

Abstract
Identification of cytokine-inducible genes is imperative for determining the mechanisms of cytokine action. A cytokine-inducible gene, mrg1 [melanocyte-specific gene (msg1) related gene], was identified through mRNA differential display of interleukin (IL) 9-stimulated and unstimulated mouse helper T cells. In addition to IL-9, mrg1 can be induced by other cytokines and biological stimuli, including IL-1α, -2, -4, -6, and -11, granulocyte/macrophage colony-stimulating factor, interferon γ, platelet-derived growth factor, insulin, serum, and lipopolysaccharide in diverse cell types. The induction of mrg1 by these stimuli appears to be transient, with induction kinetics similar to other primary response genes, implicating its role in diverse biological processes. Deletion or point mutations of either the Box1 motif (binds Janus kinase 1) or the signal transducer and activator of transcription 3 binding site-containing region within the intracellular domain of the IL-9 receptor ligand binding subunit abolished or greatly reduced mrg1 induction by IL-9, suggesting that the Janus kinase/signal transducer and activator of transcription signaling pathway is required for mrg1 induction, at least in response to IL-9. Transfection of mrg1 cDNA into TS1, an IL-9-dependent mouse T cell line, converted these cells to IL-9-independent growth through a nonautocrine mechanism. Overexpression of mrg1 in Rat1 cells resulted in loss of cell contact inhibition, anchorage-independent growth in soft agar, and tumor formation in nude mice, demonstrating that mrg1 is a transforming gene. MRG1 is a transcriptional activator and may represent a founding member of an additional family of transcription factors.
Interleukin (IL) 9 is a T cell-derived multifunctional cytokine, which acts on various cell types including T cells, B cells, mast cells, and hematopoietic progenitors (1, 2). The functions of IL-9 are mediated by the IL-9 receptor, which consists of a ligand specific α-chain (IL-9Rα) and an IL-2Rγ chain (2–4). The involvement of IL-9 in lymphomagenesis has been demonstrated by in vivo studies with transgenic mice constitutively expressing IL-9 (5). Autocrine signaling of IL-9 and IL-9R also has been suggested to be a possible mechanism for the development of certain types of Hodgkin’s disease (6). However, the mechanisms and signaling pathways for the oncogenic activity of deregulated IL-9 expression have not been identified.
Accumulated evidence has revealed that cytokine-inducible primary response genes play important roles in signal transduction controlling cell proliferation, differentiation, and transformation (7, 8). Our recent studies (9–12) have shown that the induction of c-myc and junB, two primary response genes, is a signaling event downstream of JAK/STAT (Janus kinase/signal transducer and activator of transcription; refs. 13 and 14) activation in IL-9-mediated signal transduction. To further elucidate mechanisms of IL-9 action, we have attempted to identify novel genes involved in early transcrptional events of growth factor-triggered signal transduction. We report here the identification of mrg1 [a recently cloned melanocyte-specific gene (msg1) related gene; ref. 15] as a cytokine-inducible gene. We further characterized MRG1 as a transcription factor with transformation activity.
MATERIALS AND METHODS
Differential Display and Cloning of MRG1.
Total cellular RNA was isolated from starved or IL-9-stimulated [10 ng murine IL (mIL)-9/5 × 107 cells/ml] mouse D10 G4.1 (D10) cells (16) and subjected to the differential display analysis (GenHunter, Brookline, MA) (17). A partial cDNA (299 bp) fragment that was specifically induced by IL-9 was isolated by using a 5′ primer (5′-CAGACCGTTCACTTTCAAGT-3′) and a 3′ primer (5′-TTTTTTTTTTTTGA-3′). The full-length mrg1 cDNA was isolated by screening a mouse brain cDNA library (Stratagene) by using the partial cDNA fragment obtained from differential display.
Growth Factor, Cells, and Stimulation.
Human IL (hIL)-1α (1 × 108 units/mg) was from Genzyme. hIL-6 (4 × 106 units/mg), hIL-11 (2.5 × 106 units/mg), and granulocyte/macrophage colony-stimulating factor (GM-CSF) (1.7 × 107 units/mg) were kindly provided by the Genetics Institute (Cambridge, MA). hIL-2 (2.0 × 106 units/mg), hIL-9 (1.2 × 106 units/mg), mIL-4 (1 × 107 units/mg), and mIL-9 (1.5 × 107 units/mg) were from R & D Systems. Erythropoietin (Epo) was from Amgen Biologicals. Human platelet-derived growth factor (PDGF)-AB heterodimer, human insulin, human interferon (IFN) γ (7 × 106 units/mg), and murine IFNγ (1.0 × 107 units/mg) were from GIBCO. D10 T helper lymphocytes were treated with mIL-2 (10 ng/ml), mIL-4 (10 ng/ml), or mIL-9 (10 ng/ml). Human TF1 erythroleukemic cells were treated with IL-11 (500 ng/ml), IL-6 (250 ng/ml), GM-CSF (250 ng/ml), or Epo (250 ng/ml) for 1 hr. 3T3L1 preadipocytes were stimulated with insulin (10 milliunits/ml), IL-6 (250 ng/ml), or IL-11 (500 ng/ml) as described (16, 18, 19). Rat1 fibroblasts were stimulated with insulin (10 milliunits/ml), fetal calf serum (FCS) (20% in DMEM), or PDGF (10 ng/ml). Murine 10T1/2 embryonic fibroblasts were exposed to IL-1α (10 ng/ml), and murine P388D1 macrophage-like cells were treated with lipopolysaccharide (LPS, Escherichia coli 055:B5, 10 μg/ml, Difco) or IFNγ (100 units/ml) after those cells were starved in DMEM or Basal Medium Eagle (GIBCO) supplemented with 0.5% FCS for 48 hr. Human lymphocytes (peripheral blood mononuclear cells depleted of monocytes) and monocytes were isolated as described (20). Activation of T lymphocytes was performed by treatment with phytohemagglutinin (5 μg/ml), phorbol 12-myristate 13-acetate (10 ng/ml) plus ionomycin (1 μg/ml), or 5 μg/ml of cross-linked anti-CD3 mAb OKT3. The monocytes were exposed to LPS (10 μg/ml) for different periods of time.
Construction of pFlag-IL-9Rα Mutants and Establishment of TS1 Transfectants.
The hIL-9Rα cDNA containing different C-terminal truncations, internal deletions, and point mutations were generated either by PCR or in vitro mutagenesis system (Promega). These fragments were subcloned into pRc/cytomegalovirus (CMV) vector, and the N-terminal part of these fragments was replaced by a Flag epitope-tag coding region from pFlag-CMV-1 expression vector (Eastman Kodak). These constructs were electroporated into TS1 cells (12) and then selected by G418. Cells expressing high levels of FlaghIL-9R proteins were further enriched by FACS using an anti-Flag M2 mAb (Eastman Kodak). Northern analysis and Western blotting also were carried out to ensure the equivalent expression of different forms of human IL-9Rα in transfected TS1 cells.
Cellular Localization of MRG1 Protein.
The entire coding region of mrg1 cDNA was amplified by PCR using primers containing EcoRI and BamHI sites and cloned into pEGFP (CLONTECH) such that the stop codon of mrg1 was deleted and the coding sequence continued in-frame with that of the enhanced green fluorescent protein (EGFP). The pMRG1-EGFP hybrid plasmid was transfected into NIH 3T3 cells by electroporation. Cells were grown at 37°C in DMEM plus 10% FCS for 48 hr and then examined by fluorescence microscopy to detect the distribution of MRG1-EGFP protein.
GAL4-Based Transcriptional Activation Assay.
The coding region of MRG1 was PCR-amplified by using primers containing EcoRI and XbaI sites and subsequently cloned into pM vector (CLONTECH) to generate GAL4BD-MRG1 fusion protein. The hybrid plasmid pMMRG1 and the reporter plasmid pG5CAT were cotransfected into NIH 3T3, by using pM3VP16 and pM vector transfectants as controls. Cell extracts were prepared 48 hr after transfection and a TLC assay was performed to measure the chloramphenicol acetyltransferase (CAT) activity.
Factor-Independent Growth of TS1 Cells Expressing MRG1.
TS1 cells were either untransfected or transfected with MRG1 plasmid (pMRG1), mIL-9 plasmid (pmIL-9), or pCDNA3 (vector alone) by SuperFect method (Qiagen). The transfected cells were washed twice with PBS and then diluted to 106 cells/ml in Click’s medium (Irvine Scientific) supplemented with 10% FCS and 0.1 ng/ml of mIL-9 and incubated for an additional 24 hr. Cells then were plated in 35-mm culture dishes at different densities in the presence or absence of mIL-9 (0.1 ng/ml) and G418 (0.5 mg/ml). Colony growth was scored 2 weeks after plating. IL-9-independent cell clones (MRG1-id-A, -B, -C, and -D; IL-9-id-A, and -B) were cloned from well-isolated colonies in 5- to 7-day soft agar cultures (+G418/-IL9) and maintained in Click’s medium with 10% FCS and G418. Cloning efficiency of these IL9-independent clones was analyzed by plating cells on soft agar with or without G418 and IL-9 at different densities, and colonies were scored 2 weeks later.
Bioassay of Conditioned Media.
Cells from MRG1-id-A and -B, and IL-9-id-A were washed three times with Click’s medium and then cultured at 2.5 × 105 cells/ml in the same medium containing 1%, 2%, 4%, and 10% FCS or with 0.2%, 0.4%, 0.8%, and 2% serum replacement (Sigma). Conditioned media were collected at 1, 2, 3, 4, and 5 days of culture, concentrated by using Centricon-3 (Amicon; cut-off molecular mass = 3 kDa) and reconstituted with Click’s medium to the desired fold of concentration so that the final concentration of FCS and serum replacement are 10% and 2%, respectively. Concentrated conditioned media were tested by TS1 proliferation assay as described (10).
Focus Formation Assay.
pMRG1, pCDNA3 (vector alone), or H-Ras-V12-pBabe was transfected into Rat1 cells (2 × 105 cells/10 μg plasmid in triplicate). The transfected cells were plated in a 100-mm culture dish in DMEM supplemented with 10% FCS. Cells were rinsed with medium 24 hr posttransfection, and then maintained in DMEM plus 5% FCS. The medium was changed every 3 days thereafter. The number of foci/10 μg plasmid per 2 × 105 cells was counted at 2 weeks and 4 weeks after transfection.
Colony Growth in Soft Agar.
Single cell clones of pMRG1- or pCDNA3-transfected Rat1 were isolated after G418 selection (1 mg/ml) for 2 weeks. Cells were briefly treated with trypsin-EDTA and separated by passing through a 25G 5/8 syringe needle, and then mixed with 1 ml of top agar (0.3%) and plated in triplicates on top of 1 ml of bottom agar (0.6%) in DMEM plus 20% FCS and G418 (0.5 mg/ml) at a density of 1 × 103 cells per 30-mm well. Cells were fed every 4–5 days by adding a new layer of top agar. After 2 weeks, colonies containing more than 50 cells were scored under the microscope.
Tumor Formation in Nude Mice.
To assay for oncogenicity of pMRG1 transfectants, 5 × 105 MRG1–1 cells were injected s.c. into nude mice (female, 12 weeks old; Charles River Breeding Laboratories). Mice were kept in a sterile environment and observed twice weekly for the formation of tumors. Mice that had tumors were sacrificed, and tissue was taken for histological examination.
RESULTS
Identification of mrg1 as a Cytokine-Inducible Gene.
A partial cDNA fragment induced by IL-9 was isolated by differential display using mRNAs from the IL-9-stimulated and unstimulated mouse T helper cell line, D10. Sequence analysis revealed its identity with mrg1 (15). The full-length mrg1 cDNA subsequently was cloned by hybridization screening of a mouse brain cDNA library. Northern analysis demonstrated that mrg1 is expressed in various tissues with the highest expression level in brain and testis (Fig. 1A). mrg1 is highly inducible by IL-9, as well as by other members of the IL-2Rγ chain family, including IL-2 and IL-4 (Fig. 1B). Interestingly, the induction of mrg1 is transient as the mRNA expression decreased to the basal level by 120 min, which is similar to the induction kinetics of other primary response genes (7, 21). These data clearly demonstrate that mrg1 is an IL-2Rγ chain family cytokine inducible gene.
Figure 1.

mrg1 is inducible by various biological stimuli in different cell types. (A) Expression of mrg1 transcript in mouse adult tissues. (B) Induction of mrg1 expression by cytokines in the IL-2Rγ chain cytokine family. (C) mrg1 expression in various cell types induced by different biological stimuli. Total RNAs (10 μg per lane) were separated through a 1% formadehyde-agarose gel and transferred onto a nitrocellulose membrane. The same blot was sequentially hybridized with 32P-labeled mrg1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or rRNA probes.
mrg1 Is Inducible by Various Biological Stimuli in Diverse Cell Types.
To explore the possible functions of mrg1, we further analyzed the inducibility of mrg1 in response to cytokines or stimuli other than IL-2Rγ chain cytokines. As shown in Fig. 1C, mrg1 could be activated by IL-6 and granulocyte/macrophage colony-stimulating factor, but not by IL-11 or Epo in human TF1 erythroleukemic cells after cytokine treatment for 1 hr. In addition, transient but rapid induction of mrg1 also was detected after treatment of FCS, insulin, or PDGF in Rat1 fibroblasts; IL-1-α in 10T1/2 mouse embryo fibroblasts; insulin, IL-6, or IL-11 in mouse 3T3L1 preadipocytes; LPS or INFγ in P388D1 murine macrophage-like cell line; and LPS in human monocytes. However, no significant mrg1 induction was observed in anti-CD3, phytohemagglutinin, or phorbol 12-myristate 13-acetate plus ionomycin-activated human peripheral T lymphocyte (data not shown). These results indicate that mrg1, like many primary response genes, can be induced by various biological stimuli and may function in different physiological processes.
Involvement of JAK/STAT in mrg1 Expression.
JAK/STAT activation (13, 14) is important for IL-9-mediated signal transduction (10–12). The BOX1 motif (to which JAK1 binds) and the STAT3 binding site in the cytoplasmic domain of hIL-9Rα are important for JAK1, STAT3, and JAK3 activation and growth signaling mediated by IL-9 (10, 22). Although JAK3 is associated with the IL-2Rγ chain (23), our previous study showed that mutations of IL-9Rα that decreased tyrosine phosphorylation of JAK1 also affected the tyrosine phosphorylation of JAK3, indicating that the two subunits of IL-9R may communicate with each other (10). To test whether mrg1 induction requires particular domains in IL-9Rα and/or JAK/STAT activation, wild type and mutants with deletions or point mutations in the intracellular domain of hIL-9Rα (ref. 10, indicated in Fig. 2A) were stably transfected into the mIL-9-dependent TS1 cell line. This line is normally unresponsive to hIL-9, but responsiveness can be conferred by the transfection of wild-type hIL-9Rα (3, 10). As shown in Fig. 2B, deletion of either the BOX1 motif or amino acids 338–422, which include the STAT3 binding site, abolished mrg1 induction by hIL-9. Also, point mutations of either the BOX1 motif (substitution of Pro-X-Pro with Ala-X-Ala) or the STAT3 binding site (YLPQ to YLPA) significantly reduced mrg1 induction by hIL-9 (Fig. 2B). TS1 cells transfected with these mutants previously have been shown to have decreased tyrosine phosphorylation of JAK1, JAK3, and STAT3 (10). These results therefore suggest that JAK/STAT signaling pathway is required for mrg1 induction in IL-9-stimulated cells.
Figure 2.

IL-9-induced mrg1 expression is mediated by JAK/STAT pathway. (A) Construction of pFlag-CMV-IL-9Rα plasmids containing wild-type (W) and mutated hIL-9Rα (18). Transmembrane domain (TM), BOX1 motif (BOX1), serine-rich region (SR), C-terminal truncation mutants deleting amino acids 422–522 (Daa422–522) or amino acids 338–522 (Daa338–522), internal deletion of BOX1 [Daa298–315(BOX1)] and point mutation within BOX1 motif (Paa306XPaa308 to Aaa306XAaa308, MBOX1) or STAT3 binding site (YLPQaa408 to YLPAaa408; MSTAT3) are as indicated. (B) Induction of mrg1 gene expression by hIL-9 in TS1 transfectants expressing different forms of hIL-9Rα. Cells were starved in serum-free medium for 8 hr and then stimulated with (+) or without (−) hIL-9 (30 ng/ml) for 1 hr. Ten micrograms of total RNA from unstimulated or stimulated cells was loaded to each lane. The same blot sequentially was hybridized with 32P-labeled mrg1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probes.
Cellular Localization of MRG1 Protein.
To elucidate possible functions of MRG1, we examined the cellular localization of the MRG1 protein. The mrg1 cDNA was fused in-frame into the N terminus of EGFP in the pEGFP vector (24), which expresses a green fluorescent protein to facilitate protein localization in living cells. Addition of mrg1 to the N terminus of pEGFP resulted in nuclear localization of the MRG1 fusion protein (Fig. 3A, MRG1). In contrast, the green fluorescent signal distributed throughout the cytoplasm and nuclei of cells transfected with pEGFP plasmid (Fig. 3A, Control). These results demonstrate that MRG1 encodes a nuclear protein.
Figure 3.
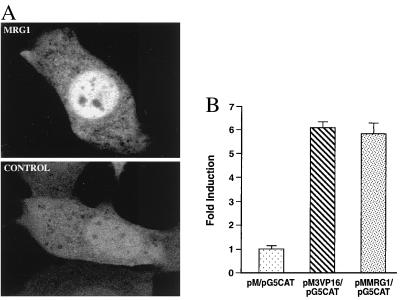
MRG1 is a nuclear protein with transcriptional activation function. (A) Cellular localization of MRG1 protein. pEGFP vector (CONTROL) or pMRG1-EGFP hybrid plasmid (MRG1) was transfected into NIH 3T3 by electroporation. Living cells were observed by fluorescence microscopy at 48 hr posttransfection. (B) Transcriptional activation of MRG1. NIH 3T3 cells were cotransfected with pM (negative control), pM3VP16 (positive control), GAL4BD-VP16 fusion protein), or pMMRG1 (GAL4BD-MRG1 fusion protein) and reporter plasmid pG5CAT. The assay for CAT activity was performed 48 hr posttransfection. All transfections were normalized according to β-galactosidase activity, which was used as an internal control for transfection. Data (X ± SEM) shown are representative of three independent experiments.
Transcriptional Activation Function of MRG1.
To test whether MRG1 could be a transcription factor, the transcriptional activity of MRG1 was tested with a GAL4-based transcriptional activation assay (25). The coding region of mrg1 cDNA was inserted into the pM expression vector to generate a GAL4-DNA binding domain-MRG1 (GAL4BD-MRG1) fusion protein. The hybrid plasmid (pMMRG1) then was cotransfected with the pG5CAT reporter plasmid (containing five copies of GAL4 DNA binding motifs and the adenovirus E1b promoter upstream from the CAT gene) into NIH 3T3 cells. As shown in Fig. 3B, GAL4BD-MRG1 strongly activated CAT expression in NIH 3T3 cells with a 6-fold increase over the GAL4BD (pM vector) alone. This level of increase is comparable to that of GAL4BD-VP16 in pM3VP16. VP16 is a well-characterized herpes simplex virus transcriptional activator. These data strongly support our hypothesis that MRG1 is a nuclear protein that functions as a transcriptional activator.
Transforming Functions of MRG1.
Many transcriptional regulators are potential targets for neoplastic transformation (8). Because overexpression of IL-9 is involved in lymphomagenesis (5, 26) and MRG1 is one of the downstream nuclear targets in the IL-9-mediated signaling pathway, we speculated that MRG1 might play a role in deregulated cell growth and oncogenesis. To address this possibility, we investigated whether overexpression of MRG1 results in cellular transformation.
TS1 cells were transfected with pMRG1 to test whether overexpression of MRG1 can convert TS1 cell from IL-9-dependent to IL-9-independent growth, one of the features of cellular transformation (27). The transfected TS1 cells were cloned in G418 containing soft agar in the presence or absence of IL-9. As a positive control, TS1 cells also were transfected with pmIL-9. As shown in Table 1, the efficiency of IL-9-independent growth in TS1 clones transfected with pMRG1 or pmIL-9, as determined by the number of G418-resistant clones that grew in the absence of IL-9, is significantly higher than that in pCDNA3 (vector alone)-transfected TS1 cells. These results indicate that IL-9-independent growth of TS1 cells can be conferred by overexpression of MRG1, suggesting a possible role of MRG1 in oncogenic transformation.
Table 1.
Efficiency of IL-9-independent growth after transfection of TS1 cells with MRG1 or mIL-9 plasmid
| Plasmid | No. of cells | Agar cloning in
|
|
|---|---|---|---|
| −mIL-9 | +mIL9 | ||
| pMRG1 | 105 | 64.7 ± 5.7 | 1.0 ± 0.0 |
| 106 | 625.7 ± 18.9 | 9.7 ± 0.6 | |
| 107 | 5,874.7 ± 61.6 | 99.0 ± 2.7 | |
| pmIL-9 | 105 | 59.3 ± 7.8 | 0.67 ± 0.6 |
| 106 | 596.3 ± 10.5 | 11.7 ± 0.6 | |
| 107 | 5,927.0 ± 77.2 | 291.3 ± 10.0 | |
| pCDNA3 | 106 | 508.0 ± 5.6 | 0 |
| 107 | 5,138.7 ± 105.0 | <0.66 ± 1.6 | |
| None | 106 | 549.7 ± 22.9 | 0 |
| 107 | 5,606.3 ± 20.6 | <0.33 ± 0.6 | |
Mean ± SEM of clones from three plates grown with G418 (pMRG1-, pmIL-9-, or pCDNA3-transfected cells) or without G418 (untransfected cells).
To elucidate whether the transformation of TS1 cells by overexpression of MRG1 is through an autocrine mechanism, four IL-9-independent cell clones (MRG1-id-A, -B, -C, and -D) from pMRG1-transfected TS1 cells were isolated, and the IL-9-independent growth at different cell densities was examined by soft agar cloning assays, in comparison with clones (IL-9-id-A and –B) derived from pmIL-9-transfected TS1 cells. As shown in Table 2, a linear relationship between the numbers of cells plated and colony growth was observed only in clones derived from pMRG1-transfected TS1 cells, suggesting that a nonautocrine mechaninsm may be involved. To further rule out the possibility that minute amounts of growth factors may be produced by these factor-independent clones, condition media from MRG1-id-A and –B grown in different concentrations of FCS or serum replacement (Sigma) were concentrated and analyzed for their ability to support the proliferation of parental TS1 cells in a thymidine incorporation assay (27). The results showed that the conditioned media collected from 1- to 5-day cultures of pMRG1 transfected IL-9-independent TS1 lines (cultured at a density of 2.5 × 105 cells/ml) were unable to support the proliferation of the parental TS1 cells, even when conditioned media were concentrated up to 10-fold (data not shown). These data suggest that the transformation of TS1 by overexpression of MRG1 is most likely mediated by a nonautocrine mechanism.
Table 2.
Cloning efficiency of individual clones derived from pMRG1- or pmIL-9-transfected TS1 cells
| Cells | No. of cells | Agar cloning in
|
|
|---|---|---|---|
| −mIL-9 | +mIL9 | ||
| MRG1-id-A | 102 | 6.0 ± 0.0* | 11.0 ± 2.7 |
| 103 | 58.0 ± 5.6 | 115.3 ± 11.9 | |
| 104 | 607.7 ± 14.2 | 1,204.3 ± 9.1 | |
| MRG1-id-B | 102 | 6.3 ± 0.6 | 10.7 ± 1.5 |
| 103 | 64.3 ± 3.2 | 111.0 ± 3.0 | |
| 104 | 629 ± 7.9 | 1,127.7 ± 16.9 | |
| IL-9-id-A | 102 | 9.7 ± 0.6 | 37.0 ± 4.4 |
| 103 | 153.7 ± 8.5 | 382.3 ± 12.0 | |
| 104 | 2,183.7 ± 46.2 | 3,767.3 ± 120.4 | |
Similar results were obtained from MRG1-id-C, MRG1-id-D, and IL-9-id-B.
Mean ± SEM of three different plates.
To further assess whether MRG1 has oncogenic potential, loss of contact inhibition was monitored by using a standard transformation assay in Rat1 cells. These cells, when transfected with pMRG1, began to form foci within 3 weeks. The frequency of focus formation was 28.67 ± 2.04/10 μg plasmid DNA per 2 × 105 cells at 4 weeks (Table 3). There was no focus formation in nontransfected Rat1 or pCDNA3-transfected Rat1 cells even beyond 4 weeks after transfection.
Table 3.
Focus formation of pMRG1-transfected Rat1 cells
| Plasmid | Foci/10μg plasmid per 2 × 105 cells |
|---|---|
| pCDNA3 | 0 (4 wk) |
| pMRG1 | 0 (2 wk) |
| 28.6 ± 2.3 (4 wk) | |
| H-Ras-V12-pBabe | 112.3 ± 4.2 (2 wk) |
| 128.0 ± 6.6 (4 wk) |
pCDNA3 and H-Ras-V12-pBabe were used as negative and positive controls, respectively. Data (X ± SEM) shown are representative of two independent experiments.
We next analyzed the loss of substrate dependence by colony growth of G418 selected single cell clones in soft agar. Four and three clones were selected from pMRG1 and pCDNA3 (vector alone)-transfected Rat1 cells, respectively. Two G418 selected pMRG1-transfected colonies, MRG1-1 and -2, which express high levels of MRG1 (based on reverse transcription–PCR, data not shown), formed colonies (>50 cells) in soft agar in 2 weeks with an average cloning efficiency of 34.2% and 33.1%, respectively. None of the G418 selected clones from vector controls grew in soft agar (Table 4).
Table 4.
Colony formation of pMRG1-transfected Rat1 clones in soft agar
| Cell clone | Clonability, % |
|---|---|
| CDNA3-1 | 0 |
| MRG1-1 | 34.2 ± 3.7 |
| MRG1-2 | 33.1 ± 4.1 |
| H-Ras-V12-pBabe-3 | 52.0 ± 6.2 |
pCDNA3 transfected Rat1 clones (CDNA3-1) and an established H-Ras-V12-pBabe-3 Rat1 clone were used as negative and positive controls, respectively. Data (X ± SEM) shown are representative of three independent experiments.
MRG1-1 cells also were injected into nude mice to test the tumorigenicity of Rat1 cells overexpressing mrg1. Although untransfected or pCDNA3-transfected Rat1 cells did not form tumors in nude mice, all four animals injected with MRG1-1 developed tumors (Table 5). These tumors appeared 11–13 weeks after injection and enlarged rapidly thereafter. Histological examination of these tumors revealed fibrosarcoma-like characteristics, reflecting the fibroblastic origin of injected Rat1 cells (Fig. 4). These results strongly suggest that mrg1 is a transforming gene with oncogenic potential.
Table 5.
Tumor formation of MRG1-1 in nude mice
Tumor formation observed 11–13 weeks after injection.
No tumor developed in 24 weeks after injection.
Figure 4.
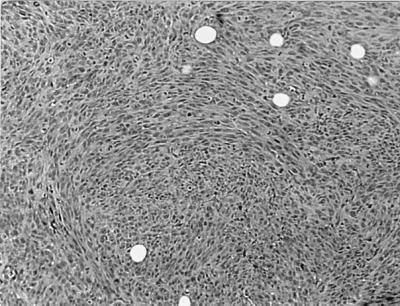
Histology of tumor in MRG1–1-injected nude mice. Photomicrograph of tumor removed from mice injected with MRG1–1 cells, and stained with hematoxylin and eosin. Histologic pattern is sarcomatoid, consistent with the connective tissue origin of the parental cells. (Magnification: ×100.)
DISCUSSION
We have identified mrg1 as a cytokine inducible gene by mRNA differential display. MRG1 is a nuclear protein with transcription activation and transformation functions. The rapid and transient expression of MRG1 is one of the earliest signaling events in cells stimulated with cytokines and biological stimuli. The fact that MRG1 can be activated by different biological stimuli in many cell types suggests that MRG1 may determine part of the biological pleiotropy and redundancy of cytokines (28, 29) and may play important roles in different physiological processes, such as hematopoiesis, immunity, and inflammation.
The expression of MRG1 induced by IL-9 appears to be mediated, at least in part, by JAK/STAT signaling pathway. Interestingly, a potential STAT binding site is localized at −720 of the 5′ flanking region of both human and mouse MRG1 (data not shown). It is possible that transcription factors other than STATs also may be involved in the expression of MRG1. Mutagenesis studies in the MRG1 promoter will be required to dissect the essential role of the STAT binding site, other cis-elements, and their corresponding binding proteins in mrg1 gene expression.
We have clearly demonstrated both in vitro and in vivo that mrg1 is a transforming gene with oncogenic potential. Our results also indicated the ability of mrg1 to transform TS1 cells appears to be mediated by a nonautocrine mechanism. Focus formation of pMRG1-transfected cells was detected more than 3 weeks after transfection, compared with 10 days in H-Ras-V12-pBabe-transfected Rat1 cells, and the focus formation frequency of pMRG1-transfected Rat1 was significantly lower than that of the H-Ras-V12-pBabe-transfected control (Table 3). In addition, tumor formation in nude mice injected with pMRG1-transfected Rat1 also was delayed compared with those injected with H-Ras-V12-pBabe-transfected Rat1 cells (data not shown). Interestingly, these results correlated with the nature of lymphomagenesis in IL-9 transgenic mice, in which constitutive overexpression of IL-9 resulted in the development of lymphoma in only about 7% of IL-9 transgenic mice (5). However, these mice are highly susceptible to chemical mutagenesis and γ-irradiation (4, 5). Because our present study showed IL-9 can induce high-level expression of mrg1, it is possible that overexpression of mrg1 in these transgenic animals may prime cells to a transformed stage such that they are more susceptible to a second transforming agent. The ability of mrg1 to cooperate with other oncogenes (30) may permit resolution of this issue.
Shioda et al. (32) have shown that msg1 and mrg1 share 75% identity at the amino acid level. The expression of msg1 is melanocyte-specific and associated with melanocyte pigmentation whereas MRG1 is ubiquitously expressed. mrg1 and msg1 contain no known functional domain but shared high sequence homology in the C-terminal region, which contains many acidic amino acid residues. It has been speculated that C-terminal acidic regions of mrg1 and msg1 may form amphipathic α-helices (15), which are present in the acidic activator domains of certain transcription factors (31). Our demonstration that MRG1 acts as a transcriptional activator, in agreement with a recent study by Shioda et al. (32), provides direct evidence for such a speculation. These studies suggest that MRG1 and MSG1 might represent members of a new family of transcription factors.
Although MRG1 appears to function as a transcriptional activator with comparable level of transcriptional activation as VP16 (Fig. 3B), this observation does not necessarily imply that MRG1 is a DNA-binding protein. Binding site selection with total genomic DNA (33) will be required to determine whether MRG1 is a DNA-binding protein, which can bind specific DNA target sequences. It is also possible that MRG1 may acquire DNA-binding capacity by forming heterodimers with other DNA-binding proteins. Further biochemical characterization of MRG1 protein and its binding partners and the identification of downstream target genes will be required to fully understand the mechanisms of transcriptional activation of MRG1.
mrg1 has been identified as a primary response gene, which is inducible by cytokines and other stimuli in many different cell types. The induction of mrg1 in response to IL-9 appears to be mediated in part through JAK/STAT activation. MRG1 is a nuclear protein with transcriptional activation and transformation activities. The fact that MRG1 may be a transcription factor opens an avenue to search for MRG1-targeted genes whose expression may determine part of the biological pleiotropy and redundancy of different biological stimuli. Finally, our finding that MRG1 has transforming potential will provide insights into our understanding of cytokine-related lymphomagenesis and other malignancies.
Acknowledgments
We thank Drs. Steve Clark, Kathy Rundell, Ann Roman, David Donner, David Leibowitz, and George Malacinski for critical reading of this manuscript; Dr. Robert Hromas and Xin-Yuan Wang for helpful discussion; Dr. Mark Marshall and Bruce Diaz for H-Ras-V12 plasmid, Rat1, and other cell lines; Dr. Monica Tsang for IL-2, IL-4, and IL-9; Ruben Sandoval Jr. for the help with fluorescence microscopy; Joann Dunn and Sean Sissons for the help with nude mouse studies; and Dr. Young J. Kim and Patricia Mantel for the help with lymphocytes/monocytes studies. This work was supported by grants from the United States Public Health Service (RO1DK43105, RO1HL48819 and RO1DK50570 to Y.-C.Y) and the Leukemia Society of America (Scholar Award to Y.-C.Y).
ABBREVIATIONS
- JAK
Janus kinase
- STAT
signal transducer and activator of transcription
- MSG1, the product of a melanocyte-specific gene
MRG1, the products of a MSG-related gene
- IL
interleukin
- IL-R
IL receptor
- hIL
human IL
- mIL
murine IL
- Epo
erythropoietin
- FCS
fetal calf serum
- EGFP
enhanced green fluorescent protein
- CAT
chloramphenicol acetyltransferase
- LPS
lipopolysaccharide
- PDGF
platelet-derived growth factor
- IFN
interferon
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Yang Y-C. Leuk Lymphoma. 1992;8:441–447. doi: 10.3109/10428199209051026. [DOI] [PubMed] [Google Scholar]
- 2.Renauld J-C, Kermouni A, Vink A, Louahed J, Van Snick J. J Leukocyte Biol. 1995;57:353–369. doi: 10.1002/jlb.57.3.353. [DOI] [PubMed] [Google Scholar]
- 3.Renauld J-C, Druez C, Kermouni A, Houssiau F, Uyttenhove C, Van Roost E, Van Snick J. Proc Natl Acad Sci USA. 1992;89:5690–5694. doi: 10.1073/pnas.89.12.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnston J A, Bacon C M, Riedy M C, O’Shea J J. J Leukocyte Biol. 1996;60:441–452. doi: 10.1002/jlb.60.4.441. [DOI] [PubMed] [Google Scholar]
- 5.Renauld J-C, Van Der Lugt N, Vink A, Van Roon M, Godfraind C, Warnier G, Merz H, Feller A, Berns A, Van Snick J. Oncogene. 1994;9:1327–1332. [PubMed] [Google Scholar]
- 6.Gruss H-J, Brach M A, Drexler H G, Bross K J, Herrmann F. Cancer Res. 1992;52:1026–1031. [PubMed] [Google Scholar]
- 7.Herschman H R. Annu Rev Biochem. 1991;60:281–319. doi: 10.1146/annurev.bi.60.070191.001433. [DOI] [PubMed] [Google Scholar]
- 8.Hunter T. Cell. 1997;88:333–346. doi: 10.1016/s0092-8674(00)81872-3. [DOI] [PubMed] [Google Scholar]
- 9.Kang L Y, Yang Y-C. J Cell Physiol. 1995;163:623–630. doi: 10.1002/jcp.1041630324. [DOI] [PubMed] [Google Scholar]
- 10.Zhu Y X, Sun H B, Tsang M L-S, Yin T, Yang Y-C. J Biol Chem. 1997;272:21334–21340. doi: 10.1074/jbc.272.34.21334. [DOI] [PubMed] [Google Scholar]
- 11.Yin T, Yang L, Yang Y-C. Blood. 1995;85:3101–3106. [PubMed] [Google Scholar]
- 12.Yin T, Keller S R, Quelle F W, Witthuhn B A, Tsang M L-S, Lienhard G E, Ihle J N, Yang Y-C. J Biol Chem. 1995;270:20497–20502. doi: 10.1074/jbc.270.35.20497. [DOI] [PubMed] [Google Scholar]
- 13.Ihle J N. Nature (London) 1994;377:591–594. doi: 10.1038/377591a0. [DOI] [PubMed] [Google Scholar]
- 14.Darnell J E, Jr, Kerr I M, Stark G R. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 15.Shioda T, Fenner M H, Isselbacher K J. Proc Natl Acad Sci USA. 1996;93:12298–12303. doi: 10.1073/pnas.93.22.12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin T, Tsang M L-S, Yang Y-C. J Biol Chem. 1994;269:26614–26617. [PubMed] [Google Scholar]
- 17.Liang P, Pardee A B. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- 18.Yin T, Taga T, Tsang M L-S, Yasukawa K, Kishimoto T, Yang Y-C. J Immunol. 1993;153:2555–2561. [PubMed] [Google Scholar]
- 19.Yin T, Miyazawa K, Yang Y-C. J Biol Chem. 1992;267:8347–8351. [PubMed] [Google Scholar]
- 20.Colotta F, Borre A, Wang J M, Tattanelli M, Maddalena F, Polentarutti N, Peri G, Mantovani A. J Immunol. 1992;148:760–765. [PubMed] [Google Scholar]
- 21.Nathans D. The Cell Cycle. Plainview, NY: Cold Spring Harbor Lab. Press; 1992. [Google Scholar]
- 22.Demoulin J-B, Uyttenhove C, Van Roost E, Delestre B, Donckers D, Van Snick J, Renauld J C. Mol Cell Biol. 1996;16:4710–4716. doi: 10.1128/mcb.16.9.4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyazaki T, Kawahara A, Fuji H, Nakagawa Y, Minami Y, Liu Z-J, Oishi I, Silvennoinen O, Witthuhn B A, Ihle J N, Taniguchi T. Science. 1994;266:1045–1047. doi: 10.1126/science.7973659. [DOI] [PubMed] [Google Scholar]
- 24.Wang S, Hazelrigg T. Nature (London) 1994;369:400–403. doi: 10.1038/369400a0. [DOI] [PubMed] [Google Scholar]
- 25.Sadowski I, Bell B, Broad P, Hollis M. Gene. 1992;118:137–141. doi: 10.1016/0378-1119(92)90261-m. [DOI] [PubMed] [Google Scholar]
- 26.Uyttenhove C, Druez C, Renauld J C, Herin M, Noel H, Van Snick J. J Exp Med. 1991;173:519–522. doi: 10.1084/jem.173.2.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katzav S, Martin-Zanca D, Barbacid M, Hedge A M, Isfort R, Ihle J N. Oncogene. 1989;4:1129–1135. [PubMed] [Google Scholar]
- 28.Kishimoto T, Taga T, Akira S. Cell. 1994;76:253–262. doi: 10.1016/0092-8674(94)90333-6. [DOI] [PubMed] [Google Scholar]
- 29.Taniguchi T. Science. 1995;268:251–255. doi: 10.1126/science.7716517. [DOI] [PubMed] [Google Scholar]
- 30.Ruley H E. Nature (London) 1983;304:602–606. doi: 10.1038/304602a0. [DOI] [PubMed] [Google Scholar]
- 31.Mitchell P J, Tjian R. Science. 1989;24:371–378. doi: 10.1126/science.2667136. [DOI] [PubMed] [Google Scholar]
- 32.Shioda T, Fenner M H, Isselbacher K J. Gene. 1997;204:235–241. doi: 10.1016/s0378-1119(97)00551-9. [DOI] [PubMed] [Google Scholar]
- 33.Blackwell T K, Weintraub H. Science. 1990;250:1104–1110. doi: 10.1126/science.2174572. [DOI] [PubMed] [Google Scholar]