

Summary
Triptolide, a diterpene triepoxide isolated from the traditional Chinese medicinal vine Trypterygium wilfordii hook f., has been shown to induce rapid apoptosis in a myriad of cancer cell lines and inhibit NFκB transactivation. To understand further the general cellular mechanisms for this therapeutically relevant natural product, binding and biological activities were assessed. Studies showed that triptolide binding was saturable, reversible, and primarily localized to cell membranes. Depletion of calcium enhanced overall binding while differentially modulating biological function. Furthermore, triptolide's structural moieties demonstrated variability in the regulation of cell death versus inhibition of NFκB transactivation. These results implicate triptolide in the manipulation of at least two distinct cellular pathways with differing requirements for calcium and effective triptolide concentration in order to elicit each particular biological function.
Introduction
Traditional Chinese herbal therapy has utilized the herb lei gong teng for centuries to treat inflammatory disorders such as rheumatoid arthritis [1, 2]. Triptolide, one of the active anti-inflammatory small molecules present in the plant extract, has been shown to possess multiple biological activities, as it also exhibits antitumor, immunosuppressive, and antifertility effects [3–8]. Although the efficacy of triptolide as an inhibitor of immune function and an inducer of tumor cell death has been well-documented, its modes of action have not been thoroughly characterized.
Multiple studies have shown that triptolide inhibits TNF-α- and IL-1β-induced transcription at a step after NFκB binding to DNA [9]. While upstream activation of the IKK complex, phosphorylation of IκBα, and NFκB nuclear translocation are not disrupted upon triptolide addition, it is postulated that triptolide may interfere with additional p65 modifications or the recruitment of a transcriptional cofactor. Triptolide additionally inhibits NF-AT, but at the level of its nuclear translocation, a process normally regulated by calcineurin and calcium flux [9]. A third transcription factor adversely regulated by triptolide is AP-1, whose nuclear translocation is blocked in response to such stimuli as PMA and the calcium ionophore ionomycin [10, 11].
In addition to its ability to attenuate the immune response, triptolide also possesses potent antitumor efficacy against leukemias, lymphomas, as well as solid tumors [5, 12]. At different concentrations, triptolide either arrests cell growth or actively induces cell death via apoptosis [13]. Moreover, the therapeutic potential of triptolide has been demonstrated by its synergistic effect with other anticancer agents [14–16], and it is currently in clinical trials for prostate cancer [13].
Structure-activity relationship studies indicate that different pharmacophores are required for the various biological effects of triptolide. While disruption of the 12,13 epoxide group or C-14 hydroxyl group of triptolide has been reported to eliminate its anti-inflammatory or immunosuppressive functions, respectively [17], the 9,11 epoxide was deemed necessary for its antitumor effect [18]. These studies raise the question of whether the various physiological effects of triptolide (i.e., anti-inflammatory, proapoptotic) are elicited by a common mechanism of action and/or interaction with a common protein target.
In this study, we first assessed the properties of triptolide binding activity in cells and discovered a link to calcium dependence. We therefore examined the effects of calcium on two discrete endpoints of triptolide's action in the cervical carcinoma cell line HeLa: induction of cell death and NFκB inhibition after TNF-α treatment. Additionally, we tested the ability of triptolide analogs to compete for binding and measured their capacity to affect these biological endpoints. Our findings demonstrate the following: (1) NFκB transcriptional inhibition, while being a calcium-independent phenomenon, is sensitive to disruption of the 12,13 epoxide and requires a higher triptolide concentration than that sufficient to induce cell death. (2) Triptolide-induced cell death is strongly affected by calcium levels and is partially dependent upon the functionality of the C-14 hydroxyl.
Results
[3H]Triptolide Binding Is Reversible and Associates with Cell Membranes
To gain insight into triptolide's mechanism of action, we sought to determine its specific binding activity by utilizing a system employing [3H]triptolide. In addition to competition with 1 μM or 10 μM unlabeled triptolide, we also measured the binding affinities of triptolide analogs (Figure 1A). These analogs have been previously described and differ structurally from triptolide in the hydrolysis of the 12,13 epoxide (2) and in formation of the ketone at the C-14 hydroxyl (3) (Figure 1A). HeLa cells readily bound [3H]triptolide during an hour-long incubation, and binding was significantly competed with pre-treatment of an excess of unlabeled triptolide (1−10 μM) (Figure 1B). At either 1 or 10 μM, both triptolide analogs showed near total displacement of [3H]triptolide (Figure 1B). The effective interaction of these analogs with a presumed triptolide binding entity led us to their use in subsequent experiments addressing the mode of triptolide's biological functions.
Figure 1. Triptolide Analogs and Their Structural Dependence to Compete for Binding.
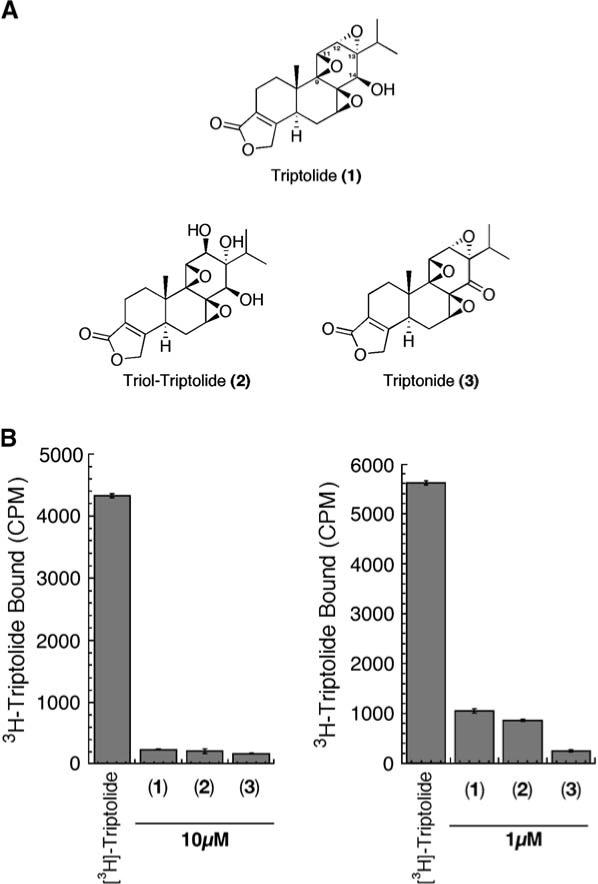
(A) Structures of triptolide and the analogs used in this study. (B) HeLa cells were treated with [3H]triptolide for 1 hr in all samples after the addition of 10 μM or 1 μM triptolide, one of its analogs, or no competition also for 1 hr. Cells were washed, total cell lysates were prepared, and samples were counted for bound [3H]triptolide activity. CPM = counts per minute by scintillation counting. n = 3; data represented as mean ± SE.
To address first the nature of triptolide binding within the cell, we determined that this interaction was reversible, as [3H]triptolide labeling was out competed by subsequent addition of excess unlabeled triptolide (Figure 2A). An additional labeling experiment followed by cellular fractionation indicated that [3H]triptolide binds predominantly to the membrane fraction (P-100) of the cell (Figure 2B). Although 25% of the total cellular counts (i.e., representing bound triptolide) were found in the cytosolic fraction, it is uncertain whether this is specific binding or simply that free triptolide had dissociated from its binding protein due to experimental manipulation during fractionation. Since our data thus far had only shown binding within whole cells, we next determined if this triptolide binding protein could be further characterized and/or enriched by its association with chromatographic reagents. HeLa cells were once again labeled with [3H]triptolide, and total cell lysates were then passed over the anion exchange resin DEAE. Batch elutions with increasing NaCl concentrations were used to disassociate interactions of variable charge. Substantial elution of [3H]triptolide was not seen in the flow-through or with several salt-free washes, and, in fact, it did not begin to elute until the addition of 0.2 M NaCl (Figure 2C). Additionally, free [3H]triptolide diluted in lysis buffer and then passed over this resin eluted in the flow-through or during salt-free washes, demonstrating that triptolide alone does not interact with this chromatographic media (data not shown). Importantly, we also observed that [3H]triptolide would only bind to intact cells in culture, but would not bind to any component of a total cellular lysate, as assessed by interaction with the DEAE resin (data not shown). Negative results for a triptolide binding interaction were also observed with DNA-cellulose and the cation exchange resin SPFF (sulphopropyl) (data not shown).
Figure 2. Triptolide Binding Is Specific, Membrane Localized, and Saturable.
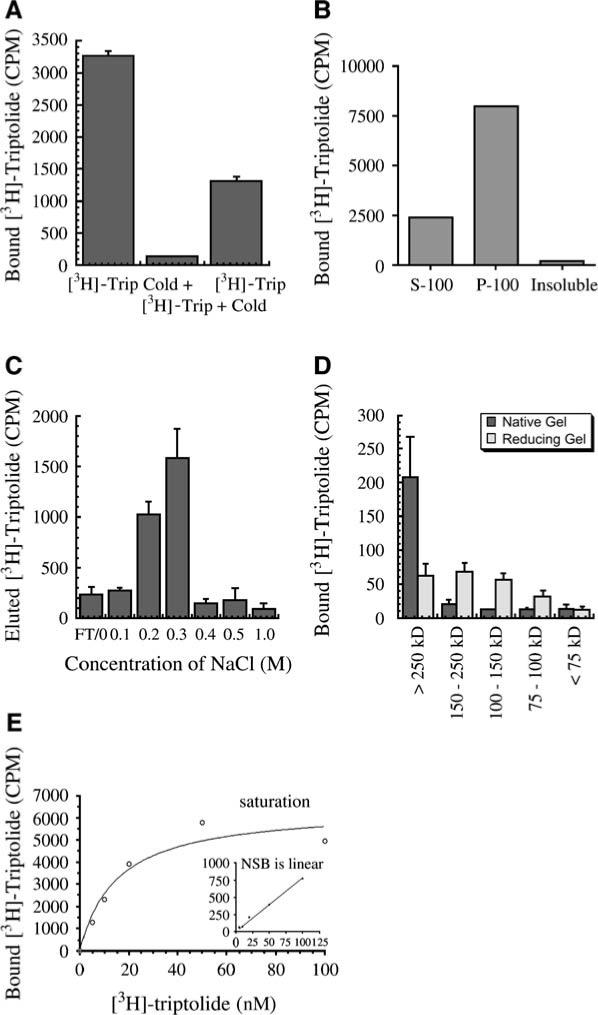
(A) HeLa cells were treated with [3H]triptolide for 1 hr in all samples. Competition of the radioligand was assessed by the addition of 1 μM unlabeled triptolide (cold) for 1 hr either before or after [3H]triptolide addition. CPM = counts per minute by scintillation counting. n = 3; data represented as mean ± SE. (B) HeLa cells were labeled with [3H]triptolide, and cellular fractions were prepared. Binding was assessed as total CPM in the cytosolic (S-100), membrane (P-100), or insoluble cellular fractions. (C) HeLa cells were labeled with [3H]triptolide for 1 hr, followed by preparation of total cellular lysates and addition to DEAE anion exchange resin. The resin was washed in batch elutions of increasing salt concentration after removal of the flow-through (FT). Each eluant was subsequently counted for [3H]triptolide activity. n = 3; data represented as mean ± SE. (D) [3H]triptolide-labeled HeLa cell P-100 fractions were run out on 8% reducing or native PAGE. Gel slices were removed by utilizing molecular weight marker designations, crushed, eluted in water, and were then counted for [3H]triptolide activity by scintillation counting. n = 2; data represented as mean ± SD. (E) HeLa cells were preincubated with 2 μM unlabeled triptolide or DMSO, followed by increasing nanomolar concentrations of [3H]triptolide for 1 hr. Specific binding was measured from scintillation counting of cellular lysates, and receptor saturation was achieved. Nonspecific binding (NSB) is shown in the inset and does not reach saturation.
Another line of evidence for direct protein interaction involved an additional separation of [3H]triptolide-labeled membrane preparations. Labeling of cells and total membrane purification by high-speed centrifugation, followed by detergent resolubilization, yielded samples that were run out on 6% polyacrylamide gels under native or denaturing conditions (the [3H]triptolide lysates were not boiled in either experimental condition). Gel slices were extracted based on molecular weight range and crushed, and water was extracted, followed by liquid scintillation. Native gel separation indicated that [3H]triptolide was bound to protein(s) above 250 kDa, while denaturing conditions showed a separation of [3H]triptolide binding in a range from 75 to greater than 250 kDa (Figure 2D). These results suggested that triptolide could be binding to a protein complex that then dissociates upon reducing conditions. These data are also supported by size-exclusion assays from total cell lysates, in which the majority of [3H]triptolide binding activity was recovered above a molecular weight of 100 kDa (data not shown).
Based upon our data described above, we next sought to determine if triptolide binding was saturable and if more than one protein (or binding site) was being targeted. Determination of the [3H]triptolide binding affinity (KD) and the binding capacity per cell (Bmax) were calculated by using a saturation plot. HeLa cells were cultured to 90% confluency and were subsequently treated with 0−100 nM [3H]triptolide for 1 hr. Nonspecific binding was defined by using 2 μM cold triptolide as competitor. Bmax was calculated to be 99 ± 19 fmol triptolide bound/105 cells of triptolide binding sites. Specific binding was found to be saturable, with a KD value of 15.5 ± 0.8 nM, while nonspecific binding was linear (nonsaturable) (Figure 2E).
Triptolide Binding Activity Is Influenced by Extracellular Calcium Concentration
To understand further the nature of triptolide's interaction within the cell, we altered cell culture conditions to examine if [3H]triptolide binding would be affected. Calcium has been shown to mediate numerous cellular functions, including transcriptional activation of NF-AT and NFκB [19–21]. Additionally, it has been established that aberrant calcium signaling can result in cell death [22, 23]. Due to triptolide's own link with NFκB and NF-AT and its propensity to induce cell death, we examined if triptolide binding could be modulated by free calcium levels. Adherent HeLa cells were cultured in the presence or absence of calcium-containing medium for 16 hr before [3H]triptolide addition. Replicate experiments confirmed that, in the absence of extracellular calcium, [3H]triptolide binding significantly increased between 2- and 4-fold, depending on cell number and density (Figure 3A). It is also noteworthy that an increase in cell density (not cell number) also increases triptolide binding regardless of calcium levels (data not shown). Additionally, specific calcium chelation by 10 μM EGTA for 1 hr in calcium-containing medium increased binding by nearly 2-fold (data not shown). These data indicate that the interaction of triptolide with its target protein(s) is potentially stabilized or enhanced when calcium levels are low.
Figure 3. Extracellular Calcium Regulates Triptolide-Mediated Binding and Cell Death Induction.
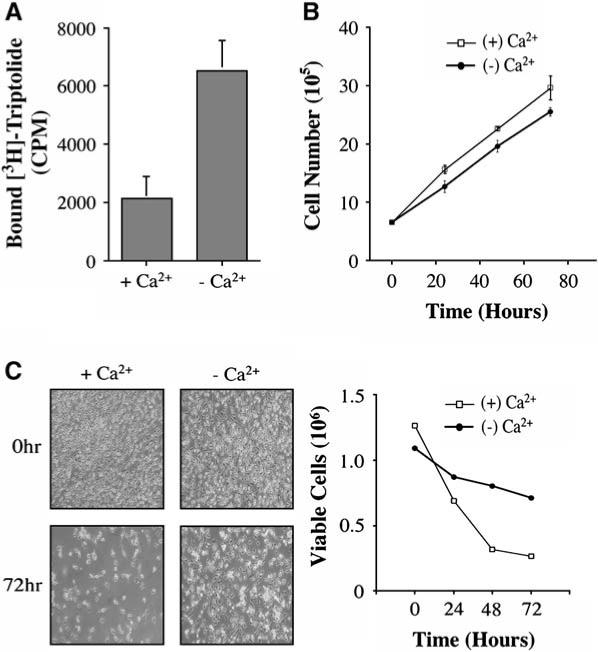
(A) HeLa cells were cultured in the presence or absence of calcium-containing media for 16 hr before the addition of 30 nM [3H]triptolide to assess binding affinity. CPM = counts per minute. n = 3; data represented as mean ± SE. (B) HeLa cells were cultured in the presence (+Ca2+) or absence (−Ca2+) of calcium-containing media over a time course of 72 hr to determine the rate of growth in each condition. n = 3; data represented as mean ± SE. (C) HeLa cells were cultured in the presence (+Ca2+) or absence (−Ca2+) of calcium-containing media plus 100 nM triptolide over a time course of 72 hr. Cells were washed with PBS, photographed, and counted by using trypan blue at 0, 24, 48, and 72 hr to assess viability. Results are representative of three separate experiments.
Triptolide-Induced Cell Death Is Delayed in the Absence of Calcium
Since extracellular calcium concentration can influence triptolide binding, we sought to determine if the presence of calcium influences the rate of triptolide-mediated apoptosis. To first establish the growth rate of HeLa in media ± calcium, cell counts were performed over a 72 hr time course. Although calcium-free media caused cells to detach more easily, the overall growth rates were similar—cell doubling occurred every 24 hr on average (Figure 3B). For triptolide experiments, cells were initially equilibrated in medium ± calcium for 16 hr before the addition of 100 nM triptolide. Cell death was assessed by trypan blue dye exclusion at 24, 48, and 72 hr after drug treatment. In the presence of calcium-containing medium, triptolide induced at least 50% cell death by 24 hr, and this trend continued through later time points (Figure 3C). In contrast, removal of calcium from the growth medium resulted in a higher proportion of viable cells (Figure 3C). Following 72 hr of triptolide addition in calcium-free media, only 35% of the cells had died, indicating that there is a significant delay in this process. These results support a role for calcium in efficient cell death induced by triptolide. There is, however, a likely secondary (albeit slower) mechanism to promote apoptosis, as the lack of calcium merely delays, but does not eliminate, cell death.
To investigate further the role of calcium in triptolide function, we utilized a system to buffer intracellular calcium levels. Various GFP-parvalbumin (PV) fusion proteins can be specifically localized to either the nucleus or the cytoplasm through a nuclear localization or exclusion signal (NLS or NES, respectively) [24]. Parvalbumin has two EF-hand calcium binding domains and can efficiently reduce the availability of free calcium in the cell [25]. HeLa cells were transiently transfected with the GFP vector control, NES-PV-GFP, or NLS-PV-GFP in calcium-containing media, and efficient expression of each construct was determined by GFP localization (Figure 4A). Normal cell growth was first assessed throughout 48 hr with each of the constructs. All transfections resulted in normal growth doubling during the course of the experiment without drug addition (Figure 4B). For triptolide experiments, cells were transfected and allowed to express the construct for 24 hr before the addition of 100 nM triptolide. After 24 and 48 hr of treatment, cells were assessed for viability. Both control GFP vector and NLS-PV-GFP showed similar apoptosis induction, whereby 50% of the cells were rounded and no longer viable after 24 hr (Figure 4C). In marked contrast, cytosolic parvalbumin (NES-PV-GFP) significantly inhibited triptolide-induced cell death at this time point (15%−20% apoptosis) (Figure 4C). This effect was transient, however, as there was complete cell death in all conditions by 48 hr (data not shown). Since the parvalbumin buffering experiments are done in the presence of extracellular calcium, the cell can operate normally, in that intracellular calcium stores may be refilled. It is reasonable to expect, then, that if calcium homeostasis is being affected (i.e., cytosolic calcium levels increase) by triptolide, then parvalbumin would eventually reach a saturation point. This might explain why the rescue from apoptosis is transient through the 24 hr time point but is lost by 48 hr. These results not only confirm the overall importance of calcium to triptolide function, but they more specifically point to cytosolic calcium levels as mediators in triptolide-induced cell death.
Figure 4. Buffering Cytosolic Calcium Can Temporarily Rescue Triptolide-Induced Cell Death.
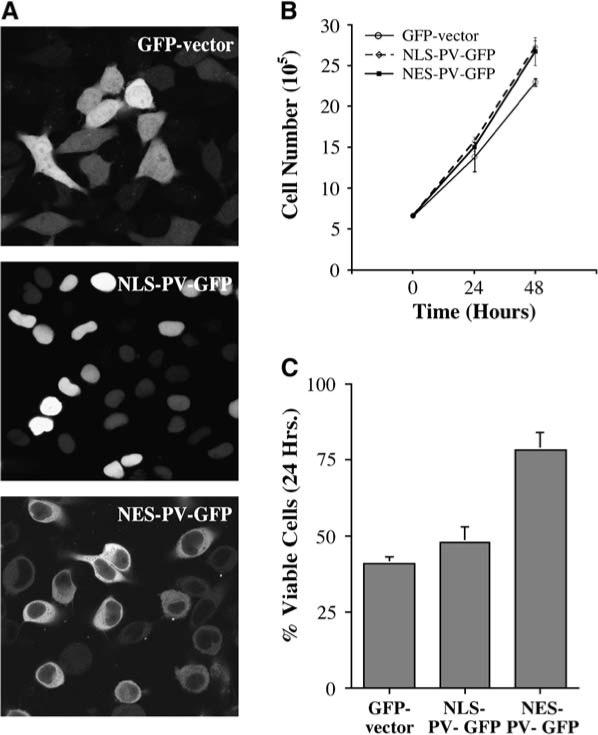
(A) HeLa cells were cultured in the presence of calcium-containing media and were transfected with one of the following constructs for 24 hr: GFP vector, NLS-parvalbumin (PV)-GFP, NES-PV-GFP. Images were acquired by confocal microscopy (40×). (B) Normal cell growth was assessed with each transient transfection construct. n = 3; data represented as mean ± SE. (C) 100 nM triptolide was added to all transfected cells, and viability was assessed after 24 hr. n = 3; data represented as mean ± SE.
Inhibition of NFκB Transactivation by Triptolide Is Calcium-Independent
Having established that triptolide induced cell death is dependent upon free calcium concentration, we next determined if this was also a requirement for the inhibition of NFκB transcription. HeLa cells were transiently transfected with the κB-luciferase reporter construct for 8 hr before being washed and then cultured in the presence or absence of calcium-containing medium for 16 hr. Triptolide (100 nM) was preincubated with cells for 1 hr before the addition of 15 ng/ml TNF-α for 5 hr. Similar profiles were seen in both the presence and absence of calcium as TNF-α induced NFκB transactivation, while triptolide effectively inhibited it (Figure 5A).
Figure 5. Inhibition of NFκB Transactivation Is Independent of the Presence of Calcium.

(A) HeLa cells were transfected with a κB-luciferase construct for all experimental conditions for 24 hr before the addition of 15 ng/ml TNF-α ± 100 nM triptolide. Cells were grown in the presence or absence of calcium-containing media for 16 hr before treatment and were then harvested for the assay after 6 hr. n = 4; data represented as mean ± SE. (B) At the same time as the κB-luciferase construct, cells were transfected with one of the following: GFP vector, NES-PV-GFP, NLSPV-GFP. They were the treated as described in (A). n = 4; data represented as mean ± SE.
As an additional experiment, transcriptional activity was also assessed by site-directed calcium buffering. HeLa cells were cotransfected with the κB-luciferase plasmid as well as one of the following constructs: GFP empty vector, NES-PV-GFP, or NLS-PV-GFP. Proper GFP localization was confirmed. Cells were grown in the presence of calcium for 24 hr before the addition of 100 nM triptolide and 15 ng/ml TNF-α. NFκB transactivation by TNF-α alone was quite high, although both NES- and NLS-parvalbumin-transfected cells showed slightly lower levels of luciferase expression as compared to the vector control. Importantly, triptolide still retained the ability to suppress NFκB transactivation in all experimental conditions (Figure 5B). These results suggest that, while efficient induction of apoptosis by triptolide is calcium-dependent, inhibition of NFκB transcriptional control is not.
Triptolide Concentration Differentially Affects Cell Death and Inhibition of NFκB
Our results have implicated reversible binding of triptolide to a potential binding protein or complex that can be regulated by calcium. To understand if triptolide function is further separable between cell death and NFκB inhibition, we examined the effect of concentration on these two endpoints. HeLa cells were cultured in the presence of 0, 10, 25, 50, or 100 nM triptolide and were separately assessed for cell death or the ability to suppress NFκB transactivation promoted by TNF-α. Following 24−48 hr of culture, viable cells were recovered and counted. Following 24 hr, triptolide concentrations from 25 to 100 nM caused greater than 50% of the cells to undergo cell death, as assessed by detachment, clumping, and the failure to exclude trypan blue dye (data not shown). After 48 hr, nearly all cells treated within this concentration range of triptolide died (Figure 6A). While untreated HeLa cells underwent two cycles of division, 10 nM triptolide inhibited cell proliferation, but did not induce cell death. This is consistent with previous studies showing that low doses of triptolide cause cell cycle arrest rather than apoptosis [13]. It is also of note that triptolide's action on the cell, which ultimately results in cell death, is initially reversible, as 3−4 hr is the minimal incubation time required for commitment to apoptosis (data not shown).
Figure 6. Triptolide Concentration Differentially Affects Viability/Growth or NFκB Inhibition.
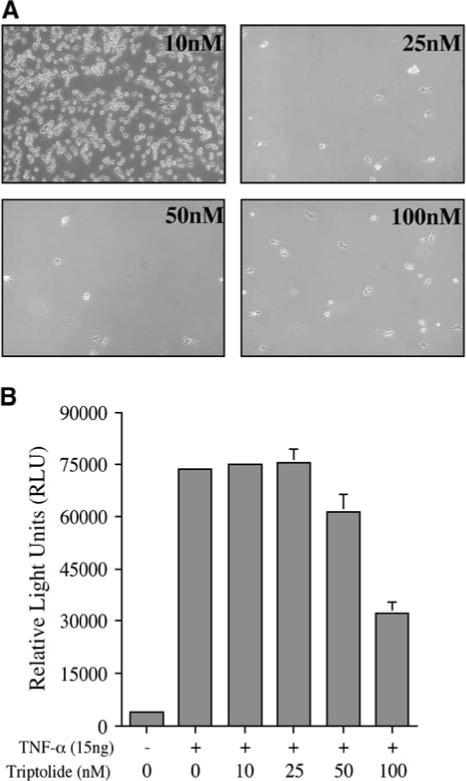
(A) HeLa cells were plated at an initial concentration of 5 × 105 and were allowed to grow 6 triptolide (10−100 nM) for 48 hr. Viable (adherent) cells were photographed under 25× bright-field microscopy, and cell death was assessed by trypan dye exclusion. Results are representative of three separate experiments. (B) After a transient transfection with the κB-luciferase reporter construct, HeLa cells were incubated with 15 ng/ml TNF-α ± triptolide (10−100 nM) for a total of 6 hr before assessing reporter activity. n = 4; data represented as mean ± SE.
NFκB transactivation was examined by using the κB-luciferase reporter construct. HeLa cells were transiently transfected and pretreated with 0−100 nM triptolide for 1 hr prior to TNF-α addition. Cells were assessed for NFκB-driven luciferase expression after an additional 5 hr of incubation, at which time TNF-α had induced transcriptional activity by approximately 15-fold in control cells. Neither 10 nM nor 25 nM triptolide inhibited TNF-α-driven transcriptional activity of NFκB (these concentrations are shown to inhibit proliferation or induce cell death, respectively), whereas 50 nM triptolide suppressed activity by 20%. The concentration of 100 nM had the most profound effect, exhibiting an average of 60% inhibition (Figure 6B). It is also of note that luciferase activity assayed after 24 hr with 10 nM triptolide + TNF-α still showed greater than a 20-fold induction (data not shown). Examination of these two biological endpoints at the same chronological time indicates that, while 10 nM is efficient at arresting cell growth, it cannot inhibit TNF-α-induced NFκB transcriptional activity. The results thus far support a divergence of triptolide-mediated functions: the pathway regulating growth arrest/death is more sensitive to triptolide and calcium than the mechanism leading to transcriptional repression of NFκB.
Triptolide Analogs Show Differential Abilities to Induce Cell Death or Inhibit NFκB Transactivation
Based on our studies examining the concentration-dependent effect of triptolide on the two measured biological endpoints, cell death and transcriptional repression, we wanted to determine how modulating triptolide's structure may also discriminate between the two pathways. For cell viability assays, HeLa cells were incubated with 0, 0.1, 1, or 10 μM of each analog or triptolide for 24 hr and then counted by using trypan blue dye exclusion. Triptolide, as shown before, effectively induced greater than 50% cell death at 0.1 μM, and there was no significant increase at the higher concentrations (Figure 7A). NFκB transcriptional inhibition was measured by using the κB-luciferase assay as previously described, after 5 hr of incubation with each analog (0−10 μM) and TNF-α addition. NFκB inhibition was greater than 60% and attenuated further as triptolide's concentration increased to 1 or 10 μM (Figure 7A).
Figure 7. Triptolide Analogs: Structural Divergence of Biological Functions.
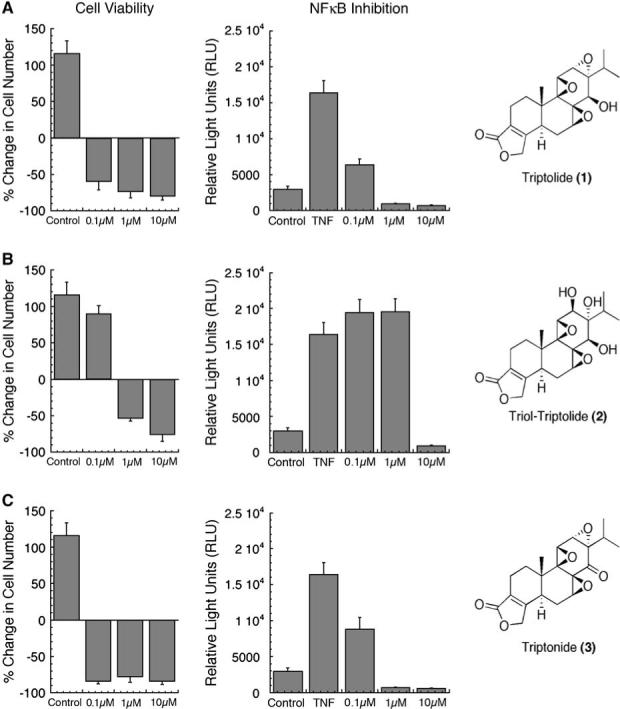
(A–C) All experiments were done with HeLa cells, and cell viability was measured 24 hr after the addition of triptolide or one of its analogs at concentration ranges of 0.1−10 μM. Cell viability was assessed by trypan dye exclusion and was recorded as the percent change in cell number from the time of triptolide or analog addition. Control cells were allowed to grow in media alone and represent a normal population doubling; n = 3. NFκB Inhibition was assessed after transfection with the κB-luciferase reporter construct and treatment with triptolide or one of its analogs (0.1−10 μM) and TNF-α for 5 hr. Control represents transfected cells without TNF-α addition. n = 4; data represented as mean ± SE. (A) Triptolide (1). (B) Triol-Triptolide (2). (C) Triptonide (3).
Upon disruption of the 12,13 epoxide in analog (2), differential effects were seen in regard to each biological endpoint. At 0.1 μM analog (2), cell viability was not different from the untreated control (Figure 7B), as compared to the ∼60% cell death observed at the equivalent concentration of triptolide.
If cells were allowed to continually grow at 0.1 μM analog (2) out to 72 hr, it became evident that there was an overall growth-suppressive effect (data not shown). Interestingly, 1 μM analog (2), a concentration that efficiently competes with triptolide for binding (Figure 1B), induced greater than 50% cell death, while having no effect on NFκB transcriptional activity (Figure 7B). It therefore appears that the mechanism of NFκB inhibition is more sensitive to the structural integrity of the 12,13 epoxide than is the cell growth/death regulatory pathway.
The most potent and biologically similar analog to triptolide was analog (3). Displacement of [3H]triptolide binding was near complete at both the 1 and 10 μM concentrations (Figure 1B). In fact, 1 μM analog (3) actually elicited a higher competitive ability than triptolide itself at the same concentration (Figure 1B). Both profiles of cell death and transcriptional repression mimicked triptolide, and there was no significant difference from 0.1 to 10 μM (Figure 7C). It is of note, however, that at 25 nM, a concentration shown to induce cell death by triptolide (Figure 6A), analog (3) had only a growth-suppressive effect (data not shown). Since competition for binding by analog (3) was so strong, these data would support the idea that triptolide-induced apoptosis at its lowest (25 nM) concentration is partially due to the functionality of the C-14 hydroxyl.
Discussion
Triptolide is a natural product isolated from the “Thunder God Vine” that has a long-standing history of medicinal value. Of the many small molecules (i.e., glycosides, terpenoids, and alkaloids) isolated from this herbal extract, triptolide has been shown to be one of the most biologically active. Its therapeutic value is high, as it has been demonstrated to inhibit inflammation (i.e., rheumatoid arthritis), reduce T cell reactivity leading to immunosuppression (i.e., lupus erythematosus or organ transplant rejection), induce apoptosis of cancerous cells, and even act as a male contraceptive. Although triptolide's predominant cytotoxic effect is largely apparent in lymphocytes and leukocytes, it is capable of inducing cell death in numerous solid tumors, including prostate [13], lung [26], breast, gastric carcinoma, and melanoma [12].
While triptolide has numerous physiological functions, there is minimal understanding of its precise mode of action. Since triptolide is a highly oxygenated diterpenoid with epoxides at the 7,8; 9,11; and 12,13 positions, it is plausible to believe that one or more is responsible for biological activity. In fact, previous research has demonstrated that the 9,11 epoxide in conjunction with the 14β hydroxyl may be responsible for triptolide's antineoplastic activity [18]. In contrast, the 12,13 epoxide has been implicated in both the anti-inflammatory and immunosuppressive effects [17]. Therefore, this limited structure-activity relationship alone would indicate that multiple cellular pathways are affected, and that triptolide could potentially interact with more than one target protein.
In this study, we sought to determine the general specificity of triptolide binding and further focused on two known endpoints of triptolide function: induction of cell death and inhibition of NFκB transactivation. To begin mechanism of action studies, we utilized [3H]triptolide as a cellular probe and discovered that binding was reversible and localized predominantly to the membrane fraction of the cell. Evidence for protein association was 2-fold: (1) a [3H]triptolide bound entity from the anion exchange resin DEAE specifically eluted upon an increasing step gradient of NaCl concentration, where free [3H]triptolide eluted in the flow-through. (2) Upon molecular weight fractionation by SDS-PAGE or size-exclusion chromatography, bound [3H]triptolide was associated with a high molecular mass (>250 kDa) that dissociated under denaturing conditions. Total cellular [3H]triptolide binding was found to be saturable, with a KD of 15.5 nM, as shown by radioligand saturation plots. However, due to limitations with volume, we could not test triptolide concentrations above 100 nM or with precision below 5 nM; due to these constraints, we cannot exclude the possibility of lower- or higher-affinity interactions that may fall beyond the testable range. Based on these data, it would be plausible that one target of triptolide binding may be a large membrane-associated protein or complex that at best supports a model of a reversibly covalent or unstable interaction. Additionally, since we discovered that the S-100 cytosolic fraction contained [3H]triptolide, we acknowledge that this may be the result of specific binding to other protein(s) or, conversely, may simply represent unbound triptolide.
Further exploration into the nature of triptolide binding led to the discovery of a calcium-dependent phenomenon: calcium depletion significantly increased triptolide binding while attenuating its proapoptotic function. Specifically, since cytosolic calcium levels are responsible for efficient triptolide-induced cell death, it would be of interest to determine if triptolide interacts with proteins sensitive to calcium flux. One hypothesis is that triptolide relies on a renewable source of calcium (such as through the endoplasmic reticulum) to initiate apoptosis by the disregulation of calcium homeostasis. In fact, as previously reported [11, 27], triptolide induces caspase activation, which could be the result of intracellular calcium overload and subsequent mitochondrial disruption.
Although calcium plays a role in triptolide-mediated apoptosis, we found that it does not influence the inhibition of NFκB transactivation, as elimination of either extracellular calcium or cell compartment-directed parvalbumin buffering had no effect. Since this divergence of a calcium requirement exists for triptolide-mediated cellular effects, it is reasonable to expect that more than one intracellular pathway is involved. However, we cannot know for certain if there is direct interaction with one versus multiple binding proteins. Triptolide may bind to a complex of proteins, thereby having the ability to manipulate multiple pathways while only interacting with one multiplex target. Alternatively, there may be multiple binding proteins with slightly higher or lower affinities that would explain the closely titrated biological effects of growth inhibition, cell death, and transcriptional suppression seen within a 40 nM span of effective concentration (10 nM, 25 nM, and 50 nM, respectively). Our receptor saturation data would support either case: (1) one protein target or complex or (2) several protein targets with similar KD values, making it difficult to pharmacologically resolve these entities.
Our limited structure-function analysis of triptolide binding and function involved the roles of the 12,13 epoxide and the 14β hydroxyl of triptolide. Previous work [17, 28] has demonstrated a link between the 12,13 epoxide and triptolide's anti-inflammatory activities by in vivo models, while we have examined the direct effect on NFκB transcription as well as binding properties of this triptolide analog (2). While 1−10 μM competition with analog (2) was just as efficient as triptolide, it was only able to suppress transcriptional activity at the highest concentration of 10 μM, thereby supporting its importance to triptolide's anti-inflammatory activity. The 12,13 epoxide was not as crucial to triptolide's role in cell survival, as all three concentrations of analog (2) had an effect in this regard: 100 nM was growth inhibitory, and 1−10 μM increased cell death to levels comparable with triptolide. Triptonide analog (3) has been independently shown to have potent anti-inflammatory, antineoplastic, and anti-male-fertility effects [29, 30], as well as being a reversible degradation product of triptolide itself [31]. Here, we have further established that analog (3) is capable of competing with triptolide for its binding sites and shares the efficiency of biological activity from 0.1 to 10 μM concentrations. However, a divergence in function was elucidated at 25 nM, the concentration at which triptolide can cause complete cell death, but analog (3) is slightly weaker, as it will only arrest the growth of cells. Therefore, triptolide's antineoplastic (pro-cell death) activity can be only modestly reduced by the formation of a ketone at C-14. These data lend further support to a divergence of triptolide function through more than one cellular pathway. In these pathways, the following are the variables regulating biological activity: (1) structural moieties of triptolide, (2) the biological concentration, and (3) the dependence upon the presence of calcium.
Although triptolide may be acting on more than one cellular process to elicit its physiological responses, one intriguing finding is that depletion of calcium can increase or stabilize triptolide binding while simultaneously weakening its apoptotic effect. This could be explained by a conformational change of a protein sensitive to free calcium concentrations (thereby facilitating binding) while at the same time being unable to elicit a functional response because that is also dependent upon calcium (cell death is delayed). One family of proteins that would meet these criteria would be the broad category of calcium channels. In members of this family, open channel conformation can be influenced by calcium concentration, and the functional effect (i.e., calcium release), again, is dependent on a renewable supply of calcium. Other possibilities may be the family of calcium pumps or sensors, as they too are sensitive to calcium flux. In contrast, we also observe that triptolide's inhibition of NFκB transactivation is not regulated by calcium. It is possible that triptolide blocks the recruitment of an essential transcriptional cofactor to the NFκB complex or inhibits further modification to NFκB directly (such as a phosphorylation event). These two pathways, however (calcium-dependent and -independent), probably converge to amplify the apoptotic response, as seen with the synergistic effect of triptolide plus TNF-α to promote apoptosis [15]. In this regard, triptolide would trigger TNF-α-induced apoptosis by inhibiting its proinflammatory transcriptional activity, while having yet another (calcium-dependent) mechanism to induce cell death as well. Removal of calcium would therefore only slow down cell death, as transcriptional repression, although slower, would ultimately result in the same fate.
Resolving triptolide's mode of action as well as potential binding proteins has proven to be complicated and a challenging endeavor. From the earliest studies elucidating the numerous cellular and physiological effects of triptolide, it was clear that this natural product had a broad spectrum of activity. The challenge lies with the intricate separation of the mechanisms that are required to inhibit inflammation, elicit immunosuppression, promote tumor cell death, and even abrogate male fertility. Future studies will require the biochemical identification of potential triptolide binding proteins as well as establishment of the biological relevance of each.
Significance
Triptolide has a broad range of therapeutic potentials ranging from attenuation of inflammation, suppression of autoimmunity, and the elimination or regression of certain tumors. Basic studies of triptolide's mechanisms of action are incomplete, and there is little understanding of how this small molecule can elicit such a broad range of effects. Utilizing [3H]triptolide as a probe, we have examined the properties of triptolide binding in the cell as well as addressed questions pertaining to two well-described biological endpoints of triptolide function: cell death and transcriptional repression of NFκB. A specific triptolide binding activity is present within intact cells, is reversible, associates predominantly with cellular membranes, and is sensitive to calcium levels. While triptolide binding increases upon extracellular calcium depletion, it is severely impaired in its ability to induce cell death. This observed calcium dependence is specific to the regulation of apoptosis, as triptolide's effect on NFκB transactivation is unaltered in the presence or absence of calcium. An overall separation of biological effects can be further discerned when triptolide is present at low nanomolar concentrations. While 10 nM is growth inhibitory and 25 nM induces cell death, neither of these concentrations can elicit transcriptional repression. Limited structure-function analysis utilizing triptolide analogs has demonstrated that, while competitive binding for triptolide interaction sites is intact, biological effects are highly dependent upon structural moieties. These findings implicate triptolide as functioning through at least two separate pathways distinguishable by calcium requirements, sensitivity to drug concentration, and preference toward structural entities. Further, we have started to characterize a specific triptolide interaction within the cell so that triptolide binding proteins may be identified in the future.
Experimental Procedures
Reagents
Triptolide was obtained from Sinobest, Inc. (China), and purity was 99%, as determined by HPLC. DMSO was used to dissolve triptolide and was then directly added into culture media for all experiments. Triptolide was tritiated by Sib Tech, Inc. (Newington, CT) and was resuspended in ethanol to a specific activity of 4−6 Ci/mmol. Purity was >95%, as confirmed by RP-HPLC on a Hypersil C18 column and by TLC on both C18 and silica gel. Epi-Triptolide/Triol-Triptolide (C.A.S. No 147852−78−6) and Triptonide (C.A.S. No 38647−11−9) were purchased from Sequoia Research Products (United Kingdom).
Cell Culture and Viability Studies
HeLa cells were incubated in DMEM or SMEM (Gibco) media + 10% FBS and were maintained at 37°C in 5% CO2 for all experiments. HeLa cell viability was assessed by trypan blue dye exclusion, as well as by morphological examination (nonviable cells were rounded and detached from culture plate).
[3H]Triptolide Labeling of HeLa Cells
Labeling studies were performed by the addition of approximately 30 nM [3H]triptolide directly into the culture media for 1 hr at 37°C. For cold competition studies, 1 μM triptolide was incubated with the cells for 1 hr before or after the addition of [3H]triptolide. Triptolide analog studies followed a similar protocol in which concentrations used for competition were either 1 or 10 μM and were added before [3H]triptolide. Medium was removed, and cells were washed × times in cold PBS. Total cell lysates were prepared (150 mM NaCl, 50 mM Tris-HCl [pH 7.4], 1 mM EDTA, 1% Triton X-100, and complete protease inhibitors [Roche]), and protein was quantitated before measuring the [3H]triptolide binding activity via liquid scintillation.
DE-52 anion exchange resin (Whatman, Inc.), a diethylaminoethyl (DEAE)-cellulose, was prepared for binding by a 1 M sodium chloride (NaCl) wash, followed by multiple washes with 0 M salt buffer (10mM HEPES [pH 7.4], 0.1 mM EDTA, 1 mM DTT, 0.1% Triton X-100). HeLa cell lysates labeled with [3H]triptolide were passed over the resin and allowed to bind at 4°C for 30 min before collecting the flow-through and subsequent washes. A step gradient from 0.0 to 1.0 M NaCl was used for protein elution. All fractions were subsequently counted by liquid scintillation.
For cellular fractionation studies, cells were allowed to swell on ice and were lysed by passage through a syringe in a hypotonic lysis buffer (10 μM Tris-HCl + complete protease inhibitors). The lysate was centrifuged at 100,000 × g, and the supernatant was saved as the S-100 cytosolic fraction. The pellet was washed and resolubilized in 1% Triton X-100 containing lysis buffer. After centrifugation, the supernatant was saved as the P-100 membrane fraction. Additionally, P-100 lysates were run out under native or reducing gel conditions without boiling. Gel slices were measured out in equal increments, and the molecular weight range of each was calculated. Each gel piece was crushed in ddH2O, followed by scintillation counting of the water extract.
[3H]Triptolide-Specific Binding
Saturation binding assays were accomplished in HeLa cells adhered on 6-well plates in DMEM + 10% FBS. All samples were at least 90% confluent at the time of addition of triptolide. Nonspecific binding of [3H]triptolide was assessed by the preincubation of 2 μM (nonlabeled) triptolide for 1 hr. After cold competition (or DMSO), 5, 10, 20, 50, or 100 nM [3H]triptolide was added into the cultures for an additional 1 hr, and then cells were lysed and counted for binding activity.
Transfection of Parvalbumin Constructs
All parvalbumin-GFP constructs and control vectors [24] were a gift of Anton Bennett (Yale University). HeLa cells were plated out on 6- or 12-well plates at a density of 5 × 105 or 1 × 105, respectively. Cells were transiently transfected with 0.5−1 μg of one of the following pcDNA3-derived plasmids for 24 hr in DMEM/10% FBS + Lipofectamine 2000 (Invitrogen): CMV-parvalbumin-GFP, CMV-NES-parvalbumin-GFP, CMV-NLS-parvalbumin-GFP. After confirmation of GFP expression and localization by microscopy, 100 nM triptolide was added into each transfected cell population (>90% transfection efficiency). Cell viability was assessed at 24 and 48 hr after triptolide addition by morphology and trypan blue dye exclusion.
NFκB Luciferase Assay
A triple κB promoter-luciferase reporter construct was a gift of Sankar Ghosh (Yale University). HeLa cells were plated at a density of 2 × 105 in 12-well plates and were transfected with 100 ng κB-luciferase plasmid plus Lipofectamine 2000 (Invitrogen) for 24 hr before the addition of 100 nM triptolide for 1 hr and 15 ng/ml recombinant (human) TNF-α (Roche) for an additional 5 hr. The transfection efficiency of HeLa cells was determined to be 80%−90%, and all samples were normalized to protein concentration. Luciferase assays were performed by using the Firefly luciferase kit as per manufacturer's protocol (Promega), and results were obtained on the Wallac Victor2 1420 Multilabel Counter (Perkin Elmer).
Acknowledgments
We would like to thank Dr. John Hines (Yale University) for helpful discussions with the radioligand binding assays. We would also like to thank Dr. Anton Bennett (Yale University) for the parvalbumin-GFP constructs and Dr. Sankar Ghosh (Yale University) for the κB-luciferase reporter. This work has been supported by the National Institutes of Health grant AI055914 (to C.M.C.) and an American Cancer Society postdoctoral fellowship (to S.J.L.).
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2−3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- 1.Su D, Song Y, Li R. Comparative clinical study of rheumatoid arthritis treated by triptolide and an ethyl acetate extract of Tripterygium wilfordii. Chung Hsi I Chieh Ho Tsa Chih. 1990;10:144–146. 131. [PubMed] [Google Scholar]
- 2.Tao X, Cush JJ, Garret M, Lipsky PE. A phase I study of ethyl acetate extract of the chinese antirheumatic herb Tripterygium wilfordii hook F in rheumatoid arthritis. J. Rheumatol. 2001;28:2160–2167. [PubMed] [Google Scholar]
- 3.Kupchan SM, Court WA, Dailey RG, Jr., Gilmore CJ, Bryan RF. Triptolide and tripdiolide, novel antileukemic diterpenoid triepoxides from Tripterygium wilfordii. J. Am. Chem. Soc. 1972;94:7194–7195. doi: 10.1021/ja00775a078. [DOI] [PubMed] [Google Scholar]
- 4.Wei YS, Adachi I. Inhibitory effect of triptolide on colony formation of breast and stomach cancer cell lines. Chung Kuo Yao Li Hsueh Pao. 1991;12:406–410. [PubMed] [Google Scholar]
- 5.Shamon LA, Pezzuto JM, Graves JM, Mehta RR, Wangcharoentrakul S, Sangsuwan R, Chaichana S, Tuchinda P, Cleason P, Reutrakul V. Evaluation of the mutagenic, cytotoxic, and antitumor potential of triptolide, a highly oxygenated diterpene isolated from Tripterygium wilfordii. Cancer Lett. 1997;112:113–117. doi: 10.1016/S0304-3835(96)04554-5. [DOI] [PubMed] [Google Scholar]
- 6.Matlin SA, Belenguer A, Stacey VE, Qian SZ, Xu Y, Zhang JW, Sanders JK, Amor SR, Pearce CM. Male antifertility compounds from Tripterygium wilfordii Hook f. Contraception. 1993;47:387–400. doi: 10.1016/0010-7824(93)90036-7. [DOI] [PubMed] [Google Scholar]
- 7.Lue Y, Sinha Hikim AP, Wang C, Leung A, Baravarian S, Reutrakul V, Sangsawan R, Chaichana S, Swerdloff RS. Triptolide: a potential male contraceptive. J. Androl. 1998;19:479–486. [PubMed] [Google Scholar]
- 8.Lu H, Hachida M, Enosawa S, Li XK, Suzuki S, Koyanagi H. Immunosuppressive effect of triptolide in vitro. Transplant. Proc. 1999;31:2056–2057. doi: 10.1016/s0041-1345(99)00262-6. [DOI] [PubMed] [Google Scholar]
- 9.Qiu D, Zhao G, Aoki Y, Shi L, Uyei A, Nazarian S, Ng JC, Kao PN. Immunosuppressant PG490 (triptolide) inhibits T-cell interleukin-2 expression at the level of purine-box/nuclear factor of activated T-cells and NF-κB transcriptional activation. J. Biol. Chem. 1999;274:13443–13450. doi: 10.1074/jbc.274.19.13443. [DOI] [PubMed] [Google Scholar]
- 10.Qiu D, Kao PN. Immunosuppressive and antiinflammatory mechanisms of triptolide, the principal active diterpenoid from the Chinese medicinal herb Tripterygium wilfordii Hook. f. Drugs R D. 2003;4:1–18. doi: 10.2165/00126839-200304010-00001. [DOI] [PubMed] [Google Scholar]
- 11.Jiang XH, Wong BC, Lin MC, Zhu GH, Kung HF, Jiang SH, Yang D, Lam SK. Functional p53 is required for triptolide-induced apoptosis and AP-1 and nuclear factor-κB activation in gastric cancer cells. Oncogene. 2001;20:8009–8018. doi: 10.1038/sj.onc.1204981. [DOI] [PubMed] [Google Scholar]
- 12.Yang S, Chen J, Guo Z, Xu XM, Wang L, Pei XF, Yang J, Underhill CB, Zhang L. Triptolide inhibits the growth and metastasis of solid tumors. Mol. Cancer Ther. 2003;2:65–72. [PubMed] [Google Scholar]
- 13.Kiviharju TM, Lecane PS, Sellers RG, Peehl DM. Antiproliferative and proapoptotic activities of triptolide (PG490), a natural product entering clinical trials, on primary cultures of human prostatic epithelial cells. Clin. Cancer Res. 2002;8:2666–2674. [PubMed] [Google Scholar]
- 14.Fidler JM, Li K, Chung C, Wei K, Ross JA, Gao M, Rosen GD. PG490−88, a derivative of triptolide, causes tumor regression and sensitizes tumors to chemotherapy. Mol. Cancer Ther. 2003;2:855–862. [PubMed] [Google Scholar]
- 15.Lee KY, Chang W, Qiu D, Kao PN, Rosen GD. PG490 (triptolide) cooperates with tumor necrosis factor-α to induce apoptosis in tumor cells. J. Biol. Chem. 1999;274:13451–13455. doi: 10.1074/jbc.274.19.13451. [DOI] [PubMed] [Google Scholar]
- 16.Chang WT, Kang JJ, Lee KY, Wei K, Anderson E, Gotmare S, Ross JA, Rosen GD. Triptolide and chemotherapy cooperate in tumor cell apoptosis. A role for the p53 pathway. J. Biol. Chem. 2001;276:2221–2227. doi: 10.1074/jbc.M009713200. [DOI] [PubMed] [Google Scholar]
- 17.Yu DQ, Zhang DM, Wang HB, Liang XT. Structure modification of triptolide, a diterpenoid from Tripterygium wilfordii. Yao Hsueh Hsueh Pao. 1992;27:830–836. [PubMed] [Google Scholar]
- 18.Kupchan SM, Shubert RM. Selective alkylation: a biomimetic reaction of the antileukemic triptolides? Science. 1974;185:791–792. doi: 10.1126/science.185.4153.791. [DOI] [PubMed] [Google Scholar]
- 19.Tomida T, Hirose K, Takizawa A, Shibasaki F, Iino M. NFAT functions as a working memory of Ca2+ signals in decoding Ca2+ oscillation. EMBO J. 2003;22:3825–3832. doi: 10.1093/emboj/cdg381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- 21.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 22.Rizzuto R, Pinton P, Ferrari D, Chami M, Szabadkai G, Magalhaes PJ, Di Virgilio F, Pozzan T. Calcium and apoptosis: facts and hypotheses. Oncogene. 2003;22:8619–8627. doi: 10.1038/sj.onc.1207105. [DOI] [PubMed] [Google Scholar]
- 23.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 24.Pusl T, Wu JJ, Zimmerman TL, Zhang L, Ehrlich BE, Berchtold MW, Hoek JB, Karpen SJ, Nathanson MH, Bennett AM. Epidermal growth factor-mediated activation of the ETS domain transcription factor Elk-1 requires nuclear calcium. J. Biol. Chem. 2002;277:27517–27527. doi: 10.1074/jbc.M203002200. [DOI] [PubMed] [Google Scholar]
- 25.Pauls TL, Cox JA, Berchtold MW. The Ca2+(−) binding proteins parvalbumin and oncomodulin and their genes: new structural and functional findings. Biochim. Biophys. Acta. 1996;1306:39–54. doi: 10.1016/0167-4781(95)00221-9. [DOI] [PubMed] [Google Scholar]
- 26.Frese S, Pirnia F, Miescher D, Krajewski S, Borner MM, Reed JC, Schmid RA. PG490-mediated sensitization of lung cancer cells to Apo2L/TRAIL-induced apoptosis requires activation of ERK2. Oncogene. 2003;22:5427–5435. doi: 10.1038/sj.onc.1206842. [DOI] [PubMed] [Google Scholar]
- 27.Liu Q, Chen T, Chen H, Zhang M, Li N, Lu Z, Ma P, Cao X. Triptolide (PG-490) induces apoptosis of dendritic cells through sequential p38 MAP kinase phosphorylation and caspase 3 activation. Biochem. Biophys. Res. Commun. 2004;319:980–986. doi: 10.1016/j.bbrc.2004.04.201. [DOI] [PubMed] [Google Scholar]
- 28.Zheng J. Screening of active antiinflammatory, immunosuppressive and antifertility components of Tripterygium wilfordii. III. A comparison of the antiinflammatory and immunosuppressive activities of 7 diterpene lactone epoxide compounds in vivo. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 1991;13:391–397. [PubMed] [Google Scholar]
- 29.Pei RJ, Qi LH, Liu XJ. Effects of triptonide on mouse immune functions. Zhongguo Yao Li Xue Bao. 1993;14:238–242. [PubMed] [Google Scholar]
- 30.Wang L, Ye W, Hui L, Liu X, Guo Y. Male contraception of triptonide and its function mechanism. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2000;22:223–226. [PubMed] [Google Scholar]
- 31.Mao YP, Tao XL, Lipsky PE. Analysis of the stability and degradation products of triptolide. J. Pharm. Pharmacol. 2000;52:3–12. doi: 10.1211/0022357001773625. [DOI] [PubMed] [Google Scholar]