

Abstract
A low-copy component of mammalian reovirus particles is μ2, an 83-kDa protein encoded by the M1 viral genome segment and packaged within the viral core. Previous studies have identified μ2 as a nucleoside triphosphate phosphohydrolase (NTPase) as well as an RNA 5′-triphosphate phosphohydrolase (RTPase), putatively involved in reovirus RNA synthesis and/or 5′-capping. Other studies have identified μ2 as a microtubule-binding protein, which also associates with the viral factory matrix protein μNS and thereby anchors the factories to cellular microtubules during infections by most reovirus strains. To extend studies of μ2 functions during infection, we tested a small interfering RNA (siRNA) directed against the M1 plus-strand RNAs of reovirus strains Type 1 Lang (T1L) and Type 3 Dearing (T3D). The siRNA strongly suppressed μ2 expression by either strain and reduced infectious yields in a strain-dependent manner. This first strain difference was genetically mapped to the M1 genome segment and tentatively assigned to a single μ2 sequence polymorphism, Pro/Ser208, which also determines a T1L-T3D strain difference in microtubule association. The siRNA-based defect in μ2 expression was rescued by plasmids, containing silent mutations in the siRNA-targeted sequence, which encoded either T1L or T3D μ2, but the growth defect was rescued only by T1L μ2. This second strain difference was also mapped to Pro/Ser208, in that swapping this one residue between T1L and T3D μ2 reversed the rescue phenotypes. Thus, the T1L-T3D strain difference in μ2-microtubule association was correlated not only with the extent of reduction in infectious yields by the siRNA but also with the extent of rescue by plasmid-derived μ2. In addition, the rescue capacity of T1L μ2 was abrogated by nocodazole treatment, providing independent evidence for the importance of μ2-microtubule association in plasmid-based rescue. In two separate cases, the results revealed functional differences between virus- and plasmid-derived μ2. Ala substitutions within the NTP-binding motif of T1L μ2 also abrogated its rescue capacity, suggesting that the NTPase or RTPase activity of μ2 is additionally required for effective viral growth.
Introduction
Mammalian orthoreoviruses (reoviruses) are nonenveloped dsRNA viruses of the family Reoviridae. Although generally mild in humans, reovirus infections of lab rodents can produce a variety of disease syndromes including hepatitis, pneumonitis, myocarditis, and encephalitis (for review, see Tyler, 2001). As a result, reovirus has provided a number of useful models for studies of viral pathogenesis and is helping to address how viruses interact with the small intestine, liver, lungs, heart, and central nervous system of infected hosts. Recently, reovirus has shown promise as an oncolytic agent (for review, see Shmulevitz et. al, 2005).
The reovirus genome comprises ~23,500 bp in ten dsRNA segments, the plus strands of which encode 11 or 12 primary translation products (for reviews of this and the following statements, see Coombs, 2006; Guglielmi et al., 2006; Nibert and Schiff 2001). Within the infectious virion, the genome segments are encased by a double-layered protein capsid. During cell entry, the virion undergoes partial disassembly and conformational changes to yield a subvirion particle called the core, which enters the host cytoplasm and mediates primary transcription, 5′-capping, and export of the viral plus-strand RNAs. These primary transcripts consecutively or alternatively serve as mRNAs for viral protein translation and templates for minus-strand synthesis to regenerate the dsRNA genome segments inside newly forming particles. Some of these newly formed particles can mediate secondary transcription, substantially amplifying the levels of the plus-strand RNAs and their encoded proteins (for review, see Zarbl and Millward, 1983). Replication of genome and assembly of new particles are thought to occur in association with cytoplasmic inclusions in infected cells, often called viral factories. The complex nature of these factories and their functions are subjects of ongoing investigations (Becker et al., 2001, 2003; Broering et al., 2002, 2004, 2005; Mbisa et al., 2000; Miller et al., 2003, 2004; Parker et al., 2002).
One of the least well understood of the reovirus proteins is μ2, an 83-kDa protein present in low copy number (20 to 24) in the viral core (Coombs, 1998; Mustoe et al., 1978). In vitro, purified μ2 demonstrates both NTPase and RTPase activities (Kim et al., 2004), and genetic evidence suggests that it may mediate one or both of these activities inside core particles (Noble and Nibert, 1997). Ala substitutions for Lys415 and Lys419 eliminate these activities of purified μ2 (Kim et al., 2004). A temperature-sensitive reovirus mutant with a lesion in the M1 genome segment encoding μ2 fails to synthesize dsRNA at nonpermissive temperature, suggesting that μ2 may have a role in the viral replicase complex (Coombs, 1996). Reassortant studies have further suggested that μ2 determines viral strain differences in core transcriptase activities (Yin et al., 1996). The μ2 protein binds both ssRNA and dsRNA in vitro (Brentano et al. 1998). In addition, μ2 binds to cellular microtubules (Kim et al., 2004) and determines strain differences in the anchoring of viral factories to microtubules in infected cells (Parker et al., 2002; Yin et al., 2004). Although there are nine amino acid differences between the μ2 proteins of our lab’s viral clones of reovirus strains T1L and T3D (Parker et al, 2002), previous studies have shown that the extent of microtubule association is specifically determined by the presence of Pro versus Ser at μ2 residue 208, with Ser208 as in T3D μ2 determining poorer microtubule association (Miller et al., 2004; Parker et al., 2002; Yin et al., 2004). Ser208 also makes the μ2 protein more prone to temperature-dependent aggregation and polyubiquitination, consistent with a folding defect (Miller et al., 2004). In anchoring the factories to microtubules, μ2 appears to act through its association with viral factory matrix protein μNS (Broering et al., 2002). Other studies have shown that μ2 is a determinant of strain differences in plaque size (Moody and Joklik, 1989), rate of viral factory formation (Mbisa et al., 2000), and sensitivity to mycophenolic acid (Hermann and Coombs, 2004), and that it also has a role in influencing viral growth and cytopathology in both cultured cells and newborn mice (Haller et. al, 1995; Matoba et al., 1991; Matoba et al., 1993; Sherry and Blum, 1994; Sherry and Fields, 1989; Sherry et. al, 1998).
siRNAs and other silencing technologies have become powerful tools for investigating mammalian RNA and protein functions (for reviews, see Cezka et. al, 2006; Mello and Conte, 2004; Sledz and Williams, 2005). They have also been used to study many viruses, including the Reoviridae family member rotavirus (Campagna et al., 2005; Cuadras et. al, 2006; Dector et al., 2002; Lopez et. al, 2005; Montero et. al, 2006; Silvestri et. al, 2004; Taraporewala et al., 2006). Of particular note is a recent rotavirus study by Taraporewala et al. (2006) combining siRNA-based knockdown of the rotavirus NSP2 protein with plasmid-based rescue of NSP2 expression to provide a new genetic system for studies of NSP2 functions in infected cells. We therefore expected that we could also utilize siRNAs to study the functions of the reovirus μ2 protein in infected cells. In fact, we would hope to extend this technology to any or all of the reovirus proteins. Towards this end, for the current study, we performed experiments using an siRNA directed against the M1 plus-strand RNA encoding the μ2 protein to suppress both μ2 expression and viral growth, coupled with plasmids encoding different forms of μ2 for mediating rescue of these defects. The results presented below indicate that this is indeed a useful approach for genetic studies of reovirus proteins, particularly as it has revealed new functional correlations with μ2-microtubule association and differences between virus- and plasmid-derived μ2.
Results
M1-si suppresses μ2 expression by reoviruses T1L and T3D
siRNA M1-si, targeting a shared sequence in the M1 plus-strand RNAs of our lab’s viral clones of reovirus strains T1L and T3D (Fig. 1a), was obtained from Dharmacon. To evaluate the effect of M1-si on expression of the M1-encoded μ2 protein of each strain, CV-1 cells were transfected with siRNA and at 24 h post-transfection (p.t.) were infected with either T1L or T3D reovirus at 10 PFU/cell. The cells were incubated for an additional 0, 24, or 48 h post-infection (p.i.) before harvest, and cell-associated proteins were prepared for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting. The blots revealed that M1-si suppressed μ2 expression to near the detection limit for both T1L (Fig. 1b) and T3D (Fig. 1c) reovirus, whereas a nonspecific control siRNA, also obtained from Dharmacon, had little effect on either virus (Fig. 1b, c). Analysis of cellular GAPDH levels suggested that there was similar loading of proteins from each sample (Fig. 1b, c). The lack of effect of the control siRNA provided initial evidence against the general importance of off-target effects in the suppression of μ2 expression by M1-si. In repeated experiments, the reduction in μ2 expression levels by M1-si was estimated to be ≥90–95% (data not shown, but see Figs. 5g, 7c, and 8d).
Fig. 1. Effect of siRNA (M1-si) targeting a shared sequence in the M1 plus-strand RNAs of reovirus strains T1L and T3D on μ2 expression levels.
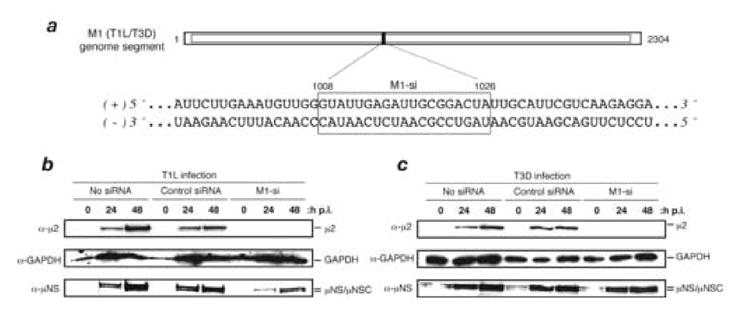
(A) The M1 genome sequence of T1L and T3D is shown in the region targeted by M1-si. The M1-si-targeted region is boxed and encompasses nucleotides 1008 to 1026. (B, C) CV-1 cells were transfected with siRNA and at 24 h p.t. were then infected with T1L (B) or T3D (C) reovirus. The cells were incubated for an additional 0, 24, or 48 h p.i., and cell-associated proteins were prepared for SDS-PAGE and immunoblotting. Protein was detected using specific antibodies against μ2, GAPDH, or μNS.
Fig 5. Effect of plasmid-based expression of T1L and T3D μ2 on M1-si-based reduction of infectious viral yields.
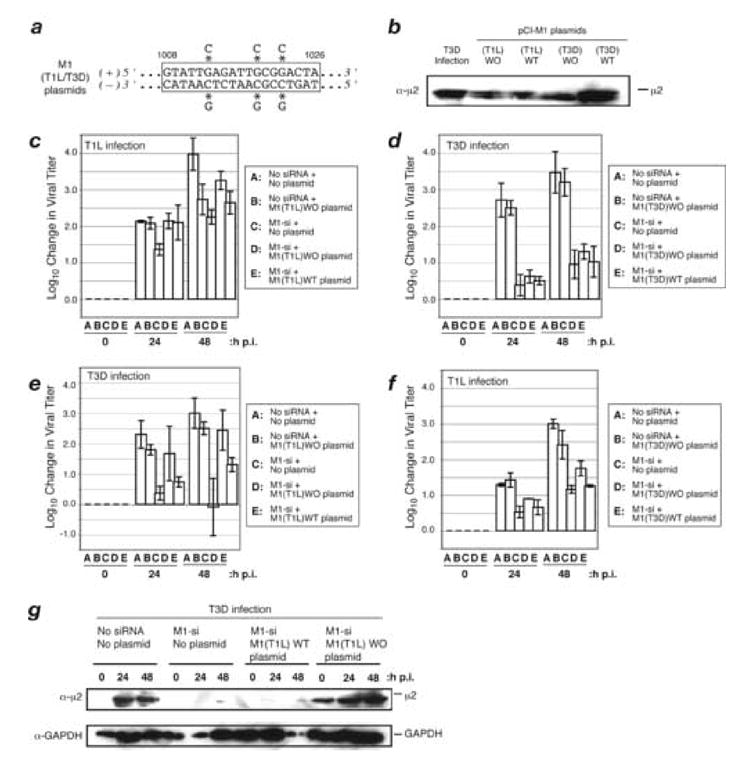
(A) Sequence of rescue plasmid encoding T1L and T3D M1 in the region targeted by M1-si. Asterisks indicate the nucleotides that were changed to encode silent (wobble) mutations. (B) CV-1 cells were transfected with 5 μg of either pCI-M1(T1L) or pCI-M1(T3D) rescue plasmids. In each case, these plasmids either contained wobble mutations in the M1-si-targeted sequence (designated as WO plasmids) or did not (designated as WT plasmids). The cells were incubated for an additional 24 h p.i., and cell-associated proteins were prepared for SDS-PAGE and immunoblotting. Protein was detected using specific antibodies against μ2. (C, D, E, F) CV-1 cells were transfected with no siRNA, M1-si, or M1-si in combination with a particular rescue plasmid, and at 24 h p.t. were then infected with T1L (C, F) or T3D (D, E) reovirus at 10 PFU/cell. Cells were harvested at 0, 24, or 48 h p.i., and viral titers were determined by plaque assays. Log10 changes in viral titer relative to time 0 are indicated. Each bar represents the average obtained from three independent experiments, and error bars indicate the standard deviations. (G) CV-1 cells were transfected with no siRNA, M1-si, or M1-si in combination with either M1(T1L)WT or M1(T1L)WO plasmid, and at 24 h p.t. were then infected with T3D reovirus. The cells were incubated for an additional 0, 24, or 48 h p.i., and cell-associated proteins were prepared for SDS-PAGE and immunoblotting. Protein was detected using specific antibodies against μ2 or GAPDH.
Fig. 7. Effect of nocodazole on complementation of M1-si-based reduction of infectious viral yields.
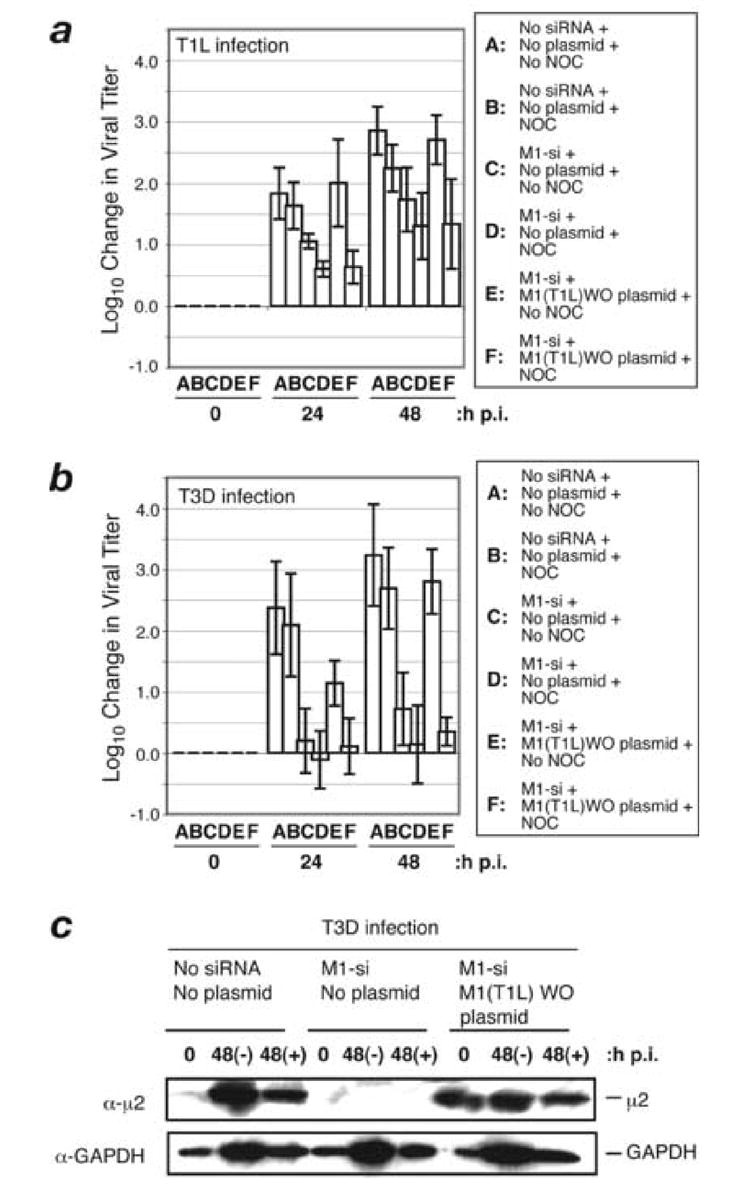
(A, B) CV-1 cells were transfected with no siRNA, M1-si, or M1-si in combination with M1(T1L)WO plasmid, and at 24 h p.t. were infected with T1L (A) or T3D (B) reovirus at 10 PFU/cell. At 6 h p.i., a subset of the infected cells were treated with 10 μM nocodazole and incubated further. Cells were harvested at 0, 24, or 48 h p.i., and viral titers were determined by plaque assays. Log10 changes in viral titer relative to time 0 are indicated. Each bar represents the average obtained from three independent experiments, and error bars indicate the standard deviations. (C) CV-1 cells were transfected with no siRNA, M1-si, or M1-si in combination with M1(T1L)WO plasmid, and at 24 h p.t. were infected with T3D reovirus at 10 PFU/cell. At 6 h p.i., a subset of the infected cells were treated with 10 μM nocodazole and incubated further. Samples were harvested at 0 and 48 h p.i., and cell-associated proteins were prepared for SDS-PAGE and immunoblotting. Protein was detected using specific antibodies against μ2 or GAPDH.
Fig. 8. Effect of NTPase or RTPase function of μ2 protein on complementation of M1-si-based reduction of infectious viral yields.
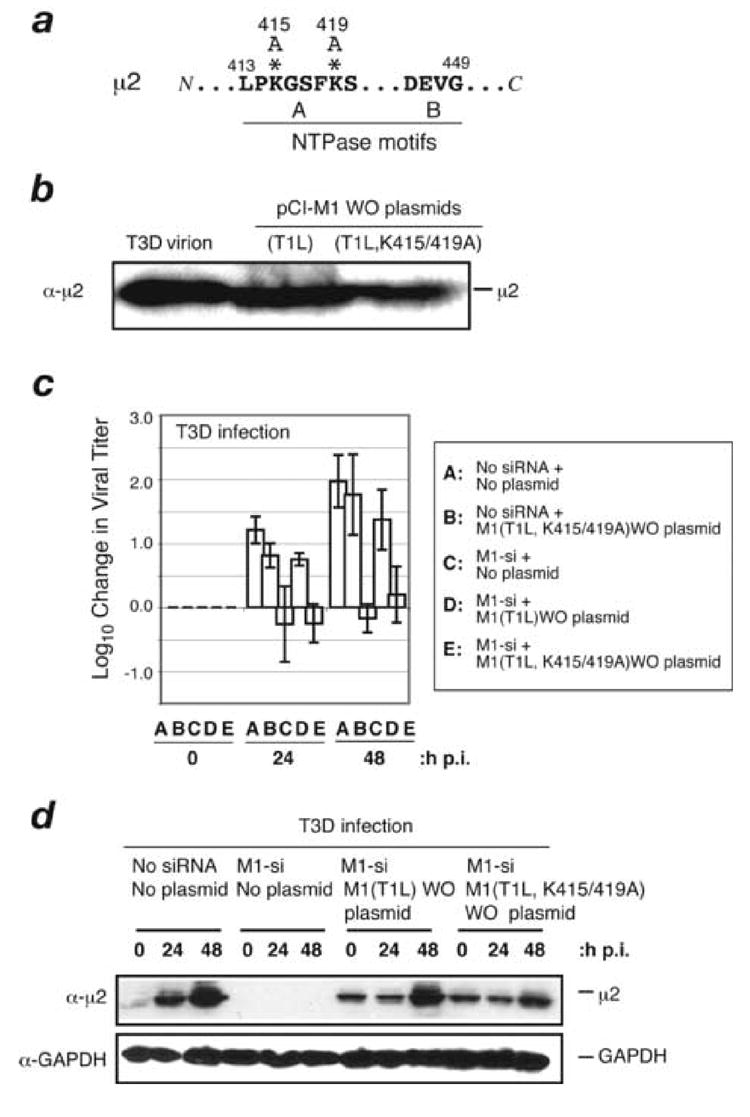
(A) Putative NTP-binding motifs in the T1L μ2 protein are shown. Asterisks indicate the two Ala-for-Lys substitutions in the mutant μ2 protein K415/419A. (B) CV-1 cells were transfected with 5 μg of M1(T1L) WO or M1(T1L,K415/419A)WO plasmid. Samples were harvested at 24 h p.i., and cell-associated protein was prepared for SDS-PAGE and immunoblotting. Protein was detected using specific antibodies against μ2. (C) CV-1 cells were transfected with no siRNA, M1(T1L,K415/419A)WO rescue plasmid, M1-si alone, M1-si plus M1(T1L)WO rescue plasmid, or M1-si plus M1(T1L,K415/419A)WO rescue plasmid, and at 24 h p.t. were infected with T3D reovirus at 10 PFU/cell. Cells were harvested at 0, 24, or 48 h p.i., and viral titers were determined by plaque assays. Log10 changes in viral titer relative to time 0 are indicated. Each bar represents the average obtained from three independent experiments, and error bars indicate the standard deviations. (D) CV-1 cells were transfected with no siRNA, M1-si, or M1-si in combination with M1(T1L)WO plasmid or M1(T1L,K415/419A)WO plasmid, and at 24 h p.t. were then infected with T3D reovirus. The cells were incubated for an additional 0, 24, or 48 h p.i., and cell-associated proteins were prepared for SDS-PAGE and immunoblotting. Protein was detected using specific antibodies against μ2 or GAPDH.
M1-si limits expression of other proteins, expansion of factories, and synthesis of genome
To explore the steps in reovirus infection at which the suppression of μ2 expression by M1-si may have subsequent effects, we examined the expression of other viral proteins. In the preceding experiments, we had in fact also analyzed the samples by immunoblotting for nonstructural protein μNS. In those samples, we found expression of μNS to be substantially reduced following M1-si treatment, though still detectable at both 24 and 48 h p.i. (Fig. 1b, c). In other experiments, immunoblotting showed that expression of structural proteins µ1 and λ2 was also greatly reduced following M1-si treatment, though again still detectable by 24 or 48 h p.i. (data not shown). To corroborate the immunoblotting results, we additionally examined cells at 24 h p.i. by immunofluorescence microscopy. In these experiments, reduced expression of the μ2 and μNS proteins (Fig. 2a, b), as well as the σNS and λ2 proteins (data not shown), of strains T1L and T3D was regularly observed following M1-si treatment. From the combination of blotting and microscopy results, we estimated that the expression levels of other viral proteins were reduced by ≥80–90% following M1-si treatment.
Fig. 2. Effect of M1-si on expression of μNS and expansion of viral factories for T1L and T3D reoviruses.
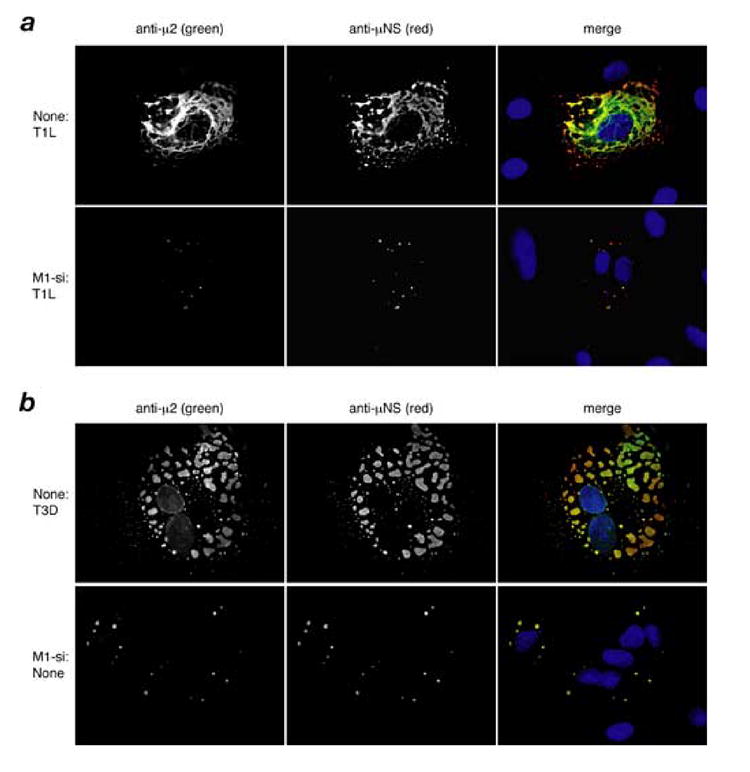
(A, B) CV-1 cells were transfected with no siRNA or M1-si, and at 24 h p.t. were then infected with T1L (A) or T3D (B) reovirus at 10 PFU/cell. At 24 h p.i., the cells were fixed and the localization of μ2 and μNS proteins were examined by immunostaining with specific antibodies to μ2 (directly conjugated to Alexa 488, green channel) and μNS (followed by secondary antibody conjugated to Alexa 594, red channel). Nuclei were counterstained with DAPI, as seen in the merge images (right column).
The microscopy approach was further used to evaluate the viral factories. Expansion of the factories by 24 h p.i. was greatly limited by M1-si (Fig. 2a, b), consistent with the reduced expression of not only μ2 but also other viral proteins. For both T1L and T3D, in the presence of M1-si, the factories remained small and globular, with the different viral proteins, though reduced in amounts, continuing to colocalize in these structures (Fig. 2a, b, and data not shown). In the case of T1L, the globular morphology of the small factories in M1-si-treated cells (Fig. 2a, bottom row) contrasted with the extensively filamentous morphology of the larger T1L factories formed in the absence of M1-si (Fig. 2a, top row). The latter morphology is explained by anchoring of the factories to cellular microtubules through the μ2 protein (Parker et al., 2002). The appearance of the T1L and T3D factories at 24 h p.i. following M1-si treatment was similar to that otherwise seen at earlier times after infection [e.g., at 6 h p.i. (Parker et al., 2002)].
Separate aliquots of cells from one of the microscopy experiments with reovirus T3D were also examined by gel electrophoresis to detect production of the viral dsRNA genome segments, reflecting minus-strand RNA synthesis coupled to new particle assembly. Production of the dsRNA segments was found to be strongly suppressed by M1-si (Fig. 3a), consistent with a block to multiplication preceding viral genome synthesis.
Fig. 3. Effect of M1-si on dsRNA synthesis and infectious viral yields.
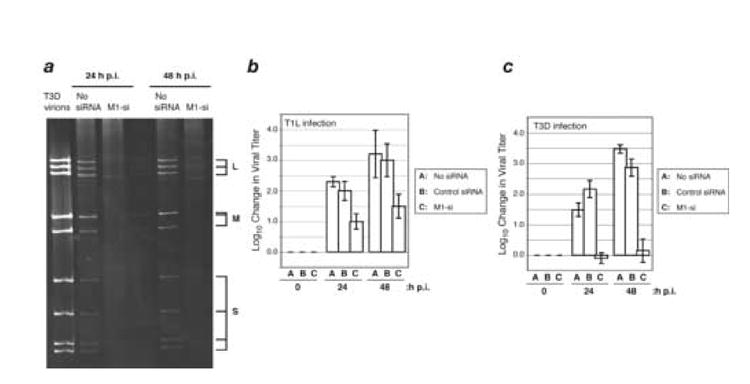
(A) CV-1 cells were transfected with no siRNA or M1-si, and at 24 h p.t. were then infected with T3D reovirus at 10 PFU/cell. Cells were incubated for 24 or 48 h p.i., and samples were prepared for gel electrophoresis to detect the viral dsRNA genome segments. L, genome segments encoding λ proteins; M, genome segments encoding μ proteins; S, genome segments encoding σ proteins. (B, C) CV-1 cells were transfected with no siRNA, nonspecific control siRNA, or M1-si, and at 24 h p.t. were then infected with T1L (B) or T3D (C) reovirus at 10 PFU/cell. Cells were harvested at 0, 24, or 48 h p.i., and viral titers were determined by plaque assays. Log10 changes in viral titer relative to time 0 are indicated. Each bar represents the average obtained from three independent experiments, and error bars indicate the standard deviations.
Based on these results, we can suggest the following with regard to the steps in infection at which the suppression of μ2 expression by M1-si may have subsequent effects. As noted in Introduction, most copies of the reovirus proteins over the course of an infection are translated from “secondary” transcripts that are synthesized by viral progeny particles following dsRNA genome synthesis within these newly formed particles (Zarbl and Millward, 1983). Thus, the limited expression of other reovirus proteins in concert with the limited expansion of the factories and the limited or absent synthesis of dsRNA genome segments is consistent with a block to infection by M1-si that precedes genome synthesis and secondary transcription. Whether “primary” transcription by infecting particles and translation of proteins from those primary transcripts are also reduced by M1-si is hard to conclude from the available data, but the routine detection of other viral protein expression and small factories may reflect that primary transcription and translation remain largely intact. Tentatively, then, we conclude that the block by M1-si follows primary translation of the viral proteins other than μ2, but precedes genome synthesis and secondary transcription. This is consistent with evidence from Coombs (1996) concerning the block to infection at nonpermissive temperature with an M1/μ2 temperature-sensitive mutant.
M1-si reduces infectious yields of T1L and T3D to differing extents
To evaluate the effect of M1-si on infectious yields of reovirus, CV-1 cells were transfected and infected as described above. The cells were incubated for an additional 0, 24, or 48 h p.i. before harvest, and infectious titers were determined by plaque assays. M1-si reduced infectious yields by more than one log10 value at 24 and 48 h for both T1L and T3D reovirus (Fig. 3b, c; compare bars A and C), whereas the nonspecific control siRNA had little or no effect (Fig. 3b, c; compare bars A and B). Interestingly, the yields of T3D were even more markedly reduced, by more than two log10 values and essentially to input levels, than were those of T1L (compare Fig. 3b and 3c, bar C). This suggests a strain difference in susceptibility of viral growth to the effect of M1-si, even though (i) the targeted RNA sequences of T1L and T3D M1 are identical (see Fig. 1a), (ii) the μ2 expression levels of both viruses were strongly suppressed by M1-si (see Fig. 1b, c), and (iii) the expression of other viral proteins and expansion of the viral factories for both viruses were greatly limited by M1-si (see Fig. 2). Further experiments were performed to confirm this strain difference and to identify its genetic basis.
Reduction in infectious yields by M1-si maps to the T1L and T3D M1 genome segments
The T1L-T3D strain difference seemed most likely to reflect an inherent property of the respective M1 genome segments or their products, which in turn modulated the targeted effect of M1-si. In that case, the strain difference should map to the M1 segment in a reassortant analysis. To investigate this possibility, we transfected CV-1 cells with or without M1-si and then infected the cells with a panel of viruses including T1L and T3D in addition to T1L-x-T3D reassortants (Brown et al., 1983; Drayna et al., 1982; Nibert et al., 1996). The cells were incubated for an additional 0 or 48 h p.i. before harvest, and infectious titers were determined by plaque assays. For each reassortant, the difference in infectious yields in the presence or absence of M1-si was calculated, and the reassortants were ranked by the extent of reduction in yields attributable to M1-si (Table 1). M1-si reduced infectious yields for those reassortants with the T3D M1 segment to a consistently greater extent than for those reassortants with the T1L M1 segment, and a statistical analysis of the data [Mann-Whitney test (Bodkin and Fields, 1989; Sherry and Fields, 1989; Zar, 1984)] revealed that origin of the M1 segment, and only the M1 segment, segregated with this phenotype to a significant degree (M1, p = 0.0016; other segments, p > 0.05) (Table 1). These results corroborate the T1L-T3D strain difference and indicate that this difference reflects an inherent property of the respective M1 segments or their products. As a consequence, they represent further evidence that the primary effect of M1-si is targeted to the M1 RNA. As noted in Introduction, the M1-encoded μ2 proteins of our lab’s T1L and T3D viral clones differ by nine amino acids including Pro/Ser208, which determines the relative capabilities for better (Pro208, T1L) versus poorer (Ser208, T3D) microtubule association, the latter accompanied by increased aggregation and polyubiquitination of μ2 (Miller et al., 2004; Parker et al., 2002; Yin et al., 2004). Thus, one possible explanation for the new strain difference observed in this study was that the poorer functionality of T3D μ2 made the T3D virus and T3D-M1-containing reassortants more sensitive to the effects of reducing μ2 levels by M1-si.
Table 1.
Genetic anaylsis of M1-si induced reduction in infectious viral yield using T1L X T3D reassortant viruses
| Virus straina | Genotypeb | Log 10 difference in yield between +/-siRNA groups at 48h c | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| L1 | L2 | L3 | M1 | M2 | M3 | S1 | S2 | S3 | S4 | ||
| EB144 | L | L | L | L | D | D | L | L | D | L | 0.50 +/- .24 |
| EB39 | L | D | D | L | D | D | D | D | D | D | 0.70 +/- .19 |
| EB87 | L | D | L | L | D | L | L | D | L | L | 0.73 +/- .19 |
| EB143 | D | L | L | L | L | L | D | L | L | L | 0.86 +/- .51 |
| H24 | L | L | L | L | L | L | L | L | L | D | 1.04 +/- .11 |
| EB140 | D | D | L | L | L | L | L | L | D | L | 1.04 +/- .61 |
| H14 | L | L | D | L | L | L | L | D | D | L | 1.10 +/- .43 |
| T1L | L | L | L | L | L | L | L | L | L | L | 1.77 +/- .39 |
| EB113 | L | L | L | D | L | L | L | L | D | L | 1.93 +/- .25 |
| EB146 | L | L | L | D | L | L | L | L | L | D | 1.97 +/- .12 |
| EB108 | L | D | L | D | L | L | L | L | D | D | 2.20 +/- .20 |
| G16 | L | L | L | D | L | L | L | D | L | L | 2.70 +/- .34 |
| T3D | D | D | D | D | D | D | D | D | D | D | 3.52 +/- .27 |
|
| |||||||||||
| P valuesd | 0.87 | 0.99 | 0.94 | 0.0016 | 0.20 | 0.47 | 0.81 | 0.83 | 0.99 | 0.38 | |
Reassortants are described in materials and methods. Strains are listed in order of increasing difference in infectious yield between M1-si treated and non-treated samples.
Parental origins of genome segments are indicated: D, T3D; L, T1L.
Mean +/- standard deviation of three determinations each.
Two tailed significance values from the Mann-Whitney test performed using instat Version 3 (Graph Pad).
Reduction in infectious yields by M1-si correlates with difference in microtubule association conferred by Pro/Ser208
Different viral clones of T3D reovirus obtained from other labs have been shown to differ in their capabilities for microtubule anchoring of the viral factories, and these differing capabilities have again been correlated with the presence of Pro versus Ser at μ2 residue 208 (Parker et al., 2002; Yin et al., 2004). For example, a T3D viral clone obtained from the Cashdollar lab (T3DC) has Pro instead of Ser at μ2 residue 208 and forms extensively microtubule-anchored factories (Fig. 4a), unlike the T3D viral clone from our lab (Parker et al., 2002). On the other hand, an independent viral field isolate, Type 3 clone 12 (T3c12), has Ser at μ2 residue 208 and does not form extensively microtubule-anchored factories (Fig. 4a) (Yin et al., 2004). Notably, both T3DC and T3c12 are identical to T1L and T3D from our lab in the M1 RNA sequence targeted by M1-si (data not shown). Upon testing T3DC and T3c12 for the effects of M1-si on viral growth as in the preceding sections, we found that T3DC behaved similarly to T1L, with infectious yields reduced by less than two log10 values at 24 and 48 h p.i., whereas T3c12 behaved similarly to our lab’s viral clone of T3D, with infectious yields reduced by more than two log10 values, and essentially to input levels, at 24 and 48 h p.i. (Fig. 4b, compare bars C and D). These results are therefore consistent with the conclusion that the strain differences in growth reduction by M1-si are correlated with the other differing properties of μ2, including microtubule association, conferred by the sequence polymorphism Pro/Ser208. Further genetic tests would be useful to confirm this conclusion, but were beyond the scope of this study.
Fig. 4. Effect of M1-si on infectious viral yields of T3DC and T3c12 reoviruses.
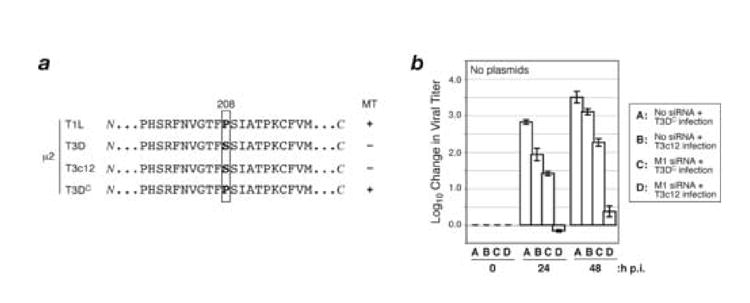
(A) Protein sequences for μ2 from reovirus strains T1L, T3D, T3DC, and T3c12. T1L and T3DC reoviruses have a Pro at μ2 residue 208 and encode μ2 proteins that strongly associate with microtubules (+); T3D and T3c12 reoviruses have a Ser at this position and encode a μ2 protein that weakly associates with microtubules (−). (B) CV-1 cells were transfected with no siRNA or M1-si, and at 24 h p.t. were then infected with T3DC or T3c12 reovirus at 10 PFU/cell. Cells were harvested at 0, 24, or 48 h p.i., and viral titers were determined by plaque assays. Log10 changes in viral titer relative to time 0 are indicated. Each bar represents the average obtained from three independent experiments, and error bars indicate the standard deviations.
Reduction of infectious yields by M1-si is rescued by plasmid encoding T1L but not T3D μ2
Previous studies with rotavirus have provided evidence that the viral plus-strand RNA molecules that serve for replication and packaging into progeny particles are largely resistant to siRNA-based restriction (Dector et al., 2002; Silvestri et. al, 2004; Taraporewala et al., 2006). The reduction in infectious yields of rotavirus is thereby largely attributable to the reduction in protein expression from the viral plus-strand RNA molecules that serve for translation (i.e., those that serve as mRNAs) and are sensitive to siRNA-based restriction. As a consequence, if the viral protein whose expression is reduced by siRNA-based restriction can be effectively supplied in trans, then yields of infectious progeny can be rescued to some degree (Taraporewala et. al, 2006). Given many similarities in the basic replication strategies of rotavirus and reovirus, both members of the family Reoviridae, we hypothesized that this same mechanism of siRNA-based reduction of infectious yields may hold true for reovirus.
To test this hypothesis, we constructed rescue plasmids to express the sequence-matched μ2 proteins of our lab’s viral clones of strains T1L and T3D, but including three silent (wobble [WO]) mutations in the RNA sequence targeted by M1-si in each M1 gene (Fig. 5a; WO mutations shown by asterisks). When transfected into CV-1 cells, these plasmids expressed the respective μ2 proteins to similarly high levels as did previously described plasmids (Parker et al., 2002) containing the fully wild-type (WT) M1 genes of these strains (i.e., again sequence-matched at the amino acid level for each respective strain, but without the WO mutations in the genes) (Fig. 5b).
To test for rescue of infectious yields, CV-1 cells were transfected with (i) either of the plasmids alone, (ii) M1-si alone, or (iii) either plasmid in combination with M1-si. Cotransfections of M1-si with either M1(T1L)WT or M1(T3D)WT were included as controls. At 24 h p.t., the cells were then infected with either T1L or T3D reovirus at 10 PFU/cell. The cells were incubated for an additional 0, 24, or 48 h p.i. before harvest, and infectious titers were determined by plaque assays. In initial trials, we matched the transfected plasmids (encoding T1L or T3D μ2) to the infecting virus (T1L or T3D), but in subsequent trials we tested each plasmid with the opposite virus as explained below. In these experiments, infectious yields from cells transfected with either of the plasmids alone were mostly similar to those from mock-transfected cells (Fig. 5c–f, compare bars A and B; greatest difference seen in panel c), suggesting that the plasmids and their products were not strongly toxic. Also, reproducing preceding results (see Fig. 3b, c), infectious yields from cells transfected with M1-si alone were reduced by substantial, though differing, extents for both T1L and T3D reovirus (Fig. 5c–f, bar C).
The results for combined transfections with rescue plasmid and M1-si were as follows. Plasmid M1(T1L)WO provided ~10-fold rescue of T1L yields by 48 h p.i. (Fig. 5c; compare bars C and D). In contrast, plasmid M1(T3D)WO provided less than 10-fold rescue of T3D yields at either 24 or 48 h p.i (Fig. 5d, compare bars C and D). Moreover, as expected, plasmids M1(T1L)WT and M1(T3D)WT (both still targeted by M1-si) provided less than 10-fold rescue of either T1L or T3D yields in the respective cases (Fig. 5c, d; compare bars C and E). Together, these initial results suggest either a defect in the capacity of plasmid-derived T3D μ2 to provide rescue in this system or a defect in the capacity of T3D virus to be rescued. To distinguish between these possibilities, we tested the capacity of plasmid encoding T1L μ2 to rescue T3D yields and the capacity of plasmid encoding T3D μ2 to rescue T1L yields. In the first case, we found that plasmid M1(T1L)WO provided greater than 100-fold rescue of T3D yields by 48 h p.i. (Fig. 5e, compare bars C and D). Interestingly, plasmid T1L(M1)WT provided greater than 10-fold rescue of T3D yields by 48 h p.i. (Fig. 5e, compare bars C and E). In contrast, we found that neither plasmid M1(T3D)WO (Fig. 5f, compare bars C and D) nor plasmid M1(T3D)WT (Fig. 5f, compare bars C and E) provided as much as 10-fold rescue of T1L yields at either 24 or 48 h p.i.
Following SDS-PAGE of samples collected in parallel with those for plaque assays, immunoblot analysis revealed that M1-si strongly suppressed expression of μ2 in these experiments, as expected from preceding results (see Fig. 1b, c), and that the WO, but not the corresponding WT, plasmids strongly counteracted this expression defect (Fig. 5g, and data not shown). We conclude from these results that the difference in rescue between T1L and T3D in the first experiments with strain-matched virus and plasmid (see Fig. 5c, d) was based in the plasmid, not the virus. We furthermore conclude that plasmid-derived T3D μ2 has a largely post-expression defect in its capacity to rescue viral growth following reduction by M1-si. The latter conclusion presents an interesting paradox, in that virus-derived T3D μ2 is functional to support the growth of T3D virus and T3D-M1-containing reassortants, whereas sequence-matched, plasmid-derived T3D μ2 is much less or not so functional in the rescue system.
Strain difference in rescue by plasmid-derived μ2 is primarily determined by Pro/Ser208
As noted above, a previous study has identified nine amino acid differences between the μ2 proteins of our lab’s viral clones of strains T1L and T3D (Parker et al., 2002). Any one of these nine differences might be responsible for the different capacities of the sequence-matched, plasmid-derived T1L and T3D μ2 proteins to rescue viral growth after M1-si treatment. The difference at μ2 residue 208 (Fig. 6a) is noteworthy, however, because it determines the relative capability of μ2 to associate with microtubules and, reciprocally, its relative proneness to undergo aggregation and polyubiquitination (Miller et al., 2004; Parker et al., 2002; Yin et al., 2004). We therefore hypothesized that Pro/Ser208 may be the primary determinant of whether plasmid-derived μ2 is competent for rescue of infectious yields in this system.
Fig. 6. Effect of μ2 amino acid Pro/Ser208 on complementation of M1-si-based reduction of infectious viral yields.
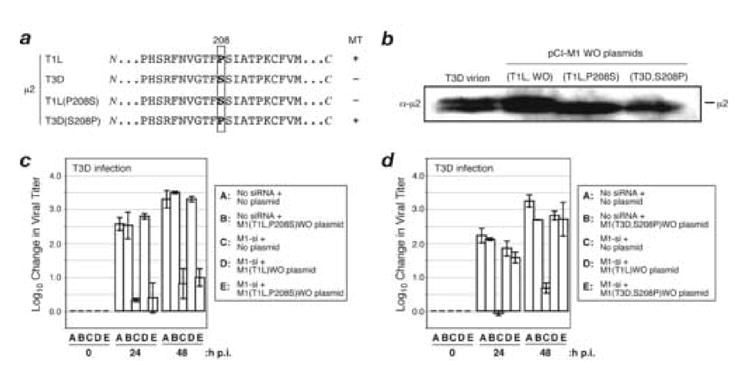
(A) Protein sequences for μ2 from reovirus strains T1L or T3D, or plasmids encoding T1L(P208S) or T3D(S208P) μ2. Box indicates the position of amino acid 208. T1L reovirus and plasmid encoding T3D(S208P) μ2 have a Pro at this position and encode a μ2 protein capable of strongly associating with microtubules (+); T3D reovirus and plasmid encoding T1L(P208S) μ2 have a Ser substituted at this position and weakly associate with microtubules (−). (B) CV-1 cells were transfected with 5 μg of either M1(T1L,P208S)WO or M1(T3D,S208P)WO rescue plasmids. The cells were incubated for an additional 24 h p.i., and cell-associated proteins were prepared for SDS-PAGE and immunoblotting. Protein was detected using specific antibodies against protein μ2. (C, D) CV-1 cells were transfected with no siRNA, rescue plasmid alone, M1-si alone, or M1-si in combination with a particular rescue plasmid, and at 24 h p.t. were infected with T3D reovirus at 10 PFU/cell. Cells were harvested at 0, 24, or 48 h p.i., and viral titers were determined by plaque assays. Log10 changes in viral titer relative to time 0 are indicated. Each bar represents the average obtained from three independent experiments, and error bars indicate the standard deviations.
To test the importance of Pro/Ser208 within the rescuing μ2 protein, we constructed plasmids that not only contained the previously utilized WO mutations in the M1-si-targeted sequence of each M1 gene (see Fig. 5a) but also encoded the reciprocal amino acids at position 208: Pro208 changed to Ser in the otherwise T1L μ2 background [T1L(P208S)] and Ser208 changed to Pro in the otherwise T3D μ2 background [T3D(S208P)]. Both of these new plasmids provided similarly high levels of μ2 protein expression following transfection of CV-1 cells (Fig. 6b). To test for rescue of infectious yields, CV-1 cells were transfected with (i) either of the new plasmids alone, (ii) M1-si alone, or (iii) either new plasmid in combination with M1-si. Cotransfections of M1-si with M1(T1L)WO were included as controls. At 24 h p.t., the cells were then infected with T3D reovirus at 10 PFU/cell. The cells were incubated for an additional 0, 24, or 48 h p.i. before harvesting, and infectious titers were determined by plaque assays. In these experiments, infectious yields from cells transfected with the new rescue plasmids alone were similar to those from mock-transfected cells (Fig. 6c, d; compare bars A and B). Also, as expected from preceding results, infectious yields of T3D from cells transfected with M1-si alone were substantially reduced (Fig. 6c, d; bar C).
The results for combined transfections with rescue plasmid and M1-si were as follows. Whereas plasmid M1(T1L)WO provided greater than 100-fold rescue of T3D yields by 48 h p.i. (Fig. 6c, compare bars C and D), plasmid M1(T1L,P208S)WO provided less than 10-fold rescue of T3D yields by either time point (Fig. 6c, compare bars C and E). On the other hand, plasmid M1(T3D,S208P)WO provided ~100-fold rescue of T3D yields by 48 h p.i. (Fig. 6d, compare bars C and E), very similar to that provided by plasmid M1(T1L)WO (Fig. 6d, compare bars C and D). In yet another experiment, we found that plasmid M1(T3D,S208P)WO also provided ~10-fold rescue of T1L yields by 48 h p.i., very similar to that provided by plasmid M1(T1L)WO (data not shown). These results obtained after swapping Pro/Ser208 into the opposite μ2 backgrounds suggest that the other eight amino acid differences between T1L and T3D μ2 play little if any role in the observed strain difference in rescue. We therefore conclude that the strain difference in capacity of plasmid-derived T1L and T3D μ2 to rescue the M1-si-based reduction of infectious yields is determined primarily by the Pro/Ser difference at residue 208.
Rescue by plasmid-based μ2 expression is microtubule dependent
Because Pro (T1L) versus Ser (T3D) at μ2 position 208 determines the relative capability of μ2 to associate with microtubules (T1L associates better than T3D) (Miller et al., 2004; Parker et al., 2002; Yin et al., 2004), one possible explanation for the difference in rescue capacity of T1L and T3D μ2 in the preceding experiments was that rescue is microtubule dependent. In other words, T1L μ2 may be better at rescuing viral growth because it is better at associating with microtubules. As a test of this possibility, we performed rescue experiments with T1L μ2 in the presence or absence of the microtubule-depolymerizing drug nocodazole.
CV-1 cells were transfected with (i) M1-si alone or (ii) rescue plasmid M1(T1L)WO in combination with M1-si, and at 24 h p.t., the cells were infected with either T1L or T3D reovirus at 10 PFU/cell. The cells were incubated for an additional 0, 24, or 48 h p.i. before harvesting, and infectious titers were determined by plaque assays. In addition, to one replicate of each sample destined for harvest at 24 or 48 h p.i., 10 μM nocodazole was added for the remainder of the growth period beginning at 6 h p.i. In these experiments, nocodazole treatment had little if any effect on infectious yields from mock-transfected cells infected with either T1L or T3D reovirus (Fig. 7a, b; compare bars A and B), consistent with previous results involving colchicine (another microtubule-depolymerizing drug) or nocodazole treatments (Dales, 1963; Spendlove et al., 1964) (J. S. L. Parker and M. L. Nibert, unpublished data). Similarly, nocodazole treatment had little if any additive effect on the reduction of infectious yields provided by transfection with M1-si alone, in the setting of either T1L or T3D infection (Fig. 7a, b; compare bars C and D). In striking contrast, however, nocodazole treatment markedly inhibited the rescue of infectious yields provided by cotransfection with plasmid M1(T1L)WO, in the setting of either T1L or T3D infection (Fig. 7a, b; compare bars D–F). Following SDS-PAGE of samples collected in parallel with those for plaque assays at the 0 and 48 h p.i. time points, immunoblot analysis revealed that treatment with nocodazole had little or no effect on the expression levels of T1L μ2 (Fig. 7c), thus indicating that the lack of rescue of infectious yields by plasmid-derived T1L μ2 in these experiments was not because of a μ2 expression defect attributable to nocodazole. We conclude from these results that the rescue of infectious yields by plasmid-derived T1L μ2 is strongly microtubule dependent at a step following μ2 expression. This conclusion presents another interesting paradox, in that virus-derived T1L μ2 is functional to support approximately normal growth of T1L virus in the presence of nocodazole whereas sequence-matched, plasmid-derived T1L μ2 is much less or not so functional in the rescue system. We furthermore infer from these results that the poor capacity of T3D μ2 to provide rescue in preceding experiments may be explained at least in part by its reduced capability to associate with microtubules.
Further test of the rescue system: importance of μ2 NTPase or RTPase function
The capacity of plasmid-based expression of T1L μ2 to rescue infection after M1-si-based reduction of infectious yields provides a general genetic approach by which sequence determinants of μ2 functions that are required for productive infection can be identified or confirmed. The preceding experiments involving plasmid-based expression of T1L, T3D, T1L(P208S), and T3D(S208P) μ2 exemplify this application. To provide further evidence for the utility of this approach, we used it to test the relevance of μ2 NTPase or RTPase function during reovirus infection. Previous studies have implicated μ2 protein in influencing in vitro enzymatic activities of reovirus core particles (Noble and Nibert, 1997; Yin et al., 1996). Results from our lab have further shown that purified μ2 possesses in vitro NTPase and RTPase activities (Kim et al., 2004), which have been mapped in part to the Lys residues at positions 415 and 419, within a putative NTP-binding motif of μ2 (Nibert and Kim, 2004; Noble and Nibert, 1997) (Fig. 8a). When these residues are mutated to Ala (Fig. 8a, asterisks), the in vitro NTPase and RTPase activities of μ2 [i.e., μ2(K415,419A)] are abrogated (Kim et al., 2004).
To address the importance of μ2 NTPase or RTPase function during reovirus infection, we incorporated changes encoding the Ala substitutions at residues 415 and 419 into the M1(T1L)WO plasmid [to produce M1(T1L,K415/419A)WO] and then tested its capacity to rescue the M1-si-based reduction of infectious yields. When transfected into CV-1 cells, this new plasmid expressed the μ2 protein to similarly high levels as M1(T1L)WO (Fig. 8b). To test for rescue of infectious yields, CV-1 cells were transfected with (i) M1(T1L,K415/419A)WO plasmid alone, (ii) M1-si alone, or (iii) either M1(T1L)WO or M1(T1L,K415/419A)WO plasmid in combination with M1-si. At 24 h p.t., the cells were then infected with T3D reovirus at 10 PFU/cell. The cells were incubated for an additional 0, 24, or 48 h p.i. before harvesting, and infectious titers were determined by plaque assays. In these experiments, infectious yields from cells transfected with the M1(T1L,K415/419A)WO alone were similar to those from mocktransfected cells (Fig. 8c, compare bars A and B). Also, as expected from preceding results, infectious yields of T3D from cells transfected with M1-si alone were substantially reduced (Fig. 8c, bar C). The results for combined transfections with rescue plasmid and M1-si were as follows. Whereas plasmid M1(T1L)WO provided greater than 10-fold rescue of T3D infection by 48 h p.i. (Fig. 8c, compare bars C and D), plasmid M1(T1L,K415/419A)WO provided less than 10-fold of T3D infection by either time point (Fig. 8c; bar E). Following SDS-PAGE of samples collected in parallel with those for plaque assays, immunoblot analysis revealed that M1-si strongly suppressed expression of T3D μ2 in these experiments (Fig. 8d), as expected from preceding results, and that both rescue plasmids counteracted this expression defect (Fig. 8d). We conclude from these results that the NTPase or RTPase function of μ2 protein newly synthesized during the course of infection is required for effective viral growth.
Discussion
siRNAs and other silencing technologies have become powerful tools for investigating RNA and protein functions in many organisms and viruses. In this study, we applied this approach to reovirus, specifically to its M1 plus-strand RNA and encoded μ2 protein, during productive infections in CV-1 cells. We targeted μ2 because it is one of the least well understood of the reovirus structural proteins and to which we have devoted a good deal of effort in trying to decipher its functions. For example, the microtubule-anchoring activity of μ2 has been established (Kim et al., 2004; Miller et al., 2004; Parker et al., 2002; Yin et al., 2004), but its significance for infection has remained unclear. The NTPase and RTPase activities of purified μ2 and the role of M1/μ2 in genetically influencing enzymatic activities of reovirus core particles have also been established, but only in vitro (Kim et al., 2004; Noble and Nibert, 1997; Yin et al., 1996); their significance for infection has remained unproven. We were therefore eager to apply the siRNA approach, in combination with a method for protein-based rescue, as a potential means for advancing our understanding of μ2 functions during reovirus infection.
It is worth emphasizing that the approach described here for rescue of infectious yields might not have worked. If the M1 plus-strand RNAs that are used for replication and packaging into infectious progeny had also been susceptible to siRNA-based degradation, then to rescue infectious yields, we would have needed to replace those molecules as well as the M1 plus-strand RNAs that are used for μ2 translation. Instead, by needing to replace only the latter, we were able to use a common expression plasmid (pCI-neo) for M1 mRNA and μ2 protein, without any effort for the RNA to be properly terminated for use in replication and packaging. Thus, our success at rescuing infectious yields by this approach provides strong evidence, as previously indicated for rotavirus (Dector et al., 2002; Silvestri et. al, 2004; Taraporewala et al., 2006), that the reovirus plus-strand RNA molecules that are used for replication and packaging into infectious progeny are much less susceptible to silencing than are the mRNAs.
The difference in susceptibility of T1L and T3D reovirus to reduction of infectious yields by M1-si, despite sharing the same target sequence, was unexpected but explainable. We propose that μ2 is one of the reovirus proteins for which a relatively low level of expression may be sufficient for supporting production of infectious progeny. We further propose that the levels of functional μ2 are in greater excess in T1L infection than in T3D infection, as reflected by the reduced microtubule association of T3D μ2 as well as by its greater tendency to undergo aggregation and polyubiquitination (Miller et al., 2004; Parker et al., 2002; Yin et al., 2004). As a result, in the setting of T3D infection, we propose that it is easier to reduce μ2 expression to levels that provide greater reduction of infectious yields. The same holds true for infections with T3D-M1-containing reassortants and T3c12. This interpretation may also help to explain why plasmid M1(T1L)WT, which remains targeted by M1-si, can support some rescue of infectious yields in certain cases. The idea would be that even the small amount of μ2 expression from this plasmid that may evade silencing would be enough to support a demonstrable increase in viral growth.
Previous results (Dales, 1963; Spendlove et al., 1964) (J. S. L. Parker and M. L. Nibert, unpublished data), reproduced in this study, have shown that the microtubule-depolymerizing drug nocodazole causes little reduction in infectious yields under normal circumstances of reovirus growth in culture. This is true both for a strain such as our lab’s viral clone of T3D, whose μ2 protein shows poor association with microtubules, and for a strain such as T1L, whose μ2 protein shows better association with microtubules (Miller et al., 2004; Parker et al., 2002; Yin et al., 2004). Thus, to date, there has been no strong evidence that μ2-microtubule association has a significance for infectious yields of reovirus in culture. Then how can we explain the nocodazole results in this report showing that, in the setting of plasmid-based rescue of T1L μ2 expression in CV-1 cells, intact microtubules are required for reovirus growth and production of infectious progeny? A possible explanation is that in the circumstance of T1L μ2 expression from plasmid-derived mRNAs, microtubule binding of μ2 is needed to ensure that μ2 can be properly folded and/or can reach the proper sites for viral replication and assembly. This would suggest in turn that T1L μ2 expression from virus-derived transcripts may be more specifically localized, in closer juxtaposition to the expression sites for other viral proteins that may assist μ2 folding and/or to the proper sites for viral replication and assembly, such that microtubule association of μ2 is not as strictly required. This explanation leaves open the question of what role μ2-microtubule association may have under the normal circumstance of μ2 expression from virus-derived transcripts, but we recognize that this role may be more evident in host tissues, such as for infection of certain cell types or spread between cells. In any case, this study provides the first example in which intact microtubules are required for μ2 function in reovirus replication.
In light of the preceding explanation for the nocadozole effect, it is possible that plasmid-derived T3D μ2 was poor at rescue of infectious yields after M1-si treatment because of its poor microtubule association capability. This explanation is consistent with the importance of Pro208, as in T1L and T3D(S208P) μ2, in supporting strong rescue of infectious yields. However, Ser208 as in T3D and T1L(P208S) μ2 is not only associated with poor microtubule association (Miller et al., 2004; Parker et al., 2002; Yin et al., 2004) and poor rescue (this study), but also with a proneness of μ2 to aggregation and polyubiquitination (Miller et al., 2004). Thus, it is also possible that plasmid-derived T3D μ2 was poor at rescue of infectious yields because less of it remains available in functional form after expression. It seems necessary to separate, if possible, the microtubule-binding and aggregation phenotypes of μ2 in order to answer this question more definitively. In any case, the results for T3D μ2 reveal another difference between virus- and plasmid-derived protein, in this case in the absence of nocodazole, in that virus-derived T3D μ2 is functional to support viral growth but plasmid-derived T3D μ2 is not so functional in the rescue system. As in the preceding paragraph, a possible explanation is that expression of μ2 from virus-derived transcripts may be more specifically localized, in closer juxtaposition to the expression sites for other viral proteins that may assist μ2 folding and/or to the proper sites for viral replication and assembly, such that the defects of virus-derived T3D μ2 are blunted.
The failure of plasmid encoding μ2(K415,419A) to rescue infectious yields of T3D following reduction with M1-si represents the first direct evidence that the μ2 NTPase or RTPase function has significance for infection. Our lab’s previous evidence with regard to these activities of μ2 has been restricted to in vitro findings (Kim et al., 2004; Noble and Nibert, 1997), as has been previous evidence from the Coombs lab implicating μ2 as a determinant of strain differences in the transcriptase activities of reovirus cores (Yin et al., 1996). In addition, the Coombs lab has provided in vivo evidence that a temperature-sensitive form of μ2 is not competent to support the synthesis of genomic dsRNA at nonpermissive temperature, implicating μ2 in the assembly or function of viral replicase particles in infected cells (Coombs, 1996). The latter results are consistent with our tentative conclusion that suppression of μ2 expression by M1-si imposes a block to reovirus infection that follows primary translation but precedes genome synthesis and secondary transcription. Combining these results, our working hypothesis is that, within infected cells, the μ2 protein is normally assembled into progeny particles, where its NTPase function is required for genome replication and/or secondary transcription, as well as for primary transcription once progeny virions are released and move on to infect new cells. In the case of transcription, the RTPase activity of μ2 may be instead or additionally required in 5′-capping of the plus-strand RNAs, i.e., for generating a diphosphorylated 5′ end on each RNA to which GMP is then linked by the guanylyltransferase λ2 (Cleveland et al., 1986; Furuichi et al., 1976; Mao and Joklik, 1991). Having successfully applied the siRNA approach, we are now in stronger position to perform a combination of future studies in vitro and in vivo to dissect the different roles of μ2 during infection.
As the current report was being finalized for initial submission, a related one from the Dermody lab was published (Kobayashi et al., 2006). In this other report, the authors describe striking new RNA interference results for the reovirus μ2, μNS, and σNS proteins, and also demonstrate rescue of infectious yields by plasmid-derived µNS. For µ2-related experiments that overlap in these two reports, the results are in strong agreement. The utility of silencing and complementation for genetic studies of reovirus has thus been established to date for two of the viral proteins.
Materials and methods
Cells, viruses, antibodies, and enzymes
CV-1 (African green monkey kidney) cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (GIBCO) containing 10% fetal bovine serum (HyClone) and 10 μg per ml of gentamicin (GIBCO). Mouse L929 cells adapted to spinner culture were maintained in Joklik’s Modified Eagle Medium (Irvine Scientific) containing 2% fetal bovine serum and 2% bovine calf serum (HyClone), plus 2 mM L-glutamine, 100 U per ml penicillin G, and 100 μg per ml streptomycin (Irvine Scientific). Reovirus strains T1L and T3D were our lab stocks derived from clones from the B. N. Fields lab. The T3D virus was the same as that designated T3DN in some other recent reports from our lab. Third-passage L929 cell lysate stocks of doubly plaque-purified reovirus clones were used for all infections. Antibodies for immunoblot detection of μ2 and μNS have been described (Broering et al., 2000; Parker et al., 2002). Antibody against GAPDH was from Santa Cruz Biotech. Microscopy studies to detect μ2 and μNS protein used anti-μ2 antibodies directly conjugated to Alexa 488 and anti-μNS antibodies followed by secondary staining with goat anti-rabbit antibodies conjugated to Alexa 594 (Miller et al., 2003; Parker et al., 2002) All antibodies were titrated to optimize signal-to-noise ratio. All enzymes were from New England Biolabs unless otherwise noted.
Rescue plasmids
pCI-M1(T1L) and pCI-M1(T3DN) [designated M1(T1L)WT and M1(T3D)WT in this report] have been described previously (Parker et al., 2002) and encode μ2 proteins with the same amino acid sequences as those of the T1L and T3D reoviruses from our lab. To obtain point mutations in the M1 gene encoding μ2, overlap PCR mutagenesis with Pfu polymerase and the QuikChange XL-site directed mutagenesis kit were employed (Stratagene). Forward primer (5′-CTTGAAATGTTGGGTATCGAGATCGCCGACTATTGCATTCGTC-3′) and reverse primer (5′-GACGAATGCAATAGTCGGCGATCTCGATACCCAACATTTCAAG-3′) were used to mutate the M1 gene by introducing silent mutations (wobble [WO] cytosines) at nucleotides 112, 118, and 121, at the same time introducing a new AciI site. AciI digests were then used to identify the clones that had the proper wobble changes. Positive candidates were then sequenced to confirm that there were no additional mutations incorporated during PCR. Positive candidates from sequencing were then digested with EcoRV and SacII to yield fragments containing the wobble mutations, which were then subcloned back into M1(T1L)WT or M1(T3D)WT to produce plasmids pCI-M1(T1L)WO and pCI-M1(T3D)WO [henceforth designated M1(T1L)WO and M1(T3D)WO in this report].
pCI-M1(T1L)-P208S and pCI-M1(T3DN)-S208P [designated M1(T1L,P208S)WT and M1(T3D,S208P)WT in this report] have been described previously (Parker et al., 2002) and encode μ2 proteins with the same amino acid sequences as those of the T1L and T3D reoviruses from our lab except for the indicated swaps of Pro and Ser at position 208. Plasmid M1(T1L,P208S)WO was made by digesting plasmid M1(T1L)WO with AflIII followed by SacII; the fragment of this sequential digest containing the wobble mutations was then inserted into the same digest of M1(T1L,P208S)WT. Plasmid M1(T3D,S208P)WO was made by digesting plasmid M1(T3D)WO with SalI, which cuts twice; the fragment of this digest containing the silent mutations was then ligated into a SalI-digested M1(T3D,S208P)WT that had been treated with calf intestinal phosphatase. The presence of the respective mutations in the two final plasmids was confirmed by sequencing.
siRNA transfection followed by reovirus infection
The following general protocol was modified from one initially developed for studies of the reovirus μNS protein (M. M. Arnold and M. L. Nibert, manuscript in preparation). Duplex siRNAs containing sequences positioned within the open reading frame of the M1 gene segment were purchased from Dharmacon, after being designed according to the company’s highest criteria score (Fig. 1a). Typically in siRNA experiments, CV-1 cells were grown to near confluency in 75-cm2 flasks. Cells were released from the flask with trypsin/EDTA solution (Irvine Scientific), harvested by pelleting at 1,500 rpm, and prepared for transfection. A 400-μl volume of siRNA and/or plasmid DNA in DMEM lacking antibiotic was added to the cells and transfected via electroporation using a Gene Pulser II device (Bio-Rad) set at 0.220 kV and high capacitance (950 μF). In most cases, 4.5μg (300 nM) of siRNA and 5μg of plasmid DNA were used for transfections. Samples were then incubated for 24 h post-transfection (p.t.) before subsequent infections. At 24 h p.t., the transfection mixture was removed from the samples and saved for future use. The cells were rinsed twice with phosphate-buffered saline (PBS) [137 mM NaCl, 3 mM KCl, 8 mM Na2HPO4 (pH 7.5)] containing 2 mM MgCl2 and then infected with T1L or T3D reovirus at 10 PFU/cell. The virus was adsorbed for 1 h at room temperature before the transfection mixture was returned to the samples. The infected cells were then incubated at 37°C for 0, 24, or 48 h before preparation for viral titer assays. Based on the titer and μ2 expression results, we generally estimated that the transfection efficiencies for siRNAs and plasmids in these experiments were respectively >90% and >30% with regard to the cells that were also infected.
Viral titer assays
Infected cells were harvested by scraping at 0, 24, or 48 h post-infection (p.i.), and the lysates were placed into 1-dram vials. The samples were then subjected to two cycles of freezing and thawing, and titered by plaque assays as described elsewhere (Middleton et al., 2002). Plaques were counted 2 to 4 days later, depending on the reovirus strain. Viral yields were calculated with the formula log10(PFU/ml)t = xd − log10(PFU/ml)t = 0, where t is time and xd is days p.i.
SDS-PAGE and immunoblotting
siRNA-transfected and/or infected cells were scraped into the medium, harvested by pelleting at 1,500 rpm, and lysed at 4°C in buffer A [10 mM HEPES-KOH, pH 7.9; 1.5 mM MgCl2; 10 mM KCl; 1X protease inhibitor cocktail (Roche Diagnostics), and 1% NP-40] for 30 min with multiple vortex steps at 5-min intervals. Following lysis, samples were prepared in SDS gel loading buffer for electrophoresis on a 10% polyacrylamide gel. Proteins were then transferred from gels onto nitrocellulose membranes, and the blots were soaked in 1X Tris-buffered saline plus Tween (TBST) (20 mM Tris, pH 7.5; 500 mM NaCl; 1% Tween-20) containing 5% milk before addition of antibodies in 1X TBST containing 1% milk. 1X TBST containing 1% milk was also used in subsequent washes. Binding of primary antibody was detected with HRP-conjugated secondary antibodies and SuperSignal Chemiluminescence reagent (Pierce) or by using alkaline-phosphatase-coupled donkey anti-rabbit (Bio-Rad) and the colorimetric reagents p-nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-indolylphosphate p-toluidine salt (Bio-Rad).
Indirect immunofluorescence microscopy and viral dsRNA synthesis
Cells to be processed for immunofluroescence microscopy were fixed for 10 min at room temperature in 2% paraformaldehyde. Fixed cells were washed with PBS and permeablized and blocked in PBS containing 1% bovine serum albumin and 0.1% Triton X-100 (PBSA-0.1%TX100). Primary antibodies were diluted in PBSA-0.1%TX100 and incubated with the cells for 45 min at 37°C. After three washes in PBS, secondary antibodies diluted in PBSA-0.1%TX100 were added and incubated with the cells for 45 min at 37°C. Cover slips were incubated with 300 nM DAPI (4,6-diamidino-2-phenylindole; Molecular Probes) in PBS for 10 min to counterstain the cell nuclei, washed in PBS, and then mounted on glass slides with Prolong (Molecular Probes). Samples were examined with a 60X, 1.4 NA objective using a Nikon TE-2000U inverted microscope equipped with phase and fluorescence optics. Images were collected digitally using a cooled, charge-coupled device camera (Hamamatsu Corp.) and analyzed with Metamorph 6.1 (Molecular Devices). All images were processed and prepared for presentation using Photoshop 5.5 (Adobe Systems, Inc.).
For dsRNA analysis, cell lysates were collected at indicated times after infection using TrizolLS reagent according to the manufacturer’s instructions (Invitrogen). Samples were heated to 60°C for 5 min and reovirus dsRNAs were separated in a 10% SDS-PAGE gel. The gel was stained with 0.5 μg/ml ethidium bromide in TAE buffer (40 mM Tris acetate, pH 8.3, 1 mM EDTA).
Nocodazole treatment
Our lab has previously reported that treatment of CV-1 cells with concentrations of nocodazole between 500 nM and 10 µM, for 1 h, resulted in complete depolymerization of cellular microtubules detected by α-tubulin staining (Parker et al., 2002). For the current studies, infected cells were treated with 10 μM nocodazole beginning at 6 h p.i. and then incubated with the drug up to 48 h p.i. as indicated.
Acknowledgments
We thank Elaine Freimont for excellent technical support. We also thank the members of our lab and John Parker for helpful discussions, and Catherine Eichwald, Cathy Miller, Ken Murray, and John Parker for helpful comments on the manuscript. This work was supported in part by NIH grants R01 AI47904 to M.L.N. and F32 AI063750 to J.C. J.C. received initial support from NIH training grant T32 AI007061. M.M.A. received additional support from NIH training grant T32 AI07245.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proofbefore it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Becker MM, Goral MI, Hazelton PR, Baer GS, Rodgers SE, Brown EG, Coombs KM, Dermody TS. Reovirus σNS protein is required for nucleation of viral assembly complexes and formation of viral inclusions. J Virol. 2001;75:1459–1475. doi: 10.1128/JVI.75.3.1459-1475.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker MM, Peters TR, Dermody TS. Reovirus σNS and µNS proteins form cytoplasmic inclusion structures in the absence of viral infection. J Virol. 2003;77:5948–5963. doi: 10.1128/JVI.77.10.5948-5963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodkin DK, Fields BN. Growth and survival of reovirus in intestinal tissue: role of the L2 and S1 genes. J Virol. 1989;63:1188–1193. doi: 10.1128/jvi.63.3.1188-1193.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentano L, Noah DL, Brown EG, Sherry B. The reovirus protein μ2, encoded by the M1 gene, is an RNA-binding protein. J Virol. 1998;72:8354–8357. doi: 10.1128/jvi.72.10.8354-8357.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broering TJ, McCutcheon AM, Centonze VE, Nibert ML. Reovirus nonstructural protein μNS binds to reovirus cores, but does not inhibit their transcription activity. J Virol. 2000;74:5516–5524. doi: 10.1128/jvi.74.12.5516-5524.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broering TJ, Arnold MM, Miller CL, Hurt JA, Joyce PL, Nibert ML. Carboxylproximal regions of reovirus nonstructural protein μNS necessary and sufficient for forming factory-like inclusions. J Virol. 2005;79:6194–6206. doi: 10.1128/JVI.79.10.6194-6206.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broering TJ, Kim J, Miller CL, Piggott CD, Dinoso JB, Nibert ML, Parker JSL. Reovirus nonstructural protein μNS recruits viral core surface proteins and entering core particles to factory-like inclusions. J Virol. 2004;78:1882–1892. doi: 10.1128/JVI.78.4.1882-1892.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broering TJ, Parker JSL, Joyce PL, Kim J, Nibert ML. Mammalian reovirus nonstructural protein μNS forms large inclusions and colocalizes with reovirus microtubule-associated protein μ2 in transfected cells. J Virol. 2002;76:8285–8297. doi: 10.1128/JVI.76.16.8285-8297.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EG, Nibert ML, Fields BN. The L2 gene of reovirus serotype 3 controls the capacity to interfere, accumulate deletions, and establish persistent infection. In: Compans RW, Bishop DHL, editors. Double-Stranded RNA Viruses. Elsevier; New York: 1983. pp. 275–288. [Google Scholar]
- Campagna M, Eichwald C, Vascotto F, Burrone OR. RNA interference of rotavirus segment 11 mRNA reveals the essential role of NSP5 in the virus replicative cycle. J Gen Virol. 2005;86:1481–1487. doi: 10.1099/vir.0.80598-0. [DOI] [PubMed] [Google Scholar]
- Cejka D, Losert D, Wacheck V. Short interfering RNA (siRNA): tool or therapeutic? Clin Sci (Lond) 2006;110:47–58. doi: 10.1042/CS20050162. [DOI] [PubMed] [Google Scholar]
- Cleveland DR, Zarbl H, Millward S. Reovirus guanylyltransferase is L2 gene product lambda 2. J Virol. 1986;60:307–311. doi: 10.1128/jvi.60.1.307-311.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs KM. Identification and characterization of a double-stranded RNA- reovirus temperature-sensitive mutant defective in minor core protein μ2. J Virol. 1996;70:4237–4245. doi: 10.1128/jvi.70.7.4237-4245.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs KM. Stoichiometry of reovirus structural proteins in virus, ISVP, and core particles. Virology. 1998;243:218–228. doi: 10.1006/viro.1998.9061. [DOI] [PubMed] [Google Scholar]
- Coombs KM. Reovirus structure and morphogenesis. Curr Top Microbiol Immunol. 2006;309:117–167. doi: 10.1007/3-540-30773-7_5. [DOI] [PubMed] [Google Scholar]
- Cuadras MA, Bordier BB, Zambrano JL, Ludert JE, Greenberg HB. Dissecting rotavirus particle-raft interaction with small interfering RNAs: insights into rotavirus transit through the secretory pathway. J Virol. 2006;80:3935–3946. doi: 10.1128/JVI.80.8.3935-3946.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dales S. Association between the spindle apparatus and reovirus. Proc Natl Acad Sci U S A. 1963;50:268–275. doi: 10.1073/pnas.50.2.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dector MA, Romero P, Lopez S, Arias CF. Rotavirus gene silencing by small interfering RNAs. EMBO Rep. 2002;3:1175–1180. doi: 10.1093/embo-reports/kvf234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi Y, Muthukrishnan S, Tomasz J, Shatkin AJ. Caps in eukaryotic mRNAs: mechanism of formation of reovirus mRNA 5′-terminal m7GpppGm-C. Prog Nucleic Acid Res Mol Biol. 1976;19:3–20. doi: 10.1016/s0079-6603(08)60905-8. [DOI] [PubMed] [Google Scholar]
- Guglielmi KM, Johnson EM, Stehle T, Dermody TS. Attachment and cell entry of mammalian orthoreovirus. Curr Top Microbiol Immunol. 2006;309:1–38. doi: 10.1007/3-540-30773-7_1. [DOI] [PubMed] [Google Scholar]
- Haller BL, Barkon ML, Vogler GP, Virgin HW., 4th Genetic mapping of reovirus virulence and organ tropism in severe combined immunodeficient mice: organ-specific virulence genes. J Virol. 1995;69:357–364. doi: 10.1128/jvi.69.1.357-364.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann LL, Coombs KM. Inhibition of reovirus by mycophenolic acid is associated with the M1 genome segment. J Virol. 2004;78:6171–6179. doi: 10.1128/JVI.78.12.6171-6179.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Parker JSL, Murray KE, Nibert ML. Nucleoside and RNA 5′-triphosphatase activities of reovirus transcriptase cofactor μ2. J Biol Chem. 2004;279:4394–4403. doi: 10.1074/jbc.M308637200. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Chappell JD, Danthi P, Dermody TS. Gene-specific inhibition of reovirus replication by RNA interference. J Virol. 2006;80:9053–9063. doi: 10.1128/JVI.00276-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez T, Camacho M, Zayas M, Najera R, Sanchez R, Arias CF, Lopez S. Silencing the morphogenesis of rotavirus. J Virol. 2005;79:184–192. doi: 10.1128/JVI.79.1.184-192.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao ZX, Joklik WK. Isolation and enzymatic characterization of protein λ2, the reovirus guanylyltransferase. Virology. 1991;185:377–386. doi: 10.1016/0042-6822(91)90785-a. [DOI] [PubMed] [Google Scholar]
- Matoba Y, Colucci WS, Fields BN, Smith TW. The reovirus M1 gene determines the relative capacity of growth of reovirus in cultured bovine aortic endothelial cells. J Clin Invest. 1993;92:2883–2888. doi: 10.1172/JCI116910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba Y, Sherry B, Fields BN, Smith TW. Identification of the viral genes responsible for growth of strains of reovirus in cultured mouse heart cells. J Clin Invest. 1991;87:1628–1633. doi: 10.1172/JCI115177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbisa JL, Becker MM, Zou S, Dermody TS, Brown EG. Reovirus μ2 protein determines strain-specific differences in the rate of viral inclusion formation in L929 cells. Virology. 2000;272:16–26. doi: 10.1006/viro.2000.0362. [DOI] [PubMed] [Google Scholar]
- Mello CC, Conte D., Jr Revealing the world of RNA interference. Nature. 2004;431:338–342. doi: 10.1038/nature02872. [DOI] [PubMed] [Google Scholar]
- Middleton JK, Severson TF, Chandran K, Gillian AL, Yin J, Nibert ML. Thermostability of reovirus disassembly intermediates, ISVPs, correlates with genetic, biochemical, and thermodynamic properties of major surface protein μ1. J Virol. 2002;76:1051–1061. doi: 10.1128/JVI.76.3.1051-1061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CL, Broering TJ, Parker JSL, Arnold MM, Nibert ML. Reovirus σNS protein localizes to inclusions through an association requiring the µNS amino-terminus. J Virol. 2003;77:4566–4576. doi: 10.1128/JVI.77.8.4566-4576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CL, Parker JSL, Dinoso JB, Piggott CD, Perron MJ, Nibert ML. Increased ubiquitination and other covariant phenotypes attributed to a strain- and temperature-dependent defect of reovirus core protein μ2. J Virol. 2004;78:10291–10302. doi: 10.1128/JVI.78.19.10291-10302.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero H, Arias CF, Lopez S. Rotavirus Nonstructural Protein NSP3 is not required for viral protein synthesis. J Virol. 2006;80:9031–9038. doi: 10.1128/JVI.00437-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody MD, Joklik WK. The function of reovirus proteins during the reovirus multiplication cycle: analysis using monoreassortants. Virology. 1989;173:437–446. doi: 10.1016/0042-6822(89)90556-4. [DOI] [PubMed] [Google Scholar]
- Mustoe TA, Ramig RF, Sharpe AH, Fields BN. Genetics of reovirus: identification of the ds RNA segments encoding the polypeptides of the µ and σ size classes. Virology. 1978;89:594–604. doi: 10.1016/0042-6822(78)90200-3. [DOI] [PubMed] [Google Scholar]
- Nibert ML, Kim J. NTPase motifs unique to turreted Reoviridae members and colti-viruses. J Virol. 2004;78:5528–5530. doi: 10.1128/JVI.78.10.5528-5530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibert ML, Margraf RL, Coombs KM. Nonrandom segregation of parental alleles in reovirus reassortants. J Virol. 1996;70:7295–7300. doi: 10.1128/jvi.70.10.7295-7300.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibert ML, Schiff LA. Reoviruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 4. Lippincott Williams and Wilkins; Philadelphia: 2001. pp. 1679–1728. [Google Scholar]
- Noble S, Nibert ML. Core protein μ2 is a second determinant of nucleoside triphosphatase activities by reovirus cores. J Virol. 1997;71:7728–7735. doi: 10.1128/jvi.71.10.7728-7735.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JSL, Broering TJ, Kim J, Higgins DE, Nibert ML. Reovirus core protein μ2 determines the filamentous morphology of viral inclusion bodies by interacting with and stabilizing microtubules. J Virol. 2002;76:4483–4496. doi: 10.1128/JVI.76.9.4483-4496.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry B, Blum MA. Multiple viral core proteins are determinants of reovirus-induced acute myocarditis. J Virol. 1994;68:8461–8465. doi: 10.1128/jvi.68.12.8461-8465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry B, Schoen FJ, Wenske E, Fields BN. Derivation and characterization of an efficiently myocarditic reovirus variant. J Virol. 1989;63:4840–4849. doi: 10.1128/jvi.63.11.4840-4849.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry B, Torres J, Blum MA. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J Virol. 1998;72:1314–1323. doi: 10.1128/jvi.72.2.1314-1323.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmulevitz M, Marcato P, Lee PWK. Unshackling the links between reovirus oncolysis, Ras signaling, translational control and cancer. Oncogene. 2005;24:7720–7728. doi: 10.1038/sj.onc.1209041. [DOI] [PubMed] [Google Scholar]
- Silvestri LS, Taraporewala ZF, Patton JT. Rotavirus replication: plus-sense templates for double-stranded RNA synthesis are made in viroplasms. J Virol. 2004;78:7763–7774. doi: 10.1128/JVI.78.14.7763-7774.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledz CA, Williams BR. RNA interference in biology and disease. Blood. 2005;106:787–794. doi: 10.1182/blood-2004-12-4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spendlove RS, Lennette EH, Chin JN, Knight CO. Effect of antimitotic agents on intracellular reovirus antigen. Cancer Res. 1964;24:1826–1833. [PubMed] [Google Scholar]
- Taraporewala ZF, Jiang X, Vasquez-Del Carpio R, Jayaram H, Prasad BVV, Patton JT. Structure-function analysis of rotavirus NSP2 octamer by using a novel complementation system. J Virol. 2006;80:7984–7994. doi: 10.1128/JVI.00172-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler KL, Clarke P, DeBiasi RL, Kominsky D, Poggioli GJ. Reoviruses and the host cell. Trends Microbiol. 2001;9:560–564. doi: 10.1016/s0966-842x(01)02103-5. [DOI] [PubMed] [Google Scholar]
- Yin P, Cheang M, Coombs KM. The M1 gene is associated with differences in the temperature optimum of the transcriptase activity in reovirus core particles. J Virol. 1996;70:1223–1227. doi: 10.1128/jvi.70.2.1223-1227.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin P, Keirstead ND, Broering TJ, Arnold MM, Parker JS, Nibert ML, Coombs KM. Comparisons of the M1 genome segments and encoded μ2 proteins of different reovirus isolates. Virol J. 2004;1:6. doi: 10.1186/1743-422X-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zar JH. Biostatistical Analysis. 2. Prentice Hall; Englewood Cliffs, N J: 1984. pp. 138–146. [Google Scholar]
- Zarbl H, Millward S. The reovirus multiplication cycle. In: Joklik WK, editor. The Reoviridae. Plenum Press; New York: 1983. pp. 107–196. [Google Scholar]