine samples were collected prior to day 1. Standard quantitative serum clinical chemistry, quantitative haematology, qualitative urinalysis, and coagulation (prothrombin time and partial thromboplastin time) assessments were measured at screening, on days −1, 5, and 10, and on follow-up. Serology was done at screening for hepatitis B surface antigen, antihepatitis C, and antihuman immunodeficiency −1 and −2 antibodies. Drug screening was done at screening and on day −1. No pharmacokinetic assessments were done as part of this study of healthy volunteers as studies in both normal and damaged gut animal models indicated that tolevamer was essentially nonabsorbed.
Statistics
No formal sample size determination was performed for this study. General study experiences in phase I trials indicate that 40 patients in a blinded randomized trial of this nature should provide sufficient data to allow initial assessment of safety and tolerability while minimizing unnecessary exposure of subjects to tolevamer potassium sodium. Data from placebo-treated subjects were pooled into one group. Descriptive summary statistics were provided when appropriate. A Wilcoxon rank sum test with P values for comparison of each dose group with placebo for adverse events was performed as well as an analysis of variance (anova) with P values for clinical laboratory data. No other formal statistical analyses were performed.
Results
Baseline demographics
Descriptive demographic statistics by treatment group and overall in the study are indicated in Table 1. Height, weight, and body mass index were similar among all treatment groups. The placebo-treated subjects were approximately 10 years older than the tolevamer-treated subjects, except for subjects in the 6 g group who, on average, were approximately 8 years younger than the subjects in the other treatment groups. All 40 subjects completed the study per protocol.
Table 1.
Summary of subject demographic characteristics
| Tolevamer | |||||||
|---|---|---|---|---|---|---|---|
| Placebo | 6 g | 9 g | 12 g | 15 g | Overall | ||
| Age (years) | Mean (SD) | 47 (20) | 30 (14) | 37 (22) | 38 (20) | 38 (22) | 38 (19) |
| Range | 18–70 | 20–58 | 19–67 | 18–63 | 20–72 | 18–72 | |
| Height (cm) | Mean (SD) | 182 (7) | 182 (5) | 182 (8) | 176 (9) | 179 (4) | 180 (7) |
| Range | 174–192 | 176–190 | 171–192 | 163–191 | 174–184 | 163–192 | |
| Weight (kg) | Mean (SD) | 83.4 (7.8) | 75.6 (11.0) | 79.3 (7.2) | 74.1 (7.2) | 80.9 (6.8) | 78.7 (8.5) |
| Range | 75.1–95.1 | 59.3–96.6 | 69.6–90.7 | 63.6–83.3 | 69.5–88.2 | 59.3–96.6 | |
| BMI (kg m−2) | Mean (SD) | 25.2 (2.9) | 22.7 (2.9) | 23.9 (2.7) | 23.9 (2.5) | 25.4 (2.0) | 24.2 (2.7) |
| Range | 20.8–30.0 | 19.1–26.8 | 19.1–27.7 | 20.5–28.2 | 23.0–28.7 | 19.1–30.0 | |
Treatment-emergent adverse events
AEs that occurred from the time each subject received the first dose of study medication until follow-up were regarded as treatment-emergent AEs (TEAEs). Twenty-eight subjects reported a total of 68 TEAEs, all of which were of mild intensity, transient, and resolved without sequelae. The frequency of occurrence of TEAEs and the number and percentage of subjects experiencing TEAEs per treatment group are indicated in Table 2. Fourteen TEAEs were reported by five subjects receiving placebo, 12 TEAEs by six subjects in the 6 g group, 19 TEAEs by six subjects in the 9 g group, seven TEAEs by six subjects in the 12 g group, and 16 TEAEs by five subjects in the 15 g group. There were no statistically significant differences in the percentage of subjects experiencing TEAEs in any of the treatment groups vs. placebo.
Table 2.
Summary of TEAEs per treatment group*
| Total (n = 40) | |||
|---|---|---|---|
| Treatment | E | N (%) | P |
| Total AEs (n = 40) | 68 | 28 (70) | – |
| Placebo (n = 8) | 14 | 5 (63) | – |
| 6 g (n = 8) | 12 | 6 (75) | 0.9545 |
| 9 g (n = 8) | 19 | 6 (75) | 0.4860 |
| 12 g (n = 8) | 7 | 6 (75) | 0.5097 |
| 15 g (n = 8) | 16 | 5 (63) | 1.0000 |
All TEAEs were of mild intensity. n, number of subjects per group; E, number of events; N (%), number and percentage of subjects experiencing AEs. P value calculated from a (Wilcoxon rank-sum) comparison of the number of TEAEs per subject for each dose group with the placebo group.
Drug-related adverse events
TEAEs related to tolevamer are summarized in Table 3A. For the overall study period from first dose until follow-up there were 29 TEAEs reported by 20 subjects that were considered by the investigator to be possibly related (25 TEAEs) or probably related (four TEAEs) to study treatment. All were gastrointestinal disorders (i.e. flatulence, loose stools, abdominal distension, abdominal pain, defaecation urgency). No dose-dependent relationship was observed. Indeed, the frequency of the most commonly reported TEAE (flatulence) was inversely related to tolevamer dose.
Table 3.
Summary of drug-related TEAEs
| Total (n = 40) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total daily dose | Placebo (n = 8) | 6 g (n = 8) | 9 g (n = 8) | 12 g (n = 8) | 15 g (n = 8) | |||||||
| System organ class/preferred term | E | N (%) | E | N (%) | E | N (%) | E | N (%) | E | N (%) | E | N (%) |
| A.Entire study period through follow-up | ||||||||||||
| Total | 29 | 20 (50) | 2 | 2 (25) | 10 | 6 (75) | 8 | 6 (75) | 2 | 2 (25) | 7 | 4 (50) |
| Gastrointestinal disorders | 29 | 20 (50) | 2 | 2 (25) | 10 | 6 (75) | 8 | 6 (75) | 2 | 2 (25) | 7 | 4 (50) |
| Abdominal distension | 3 | 1 (3) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 3 | 1 (13) |
| Abdominal pain | 2 | 2 (5) | 0 | 0 (0) | 2 | 2 (25) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) |
| Defaecation urgency | 1 | 1 (3) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 1 | 1 (13) |
| Flatulence | 18 | 16 (40) | 2 | 2 (25) | 8 | 6 (75) | 5 | 5 (63) | 2 | 2 (25) | 1 | 1 (13) |
| Loose stools | 5 | 5 (13) | 0 | 0 (0) | 0 | 0 (0) | 3 | 3 (38) | 0 | 0 (0) | 2 | 2 (25) |
| Loading dose day | Within 24 h only | |||||||
|---|---|---|---|---|---|---|---|---|
| Day 1 LD (n = 16) | Day 9 LD (n = 16) | Day 1 LD (n = 16) | Day 9 LD (n = 16) | |||||
| System organ class/preferred term | E | N (%) | E | N (%) | E | N (%) | E | N (%) |
| B.By loading dose (LD) day | ||||||||
| Total | 12 | 10 (63) | 2 | 2 (13) | 7 | 6 (38) | 2 | 2 (13) |
| Gastrointestinal disorders | 12 | 10 (63) | 2 | 2 (13) | 7 | 6 (38) | 2 | 2 (13) |
| Abdominal pain | 1 | 1 (6) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) |
| Defaecation urgency | 1 | 1 (6) | 0 | 0 (0) | 1 | 1 (6) | 0 | 0 (0) |
| Flatulence | 8 | 8 (50) | 1 | 1 (6) | 5 | 5 (31) | 1 | 1 (6) |
| Loose stools | 2 | 2 (13) | 1 | 1 (6) | 1 | 1 (6) | 1 | 1 (6) |
n, number of subjects exposed; E, number of AEs; N (%), number and percentage of subjects with AEs. Note: Each occurrence of each AE was counted, even if the same AE occurred multiple times in the same patient during one treatment. A relationship to the study medication of ‘definite’, ‘probable’, or ‘possible’ is considered to be ‘related.’
The frequency of occurrence of drug-related TEAEs and the number and percentage of subjects experiencing drug-related TEAEs after the loading dose is summarized in Table 3B. Within 24 h after administration of the day 1 and day 9 loading doses, there were seven and two drug-related TEAEs observed, respectively. TEAEs occurring within the first day of dosing and associated with the day 1 loading dose were most often episodes of mild flatulence, with defaecation urgency and loose stools also reported. The two drug-related TEAEs seen within 24 h after the day 9 loading dose were mild flatulence and loose stools. The incidence of flatulence declined over the course of the study, possibly due to habituation to the study medication.
These results suggest that three times daily oral treatment with a total daily dose of up to 15 g of liquid tolevamer for 7 days and oral treatment with a loading dose of up to 15 g of liquid tolevamer was generally safe and well-tolerated by healthy male volunteers.
Clinical laboratory parameters
No clinically significant changes in laboratory values were observed with tolevamer administration, including serum concentrations of potassium, creatinine, calcium, and magnesium, and urinary excretion of potassium. Serum potassium concentrations were not adversely affected in any group, and no dose–response was observed in urinary potassium excretion. The 24 h urinalysis demonstrated that potassium balance was achieved with this potassium sodium formulation of tolevamer at the 6, 9, and 12 g doses (Table 4, Figure 1). In the 6 g day−1 treatment group, there was no significant change from baseline until day 10 when the change from baseline was −26.3 mmol 24 h−1 (P < 0.05). In the 9 g group (phase III daily dose) there was a small increase in the mean 24 h urinary potassium excretion compared with baseline each day on study days 3 through 9, averaging 14 mmol 24 h−1 overall, and a decrease on study day 10 of 5.7 mmol 24 h−1, with none of the changes achieving statistical significance. Similar results were observed in the 12 g group, with an average increase in the mean 24 h urinary potassium excretion across all study days of 5 mmol 24 h−1vs. baseline, and no significant decrease at any time point studied. In the 15 g day−1 treatment group, the daily mean decrease in 24 h urinary potassium excretion was 9.8, 13.1, 4.2 and 21.6 mmol 24 h−1 on study days 4, 6,8, and 10, respectively (P < 0.05 for each time point), with the mean daily urinary potassium excretion in this group ranging from 83 to 104 mmol 24 h−1, and with an average decrease across all study days of 12 mmol 24 h−1vs. baseline.
Table 4.
24 h urinary potassium excretion (mmol 24 h−1)
| Treatment group | Baseline (Day 2) | Day 4 | Day 6 | Day 8 | Day 10 |
|---|---|---|---|---|---|
| Placebo | 90.4 (21.4) | 102.9 (22.7) | 105.7 (13.8) | 107.6 (11.3) | 94.4 (11.1) |
| 6 g day−1 | 106.3 (28.7) | 122.0 (24.3) | 107.1 (28.9) | 104.8 (16.2) | 80.0 (25.2)* |
| 9 g day−1 | 89.1 (21.5) | 95.0 (27.4) | 104.6 (11.7) | 104.2 (16.3) | 83.4 (14.5) |
| 12 g day−1 | 85.8 (19.3) | 93.2 (16.5) | 96.0 (12.6) | 99.4 (12.0) | 81.5 (14.9) |
| 15 g day−1 | 104.3 (9.8) | 94.5 (15.5)* | 91.1 (30.9)* | 100.1 (11.4)* | 82.7 (14.0)* |
P value <0.05 (anova). Urine samples were collected daily over the course of the 9 day treatment interval. For convenience, data from alternating days are presented as mean (SD). Statistical comparison was made of the mean change from baseline for each time postbaseline with the corresponding mean change from baseline in the placebo group.
Figure 1.
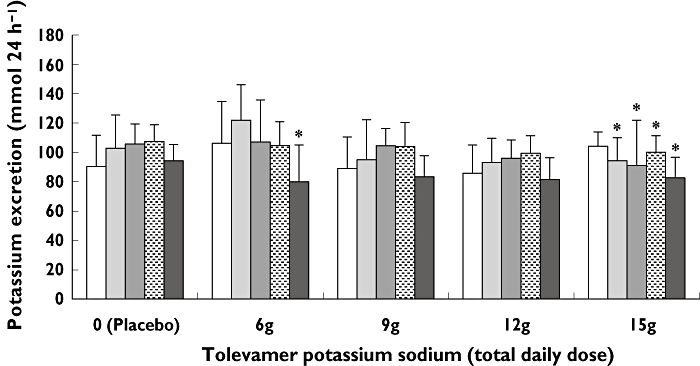
24h urinary potassium excretion (mmol 24 h−1) by treatment group at baseline and on days 4, 6, 8, and 10. A comparison was made of the mean change from baseline for each time post baseline with the corresponding mean change from baseline in the placebo group. *P value <0.05 (anova). There was a statistically significant change in urinary potassium excretion on day 10 in subjects who received 6 g day−1 tolevamer potassium and on days 4, 6, 8, and 10 in subjects who received 15 g day−1 tolevamer potassium sodium. There was no significant effect of tolevamer potassium sodium administered at 9 g day−1 or 12 g day−1. Baseline (Day 2), ( ); Day 4, (
); Day 4, ( ); Day 6, (
); Day 6, ( ); Day 8, (
); Day 8, ( ); Day 10, (
); Day 10, ( )
)
Other safety evaluations
There were no changes in vital signs or the results of physical examination.
Discussion
This study evaluated in healthy volunteers the safety and tolerability of a new oral formulation of tolevamer designed to minimize the incidence of hypokalaemia observed in previous clinical studies with tolevamer sodium. The results indicate that a potassium sodium formulation of tolevamer is generally well-tolerated over a 9 day treatment period at the doses administered, including loading doses equivalent to the full daily dose of the treatment cohort to which the subject was assigned. All drug-related TEAEs were gastrointestinal in nature, and without apparent relationship to tolevamer dose. Notably, TEAEs were generally mild, transient, and resolved without clinical sequelae. No serious AEs or deaths were reported in this study.
No clinically or statistically significant change in serum potassium was observed from baseline to end of treatment in any dose group in this study. No clinically or statistically significant changes in 24 h mean urinary potassium excretion were observed at any time point in the 9 or 12 g day−1 treatment groups. A modest reduction in 24 h mean urinary potassium excretion from baseline was observed in the 15 g group, with the average decrease across all study days of 12 mmol 24 h−1. However, as a point of reference, the daily minimum dietary potassium requirement is approximately 50 mmol, and diets containing large amounts of fruits and vegetables tend to include 200–250 mmol day−1 of potassium [29]. Therefore, no serious adverse clinical impact of tolevamer potassium sodium on potassium balance was anticipated in phase III studies designed to examine the effect of 9 g day−1 in patients with CDAD.
Tolevamer may satisfy an important unmet medical need as a new, nonantimicrobial treatment for patients with CDAD. High recurrence rates associated with antibiotic therapies represent the greatest weakness of current standards of care [2, 18–20]. Moreover, the use of both vancomycin and metronidazole has been associated with persistent carriage of C. difficile, and selection for resistant bacteria [19, 20, 30–33]. Although both antibiotics have approximately equivalent efficacy, metronidazole is more commonly used for treating patients with CDAD since it is less costly than vancomycin and since the use of vancomycin is associated with the selection of vancomycin-resistant gram-positive cocci [34–36]. However, recent observational studies indicate a relatively poor response to metronidazole therapy in patients with CDAD [21, 22]. Whereas earlier reports suggested cure rates of 90% and modest rates of recurrence with metronidazole, a recent study conducted in Houston, Texas demonstrated complete response (initial cure with no recurrence of disease) in only 50% of patients [22]. Lack of response to metronidazole was shown in this study to be associated with a higher mortality in patients who did not respond to an initial course of this drug compared with the mortality among responders (33% vs. 21%; P < 0.05) [22]. Further, symptoms and/or signs of C. difficile colitis persisted in 22% of patients for ≥10 days after initiation of metronidazole therapy, and an additional 28% responded initially but had recurrence of disease within the ensuing 90 days [22]. An even higher recurrence rate was noted in another recent observational study in a hospital in Quebec, Canada, in which the 60 day CDAD recurrence rates with metronidazole therapy more than doubled, from 21% between 1991 and 2002 to 47% between 2003 and 2004 (P < 0.001) [21].
Suboptimal responses to metronidazole may occur because it is systemically absorbed and achieves variable concentrations in the colon. Moreover, metronidazole may be poorly tolerated, causing abdominal cramps, nausea, altered taste, and headache. Long-term use is associated with serious AEs, including peripheral neuritis, pancreatitis, possible carcinogenicity, and teratogenicity [2, 37].
In conclusion, the tolevamer potassium sodium oral solution given three times per day up to a total daily dose of 15 g for 8 days and up to a 15 g loading dose was safe and well-tolerated in healthy male volunteers. Based on the encouraging results in a phase II trial with 6 g day−1 in subjects with mild-to-moderate CDAD [26, 27], and in this phase I trial, tolevamer potassium sodium underwent further clinical development as a novel nonantibiotic treatment for patients with CDAD. The recently reported results of the North American phase III trial validate the success of the phase III salt change in minimizing the risk of hypokalaemia [38]. In this randomized, double-blind study in which 543 patients were included in the full analysis data set, the adverse event rate of hypokalaemia was similar in all treatment arms, 15.2% tolevamer, 14% vancomycin and 15.1% metronidazole. Results of a second, confirmatory, phase III trial are not yet available. As the incidence and severity of CDAD increase globally, and poor outcomes are reported with standard antibiotic therapy, novel strategies such as toxin neutralization may find a role in the management of this nosocomial epidemic [5, 13, 22].
This study was sponsored by the Genzyme Corporation, Cambridge, MA. 02142.
Competing interests: none to declare.
REFERENCES
- 1.Hogenauer C, Hammer HF, Krejs GJ, Reisinger EC. Mechanisms and management of antibiotic-associated diarrhea. Clin Infect Dis. 1998;27:702–10. doi: 10.1086/514958. [DOI] [PubMed] [Google Scholar]
- 2.Pothoulakis C, LaMont JT. Clostridium difficile colitis and diarrhea. Gastroenterol Clin North Am. 1993;22:623–37. [PubMed] [Google Scholar]
- 3.Kelly CP, LaMont JT. Clostridium difficile infection. Annu Rev Med. 1998;49:375–90. doi: 10.1146/annurev.med.49.1.375. [DOI] [PubMed] [Google Scholar]
- 4.Buchner AM, Sonnenberg A. Epidemiology of Clostridium difficile infection in a large population of hospitalized US military veterans. Dig Dis Sci. 2002;47:201–7. doi: 10.1023/a:1013252528691. [DOI] [PubMed] [Google Scholar]
- 5.Valiquette L, Low DE, Pepin J, McGeer A. Clostridium difficile infection in hospitals: a brewing storm. Can Med Assoc J. 2004;171:27–9. doi: 10.1503/cmaj.1040957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilcox MH, Smyth ET. Incidence and impact of Clostridium difficile infection in the UK, 1993–1996. J Hosp Infect. 1998;39:181–7. doi: 10.1016/s0195-6701(98)90256-0. [DOI] [PubMed] [Google Scholar]
- 7.Archibald LK, Banerjee SN, Jarvis WR. Secular trends in hospital-acquired Clostridium difficile disease in the United States, 1987–2001. J Infect Dis. 2004;189:1585–9. doi: 10.1086/383045. [DOI] [PubMed] [Google Scholar]
- 8.Eggertson L, Sibbald B. Hospitals battling outbreaks of C. difficile. Can Med Assoc J. 2004;171:19–21. doi: 10.1503/cmaj.1040979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hyland M, Ofner-Agostini M, Miller M. N-CDAD in Canada: results of the Canadian nosocomial infection surveillance program 1997 N-CDAD prevalence surveillance project. Can J Infect Dis. 2001;12:81–8. doi: 10.1155/2001/304098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loo VG, Libman MD, Miller MA, Bourgault AM, Frenette CH, Kelly M, Michaud S, Nguyen T, Poirier L, Vibien A, Horn R, Laflamme PJ, Rene P. Clostridium difficile: a formidable foe. Can Med Assoc J. 2004;171:47–8. doi: 10.1503/cmaj.1040836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warny M, Pepin J, Fang A, Killgore G, Thompson A, Brazier J, Frost E, McDonald LC. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet. 2005;366:1079–94. doi: 10.1016/S0140-6736(05)67420-X. [DOI] [PubMed] [Google Scholar]
- 12.McDonald LC, Killgore GE, Thompson A, Owens RC, Kazakova SV, Sambol SP, Johnson S, Gerding DN. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med. 2005;353:2433–41. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 13.Loo VG, Poirier L, Miller MA, Oughton M, Libman MD, Michaud S, Bourgault AM, Nguyen T, Frenette C, Kelly M, Vibien A, Brassard P, Fenn S, Dewar K, Hudson TJ, Horn R, Rene P, Monczak Y, Dascal A. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med. 2005;353:2442–9. doi: 10.1056/NEJMoa051639. [DOI] [PubMed] [Google Scholar]
- 14.Bartlett JG, Peri TM. The new Clostridium difficile – what does it mean? N Engl J Med. 2005;353:2503–5. doi: 10.1056/NEJMe058221. [DOI] [PubMed] [Google Scholar]
- 15.Kelly CP, Pothoulakis C, LaMont JT. Clostridium difficile colitis. N Engl J Med. 1994;330:257–62. doi: 10.1056/NEJM199401273300406. [DOI] [PubMed] [Google Scholar]
- 16.Fiorentini C, Thelestam M. Clostridium difficile toxin A and its effects on cells. Toxicon. 1991;29:543–67. doi: 10.1016/0041-0101(91)90050-2. [DOI] [PubMed] [Google Scholar]
- 17.Rothman SW, Brown JE, Diecidue A, Foret DA. Differential cytotoxic effects of toxins A and B isolated from Clostridium difficile. Infect Immun. 1984;46:324–31. doi: 10.1128/iai.46.2.324-331.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fekety R, Shah AB. Diagnosis and treatment of Clostridium difficile colitis. JAMA. 1993;269:71–5. [PubMed] [Google Scholar]
- 19.Fekety R, McFarland LV, Surawicz CM, Greenberg RN, Elmer GW, Mulligan ME. Recurrent Clostridium difficile diarrhea: characteristics of and risk factors for patients enrolled in a prospective, randomized, double-blinded trial. Clin Infect Dis. 1997;24:324–33. doi: 10.1093/clinids/24.3.324. [DOI] [PubMed] [Google Scholar]
- 20.Yassin SF, Young-Fadok TM, Zein NN, Pardi DS. Clostridium difficile-associated diarrhea and colitis. Mayo Clin Proc. 2001;76:725–30. doi: 10.4065/76.7.725. [DOI] [PubMed] [Google Scholar]
- 21.Pepin J, Alary ME, Valiquette L, Raiche E, Ruel J, Fulop K, Godin D, Bourassa C. Increasing risk of relapse after treatment of Clostridium difficile colitis in Quebec, Canada. Clin Infect Dis. 2005;40:1591–7. doi: 10.1086/430315. [DOI] [PubMed] [Google Scholar]
- 22.Musher DM, Aslam S, Logan N, Nallacheru S, Bhaila I, Borchert F, Hamill RJ. Relatively poor outcome after treatment of Clostridium difficile colitis with metronidazole. Clin Infect Dis. 2005;40:1586–90. doi: 10.1086/430311. [DOI] [PubMed] [Google Scholar]
- 23.Gerding DN. Metronidazole for Clostridium difficile-associated disease: is it okay for mom? Clin Infect Dis. 2005;40:1598–600. doi: 10.1086/430317. [DOI] [PubMed] [Google Scholar]
- 24.Braunlin W, Xu Q, Hook P, Fitzpatrick R, Klinger JD, Burrier R, Kurtz CB. Toxin binding of tolevamer, a polyanionic drug that protects against antibiotic-associated diarrhea. Biophys J. 2004;87:534–9. doi: 10.1529/biophysj.104.041277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurtz CB, Cannon EP, Brezzani A, Pitruzzello M, Dinardo C, Rinard E, Acheson DW, Fitzpatrick R, Kelly P, Shackett K, Papoulis AT, Goddard PJ, Barker RH, Palace GP, Klinger JD. GT160-246, a toxin binding polymer for treatment of Clostridium difficile colitis. Antimicrob Agents Chemother. 2001;45:2340–7. doi: 10.1128/AAC.45.8.2340-2347.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davidson D, Peppe J, Louie T. A phase 2 study of the toxin binding polymer tolevamer in patients with C. difficile associated diarrhea [poster]. Paper presented at: First International Clostridium difficile Symposium (FICDS), May 5–8, 2004, Gozd Matuljek, Slovenia.
- 27.Louie TL, Peppe J, Watt CK, Johnson D, Mohammed R, Dow G, Weiss K, Simon S, John JF, Garber G, Chasen-Taber S, Davidson DM for the Tolevamer Study Investigator Group. Tolevamer, a novel nonantibiotic polymer, compared with vancomycin in the treatment of mild to moderately severe Clostridium difficile-associated diarrhea. Clin Infect Dis. 2006;43:411–20. doi: 10.1086/506349. [DOI] [PubMed] [Google Scholar]
- 28.Barker RH, Dagher R, Davidson DM, Marquis JK. Review article: tolevamer, a novel toxin-binding polymer: overview of preclinical pharmacology and physiochemical properties. Alimen Pharmacol Ther. 2006;24:1525–34. doi: 10.1111/j.1365-2036.2006.03157.x. [DOI] [PubMed] [Google Scholar]
- 29.Cohen JM, Kowey PR, Whelton PK, Prisant LM. New guidelines for potassium replacement in clinical practice. Arch Intern Med. 2000;160:2429–36. doi: 10.1001/archinte.160.16.2429. [DOI] [PubMed] [Google Scholar]
- 30.Fekety R, Silva J, Buggy B, Deery HG. Treatment of antibiotic-associated colitis with vancomycin. J Antimicrob Chemother. 1984;14(Suppl. D):97–102. doi: 10.1093/jac/14.suppl_d.97. [DOI] [PubMed] [Google Scholar]
- 31.Fekety R, Silva J, Kauffman C, Buggy B, Deery HG. Treatment of antibiotic-associated Clostridium difficile colitis with oral vancomycin: comparison of two dosage regimens. Am J Med. 1989;86:15–9. doi: 10.1016/0002-9343(89)90223-4. [DOI] [PubMed] [Google Scholar]
- 32.Gerding DN. Is there a relationship between vancomycin-resistant enterococcal infection and Clostridium difficile infection? Clin Infect Dis. 1997;25(Suppl. 2):S206–10. doi: 10.1086/516247. [DOI] [PubMed] [Google Scholar]
- 33.Pelaez T, Alcala L, Alonso R, Rodriguez-Creixems M, Garcia-Lechuz JM, Bouza E. Reassessment of Clostridium difficile susceptibility to metronidazole and vancomycin. Antimicrob Agents Chemother. 2002;46:1647–50. doi: 10.1128/AAC.46.6.1647-1650.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fekety R. Guidelines for the diagnosis and management of Clostridium difficile-associated diarrhea and colitis. American College of Gastroenterology Practice Parameters Committee. Am J Gastroenterol. 1997;92:739–50. [PubMed] [Google Scholar]
- 35.Johnson S, Gerding DN. Clostridum difficile-associated diarrhea. Clin Infect Dis. 1998;26:1027–34. doi: 10.1086/520276. [DOI] [PubMed] [Google Scholar]
- 36.Bartlett JG. Antibiotic-associated diarrhea. N Engl J Med. 2002;346:334–9. doi: 10.1056/NEJMcp011603. [DOI] [PubMed] [Google Scholar]
- 37.Kasten MJ. Clindamycin, metronidazole, and chloramphenicol. Mayo Clin Proc. 1999;74:825–33. doi: 10.4065/74.8.825. [DOI] [PubMed] [Google Scholar]
- 38.Louie T, Gerson M, Grimard D, Johnson S, Poirier A, Weiss K, Peppe J, Donovan J, Davidson D. for the Polymer Alternative for CDAD Treatment (PACT) Investigator. 47th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC); 2007 September 17–20; Chicago, Illinois. Conference Poster 3826.