

Abstract
The NMR assignment of 13C, 15N-labeled proteins with the use of triple resonance experiments is limited to molecular weights below ∼25,000 Daltons, mainly because of low sensitivity due to rapid transverse nuclear spin relaxation during the evolution and recording periods. For experiments that exclusively correlate the amide proton (1HN), the amide nitrogen (15N), and 13C atoms, this size limit has been previously extended by additional labeling with deuterium (2H). The present paper shows that the implementation of transverse relaxation-optimized spectroscopy ([15N,1H]-TROSY) into triple resonance experiments results in several-fold improved sensitivity for 2H/13C/15N-labeled proteins and approximately twofold sensitivity gain for 13C/15N-labeled proteins. Pulse schemes and spectra recorded with deuterated and protonated proteins are presented for the [15N, 1H]-TROSY-HNCA and [15N, 1H]-TROSY-HNCO experiments. A theoretical analysis of the HNCA experiment shows that the primary TROSY effect is on the transverse relaxation of 15N, which is only little affected by deuteration, and predicts sensitivity enhancements that are in close agreement with the experimental data.
In the standard protocol for protein structure determination by NMR spectroscopy, sequence-specific resonance assignment plays a pivotal role (1). Several different assignment strategies are available, and one of the established procedures for obtaining sequential assignments (2) involves uniform 13C/15N labeling and delineation of heteronuclear scalar couplings with triple-resonance experiments (3–7). In these experiments, the transfer of magnetization along networks of scalar-coupled spins includes long delays during which 13C and 15N magnetization evolve in the transverse plane. Fast transverse relaxation during these delays and during 1H acquisition, limits the application of triple-resonance NMR experiments with larger proteins. For experiments that exclusively correlate 1HN, 15N, and 13C, the situation has been improved by 2H-labeling of the 13CHn moieties, which eliminates dipolar 13C relaxation by the directly attached protons (8) and reduces 1HN line broadening by dipole–dipole (DD) coupling with remote protons. However, uniform deuteration also imposes stringent limitations on the structural information that can be obtained by NMR (8,9) and does not significantly reduce 15N relaxation during the delays when this spin is in the transverse plane. Here, we propose to extend the application of triple-resonance experiments by using the principle of transverse relaxation-optimized spectroscopy (TROSY) (10). TROSY suppresses transverse relaxation in 15N–1HN moieties by constructive use of interference between dipole–dipole coupling and chemical shift anisotropy (CSA) (10) and thus results in improved sensitivity of triple-resonance experiments by minimizing 15N transverse relaxation during 15N evolution periods and 1HN transverse relaxation during detection. Because part of the gain achieved with TROSY stems from the reduced T2 relaxation rate of 15N, TROSY will benefit triple-resonance experiments with deuterated as well as protonated proteins. In this paper, we present experimental schemes for the implementation of TROSY in the amide proton-to-nitrogen-to-α carbon correlation (HNCA) and amide proton-to-nitrogen-to-carbonyl carbon correlation (HNCO) experiments, which correlate the 15N–1H(i) group with 13Cα(i) and 13Cα(i − 1), and with 13CO(i − 1), respectively (11, 12). For a quantitative evaluation of the sensitivity gain that can be achieved, we compare [15N, 1H]-TROSY-HNCA with conventional HNCA (13).
METHODS
Triple-resonance experiments use coherence transfers along a network of scalar coupled 15N, 13C, and 1H spins (3–7). Thereby, coherence transfer from 15N to either 13Cα or 13CO requires long delays due to the small 1J(15N, 13Cα)- and 1J(15N, 13CO)-coupling constants. Because the 15N magnetization is in the transverse plane throughout these transfer periods, the 15N chemical shift evolution is recorded in a constant-time (ct) fashion during the magnetization transfer (14, 15), and relaxation of 15N due to DD coupling with the directly bound 1HN and to 15N CSA leads to severe loss of coherence. Using the TROSY principle (10), transverse relaxation during these critical 15N evolution periods can be efficiently suppressed. Similarly, 1HN transverse relaxation during detection due to 1HN CSA and to DD coupling with 15N can be suppressed with the use of TROSY.
In the following product operator analysis (16) of the [15N, 1H]-TROSY-HNCA experiment (Fig. 1), we describe the 13C magnetization (C) with Cartesian operators, whereas for the 1HN (H) and 15N (N) magnetizations single-transition operators (17) are used. For both 13C/15N- and 2H/13C/15N-labeled proteins, the 1H as well as the 15N steady–state magnetizations are used at the outset, and the density matrix after the first insensitive nuclei enhanced by polarization transfer (INEPT) step at time point b in Fig. 1 becomes
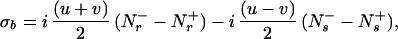 |
1 |
where the constant factors u and v represent the relative magnitudes of the steady–state 1H and 15N magnetizations (18, 19), and the single-transition operators Nr± and Ns± represent the 15N magnetization associated with the rotating frame transition frequencies ωsN = ωN + πJHN and ωrN = ωN − πJHN, respectively (20). Only the Ns− and Ns+ terms are transferred to detectable magnetization after the single transition-to-single transition polarization transfer (ST2-PT) element (18) (time points e–f in Fig. 1). During the ct period T/2 between time points b and c, the Ns± spin operators evolve due to the 15N chemical shift and the J-couplings to the two neighboring α-carbons, 13Cα(i) and 13Cα(i − 1). The resulting antiphase terms Ns± Czα(i) and Ns± Czα (i − 1) are converted to multiple-quantum coherences by the 90° (13Cα) pulse at time point c. During t2, these terms evolve due to the 13Cα chemical shift because carbonyl carbons and deuterons or protons are decoupled (Fig. 1). The 90° (13Cα) pulse at time point d completes the 13Cα evolution period. During the second half of the ct 15N evolution period, the Ns± Czα (i) and Ns± Czα (i − 1) terms again evolve due to the 15N chemical shift (21),
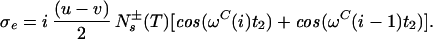 |
2 |
Two signals in the 13C(t2) dimension exhibit the intraresidual correlation ω1(15Ni)/ω2(13Ciα) modulated by cos(ωC(i)t2) and the sequential correlation ω1(15Ni)/ω2(13Ci−1α) modulated by cos(ωC(i − 1)t2). Transverse 15N relaxation during the ct period T = 1/1JNCα is given by (see Appendix):
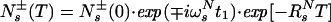 |
3 |
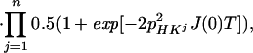 |
where the term pHKj2J(0) represents the relaxation of 15N due to DD interactions of 1HN (H) with remote protons Kj (see Appendix) and RsN is the 15N relaxation rate due to DD coupling with 1HN and to 15N chemical shift anisotropy CSA. Auto- and cross-relaxation terms due to 13Cα–15N and 13CO–15N DD coupling were neglected because they contribute <10% to the overall relaxation rate of 15N (see Appendix). The ST2-PT element (18) (Fig. 1, e-f) transfers the magnetization from 15N to 1HN (Ns+ → Hs−), resulting in the following coherence being acquired during t3:
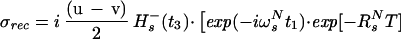 |
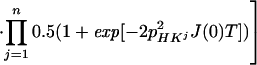 |
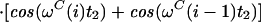 |
4 |
In Eq. 4, the transition Hs− is associated with the resonance frequency ωsH = ωH + πJHN, and the 1HN relaxation is given by Eq. 5 (10):
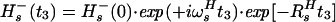 |
5 |
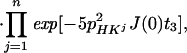 |
where RsH is the 1HN relaxation rate due to DD coupling with 15N and to 1HN CSA (10).
Figure 1.

Experimental scheme for the [15N, 1H]-TROSY-HNCA experiment. The radio-frequency pulses on 1H, 15N, 13Cα, 13CO, 2H, and 1Hα are applied at 4.7, 118, 56, 177, 3.6, and 4.7 ppm, respectively. Narrow and wide black bars indicate nonselective 90° and 180° pulses. Sine bell shapes on the lines marked 1H and 1Hα indicate selective 90° pulses. On the line marked 13CO, three selective 180° pulses are applied off-resonance with a duration of 120 μs and a Gaussian shape. The line marked PFG indicates the durations and amplitudes of pulsed magnetic field gradients applied along the z-axis: G1: 800 μs, 15 G/cm; G2: 800 μs, 9 G/cm; and G3: 800 μs, 22 G/cm. The delays are τ = 2.7 ms and T = 44 ms. The phase cycle is: φ1 = {y, −y, −x, x}; φ2 = {4x, 4(−x)}; φ3 = {−y}; φ4 = {−y}; φrec = {y, −y, −x, x, −y, y, x, −x}, with all other radio-frequency pulses applied with phase x. A phase-sensitive spectrum in the 15N(t1) dimension is obtained by recording a second FID for each t1 value, with φ1 = {y, −y, x, −x}, φ3 = {y} and φ4 = {y}, and data processing as described by Kay et al. (41). Quadrature detection in the 13Cα(t2) dimension is achieved by the States-TPPI method (42) applied to the phase φ2. The use of water flip-back pulses (43) at times a and e ensures that the water magnetization stays aligned along +z throughout both the ct period T and the data acquisition period t3. Residual transverse water magnetization is suppressed immediately before data acquisition (44). The scheme is used for two alternative experiments. For 2H-labeled proteins, 2H-decoupling during t2 is achieved with WALTZ-16 composite pulse decoupling (45) at a field strength of γB2 = 2.5 kHz. For measurements with protonated proteins, selective 1Hα-decoupling during the 13Cα(t2) evolution period is applied instead, using a DIPSI-2 decoupling scheme (46) with γB2 = 0.51 kHz. The two selective pulses on the water resonance before and after DIPSI-2 ensure the correct treatment of the water during the subsequent t1 and t2 evolution periods.
EXPERIMENTAL PROCEDURES
NMR spectra were recorded with two 23-kDa globular proteins, i.e., uniformly 2H/13C/15N-labeled gyrase-23B (95% H2O/5% D2O, pH 6.5 at 20°C) (22–24) and uniformly 13C/15N-labeled FimC (90% H2O/10% D2O, pH 5.0 at 20°C) (25). A Bruker DRX-750 spectrometer equipped with four radio-frequency channels was used. Data processing included zero-filling and sine bell filtering (26), using the program prosa (27), and the spectra were analyzed with xeasy (28). For gyrase-23B and FimC at 20°C, the isotropic rotational correlation time, τc, was estimated from the T1/T2 ratio of the backbone 15N nuclei (29) to be 15 ns.
RESULTS
The pulse sequence of Fig. 1 was applied with 2H/13C/15N-labeled gyrase-23B (22–24) and 13C/15N-labeled FimC (25). Fig. 2a shows [ω2(13C), ω3(1H)] strips from a three-dimensional (3D) [15N, 1H]-TROSY-HNCA spectrum of gyrase-23B, and Fig. 2b shows the corresponding strips from a conventional HNCA experiment (13) recorded with identical conditions. Using TROSY, all sequential 1HN-13Cα connectivities could be identified, as indicated by the broken lines (Fig. 2a), whereas with conventional HNCA no reliable sequential assignment was possible (Fig. 2b). A more quantitative assessment of the gain in signal-to-noise is afforded by the cross sections in Fig. 2a′ and b′. Data of this type were collected in Fig. 3 for the complete sequence of gyrase-23B. In [15N, 1H]-TROSY-HNCA (Fig. 3a), nearly all sequential cross peaks were present with sufficient intensity to allow sequential assignments, which was feasible only for a fraction of the sequence when conventional HNCA is used (Fig. 3b). TROSY also yielded greatly increased intensities of the intraresidual correlation peaks [ω1(15Ni)/ω2(13Ciα)/ω3(1HiN)] (Fig. 3a′ and b′). In line with theoretical considerations (10), the highest sensitivity gains were obtained for the immobilized core of the protein, with values of 2.9 for the α-helices and 3.4 for the β-sheets. The average sensitivity gain for sequential and intraresidual correlation peaks of the entire protein was 2.4-fold.
Figure 2.

Comparison of a [15N, 1H]-TROSY-HNCA recorded with the scheme of Fig. 1 and b conventional HNCA (13) using 1H DIPSI-2 decoupling with γB2 = 3.13 kHz during t1 and t2, and 15N WALTZ-16 decoupling with γB2 = 1.6 kHz during acquisition. Both experiments were recorded with a 1 mM solution of uniformly 2H/13C/15N-labeled gyrase-23B. 26(t1) × 32(t2) × 512(t3) complex points were accumulated, with t1max(15N) = 21.7, t2max(13Cα) = 6.4, and t3max(1H) = 48.7 ms. Fifty-six scans per increment were acquired, resulting in a total measuring time of 38 h per 3D spectrum. Corresponding [ω2(13C),ω3(1H)] strips from the two 3D experiments centered about the 1HN chemical shifts were taken at the 15N chemical shifts of residues 47–50. Because no decoupling during t1 and t3 is used in TROSY, the amide 1HN and 15N resonances in a are shifted in both dimensions by ≈45 Hz relative to the corresponding resonances in b. The sequence-specific assignments are indicated at the top by the one-letter amino acid symbol and the residue number in the amino acid sequence. In both spectra, dashed lines indicate sequential connectivities that could be reliably identified (see text). (a′ and b′) cross sections along the ω2(13C) dimension through the four [ω2(13C),ω3(1H)] strips at the ω3(1H) positions indicated at the bottom in a and b, respectively, where the complete chemical shift range acquired in the ω2(13C) dimension is plotted.
Figure 3.

Plots of the relative signal intensities, Irel, along the amino acid sequence of uniformly 2H/13C/15N-labeled gyrase-23B (1 mM protein concentration in 95% H2O/5% D2O at pH 6.5 and T = 20°C). (a) Sequential correlation peaks (ω2(13Ci−1α)/ω3(1HiN)) in [15N, 1H]-TROSY-HNCA. (b) Same as a measured with conventional HNCA (13). (a′) Intraresidual correlation peaks (ω2(13Ciα)/ω3(1HiN)) in [15N, 1H]-TROSY-HNCA. (b′) Same as a′ measured with conventional HNCA. Both spectra were recorded with the same experimental conditions and processed identically, as described in the text. The α-helical and β-sheet regions in the x-ray structure of gyrase-23B (22) are identified in b′ by open and filled bars, respectively.
Measurements corresponding to those in Figs. 2 and 3 also were performed with uniformly 13C/15N-labeled FimC in order to evaluate the performance of [15N, 1H]-TROSY-HNCA with protonated proteins. The experimental scheme of Fig. 1 was used with 1Hα decoupling. The average sensitivity enhancement for the entire protein was 1.5-fold, with a sensitivity gain of 1.7 for the regular secondary structures, which in FimC consist exclusively of β-sheets.
The pulse scheme of the [15N, 1H]-TROSY-HNCO experiment (Fig. 4a) was applied to 2H/13C/15N-labeled gyrase-23B with identical conditions to those described for the HNCA experiment in Fig. 2. As an illustration of the results obtained, Fig. 4b and c compares corresponding cross-sections from [15N, 1H]-TROSY-HNCO and conventional HNCO (30). From a corresponding data set to Fig. 3, the average gain in sensitivity for the entire protein was found to be 2.4-fold, with values of 2.5 and 2.9 for the α-helices and the β-sheets, respectively.
Figure 4.

(a) Experimental scheme for the [15N, 1H]-TROSY-HNCO experiment. All experimental parameters and the phase cycle are the same as in the [15N, 1H]-TROSY-HNCA scheme of Fig. 1. (b and c) Show, respectively, cross sections along the ω3(1H) dimension through four peaks of the 3D [15N, 1H]-TROSY-HNCO spectrum and the conventional 3D HNCO spectrum (30) of uniformly 2H/13C/15N-labeled gyrase-23B (1 mM protein concentration in 95% H2O/5% D2O at pH 6.5 and T = 20°C). 26(t1) × 30(t2) × 512(t3) complex points were accumulated, with t1max (15N) = 10.8, t2max(13CO) = 12.0, and t3max(1H) = 48.7 ms. Eight scans per increment were acquired, resulting in a total measuring time of 7 h per 3D spectrum. At the top of each panel, the sequence-specific assignment is indicated by the one-letter amino acid symbol and the residue number in the amino acid sequence, and the ω1(15N) and ω2(13CO) chemical shifts are indicated in parentheses.
DISCUSSION
The HNCA and HNCO measurements (Figs. 2–4) showed that significant improvement of triple resonance experiments can be achieved by suppression of transverse 15N and 1HN relaxation with TROSY. In this section, the origins of the enhanced sensitivity of [15N, 1H]-TROSY-HNCA are further analyzed.
In Eq. 5, the 1HN relaxation can be represented by a single exponential, with the rate constant
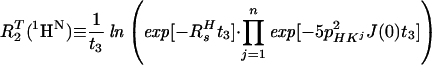 |
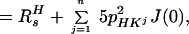 |
6 |
where R2T(1HN) denotes the transverse 1HN relaxation rate in [15N, 1H]-TROSY-HNCA. To a good approximation for the molecular size range of interest, the 15N relaxation in [15N, 1H]-TROSY-HNCA (Eq. 3) may similarly be represented by a single exponential decay, with the rate constant R2T(15N):
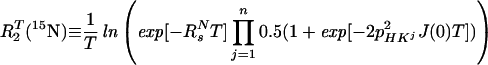 |
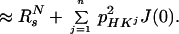 |
7 |
The approximation of Eq. 7 assumes that pHKj2J(0)T ≪ 1, which is satisfied for commonly used ct periods, T, over the τc range 1–80 ns. For the conventional HNCA experiment, corresponding transverse relaxation rates, R2C(15N) and R2C(1HN), were evaluated as the average of the relaxation rates in the individual components of the 15N and 1HN doublets, respectively. Using Eqs. 6 and 7 for a 23-kDa protein with τc = 15 ns at 750 MHz, and the corresponding formalism for conventional HNCA, one predicts for the protonated protein that TROSY yields 2.9-fold and 1.5-fold reductions of the 15N and 1HN relaxation rates, respectively, when compared to conventional HNCA (Table 1). For conventional HNCA, one expects further that deuteration reduces the 1HN relaxation rates 2.5-fold and 1.6-fold for β-sheets and α-helices, respectively, and that deuteration yields only a small reduction, by less than a factor 1.3, of the 15N relaxation rate. For [15N, 1H]-TROSY-HNCA, deuteration has approximately the same absolute effects on R2T(15N) and R2T(1HN), but because of the greatly reduced RsN and RsH rates the relative improvement is larger, i.e., up to 6.5 for 1HN and up to 2.9 for 15N (Table 1).
Table 1.
Transverse 1HN and 15N relaxation rates predicted for a 23-kDa protein at 750 MHz in [15N, 1H]-TROSY-HNCA and in conventional HNCA*
| TROSY-HNCA
|
HNCA
|
Gain†
|
|||
|---|---|---|---|---|---|
| R2T(15N)[s−1] | R2T(1HN) [s−1] | R2C(15N) [s−1] | R2C(1HN) [s−1] | AT/AC | |
| Isolated 15N–1HN group | 3.0 | 3.2 | 20.9 | 20.3 | 8.0 |
| β-sheet 13C/15N-labeled‡ | 10.6 | 41.1 | 28.5 | 58.2 | 1.8 |
| α-helix 13C/15N-labeled§ | 8.7 | 31.5 | 26.6 | 48.6 | 2.0 |
| β-sheet 2H/13C/15N-labeled¶ | 3.7 | 6.3 | 21.6 | 23.5 | 4.7 |
| α-helix 2H/13C/15N-labeled‖ | 5.0 | 13.2 | 22.9 | 30.3 | 2.9 |
15N and 1HN relaxation rates were calculated using Eqs. 6 and 7, respectively, as described in the text. The values listed for conventional HNCA are the average relaxation rates of both components of the 15N and the 1HN doublets, given by RsN and RrN, and by RsH and RrH, respectively. The following parameters were used: rHN = 1.04 Å (39), ΔσN = 155 ppm, ΔσH = 15 ppm, ΘN = 15°, ΘH = 10° (40), and τc = 15 ns.
AT and AC are the relative signal intensities obtained with [15N, 1HN]-TROSY-HNCA and conventional HNCA. The ratio AT/AC was calculated with Eq. 8, using T = 52 ms.
Remote protons considered are 1HN (i − 1), 1HN (i + 1), 1HN (j), 1Hα (i), 1Hα (i − 1), 1Hα (j), 1Hβ (i), and 1Hβ (i − 1) at distances of 4.3, 4.3, 3.3, 2.8, 2.2, 3.2, 2.5, and 3.2 Å, respectively, which are typical for an antiparallel β-sheet [i is the observed residue, (i − 1) and (i + 1) the sequential neighbors, and j indicates a long-range contact across the β-sheet (2)].
Remote protons are 1HN (i − 1), 1HN (i + 1), 1HN (i − 2), 1HN (i + 2), 1Hα (i), 1Hα(i − 1), 1Hα(i − 2), 1Hα(i − 3), 1Hα(i − 4), and 1Hβ(i) at distances of 2.8, 2.8, 4.2, 4.2, 2.6, 3.5, 4.4, 3.4, 4.2, and 2.5 Å, respectively, which are typical for an α-helix (2).
Remote protons are 1HN (i − 1), 1HN (i + 1), and 1HN (j) at 4.3, 4.3, and 3.3 Å (2).
Remote protons are 1HN (i − 1), 1HN (i + 1), 1HN (i − 2), and 1HN (i + 2) at 2.8, 2.8, 4.2, and 4.2 Å (2).
We calculated a theoretical sensitivity gain for [15N, 1H]-TROSY-HNCA relative to conventional HNCA by using Eq. 8,
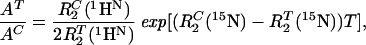 |
8 |
where AT and AC are the peak amplitudes in corresponding [15N, 1H]-TROSY-HNCA and conventional HNCA experiments, respectively. The amplitudes are linearly proportional to the 1HN relaxation rates, whereas the dependence on the 15N transverse relaxation rates is exponential because of the ct evolution in the 15N dimension (15). For a 23-kDa protein, Eq. 8 predicts an 8-fold sensitivity gain for the hypothetical isolated 15N–1HN moiety. When allowing for DD coupling with remote protons, the gain for β-sheets and α-helices amounts to 4.7 and 2.9 in deuterated proteins and to 1.8 and 2.0 in protonated proteins, respectively. Sensitivity gains measured for the 2H/13C/15N-labeled gyrase-23B were 3.4 for β-sheets and 2.9 for α-helices, and for 13C/15N-labeled FimC, a gain of 1.7 was measured for the β-sheet regions. These experimental data are in good agreement with the theoretical predictions (Table 1).
The sensitivity gain (Eq. 8) depends on the molecular size. Fig. 5 shows the relative peak amplitudes calculated for [15N, 1H]-TROSY-HNCA (bold lines) and for conventional HNCA (thin lines) as a function of the rotational correlation time, using the parameters listed in Table 1. The thick and thin solid lines indicate the peak intensities for 15N–1HN moieties located in β-sheet regions of a 2H/13C/15N-labeled protein. The dashed lines show the corresponding intensities expected for a 13C/15N-labeled protein. The rapid decrease of the peak amplitude with increasing molecular size limits the application of the conventional HNCA experiment to much smaller proteins than [15N, 1H]-TROSY-HNCA (Fig. 5). Actually, for deuterated proteins similar peak amplitudes are expected for τc = 15 ns with HNCA and τc = 50 ns with [15N, 1H]-TROSY-HNCA. For τc values above 15 ns, [15N, 1H]-TROSY-HNCA with protonated proteins is predicted to yield similar sensitivity as conventional HNCA with deuterated proteins (see Fig. 5).
Figure 5.

Relative signal amplitudes, A, calculated during the ct 15N evolution and 1H acquisition periods for [15N, 1H]-TROSY-HNCA (thick lines) and conventional HNCA (thin lines) at a magnetic field strength of 750 MHz for variable isotropic rotational correlation times, τc, in the range from 5 to 80 ns, which corresponds to protein sizes in H2O solution at 20°C from ≈7 kDa at τc = 5 ns to ≈260 kDa at τc = 80 ns. The solid and dashed lines represent the signal amplitudes, A, for a 15N–1HN moiety in a β-sheet of a deuterated and a protonated 13C/15N-labeled protein, respectively (see Table 1 for the parameters used). (Inset) An expanded plot for the range of A from 0 to 1. The dotted vertical line indicates the τc value of 15 ns estimated for the 23-kDa proteins gyrase-23B and FimC.
Similar results to those obtained here with [15N, 1H]-TROSY-HNCA are predicted for the use of [15N, 1H]-TROSY with other triple-resonance experiments that use coherence pathways with ct 15N evolution periods. For HNCO this is born out by the experiment in Fig. 4, and similar results have been obtained for HN(CO)CA (15), HNCACB (31, 32), and HN(CO)CACB (7,33) (M.S., G.W., K.P., H.S., and K.W., unpublished results). Overall, the present investigation predicts that TROSY-type triple-resonance experiments (Figs. 1 and 4) will be applicable for the assignment of significantly larger proteins than the corresponding conventional triple-resonance experiments. The predictions of Fig. 5 may serve as a platform for estimating the feasibility of resonance assignments and the instrument time needed for particular proteins in the size range 10–250 kDa. Thereby, one has to take into account that these curves can only provide a general guideline, since the CSA parameters may be somewhat variable in the individual amino acid residues (34–38).
ABBREVIATIONS
- ct
constant-time
- DD
dipole–dipole
- CSA
chemical shift anisotropy
- HNCA
amide proton-to-nitrogen-to-α carbon correlation
- HNCO
amide proton-to-nitrogen-to-carbonyl carbon correlation
- ST2-PT
single transition-to-single transition polarization transfer
- TROSY
transverse relaxation-optimized spectroscopy
- 3D
three-dimensional
Appendix
The transverse 15N relaxation rate is calculated for a four-spin system H-N-C-K representing the backbone atoms 1HN, 15N, and 13Cα and a remote proton K, with scalar couplings 1JHN, 1JNC, and JKN. 13CO, which is not considered (see below), would be treated in the same way as 13Cα. The following single spin transition basis operators (47) were selected to evaluate the transverse relaxation of the 15N single-quantum coherences that evolve in [15N, 1H]-TROSY-HNCA:
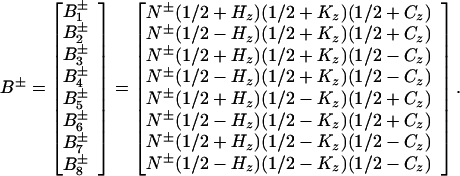 |
A1 |
In this basis, the Liouville matrix in the rotating frame has the diagonal form given by Eq. A2:
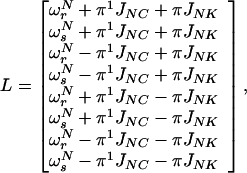 |
A2 |
with ωrN = ΩN − Ω0 + π1JHN and ωsN = ΩN − Ω0 − π1JHN, where Ω0 is the reference frequency and ΩN the Larmor frequency of N. In calculating the relaxation matrix, Γ, the DD interactions H-N, N-C, C-K, and H-K, the CSA of H and N, and all cross-correlation terms between these relaxation interactions are taken into account. Not included are the very small N-C DD interactions (48). In the slow-tumbling approximation, only terms in J(0) need to be retained:
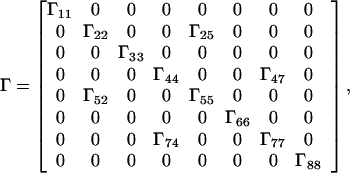 |
A3 |
where the Γii represent the relaxation rates of individual transitions of the N multiplet. Γ25, Γ52, Γ47, and Γ74 represent the cross-relaxation between different nondegenerate transitions of the N multiplet with resonance frequencies separated by π1JHN. The individual relevant matrix elements are:
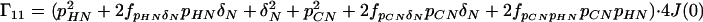 |
A4 |
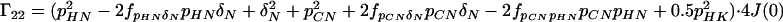 |
A5 |
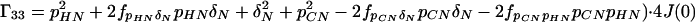 |
A6 |
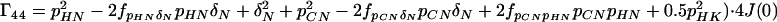 |
A7 |
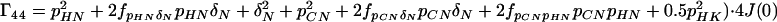 |
A8 |
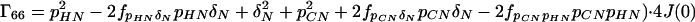 |
A9 |
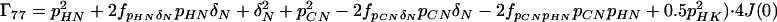 |
A10 |
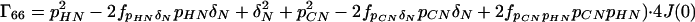 |
A11 |
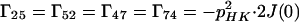 |
A12 |
fki = 0.5(3cos2Θkl − 1), pij = (2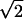 rij3)−1ћγiγj and δN = (3
rij3)−1ћγiγj and δN = (3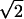 )−1 γNB0ΔσN, where ћ is the Planck constant divided by 2π, Θkl the angle between the unique tensor axes of the interactions k and l, γi the gyromagnetic ratio of spin i, rij the distance between the spins i and j, B0 the polarizing magnetic field, and ΔσN the difference between the axial and perpendicular principal components of the 15N chemical shift tensor, which is assumed to be axially symmetric. Numerical calculations using Eqs. A4–A12 showed that the terms (pCN2 ± 2fpCNδNpCNδN ± 2fpCNpHNpCNpHN)⋅4J(0) contribute <10% to the overall relaxation of 15N and can be neglected. This applies for both 13Cα and 13CO. The Eqs. A2 and A3 show that there is no interference between the group of basis operators B1±, B3±, B6±, and B8± and the other four basis operators. Thus, for these operators, the time evolution can be calculated individually, resulting in single-exponential relaxation determined exclusively by interactions within the H-N spin subsystem. Because of the pairwise linkage of B2± with B5±, and of B4± with B7±, the relaxation of the corresponding transitions is in general biexponential. However, since Γij ≪ π1JHN (i ≠ j) the off-diagonal elements can be neglected, so that a single-exponential decay is obtained for all basis operators:
)−1 γNB0ΔσN, where ћ is the Planck constant divided by 2π, Θkl the angle between the unique tensor axes of the interactions k and l, γi the gyromagnetic ratio of spin i, rij the distance between the spins i and j, B0 the polarizing magnetic field, and ΔσN the difference between the axial and perpendicular principal components of the 15N chemical shift tensor, which is assumed to be axially symmetric. Numerical calculations using Eqs. A4–A12 showed that the terms (pCN2 ± 2fpCNδNpCNδN ± 2fpCNpHNpCNpHN)⋅4J(0) contribute <10% to the overall relaxation of 15N and can be neglected. This applies for both 13Cα and 13CO. The Eqs. A2 and A3 show that there is no interference between the group of basis operators B1±, B3±, B6±, and B8± and the other four basis operators. Thus, for these operators, the time evolution can be calculated individually, resulting in single-exponential relaxation determined exclusively by interactions within the H-N spin subsystem. Because of the pairwise linkage of B2± with B5±, and of B4± with B7±, the relaxation of the corresponding transitions is in general biexponential. However, since Γij ≪ π1JHN (i ≠ j) the off-diagonal elements can be neglected, so that a single-exponential decay is obtained for all basis operators:
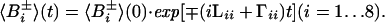 |
A13 |
The sum of the operators B2±, B4±, B6±, and B8± represents the magnetization of the TROSY 15N multiplet component, so that the transverse 15N magnetization at time t can be described by:
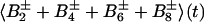 |
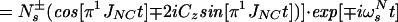 |
A14 |
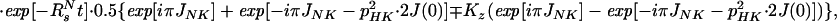 |
where Ns± = N± (1/2 − Hz) and RsN = (pHN2 − 2fpHNδNpHNδN + δN2)⋅4J(0). Eq. A14 can be further simplified by assuming that JNK = 0. Furthermore, since N± Kz does not result in an observable signal, the evolution of the 15N magnetization can be described by A15:
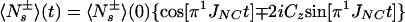 |
A15 |
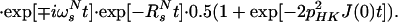 |
This treatment can be expanded to n mutually noninteracting remote protons, K1, K2, …, Kn, resulting in Eq. A16:
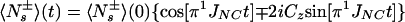 |
A16 |
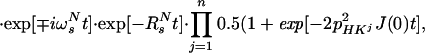 |
Eq. A16 was used for the derivation of Eq. 3 in the main text.
References
- 1.Wüthrich K, Wider G, Wagner G, Braun W. J Mol Biol. 1982;155:311–319. doi: 10.1016/0022-2836(82)90007-9. [DOI] [PubMed] [Google Scholar]
- 2.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. [Google Scholar]
- 3.Montelione G T, Wagner G. J Am Chem Soc. 1989;111:5474–5475. [Google Scholar]
- 4.Ikura M, Kay L E, Bax A. Biochemistry. 1990;29:4659–4667. doi: 10.1021/bi00471a022. [DOI] [PubMed] [Google Scholar]
- 5.Bax A, Grzesiek S. Acc Chem Res. 1993;26:131–138. [Google Scholar]
- 6.Edison A S, Abildgaard F, Westler W M, Mooberry E S, Markley J. Methods Enzymol. 1994;239:3–79. doi: 10.1016/s0076-6879(94)39003-7. [DOI] [PubMed] [Google Scholar]
- 7.Yamazaki T, Lee W, Arrowsmith C H, Muhandiram D R, Kay L E. J Am Chem Soc. 1994;116:11655–11666. [Google Scholar]
- 8.LeMaster D M. Prog Nucl Magn Reson Spectrosc. 1994;26:371–419. [Google Scholar]
- 9.Kay L E, Gardner K H. Curr Opin Struct Biol. 1997;7:722–731. doi: 10.1016/s0959-440x(97)80084-x. [DOI] [PubMed] [Google Scholar]
- 10.Pervushin K, Riek R, Wider G, Wüthrich K. Proc Natl Acad Sci USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kay L E, Ikura M, Tschudin R, Bax A. J Magn Reson. 1990;89:496–514. doi: 10.1016/j.jmr.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 12.Grzesiek S, Bax A. J Magn Reson. 1992;96:432–440. [Google Scholar]
- 13.Yamazaki T, Lee W, Revington M, Mattiello D L, Dahlquist F W, Arrowsmith C H, Kay L E. J Am Chem Soc. 1994;116:6464–6465. [Google Scholar]
- 14.Vuister G W, Bax A. J Magn Reson. 1992;98:428–435. [Google Scholar]
- 15.Wider G. Prog Nucl Magn Reson Spectrosc. 1998;32:193–275. [Google Scholar]
- 16.Sørensen O W, Eich G W, Levitt M H, Bodenhausen G, Ernst R R. Prog Nucl Magn Reson Spectrosc. 1983;16:163–192. [Google Scholar]
- 17.Ernst R R, Bodenhausen G, Wokaun A. The Principles of Nuclear Mangetic Resonance in One and Two Dimensions. Oxford: Clarendon; 1987. pp. 38–40. [Google Scholar]
- 18.Pervushin K, Wider G, Wüthrich K. J Biomol Nucl Magn Reson. 1998;12:345–348. doi: 10.1023/A:1008268930690. [DOI] [PubMed] [Google Scholar]
- 19.Pervushin K, Riek R, Wider G, Wüthrich K. J Am Chem Soc. 1998;120:6394–6400. [Google Scholar]
- 20.Farrar T C, Stringfellow T C. In: Encyclopedia of NMR. Grant D M, Harris R K, editors. Vol. 6. New York: Wiley; 1996. pp. 4101–4107. [Google Scholar]
- 21.Szyperski T, Braun D, Fernandez C, Bartels C, Wüthrich K. J Magn Reson B. 1995;108:197–203. [Google Scholar]
- 22.Lewis R J, Singh O M, Smith C V, Skarzynski T, Maxwell A, Wonacott A J, Wigley D B. EMBO J. 1996;15:1412–1420. [PMC free article] [PubMed] [Google Scholar]
- 23.Wigley D B. Annu Rev Biophys Biomol Struct. 1995;24:185–208. doi: 10.1146/annurev.bb.24.060195.001153. [DOI] [PubMed] [Google Scholar]
- 24.Maxwell A. Trends Microbiol. 1997;5:102–109. doi: 10.1016/S0966-842X(96)10085-8. [DOI] [PubMed] [Google Scholar]
- 25.Pellecchia M, Güntert P, Glockshuber R, Wüthrich K. J Biomol Nucl Magn Reson. 1998;11:229–230. doi: 10.1023/a:1008207111214. [DOI] [PubMed] [Google Scholar]
- 26.DeMarco A, Wüthrich K. J Magn Reson. 1976;24:201–204. [Google Scholar]
- 27.Güntert P, Dötsch V, Wider G, Wüthrich K. J Biomol Nucl Magn Reson. 1992;2:619–629. [Google Scholar]
- 28.Bartels C, Xia T, Billeter M, Güntert P, Wüthrich K. J Biomol Nucl Magn Reson. 1995;5:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 29.Kay L E, Torchia D A, Bax A. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 30.Muhandiram D R, Kay L E. J Magn Reson B. 1994;103:203–216. [Google Scholar]
- 31.Grzesiek S, Bax A. J Magn Reson. 1992;99:201–207. [Google Scholar]
- 32.Wittekind M, Mueller L. J Magn Reson B. 1993;101:201–205. [Google Scholar]
- 33.Grzesiek S, Bax A. J Am Chem Soc. 1992;114:6291–6292. [Google Scholar]
- 34.Tjandra N, Bax A. J Am Chem Soc. 1997;119:8076–8082. [Google Scholar]
- 35.Tessari M, Mulder F A A, Boelens R, Vuister G W. J Magn Reson. 1997;127:128–133. [Google Scholar]
- 36.Tessari M, Vis H, Boelens R, Kaptein R, Vuister G W. J Am Chem Soc. 1997;119:8985–8990. [Google Scholar]
- 37.Fushman D, Cowburn D. J Am Chem Soc. 1998;120:7109–7110. [Google Scholar]
- 38.Fushman, D., Tjandra, N. & Cowburn, D. (1998) J. Am. Chem. Soc., in press.
- 39.Roberts J E, Harbison G S, Munowitz M G, Herzfeld J, Griffin R G. J Am Chem Soc. 1987;109:4163–4169. [Google Scholar]
- 40.Wu C H, Ramamoorthy A, Gierasch L M, Opella S J. J Am Chem Soc. 1995;117:6148–6149. [Google Scholar]
- 41.Kay L E, Keifer P, Saarinen T. J Am Chem Soc. 1992;114:10663–10664. [Google Scholar]
- 42.Marion D, Ikura M, Tschudin R, Bax A. J Magn Reson. 1989;85:393–399. [Google Scholar]
- 43.Grzesiek S, Bax A. J Am Chem Soc. 1993;115:12593–12594. [Google Scholar]
- 44.Piotto M, Saudek V, Sklenar V J. J Biomol Nucl Magn Reson. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 45.Shaka A J, Keeler J, Frenkiel T, Freeman R. J Magn Reson. 1983;52:335–338. [Google Scholar]
- 46.Cavanagh J, Rance M. J Magn Reson. 1992;96:670–678. [Google Scholar]
- 47.Vold R R, Vold R R. J Chem Phys. 1976;64:320–324. [Google Scholar]
- 48.Peng J W, Wagner G. J Magn Reson. 1992;98:308–332. [Google Scholar]