

Abstract
Traditionally, four groups of factors have been identified in the etiology of temporomandibular disorder (TMD): anatomical variation in the masticatory system; psychosocial characteristics; pain in other body regions; and demographics. Orthodontic treatment has been variously cited both as a protective and harmful factor in TMD etiology. Recently, a search has begun for a genetic influence on TMD etiology. Genetic markers can be of additional value in identifying gene-environment interactions, that is, isolating population sub-groups, defined by genotype in which environmental influences play a relatively greater or lesser etiological role. This paper reviews concepts and study design requirements for epidemiological investigations into TMD etiology. Findings are presented from a prospective cohort study of 186 females that illustrate an example of gene-environment interaction in TMD onset. Among people with a variant of the gene encoding catechol-O-methyl-transferase, an enzyme associated with pain responsiveness, risk of developing TMD was significantly greater for subjects who reported a history of orthodontic treatment compared with subjects who did not (P=0.04). While further studies are needed to investigate TMD etiology, this genetic variant potentially could help to identify patients whose risk of developing TMD is heightened following orthodontic treatment, hence serving as a risk marker useful in planning orthodontic care.
INTRODUCTION
Temporomandibular disorders (TMD) are a set of painful musculoskeletal conditions of multifactorial etiology that affect an estimated 5–15 percent of adults in the US population.1 According to the US Surgeon General, TMD limits oral function, affects quality of life and incurs billions of dollars in health care costs annually.2 While it is clear that no single cause is responsible for development of TMD, there is a potentially bewildering array of factors that are thought to be involved in its etiology.3 Traditionally, the focus has been on anatomical variation in the masticatory system together with pathology and trauma of the TM joints and muscles themselves.4 Similarly, there has been a long-standing recognition that psychosocial characteristics, including depression, and stress, are associated with TMD.5 Pathological and physiological processes that involve other body regions or which produce pain in other body regions6 may also contribute to the etiology of TMD by influencing endogenous pain regulatory systems.7 Conceptual models that link all of those factors have been developed and are characterized as “biopsychosocial” models of TMD.8 Onset of TMD is also known to vary among age groups and between the sexes. A systematic review of the literature concluded that depression, pre-existing pain conditions and female sex were risk factors most consistently associated with TMD.9
Other researchers are less certain about the causes of TMD. Based on a review of this body of research, Svensson,10 concluded that “the pathophysiology and etiology of craniofacial muscle pain are not known in sufficient detail to allow causal treatment.” Other investigators have begun to search for a genetic influence on TMD etiology in what has been described provocatively as a transition in research “from chasing occlusal contacts to vulnerability alleles”.11
Orthodontic treatment has been variously cited both as a protective and harmful factor in TMD etiology. A systematic review of 31 studies drew no definitive conclusion about the relationship, and found that the data “do not indicate that traditional orthodontic treatment increased the prevalence of TMD”.12 Yet the authors of that review commented that they were hampered by variations in study design and the lack of consistent, reliable and valid diagnostic criteria for TMD among the 31 studies reviewed. For example, only eight studies used longitudinal study designs that compared treated and untreated groups, hence meeting the minimum epidemiological study design requirement for drawing causal inferences. Furthermore, six of those studies used different combinations of symptoms or signs to assess TMD, and the remaining two used an index of dysfunction rather than a case definition that is necessary to assess TMD risk per se.
This brief introduction is intended to illustrate some of the challenges that arise when attempting to review published evidence about TMD risk and factors that influence that risk. The challenges include conceptual distinctions, for example between “etiological factors” and factors that merely are “associated” with disease. Interpretations are further limited by study design, including the criteria used to assess TMD and the analytic methods used to compare risk among groups of subjects. Finally, researchers and clinicians are now becoming increasingly aware of the possibility that genetic variation may play a role in pain perception and onset of TMD.
Aims
This paper reviews risk factors for TMD and illustrates potential for the existence of gene-environment interactions that can influence TMD risk, using orthodontic treatment as an example of an environmental influence. The specific aims are: (i) to review definitions of risk and methods required for identification of risk factors; (ii) to summarize recent findings regarding genetic risk factors for TMD; and (iii) present new findings from a prospective study that examined the influences of genotype, pain amplification and psychological factors on TMD risk. In that study, approximately one half of subjects reported a history of orthodontic treatment, therefore creating an opportunity to examine the relationship between genotype, orthodontic treatment and TMD risk.
Methods for identifying risk factors
Requirement for case definition
Quantitative methods for studying TMD risk in human populations require first that a case definition be developed by which individuals can be classified either as having TMD or not having TMD. The most comprehensive case definition comes from the work of Dworkin and LeResche which led the development of the research diagnostic criteria for TMD (RDC/TMD).13 The case definition is based on specific combinations of symptoms reported by study subjects together with clinical signs detected by trained examiners who use clearly-defined methods for clinical examination, thereby evaluating pain and dysfunction in the temporomandibular tissues. RDC methods and criteria have been tested for reliability and validity, and they have been found to be superior to case definitions for TMD that are based solely on self-reported symptoms, such as responses to the question, “Have you had pain in the face, jaw, temple, in front of the ear, or in the ear?”
Quantifying risk of developing TMD
An individual’s risk of developing TMD is measured as a probability, and hence can range from zero to one. As with all diseases, TMD risk must be estimated from observations made among a population of individuals, preferably using prospective cohort studies of people who, at baseline, are known to be free of the disease. Cumulative incidence, calculated as the proportion of people in the cohort who are diagnosed with the condition during a nominated time period, represents the best estimate of an individual’s risk of developing disease. For example, in a study of 588 people enrolled in a health maintenance organization who did not report TMD at baseline, Von Korff et al14 found that 38 people (6.5%) reported TMD symptoms when interviewed three years after baseline. The “average” person in that cohort therefore had a risk of 0.065 of developing TMD symptoms over three years.
Estimates of disease risk are informative primarily for public health purposes to estimate the burden of disease in a population and likely need for health care. However, “average” disease risk is a misnomer when applied to individuals, because no individual can possess factors that influence disease in the same ratios that those factors exist in a population. For example, in the study by Von Korff et al,14 57.8% of the baseline sample was female, and TMD incidence among females was 7.7%, compared with 4.3% among the 42.2% who were males. Yet the “average” person in the sample of 588 studied by Von Korff et al14 was not 0.578 female and 0.422 male.
Identifying risk markers and risk factors for TMD
Information about disease risk becomes more relevant for individuals when it is used to provide advice about attributes of people that are associated with relatively high- or low-risk of disease onset. A widely used measure of the extent to which an attribute is associated with disease risk is relative risk, computed as the quotient of risk among people who have an attribute of interest divided by risk among people who do not have that attribute. The latter group is often referred to as the reference group. In the preceding example, female sex would have a relative risk of TMD onset equal to 0.077 ÷ 0.043 = 1.8 compared with the reference group of males.
Estimates of relative risk are informative even when the attribute is one that cannot be altered, such as the person’s age or sex. For example, knowledge that females have a 1.8-fold higher risk of developing TMD than males may support a preventive intervention for females, whereas the same intervention, if it was equally as efficacious for females and males, necessarily would prevent a smaller number of cases among males, and therefore may not be justifiable. This conclusion can be reached even if females’ increased risk of TMD relative to males was not related to sex, but instead to a factor only coincidentally related to sex, such as higher levels of educational attainment – another factor that was found by Von Korff et al14 to be associated with increased risk of TMD. Using an attribute associated with increased disease risk to target preventive care also remains justifiable if the attribute is not a cause of the disease. These non-etiological correlates of disease have been labeled “risk markers”.15
Sex should probably be classified as a risk marker for TMD because there is inconclusive evidence that sex-specific characteristics are involved in the etiology of TMD. One study demonstrated that phasic changes in female reproductive hormones contributed to heightened responsiveness to experimental pain through phases of the menstrual cycle.16 In another study of TMD patients, the severity of facial pain was found to increase during the premenstrual phase and peaked during menstruation, when estrogen levels would be lower.17 However, other studies of the relationship between use of oral contraceptives18 and TMD risk have revealed no association, and hence challenge the notion that reproductive hormones play a role in etiology of TMD.
The term “risk factor” has been used to refer to attributes that are associated statistically with elevated risk of disease and for which there is additional evidence of the attribute being a cause of the disease.15 The key distinction between risk factors and risk markers therefore is that risk factors are known to play a causal role in disease etiology. In contrast, risk markers are attributes known not to play a causal role in disease etiology or that are not clearly established in the etiology of the disease.15 The uncertainty in etiological status may arise because appropriate studies regarding biological plausibility have not been undertaken, or there may be conflicting evidence regarding biological plausibility, as noted above regarding the role of female reproductive hormones.
Some risk markers are associated statistically with disease merely because they are consequences of true etiological factors or, indeed, because they are pre-clinical signs of the condition. The finding by Von Korff et al14 that pain at other bodily sites was predictive of the risk of developing TMD symptoms may merely represent “collateral damage” that was caused by an underlying etiological factor, such as a physiological or psychological characteristic, that was also responsible for increasing TMD risk.
Information about disease risk becomes most valuable for patient care when it identifies risk factors that can be modified, either by avoiding exposure to those risk factors or by interventions that block their effects. Disease can be prevented by modifying risk factors, but not by modifying risk markers. However, risk factors cannot be identified solely by epidemiological studies: they require additional evidence regarding biological plausibility, which may come from in-vitro or animal experimental studies.15 Furthermore, to provide supporting evidence that a putative risk factor plays an etiological role in disease, epidemiological studies must be designed and analyzed in ways that address established causal criteria.19, 20 In summary, the key criteria are: (i) the study demonstrates a temporal sequence between acquisition of or exposure to the putative risk factor and onset of disease; (ii) the quality of the study in controlling bias, confounding, chance and misclassification; and (iii) the strength, consistency, and biological gradient (dose-response) of the association between putative risk factor and disease.
For many risk factors, the requirement for temporal sequence can be met only by undertaking prospective cohort studies in which disease-free people at baseline are classified as to the presence or absence of putative risk factors. The prospective nature of the study design entails waiting while disease develops, hence permitting enumeration of new cases of disease. This was the study design used by Von Korff et al.14 While there is no single method sufficient to insure all aspects of quality, bias can be reduced by careful selection of representative samples of subjects, systematic and independent measurement of risk factors and disease, and adjustment for additional risk factors that are known to play a role in disease etiology. Von Korff et al14 addressed these needs by random selection of subjects from the population of interest, use of consistent and validated questions to inquire about putative risk factors and TMD symptoms, and statistical analyses that controlled simultaneously for potential confounding. For example, they found that the number of pain conditions was associated with a statistically significant 3.7-fold elevation in odds of TMD symptoms after adjustment for subjects’ age, sex and educational attainment.14 The requirement to demonstrate consistency of associations calls for studies that are replicated in other populations. In a study of a different cohort of people recruited with non-dysfunctional TMD, widespread bodily pain reported at baseline was predictive of a statistically significant 1.9-fold elevation in odds of developing dysfunctional TMD among women, but not among men.6
While these studies have met many of the criteria required for epidemiological evaluation of causation, it is necessary to turn to other types of studies to assess the biological plausibility of a causal relationship. Such studies of TMD suggest that there is a general state of pain amplification and possible central sensitization to noxious stimuli that contributes to risk of both TMD and pain at other body sites. For example, compared with healthy controls, TMD patients have been found to have reduced pressure pain thresholds,21 lowered tolerance to muscle ischemia,22, 23 greater sensitivity to other experimentally-induced noxious stimuli, and an increase in temporal summation of pain.21, 22, 24, 25, 26 This combined evidence from epidemiological studies and experimental trials suggests that pain at other bodily sites is a risk marker for TMD, because it probably can be attributed to an underlying etiological factor, such as pain sensitivity and/or altered central pain regulatory mechanisms.
Genetic influences on TMD risk
Methods for identifying genetic influences on disease
There are only a few rare pain conditions in which single gene mutations are necessary and sufficient causes and that therefore follow Mendelian patterns of inheritance.27 Relationships between such genetic variants and disease can be investigated using family aggregation studies, where clusters of disease within genetically-related family members are analyzed.28 Twin studies represent a special class of family aggregation studies in which disease occurrence is investigated in pairs of monozygotic and dizygotic twins. Such studies permit estimation of heritability, defined as that part of the total phenotypic variance that is due to genetic variance.29
To date, family-aggregation studies have failed to identify a genetic influence on TMD,30, 31, 32 but these studies have been underpowered. Assuming that any genetic influences on TMD operate in conjunction with other etiological factors, twin studies would require several thousand subjects to have sufficient power to measure the heritability of this disorder.33 The largest twin study investigating TMD included only 494 twins31 which, as noted by the authors, permitted the identification only of those factors with heritability above 0.5. In contrast, twin studies that have identified a genetic component to chronic pain traits have employed considerably larger sample sizes. For example, estimation of heritability for common migraine, which ranges from 0.34 to 0.57, required a total of 29,717 twin pairs from six countries.34 Heritability of sciatica, which is estimated to be 0.208, required 9,365 twin pairs to detect an effect.35 Given the multifactorial nature and high prevalence of TMD coupled with the requirement for very large population sizes to detect levels of heritability below 0.5, it is not surprising that previous twin- and sib-pair-studies, have failed to identify a genetic influence on this disorder.
Most pain conditions, including TMD, are best classified as complex, multifactorial disorders that are induced and influenced by both diverse environmental factors (e.g., trauma, lifestyle and stress) and a complex array of multiple genetic polymorphisms. These genetic factors consist of many highly prevalent polymorphic genes, rather than single rare mutations, and they therefore fail to follow traditional Mendelian modes of inheritance. It is more appropriate to search for allelic association using traditional epidemiological study designs in which risk of disease is contrasted among sub-groups based on common allelic variants.33, 36, 37, 38
Recent findings demonstrating a genetic influence on pain and TMD
In 2003, Zubietta et al. reported that a common variant of the gene that codes for the enzyme catechol-O-methyl-transferase (COMT) was associated in humans with diminished activity of pain regulatory mechanisms in the central nervous system (CNS).39 Specifically, they analyzed images of the CNS, made using positron emission tomography, to demonstrate that CNS µ-opioid receptor activity during a prolonged infusion of hypotonic saline into the masseter muscle was influenced by a single nucleotide polymorphism (SNP) of chromosome 22 labeled val158met. Individuals with the val/val variant of this SNP produce COMT that has the greatest biological activity, while those who have the met/met variant produce COMT that has the lowest biological activity. The alleles are co-dominant, which means that individuals who are heterozygous (met/val) produce COMT with intermediate biological activity. COMT is a multi-substrate enzyme catalyzing methylation of catecholamines, including the neurotransmitters norepinephrine and dopamine.40 Variations in COMT activity produce compensatory effects in the CNS, including altered µ-opioid receptor activity, such that individuals with the met/met genotype who produce low-activity COMT experience reduced analgesia through µ-opioid pathways.39
We have identified variations in three additional SNPs of the gene encoding COMT that, when combined with val158met, form combinations of SNPs called haplotypes. We found that combinations of three common COMT haplotypes accounted for 11% of variability in perceptions of experimental pain in a cohort of women.41 We labeled these haplotypes as low pain sensitivity (LPS), average pain sensitivity (APS), and high pain sensitivity (HPS). The labels were based on our observed associations of these haplotypes with sensitivity to experimental pain in humans, supported additionally by the results of in vitro and in vivo experiments.41 Furthermore, during a three year prospective study of females who did not have TMD when examined at baseline, the rate of TMD onset was 2.3-fold greater for subjects who had only HPS and/or APS haplotypes compared with subjects who had one or two LPS haplotypes. Interestingly, these findings regarding COMT are consistent with a study conducted three decades ago in which patients with facial pain had above-average urinary levels of catecholamine metabolites and diminished COMT activity in erythrocytes, implicating COMT in the development of TMD.42
Genetic markers, such as SNPs or combinations of SNPs, are significant not only because they implicate enzymes and other biological mechanisms involved in the etiology of pain, but also because they have additional potential to identify population groups in which environmental influences play a relatively greater or lesser etiological role. Hence, it is conceivable that an environmental factor (e.g., depression) could be risk factor for TMD only among people who have one combination of alleles for a SNP, but not among people with alternative combinations of alleles for the same SNP. This represents a gene-environment interaction,28 meaning that the environmental influence on disease is expressed only among people with a specific genotype. Stohler has explained the clinical relevance of such interactions, noting that “symptoms should be understood as the person’s complex response trait with specific complaints being either amplified or attenuated by the unique genetic makeup and/or prior experience”.11
The remaining parts of this paper illustrate a gene-environment interaction, presenting new findings from our prospective cohort study of TMD in which we have previously identified a role for a genetic influence on TMD.41
MATERIALS & METHODS
The following data are from a three-year, prospective cohort study of TMD.41 The study was reviewed and approved by the University of North Carolina (UNC) School of Dentistry’s Committee on Investigations Involving Human Subjects.
The study population comprised female volunteers aged 18–34 years who were recruited using advertisements placed in local newspapers in central North Carolina (Figure 1). Respondents to advertisements phoned a research nurse who asked screening questions that were used to exclude from the study subjects who: (i) reported a history of TMD; (ii) were undergoing active orthodontic treatment; or (iii) reported a significant medical history (including heart disease, hypertension, severe psychiatric dysfunction, history of persistent pain conditions, and use of centrally acting medications). Prior to enrolment in the study, volunteers attended the UNC School of Dentistry ’s Neurosensory Disorders Unit for baseline clinical assessment that included physical examination of the head and neck conducted by one of two examiners. Volunteers were excluded if they were diagnosed with TMD based on the RDC case definition.13 During the baseline visit, enrolled subjects were asked if they had ever had orthodontic treatment and the answer was recorded as “yes” or “no”. There was no attempt to further categorize the type of orthodontic treatment. In addition, peripheral blood samples were collected, subjects completed psychosocial questionnaires, and subjects underwent a series of psychophysical tests to measure responsiveness to standardized noxious stimuli.43
Figure 1. Flowchart of recruitment and follow-up procedures used in the three year prospective study of TMD.
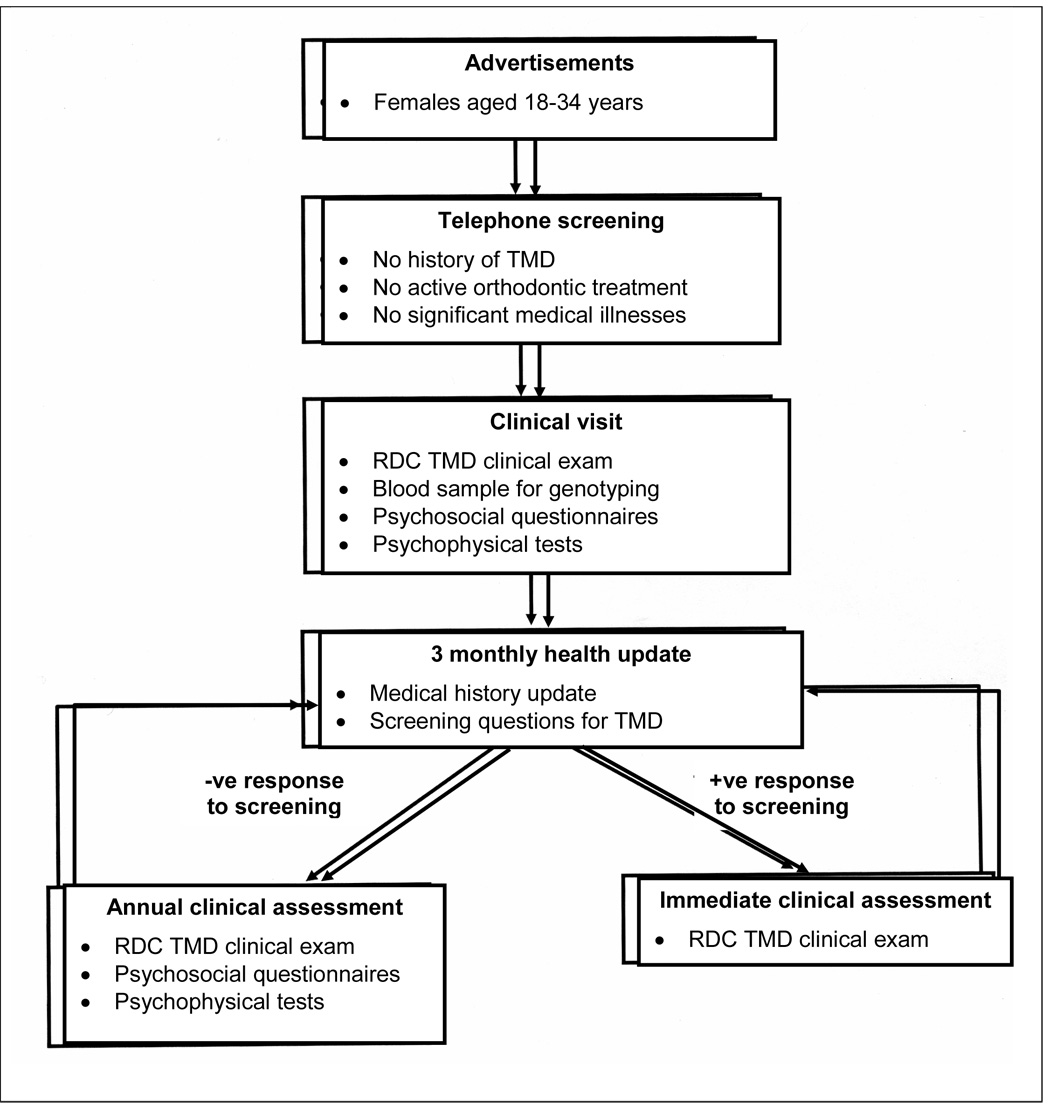
RDC/TMD = research diagnostic criteria for TMD.13
For three years after their baseline assessment, subjects were contacted every three months by research staff who administered a medical history update questionnaire (Figure 1). Any subjects responding positively to key questions about TMD symptoms were immediately recalled for a physical head/neck examination to confirm or exclude TMD. Additionally, each year all subjects were invited to attend for physical examinations of the head and neck. New cases of TMD myalgia and/or TMD arthralgia were confirmed independently by two examiners using the RDC.13 Prior to the study, the examiners achieved 90% agreement and a kappa value of 0.80 when they independently assessed 10 people who were not study subjects using the RDC criteria. During this study, there was 100% agreement between the examiners regarding the diagnoses of the incident cases.
COMT genotyping
Peripheral blood samples were used for genotyping four COMT SNPs: rs6269, rs4633, rs4818, and val158met. Haplotypes were constructed using Phase software to classify subjects into one of two groups:
Subjects were classified as having pain sensitive haplotypes if they carried only HPS haplotypes (ACCG) or APS haplotypes (ATCA) for four SNPs of COMT: rs6269, rs4633, rs4818, and val158met (respectively).
Remaining subjects were classified as having pain resistant haplotypes.
Data analysis
Incident cases were defined as people diagnosed at any follow-up visit with TMD myalgia and/or TMD arthralgia or both, as per RDC criteria.13 For this analysis, the outcome measure was cumulative incidence, computed as the number of TMD cases divided by the number of subjects who were followed for at least three months and reassessed. Cumulative incidence was computed separately for each subgroup of COMT haplotype and for subjects who did and did not report a history. Ratios of cumulative incidence were calculated to yield relative risk (RR) and its 95% confidence interval (CI) was computed using the logit estimate.44 When the 95% CI excluded the null value of 1.0, the relative risk was deemed to be statistically significant.
RESULTS
Two hundred and fifty four females volunteered to take part in the study and completed baseline sensory assessments, of whom 212 (83%) provided a blood sample and written consent for genotyping. Follow-up data about clinical TMD status were obtained from 186 subjects. Sixty two subjects with follow-up data (33.3%) had pain-sensitive haplotypes and the remaining 124 were classified as pain-resistant. Ninety-nine subjects (53.2%) reported a history of orthodontic treatment, 75 (40.3%) said they had not had orthodontic treatment but 12 subjects (6.4%) said they did not know and they were excluded from subsequent analyses.
Among the cohort of 174 people available for analysis, 15 new cases of TMD were diagnosed using RDC criteria during the follow-up period that averaged 30 months (range=8–42 months). The fifteen new cases of TMD represented a cumulative incidence of 8.6% (95% CI = 4.4% – 12.8%). Among subjects with pain-sensitive haplotypes, cumulative incidence was 15.4% compared with 5.7% among subjects classified as pain-resistant (Table 1). This yielded a statistically significant relative risk of 2.68 (95% CI = 1.03 – 7.01) comparing pain-sensitive with pain-resistant subjects. Risk of TMD was three-fold greater among people who reported a history of orthodontic treatment compared with those who did not, although the associated relative risk did not reach statistical significance, as indicated by the 95% confidence interval (0.89 – 10.35) that included unity. Two thirds (67.3%) of subjects with pain-sensitive haplotypes reported a history of orthodontic treatment compared with 52.4% for pain-resistant haplotypes, although the difference in percentages was not statistically significant (Chi-square, 1 df, P=0.07).
Table 1.
Cumulative incidence and relative risk of developing TMD
| Sub-group | No. of subjects in cohort | Cumulative incidence (% of people) | Relative risk (95% CI†) |
|---|---|---|---|
| Pain sensitive haplotypes of COMT* | 52 | 15.4% | 2.68 (1.03 – 7.01) |
| Other haplotypes of COMT | 122 | 5.7% | reference‡ |
| History of orthodontic treatment | 99 | 12.1% | 3.03 (0.89 – 10.35) |
| No history of orthodontic treatment | 75 | 4.0% | reference‡ |
| All subjects | 174 | 8.6% | |
Catechol-O-methyl-transferase (COMT) pain sensitive haplotypes are ACCG_ATCA and ATCA_ATCA for the sequence of SNPs rs6269, rs4633, rs4818, and val158met.
95% confidence interval
Reference group whose cumulative incidence forms the denominator in calculating relative risk.
When the cohort was stratified according to subjects’ COMT haplotype, there was a striking pattern of interaction. Among subjects classified with pain-resistant COMT haplotypes, the cumulative incidence of TMD was virtually identical for people who reported a history of orthodontic treatment (cumulative incidence = 6.3%) and those who did not (5.5% – Figure 2). In contrast, among subjects with pain sensitive haplotypes, TMD incidence was 22.9% for those with a history of orthodontic treatment whereas no new cases of TMD occurred for those with no history of orthodontic treatment (cumulative incidence = 0.0% – Figure 2).
Figure 2. Stratified analysis of orthodontic treatment.
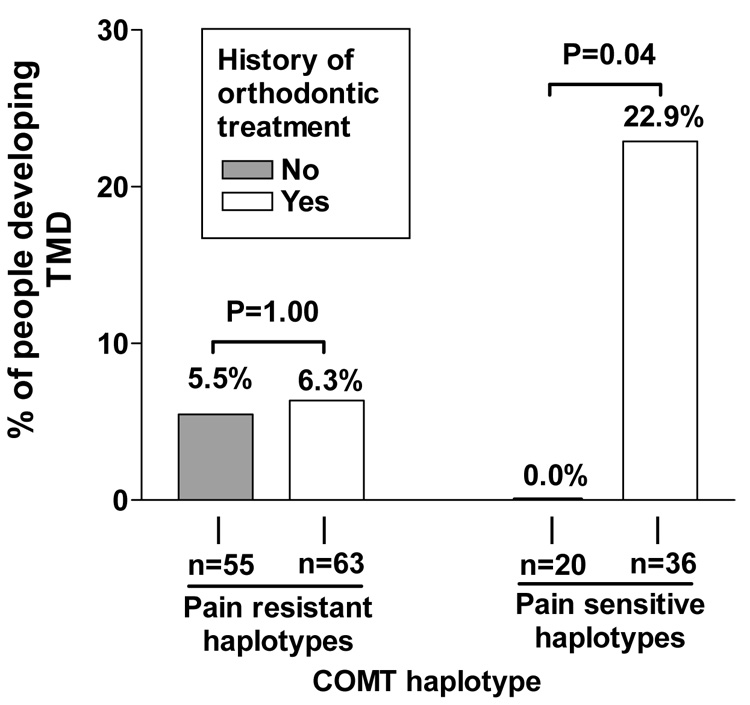
Data are from n=174 subjects genotyped for catechol-O-methyl-transferase (COMT) who completed at least one follow-up assessment in the three year cohort study and whose history of orthodontic treatment was known. The PSH (pain sensitive haplotypes) group consists of individuals carrying haplotypes ACCG or ATCA for the sequence of SNPs rs6269, rs4633, rs4818, and val158met.41 The PRH (pain resistant haplotype) group consists of individuals with other haplotype combinations. Vertical bars represent cumulative incidence, calculated as the proportion of people in each subgroup who were diagnosed with temporomandibular disorder (TMD) during follow-up. P-values are from Fisher’s exact test comparing incidence between people who a reported history of orthodontic treatment with people who reported no history of orthodontic treatment.
DISCUSSION
This analysis has illustrated an example of gene-environment interaction, demonstrating that relative risk of TMD associated with orthodontic treatment (an environmental influence) was dependent on a variant of the gene encoding COMT. Specifically, orthodontic treatment was not associated with elevated risk of TMD among people with the pain-resistant haplotypes, whereas orthodontic treatment was associated with marked elevation in risk for subjects with pain-sensitive haplotypes (Figure 2). This study met several of the study design criteria required to investigate TMD risk, including a prospective study design in which the putative risk factors (genotype and orthodontic treatment) were known to exist prior to TMD onset. New cases of TMD were diagnosed using validated RDC criteria. The number of subjects in this cohort is considerably larger than most of the studies reviewed by Kim et al12 in their review of 31 studies, and approximately twice as large as a more recent prospective cohort study of 35 year-old Swedes.45
As noted in the preceding review, demonstration of statistically significant elevation in risk is not sufficient evidence that an attribute (in this case, orthodontic treatment) is causally involved in the etiology of TMD. Additional evidence regarding biological plausibility would be needed to designate orthodontic treatment as a risk factor. In principle, there is at least some plausibility to the notion that fixed orthodontic treatment could play a causal role in TMD etiology among people who are genetically predisposed to pain. Periodic adjustments made to fixed orthodontic appliances apply forces to teeth that can cause transient discomfort or pain. In a study of Swedish children aged 12–18 years, 87% reported pain on the first evening after elastic orthodontic separators were placed between their teeth. After one day, the intensity of pain reached a maximum average of 43.7 as rated on a visual analog scale ranging from zero to 100.46 A retrospective study of 358 Chinese adults who had undergone treatment with fixed orthodontic appliances found that 91% had experienced transient pain to teeth, and among that group, 39% said that the pain was experienced with each new archwire or elastic force application.
In previous studies, we have reported that subjects with pain sensitive haplotypes of COMT have elevated responses to standardized noxious stimuli compared with people who have pain resistant haplotypes.41 Hence, it seems likely that people with pain sensitive haplotypes would have experienced relatively greater discomfort or pain when undergoing procedures used during fixed orthodontic treatment. And there is additional experimental evidence that people with genetically downregulated COMT have reduced analgesic effects of endogenous opioid systems within the central nervous system.39 Taken together, these findings provide some biological plausibility to support an interpretation that orthodontic treatment could be a risk factor for TMD.
However, important methodological features of this study should also be considered. Experience of orthodontic treatment was assessed in this study merely by asking subjects a single question, with no attempt to clarify whether or not it was fixed orthodontic treatment, duration of the treatment, or whether other treatment such a surgery was involved. Equally important were the study’s enrolment criteria, which excluded volunteers if they were undergoing active orthodontic treatment or if they had TMD at the time of recruitment. Hence, any etiological role of orthodontic treatment in this study would require that the putative causal effect of orthodontic treatment was one that persisted after completion of treatment yet which did not cause the person to develop TMD at the time of recruitment. This raises the possibility that there was yet another environmental interaction that occurred in the time between completion of orthodontic treatment and enrolment in the study.
Finally, an important criterion for causal inference is that results are replicated in other populations. To date, no other prospective studies have examined risk factors for TMD diagnosed clinically by examiners using RDC criteria that are now accepted as the “gold standard”.
On balance, therefore, it would be premature to propose that orthodontia is a risk factor for TMD, even among the sub-group of females with pain sensitive haplotypes of COMT. However, based on current evidence about biological processes involved in pain regulation, it seems plausible that there could be a subset of the population that is relatively sensitive to noxious stimuli, and for those individuals pain experienced during orthodontic treatment may interact with that pain sensitivity. Furthermore, the combined attributes of COMT pain sensitive haplotypes and the receipt of orthodontic care were useful markers to identify a sub-group with particularly high risk of developing TMD. If these findings could be replicated in other populations, this genetic marker may even help to identify patients most likely to experience pain during orthodontic treatment and for whom pain management would be an important component of treatment planning. Meanwhile, additional evidence is needed to isolate any causal role played by COMT and orthodontic treatment in development of TMD. That evidence is likely to emerge rapidly from new genetic studies, providing a wealth of knowledge that should be used, together with sound principles of study design and causal inference, to further investigate the relationship, recognizing that any causal influence may be identified only after considering gene-environment interactions.
Acknowledgments
Supported by: NIH/NIDCR DE07509, NS045685, DE007333, AR/AI-44564, NIH Intramural Grants DE00366 and AA000301, and the Comprehensive Neuroscience Program Grant USUHS G192BR-C4
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Von Korff M. Health services research and temporomandibular pain. In: Sessle BJ, Bryant PS, Dionne RA, editors. Temporomandibular disorders and related pain conditions. Seattle: IASP Press; 1995. pp. 227–236. [Google Scholar]
- 2.US Department of Health and Human Services. Oral Health in America: A Report of the Surgeon General. Rockville, MD: National Institutes of Health; National Institute of Dental and Craniofacial Research. 2000
- 3.Okeson JP, editor. Differential diagnosis and management consideration of temporomandibular disorders. Chicago: Quintessence; 1996. Orofacial Pain. Chapter 8; pp. 113–184. [Google Scholar]
- 4.Macfarlane TV, Blinkhorn AS, Davies RM, et al. Association between local mechanical factors and orofacial pain: survey in the community. J Dent. 2003;31:535–542. doi: 10.1016/s0300-5712(03)00108-8. [DOI] [PubMed] [Google Scholar]
- 5.Huang GJ, LeResche L, Critchlow CW, et al. Risk factors for diagnostic subgroups of painful temporomandibular disorders (TMD) J Dent Res. 2002;81:284–288. doi: 10.1177/154405910208100412. [DOI] [PubMed] [Google Scholar]
- 6.John MT, Miglioretti DL, LeResche L, et al. Widespread pain as a risk factor for dysfunctional temporomandibular disorder pain. Pain. 2003;102:257–263. doi: 10.1016/S0304-3959(02)00404-9. [DOI] [PubMed] [Google Scholar]
- 7.Fillingim RB, Maixner W, Kincaid S, et al. Pain sensitivity in patients with temporomandibular disorders: relationship to clinical and psychosocial factors. Clin J Pain. 1996;12:260–269. doi: 10.1097/00002508-199612000-00005. [DOI] [PubMed] [Google Scholar]
- 8.Wright AR, Gatchel RJ, Wildenstein L, et al. Biopsychosocial differences between high-risk and low-risk patients with acute TMD-related pain. J Am Dent Assoc. 2004;135:474–483. doi: 10.14219/jada.archive.2004.0213. [DOI] [PubMed] [Google Scholar]
- 9.Drangsholt M, LeResche L. Temporomandibular disorder pain. In: Crombie IK, Croft PR, Linton SJ, LeResche L, Von Korff M, editors. Epidemiology of Pain. Seattle: IASP Press; 1999. [Google Scholar]
- 10.Svensson P. Craniofacial muscle pain: review of mechanisms and clinical manifestations. J Orofacial Pain. 2001;15:117–145. [PubMed] [Google Scholar]
- 11.Stohler CS. Taking stock: from chasing occlusal contacts to vulnerability alleles. Orthod Craniofac Res. 2004;7:157–161. doi: 10.1111/j.1601-6343.2004.00291.x. [DOI] [PubMed] [Google Scholar]
- 12.Kim MR, Graber TM, Viana MA. Orthodontics and temporomandibular disorder: a meta-analysis. Am J Orthod Dentofacial Orthop. 2002;121:438–446. doi: 10.1067/mod.2002.121665. [DOI] [PubMed] [Google Scholar]
- 13.Dworkin SF, LeResche L. Research diagnostic criteria for temporomandibular disorders: review, criteria, examinations and specifications, critique. J Craniomandibular Disorders. 1992;6:302–355. [PubMed] [Google Scholar]
- 14.Von Korff M, LeResche L, Dworkin SF. First onset of common pain symptoms: a prospective study of depression as a risk factor. Pain. 1983;55:251–258. doi: 10.1016/0304-3959(93)90154-H. [DOI] [PubMed] [Google Scholar]
- 15.Beck JD. Risk revisited. Community Dent Oral Epidemiol. 1998;26:220–225. doi: 10.1111/j.1600-0528.1998.tb01954.x. [DOI] [PubMed] [Google Scholar]
- 16.Fillingim RB, Ness TJ. Sex-related hormonal influences on pain and analgesic responses. Neurosci Biobehav Rev. 2000;24:485–501. doi: 10.1016/s0149-7634(00)00017-8. [DOI] [PubMed] [Google Scholar]
- 17.LeResche L, Mancl L, Sherman JJ, et al. Changes in temporomandibular pain and other symptoms across the menstrual cycle. Pain. 2003;106:253–261. doi: 10.1016/j.pain.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Marbach JJ, Lennon MC, Dohrenwend BP. Candidate risk factors for temporomandibular pain and dysfunction syndrome: psychosocial, health behavior, physical illness and injury. Pain. 1988;34:139–151. doi: 10.1016/0304-3959(88)90159-5. [DOI] [PubMed] [Google Scholar]
- 19.Hill AB. The environment and disease: association or causation. Proc R Soc Med. 1965;58:295–300. doi: 10.1177/003591576505800503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.United States Department of Health, Education and Welfare. Washington DC: Public Health Service; Smoking and Health: Report of the Advisory Committee to the Surgeon General. 1964
- 21.Maixner W, Fillingim R, Sigurdsson A, et al. Sensitivity of patients with painful temporomandibular disorders to experimentally evoked pain: evidence for altered temporal summation of pain. Pain. 1998;76:71–81. doi: 10.1016/s0304-3959(98)00028-1. [DOI] [PubMed] [Google Scholar]
- 22.Maixner W, Sigurdsson A, Fillingim R, et al. Regulation of acute and chronic orofacial pain. In: Fricton JR, Dubner RB, editors. Orofacial Pain and Temporomandibular Disorders. New York: Raven Press, Ltd; 1995. pp. 85–102. [Google Scholar]
- 23.Kashima K, Rahman OIF, Sakoda S, et al. Increased pain sensitivity of the upper extremities of TMD patients with myalgia to experimentally-evoked noxious stimulation: possibility of worsened endogenous opioid systems. Cranio-the Journal of Craniomandibular Practice. 1999;17:241–246. doi: 10.1080/08869634.1999.11746100. [DOI] [PubMed] [Google Scholar]
- 24.Maixner W, Fillingim R, Booker D, et al. Sensitivity of patients with painful temporomandibular disorders to experimentally evoked pain. Pain. 1995;63:341–351. doi: 10.1016/0304-3959(95)00068-2. [DOI] [PubMed] [Google Scholar]
- 25.Sarlani E, Greenspan JD. Evidence for generalized hyperalgesia in temporomandibular disorders patients. Pain. 2003;102:221–226. doi: 10.1016/S0304-3959(03)00095-2. [DOI] [PubMed] [Google Scholar]
- 26.Sarlani E, Grace EG, Reynolds MA, et al. Evidence for up-regulated central nociceptive processing in patients with masticatory myofascial pain. J Orofac Pain. 2004;18:41–55. [PubMed] [Google Scholar]
- 27.Nagasako EM, Oaklander AL, Dworkin RH. Congenital insensitivity to pain: an update. Pain. 2003;101:213–219. doi: 10.1016/S0304-3959(02)00482-7. [DOI] [PubMed] [Google Scholar]
- 28.Khoury MJ. Genetic epidemiology. In: Rothman KJ, Greenland S, editors. Modern Epidemiology. 2nd ed. Philadelphia: Lippincott: Williams and Wilkins; 1998. pp. 609–621. [Google Scholar]
- 29.Griffiths AJF, Gellant W, Lewontin RC, et al. Integrating genes and genomes. 2nd Ed. New York: W Freeman and Co.; 2002. Modern genetic analysis; p. 600. [Google Scholar]
- 30.Raphael KG, Marbach JJ, Gallagher RM, et al. Myofascial TMD does not run in families. Pain. 1999;80:15–22. doi: 10.1016/s0304-3959(98)00180-8. [DOI] [PubMed] [Google Scholar]
- 31.Michalowicz BS, Pihlstrom BL, Hodges JS, et al. No heritability of temporomandibular joint signs and symptoms. J Dent Res. 2000;79:1573–1578. doi: 10.1177/00220345000790080801. [DOI] [PubMed] [Google Scholar]
- 32.Heiberg A, et al. Myofascial pain dysfunction (MPD) syndrome in twins. Community Dent. Oral Epidemiol. 1980;8:434–436. doi: 10.1111/j.1600-0528.1980.tb01323.x. [DOI] [PubMed] [Google Scholar]
- 33.Risch NJ. Searching for genetic determinants in the new millennium. Nature. 2000;405:847–856. doi: 10.1038/35015718. [DOI] [PubMed] [Google Scholar]
- 34.Mulder EJ, Van Baal C, Gaist D, et al. Genetic and environmental influences on migraine: a twin study across six countries. Twin Res. 2003;6:422–431. doi: 10.1375/136905203770326420. [DOI] [PubMed] [Google Scholar]
- 35.Heikkila JK, Koskenvuo M, Heliovaara M, et al. Genetic and environmental factors in sciatica. Evidence from a nationwide panel of 9365 adult twin pairs. Ann Med. 1989;21:393–398. doi: 10.3109/07853898909149227. [DOI] [PubMed] [Google Scholar]
- 36.Jones HB. The relative power of linkage and association studies for the detection of genes involved in hypertension. Kidney Int. 1998;53:1446–1448. doi: 10.1046/j.1523-1755.1998.00925.x. [DOI] [PubMed] [Google Scholar]
- 37.Tsuchiya N, Ohashi J, Tokunaga K. Variations in immune response genes and their associations with multifactorial immune disorders. Immunol Rev. 2002;190:169–181. doi: 10.1034/j.1600-065x.2002.19013.x. [DOI] [PubMed] [Google Scholar]
- 38.McPeek MS. Optimal allele-sharing statistics for genetic mapping using affected relatives. Genet Epidemiol. 1999;16:225–249. doi: 10.1002/(SICI)1098-2272(1999)16:3<225::AID-GEPI1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 39.Zubieta JK, Heitzeg MM, Smith YR, et al. COMT val158met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240–1243. doi: 10.1126/science.1078546. [DOI] [PubMed] [Google Scholar]
- 40.Mannisto PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev. 1999;51:593–628. [PubMed] [Google Scholar]
- 41.Diatchenko L, Slade GD, Nackley AG, et al. Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Hum Mol Genet. 2005;14:135–143. doi: 10.1093/hmg/ddi013. [DOI] [PubMed] [Google Scholar]
- 42.Marbach JJ, Levitt M. Erythrocyte catechol-O-methyltransferase activity in facial pain patients. J Dent Res. 1976;55:711. doi: 10.1177/00220345760550043801. [DOI] [PubMed] [Google Scholar]
- 43.Bhalang K, Sigurdsson A, Slade GD, et al. Associations among four modalities of experimental pain in women. J Pain. 2005;6:604–611. doi: 10.1016/j.jpain.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 44.Breslow NE, Day NE. Statistical methods in cancer research. Lyon: International Agency for Research on Cancer; 1987. [Google Scholar]
- 45.Egermark I, Magnusson T, Carlsson GE. A 20-year follow-up of signs and symptoms of temporomandibular disorders and malocclusions in subjects with and without orthodontic treatment in childhood. Angle Orthod. 2003;73:109–115. doi: 10.1043/0003-3219(2003)73<109:AYFOSA>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 46.Bergius M, Berggren U, Kiliaridis S. Experience of pain during an orthodontic procedure. Eur J Oral Sci. 2002;110:92–98. doi: 10.1034/j.1600-0722.2002.11193.x. [DOI] [PubMed] [Google Scholar]