

Abstract
There is a significant body of experimental evidence that a rise in intracellular reactive oxygen species (ROS) contributes to senescence. Here we review experiments where entry into senescence has been evaluated in cells whose intracellular ROS levels have been modulated by growth in either high or low ambient oxygen concentrations, or where the cellular antioxidant status has been perturbed. In addition, we discuss the observations that senescence triggered by oncogene expression also appears to be in part mediated by a rise in ROS levels. Finally, we discuss the emerging evidence that in vivo senescence might also be triggered by a rise in cellular oxidant levels. Although these data tend to support a role for ROS in mediating senescence, significant questions remain as to whether ROS act in a random or specific fashion and what precise oxidant species acts as the potential senescence trigger.
Since the introduction of the ‘Free Radical Theory of Aging’ by Harman over 50 years ago, oxidants have been inextricably linked to aging. Senescence which many-but not all- view as a cellular corollary to organismal aging, would appear to represent an interesting and testable model for the role of ROS in the aging process. In many ways experiments performed over the last several decades have validated the link between oxidants and senescence. As we will review, increasing intracellular oxidants by altering ambient oxygen concentrations or lowering antioxidant levels accelerates the onset of senescence while lowering ambient oxygen or increasing ROS scavenging appears to delay senescence. In addition, conditions that induce senescence often appear to be accompanied by a rise in intracellular ROS levels and perhaps most straightforwardly, treating cells exogenously with certain hydrogen peroxide concentrations can trigger entry into a senescent-like state. With that said the precise proof that ROS cause rather than correlate with entry into senescence and the relevant intracellular molecular senescent-inducing pathways that oxidants trigger remain relatively elusive. Here we review those experiments that link a rise in ROS levels to entry into senescence. In particular we will review three areas. First we will discuss experiments that have manipulated intracellular ROS levels to ascertain the subsequent effects on senescence. Next we will discuss the role of oxidants in oncogene-induced senescence. Finally, we will review the relatively recent evidence for a potential role of altered ROS levels in the induction of in vivo senescence.
Ambient Oxygen and Cellular Senescence
The observations by Hayflick that normal human diploid cells have a finite number of population doublings [1] provided the impetus for many other laboratories to begin to define the genetic and environmental factors regulating in vitro senescence. There was considerable concern initially that senescence was not an intrinsic property of cells and that it was the artificial nature of tissue culture systems that was in some way responsible for limiting the growth of cells. For instance, many were concerned that perhaps there was some essential ingredient needed for cell growth that was missing from the standard tissue culture growth cocktail. Indeed, early experiments suggested that the addition of hydrocortisone, additional bovine serum albumin or vitamin E might prolong cellular lifespan. It was also realized that the growth of cells in culture took place in an abnormally high oxygen concentration. Most cells in our body are exposed to oxygen concentration closer to 5% rather than the 20% of ambient air. Using this knowledge, Packer and his colleagues provided the first systematic evaluation of the role of ambient oxygen on in vitro senescence [2]. Using the well established human diploid fibroblast strain WI-38 first established by Hayflick, Packer was able to show that the cumulative number of population doublings was not a fixed number but rather was influenced by the amount of ambient oxygen. Compared to cells grown in the standard tissue culture environment of 20%, cells grown in 10% oxygen had considerably more population doublings (on average 20–30% more) while cells grown in a 50% oxygen environment had a similarly reduced in vitro lifespan. These results suggested that oxidative damage might be limiting for cell growth in culture.
In recent times, there has been considerable refinement in our ability to define the molecular events occurring with oxygen exposure. Using primary mouse cells with an integrator ‘mutation reporter’ a systematic evaluation of DNA damage occurring at 20% versus 3% oxygen has been performed [3]. Interestingly, growth of mouse embryonic fibroblasts (MEFs) in culture appeared to increase the frequency of genomic rearrangement 3-fold compared to what was observed when these cells were maintained within the embryo. This increased genomic instability occurred at both 3% and 20% oxygen, again giving ammunition to those investigators who worry about the potential artifactual nature of tissue culture manipulations. Additional experiments also demonstrated that MEFs grown in the standard 20% oxygen exhibited 3–4 fold higher levels of oxidatively damaged DNA than cells grown at 3% [4]. Interestingly, while mouse cells grown at 20% oxygen underwent senescence, those same cells when maintained at 3% oxygen did not [4]. Furthermore, mouse cells grown at 20% oxygen exhibit more damage than human cells that are cultured at this same oxygen concentration suggesting important species differences in oxygen sensitivity. Clearly, another important difference between mouse and human cells is the length of their telomeres. While telomere attrition is not felt to be limiting for the growth of primary mouse cells, the lost of telomere length is felt to be the predominant mechanism for human replicative cell senescence. In this regard it is interesting to note that telomere shortening is also dependent on ambient oxygen concentration. For instance, evidence suggest that under normoxic conditions, telomeres shorten by roughly 90 base pairs per population doubling, while the same cells grown in 40% oxygen reduce their telomeres at a rate of nearly 500 base pairs per division [5]. These results provide a molecular explanation of the early observation that ambient oxygen regulated replicative senescence of human cells.
The results discussed above suggest that oxygen concentration limits cellular lifespan in vitro. Most investigators believe that oxygen itself was not directly responsible for these effects. Studies with isolated mitochondria had detailed that the production of reactive oxygen species (ROS) increased as the concentration of ambient oxygen rose [6]. Therefore, it has been widely assumed that altering ambient oxygen was a surrogate for increasing the intracellular level of ROS secondary to aerobic respiration. It seemed reasonable to test this notion by directly exposing cells to oxidative stress. Such analysis was performed initially by Ames and colleagues. Using a variety of primary human lines these investigators exposed cells to sub-lethal concentrations of hydrogen peroxide [7–9]. Such exposure produced a senescent-like state wherein the treated cells remained viable but became unresponsive to growth factor or serum stimulation [7]. Molecular analysis of these hydrogen peroxide treated cells revealed that oxidant exposure induced a predominant G1 arrest with an increase in p53 protein levels and increased p53 activity, including the subsequent increased expression of the p53-dependent cell cycle inhibitor, p21[9].
Another link between intracellular ROS levels and cellular senescence came from observations where the levels of intracellular antioxidants were varied. For instance, the addition of ipophillic spin trapping chemical antioxidant α-phenyl-t-butyl nitrone (PBN), extended the lifespan of human cells in culture [8]. Similar experiments with protein antioxidants have been done. For instance, human fibroblasts engineered to have increased expression of extracellular superoxide dismutase (SOD) showed an extension of lifespan under both normoxic and hyperoxia conditions [10]. This extension appeared to be predominantly through a reduction in the rate of telomere attrition. The converse experiments have also been performed. For instance, RNAi mediated knockdown of SOD1 (the copper-zinc cytosolic form of the enzyme) in primary human fibroblasts induced a rapid p53-mediated senescent arrest [11]. Indeed, knockdown of SOD1 in p53-deficient human fibroblasts did not induce arrest, while in certain transformed cells, SOD1 inhibition appeared to trigger cell death. In contrast to these results, in certain cell types such as the human colorectal cancer cell line HCT116, overexpression of SOD2 (the manganese containing mitochondrial localizing form) induced growth arrest and an increase in senescence [12]. Again, these effects appeared to require the induction of p53. This discrepancy points out a number of unresolved issues including how normal and transformed cells might perceive and respond to an increase in ROS and whether the molecular events required for senescence differ in these two types of cells. In addition, also unresolved is the exact intracellular species that mediates-directly or indirectly- the induction of senescence. For instance, the enzymatic activity of SOD is to convert superoxide to hydrogen peroxide. Nonetheless, it remains controversial and difficult to prove exactly how steady state levels of superoxide and hydrogen peroxide are perturbed when the levels of SOD1 or SOD2 are either increased or decreased. These concerns were further magnified by observations in whole organisms. For instance, animals that are haploinsufficient for SOD2 do not seem to have a shortened lifespan, although there appears to be an increase in DNA damage and a corresponding increase in cancer predisposition [13]. In contrast, overexpression of the hydrogen peroxide scavenger catalase, especially within the mitochondrial compartment does extend the maximal lifespan of transgenic mice [14]. Thus the exact oxidant species-if any- that regulates in vitro senescence or in vivo aging remains unclear, although most evidence would suggest hydrogen peroxide is the primary mediator.
Oncogene-Induced Senescence: A role for ROS
As noted above, senescence can be induced simply by growing cells in culture (replicative senescence) or by subjecting cells to non-lethal stress such as hydrogen peroxide. A significant insight into the potential physiological relevance of senescence came with the observation that primary cells with high level expression of the Ras oncogene underwent rapid senescence [15]. These results were unexpected because the property of oncogenes was routinely associated with uninhibited and increased growth rates. Nonetheless, the basis for this prejudice was based largely on work where the oncogene was expressed in immortalized cell type such as NIH 3T3 cells that do not undergo senescence. In contrast, in normal diploid fibroblasts, Ras expression led to a rapid, telomere-independent growth arrest characterized by increased p16 expression as well as increased p21 expression [15]. Subsequent to these observations it was demonstrated that in primary cells, increased Ras expression results in an increase in intracellular ROS levels [16]. This rise in ROS appears to be important for inducing cellular senescence because culturing Ras expressing cells in either low oxygen or treating these cells with a hydrogen peroxide scavenging agent such as N-acetylcysteine (NAC) blocked Ras-induced senescence [16].
It remains unclear exactly how ROS function in the case of Ras-induced senescence, as well as how Ras expression triggers a rise in ROS levels within cells. For the latter question, one potential candidate is 5-lipoxygenase (5LO). This enzyme generates ROS in the process of its enzymatic conversion of arachidonic acid to leukotrienes. In cells engineered to overexpress Ras, 5L0 activity increases [17]. Similarly, simple 5L0 overexpression promotes senescence in a redox dependent, p53/p21 mediated fashion [17]. Other potential downstream mediators have also been implicated. Similar to diploid fibroblasts, in human ovarian surface epithelial cells, expression of an activated Ras gene induces a rapid growth arrest. Investigators using this experimental model performed an RNAi screen to characterize genes, which when inactivated, allowed the cells to overcome Ras-induced growth arrest. Using this approach it was determined that the MINK kinase was required to enforce Ras-induced growth arrest [18]. MINK is an upstream regulator of p38 MAPK, a known stress activated pathway. Interestingly, in these cells, the activation of MINK by Ras requires the generation of ROS. Another group has implicated Ras-induced repression of sestrin as an important mechanism for the Ras-induced ROS levels [19]. Sestrins are in turn modulators of peroxiredoxins, an important class of intracellular protein antioxidants. These investigators also noted an important connection between Ras, ROS and p53 activity. The ability of Ras to induce increases in ROS levels was greatly influenced by the presence or absence of p53, with sustained oxidant levels (> 7days) only achieved in cells expressing Ras but lacking p53. The intersection of Ras, ROS, and p38 MAPK was also made clearer by a recent study suggesting Ras-induced ROS levels activate p38-α MAPK and in normal cells can induce senescence while highly tumorgenic cells have inactivated this redox-sensitive pathway [20].
Expression of an activated form of Ras is not the only genetic way of inducing rapid senescence in a seemingly ROS-dependent fashion. Recently, using a human cell line expressing a temperature sensitive mutant of the simian virus 40 large T antigen that binds and inactivates both p53 and pRb, it was demonstrated that engagement of the p16INK4a/Rb pathway also induces a rise in ROS levels. In this system, redox-dependent activation of PKCδ provided a positive feedback loop for continuous ROS generation [21]. These investigators further demonstrated that persistent ROS increases were required for the inhibition of cytokinesis observed in senescent cells. Another example is the role of Akt in endothelial cells. In this cell type, continuous overexpression of Akt results in a senescence-like growth arrest [22]. This appears to occur through Akt’s regulation of the downstream transcription factor Foxo3a. Foxo proteins are members of the well conserved Forkhead family of transcriptional regulators. In C.elegans the Foxo3a ortholog is called DAF-16 and a variety of evidence suggests that DAF-16 activity can regulate the overall lifespan of the worm. Interestingly, the transcriptional targets of DAF-16 include many genes involved in ROS metabolism [23]. This role in antioxidant gene transcription is evolutionary well conserved since observations suggest that both SOD and catalase are regulated by mammalian Foxo3a [24, 25].
Senescence, ROS and Disease
The original ‘free radical theory of aging’ postulated that ROS contributed not only to the rate we age but also served as the impetus for a number of age-related diseases [26]. There are numerous studies suggesting that oxidants might contribute to a wide spectrum of human maladies including cancer, cardiovascular disease and a host of neurodegenerative conditions [6]. There is also a growing body of literature suggesting that senescence within progenitor cells or resident mature cells might contribute to the aging or disease phenotype. Increasingly there is also the notion that dysregulated production of ROS might drive cells into premature senescence in vivo and that the accumulation of these senescent cells within tissues might inhibit normal tissue homeostasis.
Because the hematopoietic stem cell (HSC) system is perhaps the most widely studied and best characterized in vivo system of stem and progenitor cells, a number of recent studies have provided an important link between ROS and stem cell self-renewal. Stem cells can by definition give rise to differentiated progenitors as well as to new stem cells. This latter property is called self-renewal and is essential to maintain and replenish stem cell numbers. Several recent studies have suggested that elevated ROS within the stem cell compartment lead to a rapid decline in stem cell self-renewal. For instance deletion of the ataxia telangiectasia mutated (Atm) kinase results in increased ROS levels within the HSC compartment. This oxidative stress results in a very rapid (months) decline in HSC number and function [27]. When Atm−/− mice are treated with antioxidants the defect in stem cell self-renewal is rescued. Similarly, deletion of all three Foxo genes within HSCs also leads to a rise in ROS within these stem cells [28]. This rise appears to be related to the role, as discussed above, of Foxo transcription factors to regulate antioxidant defenses. Again, this rise in ROS levels within the HSC compartment results in a block in stem cell self-renewal. In each of these cases, it is currently not entirely clear the full range of biological responses to the observed rise in ROS levels. In the case of the triple Foxo deletions there was an observed increase in apoptosis. However it seems probable that in some cases the rise in ROS within the HSC compartment might also accelerate stem/progenitor cell senescence.
Another potential example of ROS mediating senescence in vivo comes from studies of the vasculature. There are a number of clearly identifiable risk factors for atherosclerosis including diabetes, high cholesterol and elevated blood pressure. A long standing notion is that many if not all of the identified risk factors contribute to vascular disease by inducing redox stress within the vessel wall [29]. Perhaps it is not surprising that direct analysis of animal and human atherosclerotic plaque has revealed that diseased arterial segments exhibit an accumulation of senescent cells [30]. In culture, known risk factors such as oxidized LDL [31], or suspected risk factors such as homocysteine [32] appear to accelerate endothelial cell senescence in a redox dependent fashion. Conversely, agents such as aspirin that are known to be clinically beneficial appear to reduce oxidative stress within endothelial cells and hence delay the onset of senescence [33]. In addition, the level of circulating endothelial progenitor cells (EPCs), a presumed bone marrow derived cell that appears to maintain vascular homeostasis, are reduced in patients with known risk factors [34]. The EPC obtained from patients with multiple risk factors also exhibited senescence in vitro at an accelerated rate [34]. These and other data suggest that oxidative stress within the stem and progenitor compartment might contribute to accelerated senescence and explain the sharp age-dependent relationship of a number of human diseases.
Summary
On a cellular level there is abundant evidence that a rise in ROS levels contributes to senescence. Nonetheless, multiple questions remain. One unresolved issue is whether there is a gradient of ROS-induced effects. This notion was first inferred from the addition of exogenous hydrogen peroxide where it was clear that in some cells, very high doses of hydrogen peroxide (>1 mM) induced cell death, moderate doses (100–500 µM) induced senescence, while lower doses either were without effect or were actually mitogenic. This diverse biological spectrum of activity to ROS is puzzling and the exact molecular basis for these divergent phenotypes is largely unexplained. An additional question is how ROS participate in inducing senescence. Two not necessarily mutually incompatible scenarios exist. The first possibility is that ROS induce random damage to cellular components and therefore act as non-specific mediators of senescence. The second explanation is that ROS activate specific pathways within cells. Both scenarios are consistent with the observations of extending cellular lifespan under low oxygen conditions or for that matter for many of the observations with oncogene induced senescence. We have in general supported the concept that ROS function as messenger molecules [35] activating specific redox-dependent targets and that it is the activation of these targets that induces senescence and not the level of ROS per se. Others have disagreed and have made very cogent arguments for why oxidants may function solely in a random and non-specific fashion [36]. By whatever mechanism, the growing interest in in vivo senescence as a predictor or contributor to age-related diseases suggests that understanding the role of ROS in these processes will have important implications.
Figure.
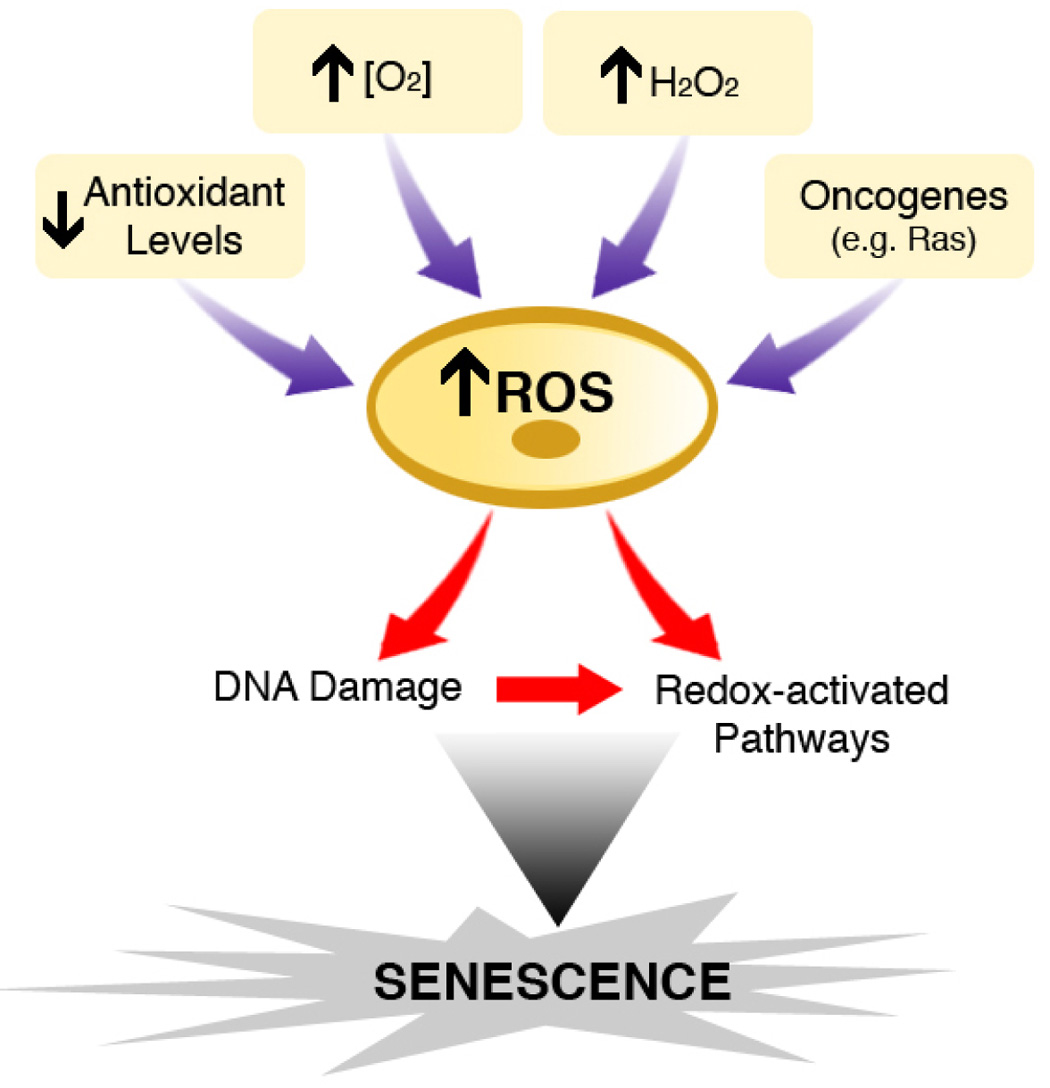
ROS as a common mediator of cellular senescence. Various extrinsic or intrinsic manipulations including changing the intracellular antioxidant status, raising ambient oxygen concentration, directly exposing cells to hydrogen peroxide or the expression of certain oncogenes, all appear to produce a rise in intracellular ROS levels. This rise in ROS within cells accompanies and potentially triggers senescence entry. As discussed in the text, oxidants may directly activate certain redox-sensitive pathways linked to senescence. Alternatively, oxidants might non-specifically induce a spectrum of damage to cellular components (e.g. DNA) that directly leads to senescence or this damage might induce senescence by secondarily activating important intracellular pathways, such as the DNA damage response.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hayflick L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 2.Packer L, Fuehr K. Low oxygen concentration extends the lifespan of cultured human diploid cells. Nature. 1977;267:423–425. doi: 10.1038/267423a0. [DOI] [PubMed] [Google Scholar]
- 3.Busuttil RA, Rubio M, Dolle ME, Campisi J, Vijg J. Oxygen accelerates the accumulation of mutations during the senescence and immortalization of murine cells in culture. Aging Cell. 2003;2:287–294. doi: 10.1046/j.1474-9728.2003.00066.x. [DOI] [PubMed] [Google Scholar]
- 4.Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–747. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.von Zglinicki T, Saretzki G, Docke W, Lotze C. Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: a model for senescence? Exp Cell Res. 1995;220:186–193. doi: 10.1006/excr.1995.1305. [DOI] [PubMed] [Google Scholar]
- 6.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Chen Q, Ames BN. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc Natl Acad Sci U S A. 1994;91:4130–4134. doi: 10.1073/pnas.91.10.4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Q, Fischer A, Reagan JD, Yan LJ, Ames BN. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc Natl Acad Sci U S A. 1995;92:4337–4341. doi: 10.1073/pnas.92.10.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen QM, Bartholomew JC, Campisi J, Acosta M, Reagan JD, Ames BN. Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem J. 1998;332:43–50. doi: 10.1042/bj3320043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serra V, von Zglinicki T, Lorenz M, Saretzki G. Extracellular superoxide dismutase is a major antioxidant in human fibroblasts and slows telomere shortening. J Biol Chem. 2003;278:6824–6830. doi: 10.1074/jbc.M207939200. Epub 2002 Dec 9. [DOI] [PubMed] [Google Scholar]
- 11.Blander G, de Oliveira RM, Conboy CM, Haigis M, Guarente L. Superoxide dismutase 1 knock-down induces senescence in human fibroblasts. J Biol Chem. 2003;278:38966–38969. doi: 10.1074/jbc.M307146200. Epub 2003 Jul 18. [DOI] [PubMed] [Google Scholar]
- 12.Behrend L, Mohr A, Dick T, Zwacka RM. Manganese superoxide dismutase induces p53-dependent senescence in colorectal cancer cells. Mol Cell Biol. 2005;25:7758–7769. doi: 10.1128/MCB.25.17.7758-7769.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR, Alderson NL, Baynes JW, Epstein CJ, Huang TT, Nelson J, Strong R, Richardson A. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics. 2003;16:29–37. doi: 10.1152/physiolgenomics.00122.2003. [DOI] [PubMed] [Google Scholar]
- 14.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. Epub 2005 May 5. [DOI] [PubMed] [Google Scholar]
- 15.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 16.Lee AC, Fenster BE, Ito H, Takeda K, Bae NS, Hirai T, Yu ZX, Ferrans VJ, Howard BH, Finkel T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem. 1999;274:7936–7940. doi: 10.1074/jbc.274.12.7936. [DOI] [PubMed] [Google Scholar]
- 17.Catalano A, Rodilossi S, Caprari P, Coppola V, Procopio A. 5-Lipoxygenase regulates senescence-like growth arrest by promoting ROS-dependent p53 activation. Embo J. 2005;24:170–179. doi: 10.1038/sj.emboj.7600502. Epub 2004 Dec 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nicke B, Bastien J, Khanna SJ, Warne PH, Cowling V, Cook SJ, Peters G, Delpuech O, Schulze A, Berns K, Mullenders J, Beijersbergen RL, Bernards R, Ganesan TS, Downward J, Hancock DC. Involvement of MINK, a Ste20 family kinase, in Ras oncogene-induced growth arrest in human ovarian surface epithelial cells. Mol Cell. 2005;20:673–685. doi: 10.1016/j.molcel.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 19.Kopnin PB, Agapova LS, Kopnin BP, Chumakov PM. Repression of sestrin family genes contributes to oncogenic Ras-induced reactive oxygen species up-regulation and genetic instability. Cancer Res. 2007;67:4671–4678. doi: 10.1158/0008-5472.CAN-06-2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11:191–205. doi: 10.1016/j.ccr.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, Nakayama KI, Ide T, Saya H, Hara E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol. 2006;8:1291–1297. doi: 10.1038/ncb1491. Epub 2006 Oct 8. [DOI] [PubMed] [Google Scholar]
- 22.Miyauchi H, Minamino T, Tateno K, Kunieda T, Toko H, Komuro I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. Embo J. 2004;23:212–220. doi: 10.1038/sj.emboj.7600045. Epub 2004 Jan 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murphy CT. The search for DAF-16/FOXO transcriptional targets: approaches and discoveries. Exp Gerontol. 2006;41:910–921. doi: 10.1016/j.exger.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 24.Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295:2450–2452. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- 25.Kops GJPL, Dansen TB, Polderman PE, Saarloos I, Wirtz KWA, Coffer PJ, Huang T-T, Bos JL, Medema RH, Burgering BMT. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 26.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 27.Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, Ikeda Y, Mak TW, Suda T. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 28.Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, Armstrong SA, Passegue E, DePinho RA, Gilliland DG. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 29.Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2005;25:29–38. doi: 10.1161/01.ATV.0000150649.39934.13. [DOI] [PubMed] [Google Scholar]
- 30.Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007;100:15–26. doi: 10.1161/01.RES.0000256837.40544.4a. [DOI] [PubMed] [Google Scholar]
- 31.Breitschopf K, Zeiher AM, Dimmeler S. Pro-atherogenic factors induce telomerase inactivation in endothelial cells through an Akt-dependent mechanism. FEBS Lett. 2001;493:21–25. doi: 10.1016/s0014-5793(01)02272-4. [DOI] [PubMed] [Google Scholar]
- 32.Xu D, Neville R, Finkel T. Homocysteine accelerates endothelial cell senescence. FEBS Lett. 2000;470:20–24. doi: 10.1016/s0014-5793(00)01278-3. [DOI] [PubMed] [Google Scholar]
- 33.Bode-Boger SM, Martens-Lobenhoffer J, Tager M, Schroder H, Scalera F. Aspirin reduces endothelial cell senescence. Biochem Biophys Res Commun. 2005;334:1226–1232. doi: 10.1016/j.bbrc.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 34.Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 35.Colavitti R, Finkel T. Reactive oxygen species as mediators of cellular senescence. IUBMB Life. 2005;57:277–281. doi: 10.1080/15216540500091890. [DOI] [PubMed] [Google Scholar]
- 36.Passos JF, Von Zglinicki T. Oxygen free radicals in cell senescence: are they signal transducers? Free Radic Res. 2006;40:1277–1283. doi: 10.1080/10715760600917151. [DOI] [PubMed] [Google Scholar]