

Abstract
Cardiac fibrillation is one of the most important causes of morbidity and mortality in the developed world, but its mechanisms are still a matter of debate. Not only is the role of certain ion channels currently being unraveled, but also investigators are starting to gather evidence for the dynamics and molecular mechanisms of both atrial (AF) and ventricular (VF) fibrillation in humans. In this brief review, we evaluate the available evidence for the separate roles played by individual sarcolemmal ion channels in AF and VF, assessing the clinical relevance of such findings. Importantly, while human data support the idea that rotors are a crucial mechanism for fibrillation maintenance in both the atria and the ventricles, there are clear inherent differences between the two chamber types, particularly in regards to the role of specific ion channels in fibrillation. But there are also similarities. This knowledge, together with new information on the changes that take place during disease evolution and between structurally normal and diseased hearts may enhance our understanding of fibrillatory processes pointing to new pharmacological or interventionist approaches to improve disease outcomes.
Keywords: atrial fibrillation, ventricular fibrillation, reentry, ionic mechanisms
Introduction
Fibrillation is defined as turbulent cardiac electrical activity whereby propagation of electrical waves through the heart is disrupted, with consequent production of wavelets, high frequency rotating waves, and inability of the myocardium to contract. Atrial fibrillation (AF) is the most common sustained arrhythmia,1 whereas ventricular fibrillation (VF) and reentrant tachycardia (VT) are the leading immediate causes of sudden cardiac death.2 Until recently, both AF and VF were assumed to result from a completely unpredictable process with random propagation of multiple wavelets.3 However, lately, the classic Lewis idea that fibrillation results from the activity of a small number of rapidly firing reentrant circuits (rotors; reentrant driving sources; see Table 1 for Glossary) that give rise to wavefront fractionation (fibrillatory conduction) has begun to regain momentum.3
Table 1.
Glossary
| Core: Center of spiral wave rotation whose diameter is established by the trajectory of the pivoting phase singularity. |
| Driver: the high frequency source that maintains fibrillatory activity. The driver may be reentrant (i.e., a rotor) or may be a focus of automatic pacemaker or triggered (early afterdepolarizations, EADs; delayed afterdepolarizations) activity. |
| Dominant frequency the average number of local activations per second as measured by the highest peak in the power spectrum, obtained by the fast Fourier transform within a limited frequency range. |
| Fibrillatory conduction: form of propagation of the rapidly successive wave fronts that emanate from a high frequency reentrant sources as they encounter and interact with anatomical and/or functional obstacles in their path, leading to fragmentation and wavelet formation. |
| Phase singularity: also known as a singularity point. It is a point in two- or three-dimensional cardiac muscle where al phases of the excitation-recovery cycle crowd together. The phase singularity is the pivot point of functional reentrant activity |
| Rotor: The organizing source (driver) of functional reentrant activity. It is the structure immediately surrounding the pivot of a rotating wave in two or three dimensions. Here rotor and reentry are used interchangeably. It may be argued that functional reentry (i.e., a rotor) and anatomical reentry (i.e., reentry around an anatomical obstacle) may have the same underlying mechanism. In fact, when a rotor that drifts across excitable cardiac tissue encounters and interacts with an unexcitable obstacle (e.g., a scar or a natural arterial orifice) it anchors to it and begins to rotate around it. Thus, functional reentry may turn into anatomical reentry. |
| Spiral wave: The wave of excitation emitted by a rotor and whose front is an involute spiral with increasing convex curvature toward the rotation center. |
| Turbulence: Disrupted propagation of electrical waves that produces wavebreaks, wavelets and rotors. |
| Wavebreak: When an electrical wave front of excitation hits a functional or anatomical obstacle it can break, leading to the formation of “phase singularities” at the broken endpoints of the daughter wavelets (see Figure 1). The resulting dynamics depend on the excitability of the medium. When the excitability is low, the wave would shrink, resulting in two-dimensional decremental conduction. At an appropriate level of higher excitability, the broken ends curve and the wavelets begin to rotate. The phenomenon is similar to eddy formation in water. |
Experimentally, optical mapping studies in atria and/or ventricles have shown a high degree of spatiotemporal organization during fibrillation.3,4 In both atria and ventricles, rotors seem to be the drivers that maintain fibrillation, at least in some cases. Rotors may form when a wave breaks upon interacting with structural and/or electrophysiological heterogeneities in its path (Figure 1).3,4 In fact, it is now certain that wavebreak, leading to wavelet formation and rotor initiation (see Table 1), is the hallmark of any proposed explanation for AF or VF maintenance.5 As illustrated in Figure 2, when a rotor is formed, the rapidly succeeding wavefronts emanating from it propagate throughout the cardiac muscle and interact with anatomical and/or functional obstacles, causing fragmentation and new wavelet formation; i.e., fibrillatory conduction.3,4 Wavelets may undergo decremental conduction or may be annihilated by collision with another wavelet or a boundary, or may form new sustained rotors, resulting in a never-ending frequency-dependent fragmentation of wave fronts into what appears to be multiple independent wavelets.5,6 Accordingly, fibrillation is not totally unpredictable, but may be considered to have both deterministic and stochastic components; i.e., highly organized and periodic rotors as drivers; and randomly distributed fragmentation of the resulting wavefronts (fibrillatory conduction) leading to the formation of multiple wavelets. Therefore, combining the rotor hypothesis with the multiple wavelet hypothesis offers a step closer to the understanding of the fibrillatory process. In other words, for multiple, randomly propagating wavelets to exist, there must be a source (e.g., a rotor) generating wavefronts at an exceedingly rapid rate. The interaction of those wavefronts with obstacles in their path results in the shedding of the wavelets. Therefore, irrespective of the underlying source, wavebreak and reentry account for the complex patterns of propagation, wavelet formation, and apparent aperiodic activity that characterize cardiac fibrillation. The manner in which different membrane ionic currents contribute to wavebreak, rotor stabilization and wave fragmentation is now being successfully investigated at various levels of integration, from the molecule to the human patient.
Figure 1.
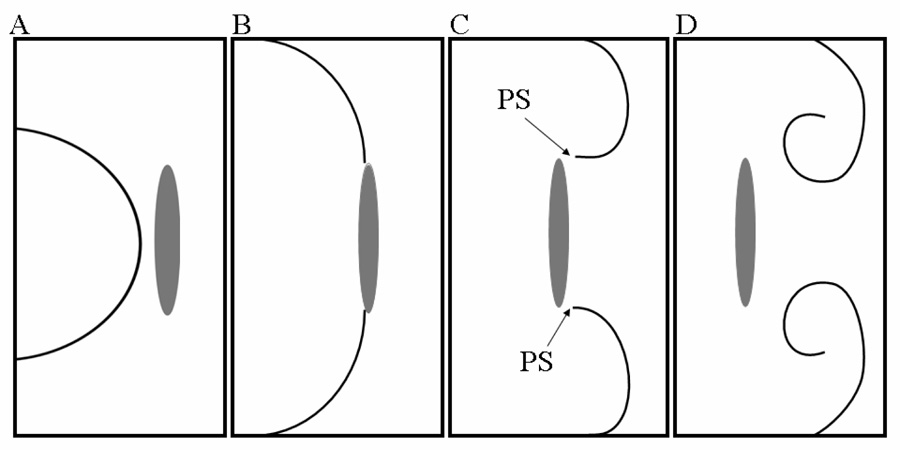
Wavebreak formation. A. An electrical wavefront moves toward an anatomical obstacle. B. The wavefront attaches to the obstacle and begins to circumnavigate it. C. Under appropriate conditions of excitability, the wavefront breaks into two daughter wavelets that detach from the obstacle, with the consequent formation of a phase singularity (PS) at each broken end. D. The two wavelets curl around their respective PSs and begin to rotate inscribing a figure-8 pattern.
Figure 2.
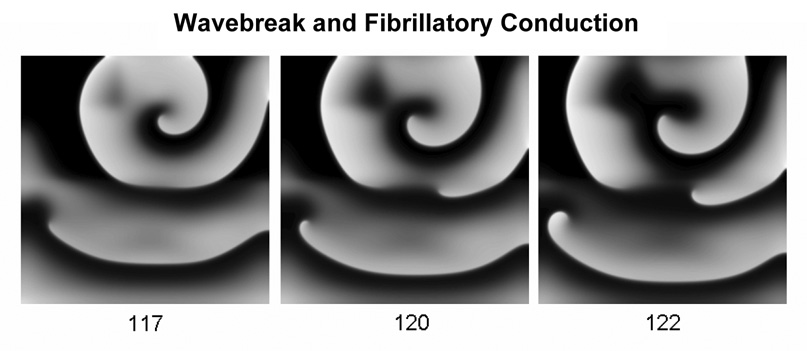
Rotors and fibrillatory conduction. Waves emanating from a high-frequency rotor undergo fibrillatory conduction in a computer model of cardiac ventricular myocytes. Numbers under each frame are milliseconds.
Here we first provide an update on the underlying ionic mechanisms for reentry and fibrillatory conduction. We also briefly review the known roles of individual ionic currents in AF and VF. Then, we discuss differences and similarities between fibrillation in atria and ventricles, and assess the clinical evidence supporting the existence of rotors in patients. Finally, we consider the changes in fibrillatory dynamics induced by myocardial remodeling secondary to chronic fibrillation, ischemia or cardiac diseases.
Ionic Bases of Functional Reentry in Normal Myocardium
The role played by individual ionic currents in rotor dynamics in the structural normal myocardium has been reviewed elsewhere.7 Below we discuss the most salient features of the manner in which each current affects wave propagation dynamics during fibrillation and the consequences of blocking that current in relation to the final AF/VF outcome. A summary of the importance of each current in rotor dynamics can be found in Table 2.
Table 2.
Effects of each ionic current in rotor dynamics
| Current(s) | Action | Frequency | CV | APD | WL | Core Size | Stability |
|---|---|---|---|---|---|---|---|
| INa | Inhibition | ↓ | ↓ | ↓ | ↓ | ↑ | ↑ |
| ( IK1; IK,Ach) | Enhancement | ↑ | ↓ (near the core) | ↓ | ↓ | ↑ | ↑ |
| ICa,L | Inhibition | ↓↑ | ? | ↓ | ↓ | ↓↑ | ↑ |
| Ito, IKur | Inhibition | ? | ? | ↑(plateau) | ↑ | ? | ↓? |
| IKs, | Inhibition | ↓= | ? | ↑ | ↑ | ? | ↑ |
| IKr | Inhibition | ? | ? | ↑ | ↑ | ? | ? |
Importance of the INa/IK1 Balance in Controlling Rotor Frequency and Stability
Biological and numerical experiments conducted over the last several years have provided strong support for the idea that the interplay between the voltage-gated tetrodotoxin (TTX)-sensitive sodium current (INa) and the inward rectifier current (IK1), which is crucial for the control of normal cardiac excitability, also controls the stability and frequency of reentry7–9 in structurally normal atrial and ventricular tissues. As shown by the cartoon in panel A of Figure 3, during reentry, the wavefront adopts a spiral shape with increasing curvature toward the center of rotation (core); its conduction velocity (CV) slows gradually toward the center, reaching a critical value at the immediate perimeter of the core. As a result, a mismatch is established between the depolarizing current (mainly INa) supplied by the wavefront and the electrotonic current that is controlled mainly by IK1 and required to depolarize resting cells inside the core, which thus remains unexcited.9,10 Consequently, a voltage gradient develops between the unexcited core and the neighboring excited cells, which abbreviates action potential duration (APD; panel B). Hence, the wavelength (WL) near the core is much shorter than that far away from the core. This was clearly demonstrated recently in a transgenic mouse heart in which upregulation of IK1 accelerated the final phase of action potential (AP) repolarization.9
Figure 3.
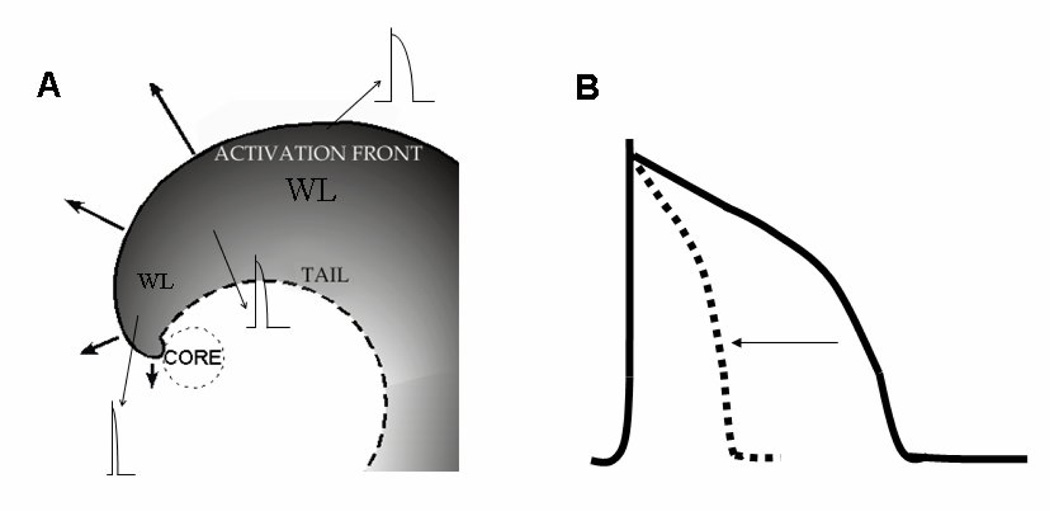
Electrotonic effects of the center of rotation (core) on conduction velocity (CV) action potential duration (APD) and wavelength (WL). During reentry the wavefront adopts a spiral shape with increasing curvature toward the core. A. During sustained reentry, the CV of the wave front slows gradually toward the center (solid arrows), reaching a critical value at the immediate perimeter of the core. As a result, the core remains unexcited. Consequently, a voltage gradient develops between the unexcited core and the neighboring active cells. B. Electrotonic currents between the core and its immediate neighbors shorten APD near the core (broken trace and horizontal arrow). Hence, wavelength (WL= CV × APD) near the core is much shorter than WL far away from the core.
As illustrated by the top panels in Figure 4, in addition to shortening APD, IK1 overexpression induced a relative hyperpolarization (top panels). During sustained reentry this effect augmented the voltage gradient established between resting cells in the core and the active cells in its immediate surroundings. Consequently, there was an increase in the electrotonic currents that flowed continuously between resting and active cells, which further contributed to hasten the repolarization of the active cells and also to reduce CV very near the core. As demonstrated by Noujaim et al,9 the increased gradient and electrotonic currents, together with a relative hyperpolarization during the excitable gap induced by the IK1 overexpression contributed to a steeper rise in the local CV as a function of the distance from the core. As a consequence, a faster, more stable rotor with a shorter WL was established in transgenic, compared to wildtype hearts (Figure 4 bottom panels). The relevance of such mouse heart studies to human VF has been highlighted by the recent demonstration that the turbulence that arises during fibrillatory activity is organized into spiral vortices, no matter what species of mammal is experiencing the VF.11 Moreover across animal species – from mice and guinea pigs to sheep, humans and horses – the frequency of VF activity can be scaled using a universal formula that relates frequency to the fourth power of body mass. So too can the size of the core of the electrical rotors.11
Figure 4.
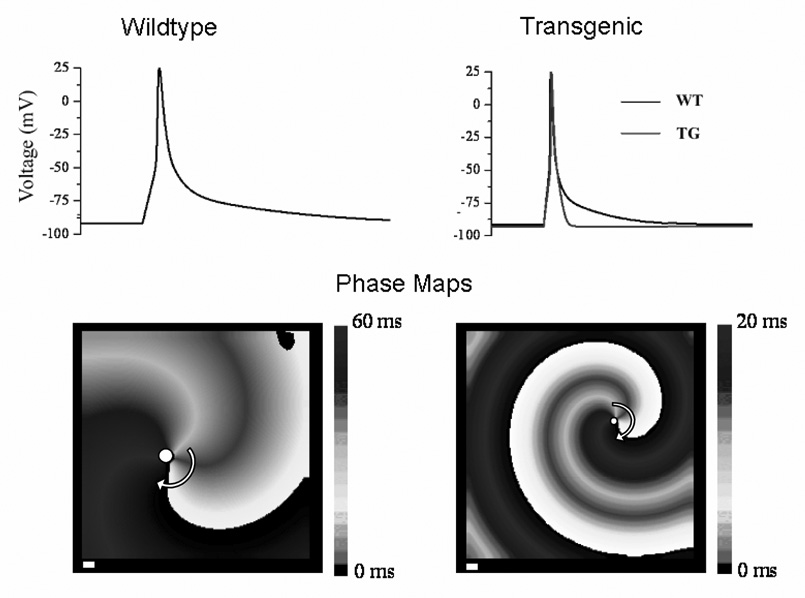
Consequences of transgenic overexpression of IK1 on action potential characteristics and reentry wavelength and frequency. Top; Left, simulated action potential of wildtype (WT) mouse ventricular myocyte; right, simulated action potential of transgenic (TG) myocyte (grey) superimposed on WT (black). Note significant acceleration of phase-3 repolarization in TG with respect to WT. Bottom, simulation of reentry in WT and TG sheets of ventricular myocytes. Overexpression of IK1 reduces the spatial extension of the excited state (wavelength) and increases the rotation frequency (WT, 13 Hz; TG, 40 Hz). Curved arrow shows rotation direction; white dot indicates position of the core. Modified from Noujaim et al9 with permission.
Ionic Basis of Fibrillatory Conduction: Role of IKs
The molecular mechanisms of wavebreak leading to fibrillatory conduction remain poorly understood. On the other hand, it has been known for some time that myocyte activation failure can occur at stimulation frequencies at which INa has had enough time to recover after previous excitation.12 This phenomenon, known as post-repolarization refractoriness, was demonstrated to be a slow process that could greatly outlast full repolarization.13 Since the deactivation kinetics of the slow component of the delayed rectifier current, IKs, are an important determinant of post-repolarization refractoriness,12 it seems reasonable to hypothesize that the spatially distributed wavebreaks and intermittent block processes that are frequently observed at the exceedingly high frequencies of cardiac fibrillation are the result, in part, of the inherent spatial heterogeneities in IKs distribution. To our knowledge, the role of the delayed rectifier currents in rotor dynamics and fibrillation frequency has received very little attention.
The recent study by Muñoz et al provides intriguing insight into what that role might be.14 They used neonatal cardiomyocyte monolayers to investigate the consequences of the overexpression of the slow component of the delayed rectifier potassium current, IKs, on excitation, propagation, and the dynamics of reentrant activation.14 They hypothesized that overexpression of IKs would contribute to wavebreak formation and facilitate fibrillatory conduction. They performed optical mapping experiments usingmonolayers infected with an adenovirus carrying the genomic sequences of KvLQT1 and minK (molecular correlates of IKs) and littermate controls infected with a GFP adenovirus. APD was significantly shorter in IKs versus controlmonolayers at all rotation frequencies. Moreover, contrary to what happens with IK1 overexpression (see above), during reentry, CV as a function of distance from the core had a significantly less pronounced increase in the IKs monolayers, consistent with a less excitable preparation.14 As a result, WLs in the latter were significantly shorter than in littermate controls at all rotation frequencies. Consequently, stable rotors occurred in both groups, but the IKs group had slightly higher rotation frequencies, lower CV, and shorter APD than the control. Most important, unlike control monolayers, the waves emanating from rotors in the IKs preparations frequently underwent wavebreak, fibrillatory conduction and formation of new, short-lived rotors.7 Moreover, the density of wavebreaks increased with time, as long as a stable source sustained thefibrillatory activity. None of the controls showed any wavebreaks other than the original rotor giving rise to sustained reentrant activity. Overall, the results by Muñoz et al14 provided the first demonstration at the molecular level that IKs involvement in post-repolarization refractoriness can lead to wavebreak formation and fibrillatory conduction. Also, fibroblasts interacting with myocytes have been shown to produce fibrillatory conduction patterns.15 These data also opened a new approach to investigate the specific roles of other sarcolemmal channels in the mechanisms of turbulent cardiac excitation during both AF and VF.
Ionic Current Modifications and A trial Fibrillation
Numerical experiments using a 2D ionic model of cardiac excitation have predicted that inhibition of INa induces rotor and AF termination by three different mechanisms: i) enlargement of the center of rotation (core); ii) decreased ability of the rotor to anchor to functional obstacles, with increasing meander and subsequent extinction at boundaries; and iii) reduction in the number of secondary wavelets that generate new primary rotors.8
The currents implicated in the plateau phase of the AP have also been suggested to prevent or stop AF;16 the elevation of the plateau phase seemed more important than the APD90, in accordance with experimental evidence where selective blockade of the atriumspecific, ultra-rapid delayed rectifier K+ current (IKur) and the transient outward K+ current (Ito) by AVE0118 effectively terminated AF in a goat model.17 The rotor was terminated when producing selective blockade (90%) of IKur or Ito, but not the fast or slow delayed rectifier K+ currents (IKr and IKs).16 Remarkably, prolongation of the APD at the plateau led to tip meander and eventual wavebreak and rotor termination. The study also suggested that rotors cannot be terminated by blockade of either IKr or IKs alone, but the combined blockade of both currents terminated the rotor as the prolongation of APD90 exceeded a critical value. Other studies confirm the importance of IKur in the AP of the human atria.18
As discussed above, the inward rectifying currents appear to be a major factor in the control of the dynamics of fibrillation. IK1 augmentation has been proposed to increase AF frequency.16 Importantly, the acetylcholine (ACh) sensitive current IK,ACh, is particularly important in atrial tissue and can produce hyperpolarization (removing voltage-dependent Na+ current inactivation) and AP shortening. These effects provide greater source current for impulse conduction and allow for more rapid rotation, stabilizing atrial reentry and AF.19
Ionic Current Modifications and Ventricular Fibrillation
Inhibition of INa in the ventricle reduces excitability and CV.7,9 During reentry, this gives rise to a smaller WL, larger core size and consequently slower rotation frequency and less wavefront-wavetail interactions. Hence, in the isolated rabbit heart treated with TTX the reduced excitability tripled the area of the core, reduced the rotation frequency and facilitated 1:1 conduction, reducing the amount of fibrillatory conduction (see Noujaim et al7 and references therein). Under these conditions, spiral-wave stabilization led to a transition from VF to monomorphic VT. Alternatively, another consequence of reducing ventricular excitability is that the increased core size may facilitate interaction with structural heterogeneities in the myocardium, leading to increased meandering and subsequent arrhythmia termination.7 However, other data have been reported, showing that INa blockade is not sufficient to terminate VF, but instead the reduction in excitability would increase the vulnerability to reentry by increasing post-repolarization refractoriness, creating a proarrhythmic substrate that could explain the deleterious clinical effects of class I antiarrhythmic drugs (see Noujaim SF et al7 for review)
Blocking the L-type calcium current (ICa-L) with verapamil has been shown to stabilize reentry and convert VF to VT by reducing the appearance of singularity points, rotor frequency (due to an increased core radius and reduced APD) and wavefront fragmentation.7,9 In contrast, other authors have reported that verapamil transiently increases VF dominant frequency (DF) and reduces core size; these discrepancies have been attributed to methodological differences.
Much less is known about the main repolarizing currents implicated in phase 1 and 2 of the ventricular AP. There is evidence for the implication of Ito in Wenckebach rhythms in the rabbit heart. The greater density of Ito in the canine epicardium sets the appropriate conditions for reentry in the presence of INa and ICa-L blockade or other diseases, like Brugada syndrome, and this Wenckebach-like activity has been related to the presence of fibrillatory conduction (see Noujaim et al and references therein7,9).
The delayed rectifier currents, IKr and IKs, which activate relatively slowly, are unlikely to have a large influence near the organizing center (i.e., the core) of reentry.14 Experimental studies using delayed rectifier blockers have suggested that the resulting APD prolongation reduces VF frequency and increases organization.7 Interestingly, APD change alone was not a determinant mechanism, since shortening the APD with the calcium channel blocker bretylium also led to frequency reduction and to increased VF organization. Studies employing sotalol and dofetilide in rabbit and cat hearts, respectively, reported a decreased VF frequency and a reduction in the complexity of the arrhythmia, although conflicting results have also been reported.7 Finally, based on the recent results of Muñoz et al in the monolayers14 it is tempting to speculate that, in the ventricles, IKs plays an important role in the ionic mechanisms of post-repolarization refractoriness, wavebreak formation and fibrillatory conduction, with important implications in tachyarrhythmias. This hypothesis, however, requires further validation.
Much more information is available regarding the inward rectifying potassium current, IK1, in the ventricles. In the guinea pig heart, regional differences in the distribution of Kir2.x protein channels provide a feasible ionic mechanism for the preferential localization and of high frequency rotors in the left ventricle (LV) with fibrillatory conduction to the right ventricle (RV).7 As inferred from the mouse heart studies discussed above,9 the larger IK1 stabilizes reentry in the LV by increasing excitability and CV during reentry, thus reducing wavefront-wavetail interactions.16 Selective IK1 blockade with Ba2+ resulted in a concentration-dependent reduction of the rotation frequency.3
AF and VF: Two Different Beasts with Common Features
There are significant differences between the atria and the ventricles in terms of their specific anatomical structure, ion channel makeup and gene expression. Yet there are striking similarities in their global electrical behavior during fibrillation. For example, rotors are a common feature of AF and VF. In fact rotors are common to many biological, chemical and physical excitable media and their dynamics have been the subject of intense research throughout the scientific world. In the heart, not only the dynamics of rotors and spiral waves are similar, but also both chambers share the presence of a left-to-right gradient of DFs, suggesting that the left heart plays the leading role in maintaining fibrillation. Below we discuss some of the common features of both arrhythmias as well as their major differences.
The dynamics of AF and VF maintenance are similar
The importance of rotors in AF and VF maintenance in patients seems clear, even when the exact mechanism is not completely understood.4,20 Rotors are essential players in both arrhythmias, they may exist alone as stationary high-frequency mother rotors that generate wavefronts that fractionate and disorganize in its periphery. They may also manifest as drifting rotors or even as rotors that rapidly die off leaving multiple offspring wavelets that originate new short-lived rotors and new wavelets.3–5 While specific ionic currents implicated in AF maintenance may be vastly different from those responsible for VF, their dynamics of wave propagation show many similarities.
The importance of anatomical structure
Clearly, the anatomy of the atria is very different from that of the ventricles. However, the atrial and ventricular muscles share common features, particularly in regards to their structural and electrophysiological heterogeneities, both of which represent excellent substrates for sustained reentry. As such, the specific anatomical structure of the cardiac chambers is likely to be a crucial factor in determining the ultimate fibrillatory behavior. For example, in the atria, Gray et al. showed that the crista terminalis and the pectinate muscles are sites of preferential propagation whose frequency dependence enabled disparity between endocardial and epicardial activation as well as reentry.21 The anatomy of the tissue is also responsible for the fragmentation of waves and conduction block. For instance, studies in isolated sheep hearts have shown that the interface between Bachmann's bundle and the branching pectinate muscles of the right atrial appendage. These regions present abrupt changes in their thickness (bigger than 1 mm) and frequency dependent sink-to-source mismatch. Here, spatially distributed intermittent block of wavefronts makes the right atrium (RA) incapable of activating 1:1 in response to impulses traveling from the left atrium (LA) at frequencies higher than ~6.8 Hz (so-called “breakdown frequency”), even if the input frequency is highly periodic.6 Recently, high stimulation frequency of the pulmonary veins originates wavebreaks in areas of abrupt fiber orientation and wall thickness in the LA posterior wall.22
Similarly, the heterogeneity of the ventricular anatomy is likely to play an important role in rotor dynamics. For example, the thicker left ventricular wall may manifest the complex dynamics of 3D scroll waves much more readily than the thinner right ventricle and the two atrial walls. In this regard, Kim et al suggested that sink-to-source mismatch between areas with different thickness in the ventricle may serve to anchor rotors23 and these rotors may span the thickness of the ventricular wall. For instance, the papillary muscles in the LV may help to stabilize rotors.24
Ionic current heterogeneities are involved in AF and VF dynamics
Electrically, the atria and ventricles have common features, but also large differences, particularly in regards to the role of the specific ion channels that might be involved in the mechanism of fibrillation maintenance, as well as the spatial distribution of those channels in the cardiac chambers, which may be important for the understanding of fibrillation pathophysiology. AF and VF share the same consequences of INa inhibition: reduction of excitability and slowing of reentry, but promotion of rotor meandering and, eventually, rotor annihilation by interaction with an unexcitable obstacle.7,8 The stabilizing role of inward-rectifying currents is also similar for AF and VF. An increased IK1 plays a stabilization role in both AF and VF, but IK,ACh is also very important in AF, while it has not been proven to be so relevant during VF. On the other hand, the differential expression of these currents partially explains the frequency gradient existing between the left and the right heart in both chambers. IK1 is preferentially expressed in the LV (as compared to the right),3 while IK,ACh density is larger in the LA.19 This may explain in part the already traditional concept that cholinergic AF maintenance in the normal heart is attributed to the heterogeneous distribution of vagal innervation and muscarinic ACh receptors. Sarmast et al19 hypothesized a heterogeneous response to cholinergic input through a larger IK,ACh activation in the LA than in the RA, setting the stage for the development of AF by means of two mechanisms: an abbreviation of the APD (increased reentry frequency); and an increase in the resting membrane conductance (reduced space constant and conduction velocity). In point of fact, in ACh-mediated AF, higher activation frequencies in the LA were associated with a larger density of IK,ACh partially due to a greater abundance of Kir3.x channels.19
With regards to the ventricles, the differential expression of IK1 that has been observed in the guinea pig heart between the LV and the RV may be a cause for the DF differences observed between the two chambers. As discussed above, during VF IK1 acts as a reentry stabilizer,9 so reducing IK1 specifically in the ventricles might be an attractive option for rotor termination. While the IK1 and INa interplay determines the cell excitability, the delayed rectifiers may play a role in fibrillatory conduction. Indeed, IKs has been implicated in wavebreak and singularity point formation, although their slow activation kinetics suggested a relatively small role in rotor frequency and stability.14 For AF, as well as for VF, the role of the main phase 1 and 2 repolarizing currents, Ito and the exclusively atrial IKur, remains to be elucidated.
Importance of Ischemia and of Structural Heart Disease
Little is known about the effects of ischemia on rotor dynamics in the atria, although some evidence exists for an increased complexity of AF during acute ischemia.25 In the ventricles, acute regional ischemia plays a dual role in the maintenance of VF, decreasing the incidence of wavebreaks in the ischemic zone while increasing it in the ischemic border zone. This suggests a predominant role of fixed heterogeneities in the formation of wavebreaks during VF in acute regional ischemia.26 Similarly chronic ischemia models have demonstrated that the ischemic border zone forms a substrate for the stabilization of sustained reentry.27 Aside from ischemia, the importance of the underlying structural disease, which may be obviously different in AF and VF, should be, and has recently become the focus of attention, because rotor dynamics and its underlying ionic mechanisms are likely to depend on the disease or disease stage.28 Disease-induced heterogeneity may be responsible for the widely different scenarios (i.e., single stationary, single drifting, short-lived rotors, multiple wavelets) that have given birth to different theories of fibrillation. It is quite possible that such scenarios actually represent different manifestations of the same phenomenon modulated by the particular disease condition of the heart.
Clinical Perspective
Rotors in the Human Heart
To date, most studies that have enhanced our understanding of the molecular and/or ionic mechanisms of sustained fibrillation have been performed in animal models.3,4 More time has been necessary to confirm these theories in patients due to technical an ethical issues. Recent studies have reported the presence of rotors during human fibrillation. Here we present some of the available evidence of the presence of rotors in the human heart.
Are There Rotors in the Human Atrium?
To our knowledge, no direct evidence of electrical rotors has been obtained in the human atria, although there have been many speculations and inferences based on electrophysiological and pharmacological studies. Repetitive activity with variable cycle length was found in the LA and RA of AF patients.29,30 Other studies31,32 have demonstrated unstable reentrant circuits or driver activity, which could be interpreted as causing fibrillatory conduction. A strong argument has been made that most patients with AF may have a focal mechanism as the initiating cause of the arrhythmia.33 Yet reentry secondary to a wavebreak is by no means out of the question as an AF trigger. It is also likely that, in at least some AF patients, a rotor or small number of rotors are the drivers that maintain the arrhythmia.4 Recently Atienza et al33 demonstrated that intravenous infusion of an adenosine (ADO) bolus accelerated drivers of human AF. ADO increases potassium conductance through activation of the same inward rectifier channels that are targeted by acetylcholine (IK,ACh). As such ADO infusion should shorten AP duration and refractoriness and reduce excitability and automaticity.34 Thus, as expected from a reentrant mechanism of AF maintenance, ADO infusion increased the DF primarily at sites that activated at the highest rate at baseline. Those results strongly suggested that the arrhythmia is maintained by reentrant sources. Moreover, Sanders et al35 identified localized sites of high-frequency activity during AF in humans with different distributions in paroxysmal and permanent AF. In another study, 9 patients with chronic AF the atrial electrogram showed an area of regular, rapid rhythm, consistent with the possibility that a driver causing fibrillatory conduction is one mechanism of AF in these patients.36 Finally, in the study of Atienza et al,33 ADO infusion in persistent AF patients increased local DFs only in the high RA, without significant changes at other atrial sites. Those data suggested that persistent AF also is maintained by high-frequency reentrant sources.
Rotors in the Human Ventricle
Fibrillatory wavefronts in the human ventricles are large, so they must be highly organized to avoid drifting and annihilation.37,38 Nash et al39 detected large wavefronts in the ventricular epicardium, generated by few reentrant sources, and at times, they observed only one source. Reentry was frequently long lasting and followed by periods with more complex patterns consistent with wandering wavelet activation, which was attributed to a combined mechanism involving rotors and multiple independent wavelets.39 Unfortunately, direct optical mapping for the assessment of rotors in human VF has been limited by spatial resolution. Recently, the submillimetric resolution obtained by Nanthakumar et al40 allowed detection of reentrant wavefronts in human VF, providing the first direct demonstration of phase singularities, wavebreaks and rotor formation in severely diseased, explanted human hearts. Importantly, they found also wavefronts as large as the entire vertical length of the optical field, which suggested a high degree of organization. New findings from simultaneous epicardial and endocardial multielectrode mapping in patients with dilated cardiomyopathy and tetralogy of Fallot,41,42 suggested that during induced VF episodes, stable reentrant wavefronts occur in the endocardium and the epicardium. The same authors demonstrated a stable source in the endocardium, with a highly organized pattern in the local electrogram and a simultaneous and disorganized pattern in the epicardium (breakthroughs), consistent with the idea of 3- dimensional scroll waves.42 Thus, the short-lived rotors on the epicardial and/or endocardial surfaces are thought to be manifestations of a scroll wave organized along the fiber orientation within the wall. Massé et al also observed variable block patterns in wavefront transmission, resulting in disorganized activity and wavefront fragmentation.42
Electrical and Structural Remodeling and Rotors
Unfortunately, the already complex picture of AF and VF mechanisms is further complicated by the electrical and structural remodeling processes that accompany and help to maintain fibrillation. This hypothesis is supported by some data from patients, although much remains to be done to relate the remodeling process to the changes in AF and VF dynamics. In the atria, the study by Atienza et al, using ADO infusion showed that in persistent AF patients the drug increased the local DF in the high RA only, while it increased DF in the pulmonary vein-atrial junction, the distal coronary sinus, and the RA of patients with paroxysmal AF.33 Thus, AF-induced atrial remodeling somehow modifies the sensitivity of channels responsible for IK,ACh (IADO) to the purinergic agonist, the end result being a complete shift in the location of the AF drivers and the DFs hierarchy. These data are in agreement with the different frequency gradients,43 and spatiotemporal organization in paroxysmal AF patients compared with persistent AF patients.44
Parasympathetic signaling is altered in human atrial myocytes obtained from chronic AF patients. Data indicate that the short APD and reduced APD response to changes in activation frequency that characterize chronic AF correlated with reduced ICa-L and increased IK1 and IK,ACh.25 In chronic AF IK,ACh develops agonist-independent constitutive activity, likely resulting from abnormal channel phosphorylation by PKC,45 contributing to the increased resting membrane conductance produced by the IK1 increase (see below). The basal current was higher in chronic AF only, whereas the muscarinic receptor-activated IK,ACh was smaller both in paroxysmal and chronic AF, which can be understood as a smaller ACh-sensitive current.45 This decrease of the response to ACh could also explain the reduced response to ADO observed in persistent AF in patients.33 The increased basal agonist-independent current would result in rotor stabilization, acceleration of reentry, and spread of the DF sites from the PV to other non-PV locations and disappearance of LA to RA frequency gradients. Since antiarrhythmic drugs like amiodarone, flecainide, quinidine, and verapamil are also IK,ACh inhibitors, it cannot be ruled out that their therapeutic effectiveness results, at least partially, from inhibition of this constitutively active current in AF patients.46 Thus, a basal increase of the IK,ACh may represent a target for development of new therapy in AF.19,45,46 However, Brundel et al. demonstrated that mRNA and protein levels of Kir3.1–3.4 were significantly reduced in chronic AF patients.47
Structural remodeling has been shown to interfere with rotor behavior. For example, In AF, fibrosis secondary to chronic heart failure has been shown to shift the pattern from a stable rotor to multiple unstable rotors and wavebreaks28 despite producing a significant slowing of rotor frequency. With regards to the ventricles, it has been shown experimentally that the dynamics of VF in the presence of heart failure are different from those in the normal heart. Heart failure remodeling decreases VF rate and increases VF organization.48 Acute stretch partially reverses these effects by a mechanism that is independent of remodeling. Interestingly, the effects of acute ischemia on VF dynamics are significantly attenuated in heart failure compared to normal hearts. Thus, the role of the structural substrate arises as a key factor differentiating AF and VF in structurally normal versus moderately or severely remodeled hearts.
Conclusions
While much work remains to be done, we have come a long way in our understanding of the dynamics and mechanisms of the two most complex and dangerous arrhythmias seen in clinical practice. With all this new knowledge at hand, several interventional (e.g., new defibrillating algorithms, new ablation procedures) and pharmacological strategies are possible and continue to arise resulting in clinical benefit for the patient. For example, we now know how certain ion channel modifications at the single cell level affect rotor dynamics and often terminate their activity. Perhaps by integrating this knowledge with a better understanding of the molecular structure of atrial specific ion channels (e.g., IKur) toward the development a novel generation of antiarrhythmic approaches that target specific ion channel protein domains, one should be able to benefit the patient at risk not only by terminating an existing AF episode, but more importantly prevent a new episode. However, the fact remains that millions of people around the world still suffer the consequences of AF, including stroke, and millions more die suddenly and prematurely as a result of VF. Arguably, fundamental research leading to a better understanding mechanisms of AF and VF as well as their similarities and differences, from their molecular mechanisms to their clinical manifestation, may help to improve our understanding of these two devastating conditions.
Acknowledgements
We thank Dr. Sandeep Pandit for helpful comments and suggestions. Supported by NHLBI Grants PO1 HL039707, PO1 HL087226, RO1 HL060843 (JJ); CICYT (SAF2005-04609), Ministerio de Sanidad y Consumo, Instituto de Salud Carlos III (Red HERACLES RD06/0009), Sociedad Española de Cardiología, and Fundación LILLY (MV); Sociedad Española de Cardiología and Health Comission of the Asturias Regional Government (DC).
Abbreviations
- Ach
Acetylcholine
- AF
A trial fibrillation
- AP
Action potential
- APD
Action potential duration
- CV
Conduction velocity
- DF
Dominant frequency
- ECG
Electrocardiogram
- GFP
Green fluorescent protein
- LA
Left atrium
- LV
Left ventricle
- RA
Right atrium
- RV
Right ventricle
- TTX
Tetrodotoxin
- VF
Ventricular fibrillation
- VT
Ventricular tachycardia
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22:983–988. doi: 10.1161/01.str.22.8.983. [DOI] [PubMed] [Google Scholar]
- 2.Zipes DP, Wellens HJ. Sudden cardiac death. Circulation. 1998;98:2334–2351. doi: 10.1161/01.cir.98.21.2334. [DOI] [PubMed] [Google Scholar]
- 3.Jalife J, Berenfeld O. Molecular mechanisms and global dynamics of fibrillation: an integrative approach to the underlying basis of vortex-like reentry. J Theor Biol. 2004;230:475–487. doi: 10.1016/j.jtbi.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 4.Jalife J, Berenfeld O, Mansour M. Mother rotors and fibrillatory conduction: a mechanism of atrial fibrillation. Cardiovasc Res. 2002;54:204–216. doi: 10.1016/s0008-6363(02)00223-7. [DOI] [PubMed] [Google Scholar]
- 5.Jalife J, Pandit SV. Ionic mechanisms of wavebreak in fibrillation. Heart Rhythm. 2005;2:660–663. doi: 10.1016/j.hrthm.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 6.Berenfeld O, Zaitsev AV, Mironov SF, et al. Frequency-dependent breakdown of wave propagation into fibrillatory conduction across the pectinate muscle network in the isolated sheep right atrium. Circ Res. 2002;90:1173–1180. doi: 10.1161/01.res.0000022854.95998.5c. [DOI] [PubMed] [Google Scholar]
- 7.Noujaim SF, Auerbach D, Jalife J. Ventricular fibrillation: Dynamics and ion channels determinants. Circulation journal. 2007;71(SupplA):A1–A11. doi: 10.1253/circj.71.a1. [DOI] [PubMed] [Google Scholar]
- 8.Kneller J, Kalifa J, Zou R, et al. Mechanisms of atrial fibrillation termination by pure sodium channel blockade in an ionically-realistic mathematical model. Circ Res. 2005;96:e35–e47. doi: 10.1161/01.RES.0000160709.49633.2b. [DOI] [PubMed] [Google Scholar]
- 9.Noujaim SF, Pandit SV, Berenfeld O, et al. Up-regulation of the inward rectifier K+ current (I K1) in the mouse heart accelerates and stabilizes rotors. J Physiol. 2007;578:315–326. doi: 10.1113/jphysiol.2006.121475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jalife J. Ventricular fibrillation: mechanisms of initiation and maintenance. Annu Rev Physiol. 2000;62:25–50. doi: 10.1146/annurev.physiol.62.1.25. [DOI] [PubMed] [Google Scholar]
- 11.Noujaim SF, Berenfeld O, Kalifa J, et al. Universal scaling law of electrical turbulence in the mammalian heart. PNAS. 2007;104:20985–20989. doi: 10.1073/pnas.0709758104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delmar M, Michaels DC, Jalife J. Slow recovery of excitability and the Wenckebach phenomenon in the single guinea pig ventricular myocyte. Circ Res. 1989;65:761–774. doi: 10.1161/01.res.65.3.761. [DOI] [PubMed] [Google Scholar]
- 13.Milberg P, Tegelkamp R, Osada N, et al. Reduction of dispersion of repolarization and prolongation of postrepolarization refractoriness explain the antiarrhythmic effects of quinidine in a model of short QT syndrome. J Cardiovasc Electrophysiol. 2007;18:658–664. doi: 10.1111/j.1540-8167.2007.00813.x. [DOI] [PubMed] [Google Scholar]
- 14.Munoz V, Grzeda KR, Desplantez T, et al. Adenoviral expression of IKs contributes to wavebreak and fibrillatory conduction in neonatal rat ventricular cardiomyocyte monolayers. Circ Res. 2007;101:475–483. doi: 10.1161/CIRCRESAHA.107.149617. [DOI] [PubMed] [Google Scholar]
- 15.Zlochiver S, Muñoz V, Oxford EM, et al. Biphasic Effect of Electrotonic Fibroblast-Myocyte Coupling on 2D Wave Conduction Velocity and Reentry. Circulation. 2007;116:II_275. Abstract. [Google Scholar]
- 16.Pandit SV, Berenfeld O, Anumonwo JM, et al. Ionic determinants of functional reentry in a 2-D model of human atrial cells during simulated chronic atrial fibrillation. Biophys J. 2005;88:3806–3821. doi: 10.1529/biophysj.105.060459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blaauw Y, Gogelein H, Tieleman RG, et al. "Early" class III drugs for the treatment of atrial fibrillation: efficacy and atrial selectivity of AVE0118 in remodeled atria of the goat. Circulation. 2004;110:1717–1724. doi: 10.1161/01.CIR.0000143050.22291.2E. [DOI] [PubMed] [Google Scholar]
- 18.Wettwer E, Hala O, Christ T, et al. Role of IKur in controlling action potential shape and contractility in the human atrium: influence of chronic atrial fibrillation. Circulation. 2004;110:2299–2306. doi: 10.1161/01.CIR.0000145155.60288.71. [DOI] [PubMed] [Google Scholar]
- 19.Sarmast F, Kolli A, Zaitsev A, et al. Cholinergic atrial fibrillation: I(K,ACh) gradients determine unequal left/right atrial frequencies and rotor dynamics. Cardiovasc Res. 2003;59:863–873. doi: 10.1016/s0008-6363(03)00540-6. [DOI] [PubMed] [Google Scholar]
- 20.Ideker RE, Rogers JM. Human ventricular fibrillation: wandering wavelets, mother rotors, or both? Circulation. 2006;114:530–532. doi: 10.1161/CIRCULATIONAHA.106.644765. [DOI] [PubMed] [Google Scholar]
- 21.Gray RA, Pertsov AM, Jalife J. Incomplete reentry and epicardial breakthrough patterns during atrial fibrillation in the sheep heart. Circulation. 1996;94:2649–2661. doi: 10.1161/01.cir.94.10.2649. [DOI] [PubMed] [Google Scholar]
- 22.Klos M, Zlochiver S, Mironov S, et al. Role of the Posterior Left Atrial Septo-Pulmonary Bundle in Wavebreak Formation During Atrial Fibrillation by Pulmonary Veins Impulses. Circulation. 2006;114:II_331. [Google Scholar]
- 23.Kim YH, Yashima M, Wu TJ, et al. Mechanism of procainamide-induced prevention of spontaneous wave break during ventricular fibrillation. Insight into the maintenance of fibrillation wave fronts. Circulation. 1999;100:666–674. doi: 10.1161/01.cir.100.6.666. [DOI] [PubMed] [Google Scholar]
- 24.Wu TJ, Lin SF, Baher A, et al. Mother rotors and the mechanisms of D600-induced type 2 ventricular fibrillation. Circulation. 2004;110:2110–2118. doi: 10.1161/01.CIR.0000143834.51102.91. [DOI] [PubMed] [Google Scholar]
- 25.Nattel S, Maguy A, Le Bouter S, et al. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007;87:425–456. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- 26.Zaitsev AV, Guha PK, Sarmast F, et al. Wavebreak formation during ventricular fibrillation in the isolated, regionally ischemic pig heart. Circ Res. 2003;92:546–553. doi: 10.1161/01.RES.0000061917.23107.F7. [DOI] [PubMed] [Google Scholar]
- 27.Wit AL, Dillon SM, Coromilas J, et al. Anisotropic reentry in the epicardial border zone of myocardial infarcts. Ann N Y Acad Sci. 1990;591:86–108. doi: 10.1111/j.1749-6632.1990.tb15083.x. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka K, Zlochiver S, Vikstrom KL, et al. Spatial distribution of fibrosis governs fibrillation wave dynamics in the posterior left atrium during heart failure. Circ Res. 2007;101:839–847. doi: 10.1161/CIRCRESAHA.107.153858. [DOI] [PubMed] [Google Scholar]
- 29.Sueda T, Nagata H, Shikata H, et al. Simple left atrial procedure for chronic atrial fibrillation associated with mitral valve disease. Ann Thorac Surg. 1996;62:1796–1800. doi: 10.1016/s0003-4975(96)00613-3. [DOI] [PubMed] [Google Scholar]
- 30.Wu TJ, Doshi RN, Huang HL, et al. Simultaneous biatrial computerized mapping during permanent atrial fibrillation in patients with organic heart disease. J Cardiovasc Electrophysiol. 2002;13:571–577. doi: 10.1046/j.1540-8167.2002.00571.x. [DOI] [PubMed] [Google Scholar]
- 31.Cox JL, Canavan TE, Schuessler RB, et al. The surgical treatment of atrial fibrillation. II. Intraoperative electrophysiologic mapping and description of the electrophysiologic basis of atrial flutter and atrial fibrillation. J Thorac Cardiovasc Surg. 1991;101:406–426. [PubMed] [Google Scholar]
- 32.Konings KT, Smeets JL, Penn OC, et al. Configuration of unipolar atrial electrograms during electrically induced atrial fibrillation in humans. Circulation. 1997;95:1231–1241. doi: 10.1161/01.cir.95.5.1231. [DOI] [PubMed] [Google Scholar]
- 33.Atienza F, Almendral J, Moreno J, et al. Activation of inward rectifier potassium channels accelerates atrial fibrillation in humans: evidence for a reentrant mechanism. Circulation. 2006;114:2434–2442. doi: 10.1161/CIRCULATIONAHA.106.633735. [DOI] [PubMed] [Google Scholar]
- 34.Belardinelli L, Shryock JC, Song Y, et al. Ionic basis of the electrophysiological actions of adenosine on cardiomyocytes. FASEB J. 1995;9:359–365. doi: 10.1096/fasebj.9.5.7896004. [DOI] [PubMed] [Google Scholar]
- 35.Sanders P, Berenfeld O, Hocini M, et al. Spectral analysis identifies sites of high-frequency activity maintaining atrial fibrillation in humans. Circulation. 2005;112:789–797. doi: 10.1161/CIRCULATIONAHA.104.517011. [DOI] [PubMed] [Google Scholar]
- 36.Sahadevan J, Ryu K, Peltz L, et al. Epicardial mapping of chronic atrial fibrillation in patients: preliminary observations. Circulation. 2002;110:3293–3299. doi: 10.1161/01.CIR.0000147781.02738.13. [DOI] [PubMed] [Google Scholar]
- 37.Nanthakumar K, Walcott GP, Melnick S, et al. Epicardial organization of human ventricular fibrillation. Heart Rhythm. 2004;1:14–23. doi: 10.1016/j.hrthm.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 38.Walcott GP, Kay GN, Plumb VJ, et al. Endocardial wave front organization during ventricular fibrillation in humans. J Am Coll Cardiol. 2002;39:109–115. doi: 10.1016/s0735-1097(01)01696-5. [DOI] [PubMed] [Google Scholar]
- 39.Nash MP, Mourad A, Clayton RH, et al. Evidence for multiple mechanisms in human ventricular fibrillation. Circulation. 2006;114:536–542. doi: 10.1161/CIRCULATIONAHA.105.602870. [DOI] [PubMed] [Google Scholar]
- 40.Nanthakumar K, Jalife J, Masse S, et al. Optical mapping of Langendorff-perfused human hearts: establishing a model for the study of ventricular fibrillation in humans. Am J Physiol Heart Circ Physiol. 2007;293:H875–H880. doi: 10.1152/ajpheart.01415.2006. [DOI] [PubMed] [Google Scholar]
- 41.Wu TJ, Ong JJ, Hwang C, et al. Characteristics of wave fronts during ventricular fibrillation in human hearts with dilated cardiomyopathy: role of increased fibrosis in the generation of reentry. J Am Coll Cardiol. 1998;32:187–196. doi: 10.1016/s0735-1097(98)00184-3. [DOI] [PubMed] [Google Scholar]
- 42.Masse S, Downar E, Chauhan V, et al. Ventricular fibrillation in myopathic human hearts: mechanistic insights from in vivo global endocardial and epicardial mapping. Am J Physiol Heart Circ Physiol. 2007;292:H2589–H2597. doi: 10.1152/ajpheart.01336.2006. [DOI] [PubMed] [Google Scholar]
- 43.Lazar S, Dixit S, Marchlinski FE, et al. Presence of left-to-right atrial frequency gradient in paroxysmal but not persistent atrial fibrillation in humans. Circulation. 2004;110:3181–3186. doi: 10.1161/01.CIR.0000147279.91094.5E. [DOI] [PubMed] [Google Scholar]
- 44.Saksena S, Skadsberg ND, Rao HB, et al. Biatrial and three-dimensional mapping of spontaneous atrial arrhythmias in patients with refractory atrial fibrillation. J Cardiovasc Electrophysiol. 2005;16:494–504. doi: 10.1111/j.1540-8167.2005.40531.x. [DOI] [PubMed] [Google Scholar]
- 45.Voigt N, Friedrich A, Bock M, et al. Differential phosphorylation-dependent regulation of constitutively active and muscarinic receptor-activated IK,ACh channels in patients with chronic atrial fibrillation. Cardiovasc Res. 2007;74:426–437. doi: 10.1016/j.cardiores.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 46.Dobrev D, Friedrich A, Voigt N, et al. The G protein-gated potassium current I(K,ACh) is constitutively active in patients with chronic atrial fibrillation. Circulation. 2005;112:3697–3706. doi: 10.1161/CIRCULATIONAHA.105.575332. [DOI] [PubMed] [Google Scholar]
- 47.Brundel BJ, Van Gelder IC, Henning RH, et al. Ion channel remodeling is related to intraoperative atrial effective refractory periods in patients with paroxysmal and persistent atrial fibrillation. Circulation. 2001;103:684–690. doi: 10.1161/01.cir.103.5.684. [DOI] [PubMed] [Google Scholar]
- 48.Moreno J, Zaitsev AV, Warren M, et al. Effect of remodelling, stretch and ischaemia on ventricular fibrillation frequency and dynamics in a heart failure model. Cardiovasc Res. 2005;65:158–166. doi: 10.1016/j.cardiores.2004.09.006. [DOI] [PubMed] [Google Scholar]