

Abstract
Abnormalities of calcium, phosphorus and magnesium homeostasis are common, and collectively are called disorders of mineral metabolism. Normal homeostatic regulation maintains serum levels, intracellular levels, and optimal mineral content in bone. This regulation occurs at three major target organs, the intestine, kidney and bone, principally via the complex integration of two hormones, parathyroid hormone and vitamin D. An understanding of normal physiology is necessary to accurately diagnose and treat disorders of mineral metabolism and will be briefly reviewed before discussing the differential diagnosis and treatment of specific disorders.
Keywords: Calcium, Phosphorus, Magnesium, Parathyroid Hormone, Vitamin D
NORMAL PHSIOLOGY
Parathyroid Hormone
A decrease in ionized calcium stimulates a release of parathyroid hormone (PTH), which maintains calcium homeostasis by 1) increasing bone mineral dissolution, thus releasing calcium and phosphorus, 2) increasing renal reabsorption of calcium and excretion of phosphorus, and 3) enhancing the gastrointestinal absorption of both calcium and phosphorus indirectly through its effects on the synthesis of 1,25(OH)2D (calcitriol). In healthy subjects, this increase in serum PTH level in response to hypocalcemia effectively restores serum calcium levels and maintains normal serum phosphorus levels. PTH also enhances the conversion of calcidiol [25(OH)-vitamin D] to calcitriol, which in turns decreases PTH secretion at the level of the parathyroid glands completing the typical endocrine feedback loop. In primary hyperparathyroidism, PTH is secreted from adenomatous glands without regard to physiologic stimuli. In secondary hyperparathyroidism, PTH is secreted as a normal response, but to abnormal stimuli. Tertiary hyperparathyroidism is a term used to describe glands from patients with secondary hyperparathyroidism that have become adenomatous, and therefore unresponsive to stimuli. Once in the circulation, PTH binds to PTH receptors that are located throughout the body. Thus, disorders of PTH excess or insufficiency not only affect serum levels of calcium and phosphorus, but also lead to bone, cardiac, skin, neurologic and other systemic manifestations.
PTH is cleaved to an 84 amino acid protein in the parathyroid gland, where it is stored with fragments in secretory granules for release. Once released, the circulating 1–84 amino acid protein has a half-life of 2–4 minutes. It is then cleaved into N-terminal, C-terminal, and mid-region fragments of PTH, which are metabolized in the liver and kidney16. PTH secretion occurs in response to hypocalcemia, hyperphosphatemia, and calcitriol deficiency and is inhibited by severe hypomagnesemia. The extracellular concentration of ionized calcium is the most important determinant of minute-to-minute secretion of PTH from stored secretory granules in response to hypocalcemia, sensed by the calcium sensing receptor. This calcium sensing receptor (CaR) has now been sequenced and cloned and is a member of the G-protein receptor superfamily, with a 7 membrane-spanning domain. Genetic defects can lead to syndromes of hypercalcemia and hypocalcemia7. The CaR has also been localized to the thyroid C-cells and the kidney, predominantly in the thick ascending limb where it controls renal excretion of calcium in response to changes in serum calcium8,17.
The major difficulty in accurately measuring PTH is the presence of circulating fragments, particularly in the presence of chronic kidney disease (CKD) where normal fragments of metabolism are not excreted16. Initial measurements of PTH using C-terminal assays, by the N-terminal assays detected inactive metabolites. The development of a two-site antibody test (commonly called “INTACT” assay) improved the detection of entire length (1–84 or active) PTH molecules. In this assay, a capture antibody binds to the N-terminus and a second antibody binds to the C-terminus12. Unfortunately this assay still detects some fragments. Nonetheless, this type of assay is the most commonly used throughout the world. However, assays are also available that truly only detect the 1–84 amino acid full length molecule. This array of PTH assays can lead to confusion in the diagnosis of disorders of parathyroid hormone, especially in patients with kidney disease when these various fragments may be circulating.
Vitamin D
Vitamin D (Figure 2) is called a ‘vitamin’ because of its exogenous source, predominately from oily fish in the form of vitamin D2 and vitamin D3. However, it is really a hormone, synthesized by the skin and metabolized to an active hormone, calcitriol, by the kidney which then acts throughout the body. In the skin, 7-dehydrocholesterol is converted to vitamin D3 in response to sun light, a process that is inhibited by sunscreen of SPF 8 or greater. Once in the blood, vitamin D2 and D3 from diet or skin bind with vitamin D binding protein, and carried to the liver where they are hydroxylated to yield 25(OH)D, often called calcidiol. Calcidiol is then converted in the kidney to 1,25(OH)2D by the action of 1α-hydroxylase (CYP27B1). The CYP27B1 in the kidney is regulated by nearly every hormone involved in calcium homeostasis, and its activity is stimulated by PTH, estrogen, calcitonin, prolactin, growth hormone, low calcium and low phosphorus. Its activity is inhibited by calcitriol, thus providing the ‘feedback’ loop that regulates calcitriol synthesis.
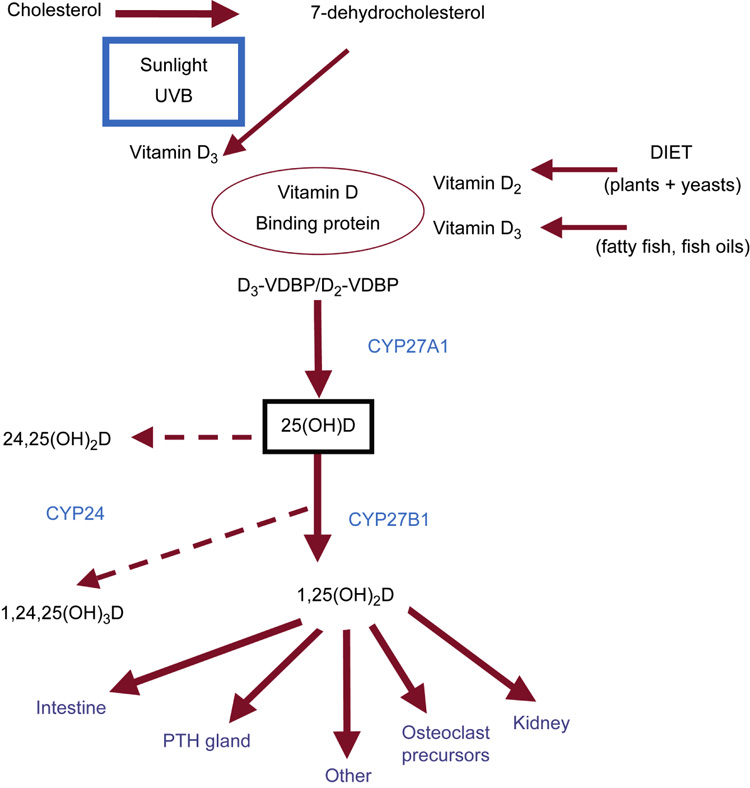
Figure 2.
Overview of vitamin D metabolism. Vitamin D is obtained from dietary sources, and is metabolized via ultraviolet light from 7-DHC in the skin. Both sources (diet and skin) of Vitamin D2 and vitamin D3 bind to vitamin D binding protein (VDBP) and circulate to the liver. In the liver, vitamin D is hydroxylated by CYP27A1 to 25(OH)D, commonly referred to as calcidiol. Calcidiol is then further metabolized to calcitriol by the 1-α-hydroxylase enzyme (CYP27B1) at the level of the kidney. The active metabolite 1,25(OH)2D (calcitriol) acts principally on the target organs of intestine, parathyroid (PTH) gland, bone cell precursors, and the kidney. Calcitriol is metabolized to the inert 1,24,25(OH)3D through the action of the 24,25-hydroxylase enzyme (CYP24). Calcidiol is similarly hydroxylated to 24,25(OH)2D. From Moe SM, Sprague S: Mineral Bone Disorders in Chronic Kidney Disease. In Brenner B (ed): The Kidney, 8th ed. Philadelphia: Saunders, 2008, Vol 2, p 1784 with permission.
Calcitriol, (1,25(OH)2D) circulates with Vitamin D binding protein, and enters the target cell to interacts with its nuclear Vitamin D receptor (VDR). This complex then combines with the retinoic acid X receptor to form a heterodimer, which in turn interacts with the Vitamin D response element (VDRE) on the target gene. The most important function is exerted on the small intestine, where calcitriol regulates the intestinal absorption of calcium and, to a lesser degree, phosphorus9, and inhibits PTH synthesis at the parathyroid gland. However, there is now evidence that the VDRE is on multiple genes, the VDR in multiple organs, and even 1-alpha hydroxylase activity in extra-renal tissues11. These effects, mostly autocrine or paracrine (local tissue level) are responsible for the non endocrine (non-mineral related) effects of vitamin D, including cell differentiation and proliferation, immune function, and fighting of infections. These effects have led to its therapeutic use in cancer and skin disorders11,21.
Phosphatonins
This is a group of proteins that have been identified in genetic and cancer related disorders of renal phosphate wasting. Fibroblast growth factor 23 (FGF23) is produced by tumors from patients with tumor induced osteomalacia, and corresponding genetic defects were identified in autosomal dominant hypophosphatemic rickets (ADHR). FGF23 is made in osteocytes in bone, and acts in the kidney to excrete urinary phosphorus, and inhibits the conversion of calcidiol to calcitriol. Levels of FGF23 are very elevated in patients with CKD, presumably due to net phosphate retention.5,38. Another factor, secreted frizzled-related protein 4 (FRP4) also can induce renal phosphate wasting38 It is now clear that these factors are important in non-PTH mediated urinary phosphate wasting, but their role of phosphatonins in normal homeostasis is not yet completely clear.
CALCIUM
Serum calcium levels are tightly controlled within a narrow range, usually 8.5–10.5 mg/dL (2.1–2.6 mmol/L). However, the serum calcium level is a poor reflection of overall total body calcium, as serum levels are only 0.1–0.2% of extracellular calcium, which in turn is only 1% of total body calcium. The remainder of total body calcium is stored in bone. Ionized calcium, generally 40% of total serum calcium level is physiologically active, while the non-ionized calcium is bound to albumin or anions such as citrate, bicarbonate and phosphorus. In the presence of hypoalbuminemia, there is a relative increase in the ionized calcium relative to the total calcium, thus total serum calcium may underestimate the physiologically active (ionized) serum calcium. A commonly utilized formula for estimating the ionized calcium from total calcium is to add 0.8 mg/dl for every 1 mg decrease in serum albumin below 4 mg/dl.
Serum levels of ionized calcium are maintained in the normal range by inducing increases in the secretion of PTH (Figure 1). PTH acts to increase bone resorption, increase renal calcium reabsorption, and increases the conversion of 25(OH)D to 1,25(OH)2D in the kidney, thereby increasing gastrointestinal calcium absorption. Individuals with normal kidney function have protection against calcium overload by virtue of their ability to increase renal excretion of calcium and reduce intestinal absorption of calcium by actions of PTH and 1,25(OH)2D. Calcium absorption across the intestinal epithelium occurs in both a vitamin D dependent mechanism, and a vitamin D independent or passive, concentration dependent pathway. In the kidney, the majority (60–70%) of calcium is reabsorbed passively in the proximal tubule driven by a gradient that is generated by sodium and water reabsorption. In the thick ascending limb, another 10% of calcium is reabsorbed via paracellular transport. Finally, at the distal convoluted tubule, the connecting tubule, and the initial portion of the cortical collecting duct another 10% of calcium reabsorption occurs. It is also primarily through these latter distal segments of the kidney where the regulation of urinary calcium excretion occurs24. As detailed below, the treatment of hypercalcemia includes volume expansion to reduce the salt driven proximal reabsorption and loop diuretics which block the paracellular thick ascending limb transport.
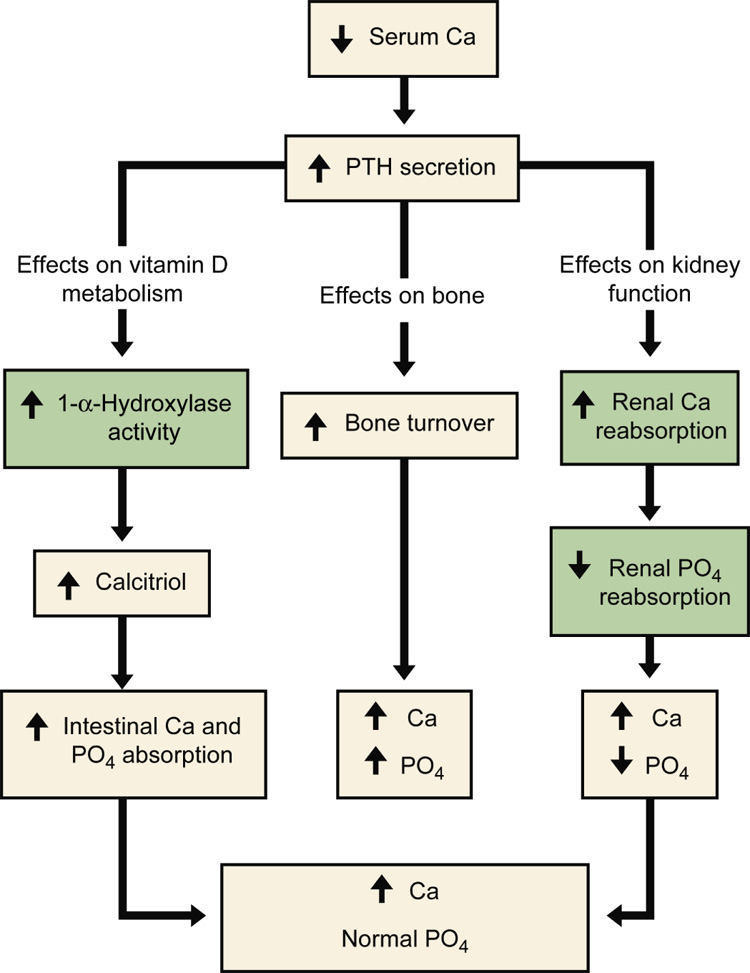
Figure 1.
Normal homeostatic response to hypocalcemia. In the presence of hypocalcemia, parathyroid hormone (PTH) secretion is increased. PTH acts on three target organs. PTH works level at the intestine indirectly by first increasing the 1-α-hydroxalase activity in the kidney. This enzyme converts calcidiol to calcitriol, which then increases intestinal absorption of both calcium and phosphorus. In bone, parathyroid hormone increases bone turnover, resulting in a release of calcium and phosphorus from bone. Lastly, PTH works directly on the kidney to increase renal calcium reabsorption and decrease renal phosphorus reabsorption. The net effect is a rise in serum calcium but no net change in serum phosphorus. The gray boxes indicate homeostatic steps in the kidney that are impaired in the setting of kidney disease. From Moe SM, Sprague S: Mineral Bone Disorders in Chronic Kidney Disease. In Brenner B (ed): The Kidney, 8th ed. Philadelphia: Saunders, 2008, Vol 2, p 1784 with permission.
Phosphorus
Inorganic phosphorus is critical for numerous normal physiologic functions including skeletal development, mineral metabolism, energy transfer through mitochondrial metabolism, cell membrane phospholipid content and function, cell signaling, and even platelet aggregation. Because of its importance, normal homeostasis maintains serum concentrations between 2.5 to 4.5 mg/dl (0.81 to 1.45mmol/L). The terms phosphorus and phosphate are often used interchangeably, but the term phosphate actually means the inorganic freely available form (HPO4 −2 to H2PO4 −1). However, most laboratories report this measurable, inorganic component as phosphorus. For simplicity we will refer to this measurable component as phosphorus for the remainder of this chapter.
Total adult body stores of phosphorus is approximately 700 g, of which 85% is contained in bone in the form of hydroxyapatite [(Ca)10(PO4)6(OH)2]. Of the remaining, 14% is intracellular, and only 1% is extracellular. Of this extracellular phosphorus, 70% is organic and contained within phospholipids, and 30% is inorganic, 15% is protein bound, and the remaining 85% is either complexed with sodium, magnesium, or calcium or circulates as the free monohydrogen or dihydrogen forms. It is this latter 0.15% of total body phosphorus (15% of extracellular phosphorus) that is freely circulating and measured. At pH of 7.4, it is in a ratio of about 4:1 HPO4 −2 to H2PO4 −1. For that reason, phosphorus is usually expressed in mmol rather than meq/L. Thus, similar to calcium, serum measurements only reflect a minor fraction of total body phosphorus, and therefore do not consistently reflect total body stores.
The recommended daily allowance (RDA) for phosphorus is 800 mg/day but the average American diet ingests approximately 1000–1400 mg phosphorus per day. Approximately 2/3 of the ingested phosphorus is excreted in the urine, and the remaining 1/3 in stool and this phosphorus excretion is highly dependent on kidney function. Many pre-packaged, fast food, and dark (cola) beverages contain extra phosphorus as preservative and thus it is difficult to accurately predict dietary intake based on the food type alone. In general, foods high in protein and dairy products contain the most phosphorus, whereas fruits and vegetables contain the least. Animal or synthetic protein has more bioavailable phosphorus than soy or grain based protein. Between 60 and 70% of dietary phosphorus is absorbed by the gastrointestinal tract, in all intestinal segments. Phosphorus absorption is dependent on both passive transport related to the concentration in the intestinal lumen (i.e. increased after a meal) and active transport stimulated by calcitriol42. Medications or foods that bind phosphorus (antacids, phosphate binders, calcium) can decrease the net amount of phosphorus absorbed by decreasing the free phosphate for absorption. In the kidney, approximately 70–80% of the filtered load of phosphorus is reabsorbed in the proximal tubule which serves as the primary regulated site of the kidney. The remaining approximately 20–30% is reabsorbed in the distal tubule14.
When serum phosphorus levels decrease, there is a stimulation of the 1-alpha hydroxylase enzyme in the kidneys, thereby increasing conversion of calcidiol to calcitriol which in turn increases intestinal phosphorus absorption. There is also a reduction in urinary phosphorus excretion. In the presence of hyperphosphatemia, there is a rapid increase in urinary excretion of phosphorus mediated by the serum phosphorus level, PTH, and most likely FGF2337. There is a rapid response of the kidneys to excrete urinary phosphorus after dietary ingestion such that sustained hyperphosphatemia is clinically not seen without kidney disease. Although the effects are more minor, renal phosphorus excretion is also increased by volume expansion, metabolic acidosis, glucocorticoids and calcitonin, and decreased by growth hormone and thyroid hormone31
Magnesium
The normal homoestasis for magnesium is less studied but there has been recent increased interest. Magnesium is critical for normal ATP (adenosine triphosphate) function and glucose metabolism and therefore has widespread cellular effects. Magnesium is also important in cellular cytoskeleton contraction and at the myoneural junction, and therefore can alter skeletal and cardiac muscle function. Magnesium is the second must abundant intracellular cation, with 67% of total body stores found in bone, 31% intracellular, and only 2% in the extracellular (measurable space). Normal serum levels are 1.5 to 2.5 meq/L, and approximately 30% is bound to albumin. Similar to the other divalent mineral ions, less than 1% of the total body magnesium is in the extracellular space (and therefore measurable) and thus levels do not accurately reflect total body stores22. Unfortunately, there is no ‘ionized’ magnesium test clinically available to diagnose deficiency. Magnesium is absorbed via intestinal epithelial channels in the intestine in a non-vitamin D dependent process. At the kidney, magnesium is reabsorbed along with calcium in a paracellular manner in the thick ascending limb, and via specific magnesium transport channels in the distal tubule. Although there is biologic plausibility of a magnesium sensing hormone, at this time there is no evidence for one. Thus, the major regulator of magnesium is the serum concentration itself43.
BONE
The majority of the total body stores of calcium and phosphorus are located in bone in the form of hydroxyapatite [Ca10(PO4)6(OH)2]. Trabecular (cancellous) bone is located predominately in the epiphyses of the long bones, and cortical (compact) bone is in the shafts of long bones. Bone consists principally (90%) of highly organized cross-linked fibers of type I collagen; the remainder consists of proteoglycans, and “non-collagen” proteins. Osteoclasts are the bone resorbing cells and derive from circulating hematopoietic cells, and osteoblasts are the bone forming cells that derive from the marrow. Bone is a dynamic organ and remodels or turns over in response to hormones, cytokines, and changes in mechanical forces. The control of bone remodeling is highly complex occurring in very distinct phases: 1) osteoclast resorption, 2) reversal, 3) pre-osteoblast migration and differentiation, 4) osteoblast matrix (osteoid or unminerlized bone) formation, 5) mineralization, 6) quiescent stage. At any one time, less than 15–20% of the bone surface is undergoing remodeling, controlled via the osteoprotegerin pathway by nearly every cytokine and hormone including PTH and calcitriol as well as inflammatory cytokines20. Thus, alterations in bone remodeling can affect calcium and phosphorus homeostasis.
DISORDERS OF MINERAL METABOLISM
HYPERCALCEMIA
Ionized calcium represents the biologically active fraction of total calcium. Hypercalcemia occurs when the level of serum ionized (not total) calcium increases. it can be estimated using the total serum calcium and albumin concentrations. A difference of 1 g/dl of albumin in either direction will influence serum ionized calcium by 0.8–1.0 mg/dl in the same direction. This estimation is not always accurate and in certain circumstances, such as paraprotieneamic blood disorders, hyperphosphatemia, and the presence of acid-base disorders, measuring serum ionzed calcium will be more helpful.
Clinical manifestations of hypercalcemia
The severity of symptoms caused by hypercalcemia will depend on the magnitude of hypercalcemia and the rate of rise in serum calcium. Gastrointestinal symptoms are the most common, including nausea, vomiting, constipation, and abdominal pain. Patients may also complain of difficulty concentrating, fatigue, lethargy, and muscle weakness. Hypercalcemia can induce a nephrogenic diabetes insipidus with resulting polyuria that worsens the hypercalcemia because of volume depletion. Cardiovascular effects include hypertension and shortening of the QT interval on the electrocardiogram. Although cardiac arrhythmias are rare, they are more likely with digitalis toxicity. Importantly, hypercalcemia, especially in the setting of volume depletion can lead to acute renal failure.
Differential Diagnosis of Hypercalcemia
The most common causes of hypercalcemia are malignancy and hyperparathyroidism. In most studies, these two diagnoses account for over 80% of cases4,39. The remaining causes are shown in List 1 and a few discussed in more detail below.
Malignancy
In recent years malignancy has surpassed primary hyperparathyroidism as the most common cause of hypercalemia. Its presence in cancer patients confers a poor prognosis. Depending on the type of malignancy, hypercalcemia can result from 1) direct invasion of bone due to metastatic disease (Local Osteolytic Hypercalcemia; LOH), 2) by production of circulating factors that stimulate osteoclastic resorption of bone or 3) increased production of calcitriol which stimulates gastrointestinal absorption of calcium. LOH occurs most commonly with breast cancer and multiple myeloma in which tumor cells within the bone marrow space produce a variety of inflammatory cytokines collectively referred to as osteoclast activating factors leading to net bone resorption and hypercalcemia20. Humoral hypercalemia of malignancy is caused by secretion of parathyroid hormone related peptide (PTHrP) by malignant tumor cells. PTHrP bears similarity to PTH only in the initial 8 amino acid sequence, but its binding to the PTH receptor is essentially the same leading to increased bone turnover and hypercalcemia. Specific assays are available to distinguish circulating levels of PTHrP from PTH. Various lymphoid tumors, most notably Hodgkin’s lymphoma, have been shown to synthesize large quantities of calitriol which acts directly on the intestines and kidneys to increase calcium absorption and reabsorption leading to hypercalcemia.
Hyperparathyroidism
The incidence of hyperparathyroidism has declined over the last 30 years, but it is still the second most common cause of hypercalcemia. In the majority of cases, it is caused by a benign adenoma of a single parathyroid gland that autonomously secretes PTH, or primary hyperparathyroidism. It may also be sporadic, familial or inherited as a component of the constellation of multiple endocrine neoplasia (MEN). The elevation in PTH results in increased intestinal reabsorption of calcium by stimulating the production of calcitriol, increased osteoclastic bone resorption, and increased renal tubular reabsorption of calcium. With the elevation in serum calcium and therefore the amount of c filtered calcium, the kidney may not be able to reabsorb all the calcium leading to hypercalcuria and nephrolithiasis.
Secondary hyperparathyroidism is due to diffuse hyperplasia of parathyroid glands in response to ongoing stimuli such as hypocalcemia or hyperphosphatemia. Secondary hyperparathyroidism can also cause hypercalcemia when severe due to increased bone resorption. However, most patients with CKD stages 3–5 (estimated GFR 15 ml/min) are hypocalcemic due to the low levels of calcitriol. Tertiary hyperparathyroidism is a term used to describe hyperplastic glands that become adenomatous, and therefore unresponsive, over time. This is usually seen in CKD after years of secondary hyperparathyroidism32. With a kidney transplant, the PTH continues to be secreted leading to hypercalcemia33.
Vitamin D excess
Vitamin D is activated to calcitriol in the kidneys and markedly increases intestinal calcium absorption. Toxicity can result from excessive exogenous intake of native vitamin D supplements (ergocalciferol and cholecalciferol) but this is actually quite rare, as these forms of vitamin D require conversion in the kidney to calcitriol via 1-alpha hydroxylase whose activity is tightly regulated by calcium levels. In contrast, the administration of calcitriol, or other vitamin D analogs (paricalcitol, doxercalciferol) will not have internal regulation and may lead to hypercalcemia. These drugs are most commonly given to treat secondary hyperparathyroidism in CKD. Another source of calcitriol is production by non-kidney tissue such as lymphoma and granulomas. Granulomatous diseases such as sarcoidosis, tuberculosis, and leprosy are thought to cause hypercalcemia by increased production of calcitriol by monocytes and macrophages that possess 1-alpha hydroxylase activity15.
Familial hypocalciuric hypercalcemia
The extracellular calcium sensing receptor (CaSR) has been found in many tissues, most heavily expressed in the parathyroid gland and on the basolateral aspect of the renal tubular epithelial cells. Inactivating mutations of the CaSR is responsible for familial hypocalciuric hypercalcemia. It is a rare hereditary disease with autosomal dominant transmission. Calcium is unable to activate the receptor in the renal tubules, leading to increased renal reabsorption of calcium into the blood from the tubular fluid and hypocalcuria usually with urine calcium excretion < 100 mg/day. Because this mutation may also affect the receptor at the level of the parathyroid gland, PTH may be slightly elevated out of proportion for the serum calcium. Other clues pointing to this diagnosis include a family history of asymptomatic hypercalcemia36.
The approach to the patient with hypercalcemia
A logical approach to the patient with hypercalcemia is to formulate a differential diagnosis based on the physiology of calcium homeostasis (Box 1). Approaching a patient in this manner allows the clinician to order the appropriate diagnostic studies.
Box 1
Causes of Hypercalcemia
Malignancy
Local Osteolytic Hypercalcemia
Humoral Hypercalcemia of Malignancy (PTHrp)
Hematologic Malignancies (ectopic calcitriol synthesis)
Hyperparathyroidism
Thyrotoxicosis
Granulomatous diseases
Drug induced
Vitamin D
Thiazide diuretics
Estrogens and antiestrogens
Androgens (breast cancer therapy)
Vitamin A
Lithium
Immobilization
Total parenteral nutrition
Kidney Disease (acute and chronic, usually from medications)
Parathyroid Glands
In all patients with hypercalcemia, the PTH level should be suppressed. Thus, interpretation of a PTH level must always be done in conjunction with a simultaneous calcium level. For example, if a serum calcium level is 11.5, and the PTH is 50 pg/ml there is an inappropriate circulating level of PTH as it should be very suppressed. Thus despite the PTH in the normal range (usually 10–65 pg/ml), it is indicative of hyperparathyroidism. However, if the calcium level was 8.0 mg/dl this level of PTH would be normal. PTH also increases urinary phosphorus excretion such that an elevated PTH, with hypercalcemia and low normal or low phosphorus is essentially diagnostic of primary hyperparathyroidism. A nuclear medicine sestamibi scan may be helpful in localizing an adenomatous gland18. However, there is a high risk of false negative scans, and a well trained surgeon can usually locate the enlarged gland. Rarely, glands are found in the mediastinum. In patients with CKD, secondary hyperparathyroidism is usually associated with hyperphosphatemia. For these patients, no imaging to localize enlarged glands is needed prior to any surgery. Rarely, malignancy can occur in the parathyroid gland cells, resulting in severe hyperparathyroidism and hypercalcemia.
Bone
Hypercalcemia at the level of the bone occurs due to enhanced bone turnover (net bone resorption greater than bone formation) induced by local tumor invasion or as a result of increased secretion of hormonal factors by the tumor cells (PTHrP, calcitriol, and excess secretion of PTH)40. Alternatively, immobilization may lead to release of calcium from the bone, especially in the setting of excess turnover such as Paget’s disease. However, immobilization should be a diagnosis of exclusion and is actually very rare41. The diagnostic studies for bone induced hypercalcemia include PTHrp, urine and serum protein electrophoresis to diagnose multiple myeloma, and an alkaline phosphatase level which is markedly elevated in Paget’s disease and other high bone turnover diseases.
Intestine
Enhanced intestinal absorption of calcium can occur in conditions resulting in elevated circulating levels of calcidiol or calcitriol. This can occur as a result of vitamin D toxicity with very high calcidiol levels, calcitriol therapy in patients with secondary hyperparathyroidism, calcitriol secreting granulomatous diseases and lymphomas, and with hyperparathyroidism which in turn increases calcitriol synthesis. In addition, excess calcium ingestion, especially with alkali, can lead to hypercalcemia. In the past this was called milk-alkali syndrome named for the combination of therapies used to treat peptic ulcer disease before the advent of H2 blockers. However, it is now rarely observed. In order to detect vitamin D toxicity, both a calcidiol and calcitriol level should be measured. In the setting of exogenous vitamin D intake, calcidiol levels will be high, and calcitriol levels normal to high. In the setting of granulomatous production, calcitriol levels will be high, with any level of calcidiol (usually low normal).
Kidneys
In the setting of volume depletion, serum calcium levels will rise and thus may lead to mild hypercalcemia. Thiazide diuretics block sodium reabsorption and enhance calcium reabsorption in the distal tubule leading to an increase in serum calcium and a reduction in urinary calcium excretion. These effects are used to treat hypercalcuria in patients with nephrolithiasis. However, in most cases, the rise in calcium in response to thiazide diuretics does not result in frank hypercalcemia. When thiazides do induce hypercalcemia in healthy individuals, there is often previously undetected mild hyperparathyroidism or significant volume depletion.
The urinary calcium excretion may help differentiate hyperparathyroidism from familial hypocalciuric hypercalcemia. In the setting of primary hyperparathyroidism the urinary calcium/creatinine ratio is usually greater than 0.2 (mg/mg), whereas in patients with familial hypocalcuria hypercalcemia, the urinary calcium/creatinine ratio is < 0.01 mg/mg. Ideally a 24 hour urine collection should be measured, but a random or spot collection may sometimes be useful to differentiate primary hyperparathyroidism from familial hypocalcuria hypercalcemia4.
Treatment
The ultimate goal of therapy is to treat the underlying cause of hypercalcemia. However, patients presenting with acute symptoms of hypercalcemia require immediate treatment to reduce the serum levels of calcium. The safest and most effective treatment in patients with reasonable cardiac and renal function is intravenous volume resuscitation with normal saline, which reduces the drive for proximal tubular reabsorption of salt and water (and calcium). Most patients with symptomatic hypercalcemia are volume depleted because of the polyuria induced by hypercalcemia. In more severe cases, very aggressive volume resuscitation with normal saline at 200–500 ml/hour may be required. Once volume expansion is achieved, the patient should receive intravenous furosemide or other loop diuretics which block the Na/K/2CL exchanger in the thick ascending limb of Henle. This in turn creates a favorable electrochemical gradient for passive (paracellular) calcium reabsorption. It is important to remember that patients must be adequately hydrated prior to giving the diuretic since the urinary output in response to a given dose of furosemide is difficult to predict. Accurate assessment of intake and output is critical to optimize this treatment approach and the primary reason for lack of responsiveness to volume repletion/lasix is inadequate volume of saline.
If these conservative treatments fail to restore hypercalcemia, pharmacologic options should be utilized. Intravenous bisphosphonates are very effective for the treatment of hypercalcemia. In the United States, two bisphosphonates, pamidronate (60–90 mg IV over 4 hours) and zoledronate (4 mg over 15 minutes) have been approved for the acute treatment of malignancy associated hypercalcemia39. These agents block osteoclast mediated bone resorption by inducing osteoclast apoptosis and will also lower serum calcium. A clinical response takes 2–4 days to occur and the nadir in serum calcium occurs within 4–7 days. Very rare side effects of intravenous bisphosphonates are acute renal failure and ostoenecrosis of the jaw. Another treatment option is calcitonin (4–12 u IM or SQ every 12 hours for 2–3 days). Calcitonin has the advantage of rapid reduction of serum calcium, but its use is limited by short duration of action and the generation of tachyphylaxis. Glucocorticoids are effective first line agents along with saline diuresis in conditions in which the hypercalcemia is mediated by elevated circulating levels of calcitriol in granulomatous disorders or lymphoma, usually given orally beginning at 40–60 mg per day.
The approach to the patient with hyperparathyroidism is more controversial4. In primary hyperparathyroidism, intervention may only be indicated with symptoms (nephrolithiasis, lethargy, fatigue). When to intervene was the topic of a National Institutes of Health consensus conference in 20023. This group felt that patients should undergo surgical removal of the parathyroid gland if the serum calcium was 1.0 mg/dl greater than the laboratory upper limits of normal, the urine calcium excretion > 400 mg/day, the creatinine clearance reduced by 30% or more, a dual Xray absorptiometery (DXA) T-score of −2.5 at any major site, or age < 50. A new alternative to surgical parathyroidectomy is the use of cinacalcet (Sensipar®), a Calcimimetic. This new drug is an allosteric activator of the calcium sensing receptor, “mimicking” higher levels of calcium, which decrease PTH secretion and is approved for the treatment of secondary hyperparathyroidism. However, it may also be effective for the treatment of primary hyperparathyroidism with a starting dose of 30 mg twice daily35. The treatment of secondary hyperparathyroidism is beyond the scope of this chapter, but requires the inhibition of the aberrant stimuli by reduction of serum phosphorus with phosphate binders, the administration of calitriol or its derivatives, and/or cinacalcet13.
HYPOCALCEMIA
Clinical laboratories routinely report total calcium. However, true hypocalcemia is due to a low ionized calcium concentration. In patients with hypoalbuminemia, there will be a decrease in the total calcium, but not a decrease in the ionized calcium. Thus, before a diagnosis of hypocalcemia can be made, one should first estimate the corrected calcium by adding 0.8 mg/dl to the total calcium for every 1 mg decrease in the serum albumin below 4 mg/dl. If hypo calcemia persists, then an ionized calcium should be measured. In patients with excess citrate (from blood transfusions) or acute administration of bicarbonate, the percentage of calcium that is bound to these negatively charged ions increases reducing the free ionized calcium, usually with only a minimal change in total calcium. Acute respiratory alkalosis will also lower the ionized calcium. A decrease in the hydrogen ion concentration will free up binding sites on albumin, leading to increased protein binding of ionized calcium, and a decrease in the ionized component. Thus, hypoalbuminemia would not result in symptomatic hypocalcemia as there is no true change in the ionized calcium, whereas citrate and acute respiratory alkalosis would lead to symptoms due to a real change in ionized calcium.
Clinical manifestations of hypocalcemia
Most patients with mild hypocalcemia are asymptomatic, but large or abrupt changes in ionized calcium may lead to symptoms. The most specific symptom is perioral numbness and carpopedal spasms of the hands and feet. In some patients this may progress to tetany. This increased neuromuscular reactivity can be tested by Chvostek’s sign and Trousseau’s sign. Chovosteks sign is tested by tapping on the facial nerve near the temporal mandibular joint and watching for grimacing caused by spasm of the facial muscles. Trousseau’s sign is tested by inflating a blood pressure cuff above the systolic blood pressure for 3 minutes and watching for spasm of the outstretched hand. Of these two signs, Trousseau’s is more specific. It is recommended that both clinical signs be confirmed with a measurement of ionized calcium.
Differential Diagnosis of hypocalcemia
Once hypoalbumenemia has been ruled out, there are many causes of hypocalcemia. The diagnosis can usually be made by measuring calcidiol levels and intact PTH46. These can be best thought of by mechanism:
Vitamin D deficiency
Vitamin D, once activated to calcitriol is the primary determinant of intestinal calcium absorption. Individuals may be vitamin D deficient from poor absorption of dietary sources of vitamin D such as in malabsorption, short bowel, and poor nutrition; abnormal conversion of calcidiol to calcitriol in the liver in cirrhosis and with some drugs; and from decreased renal conversion of calcidiol to calcitriol in chronic kidney disease stages 3–5. These patients will have low vitamin D levels and an increase in PTH.
Hypoparathyroidism
A deficiency or inactivity of PTH will result in hypocalcemia. This may be due to the inadvertent removal of the parathyroid gland during thyroid surgery or damage with radiation, congenital defects, or autoimmune disease. These patients will have an inappropriately low PTH for the low calcium. In the absence of PTH, the only mechanism to increase serum calcium is via intestinal absorption by the administration of vitamin D (usually in the active form, calcitriol) and oral calcium. Hypomagnesemia may also cause a relative inactivity of PTH30.
Pseudohypoparathyroidism
This term describes a group of rare disorders characterized by hypocalcemia and hypophosphatemia but elevated PTH, indicating unresponsiveness to PTH at the tissue level. The magnesium and calcidiol levels should be normal. The intravenous administration of PTH normally results in increased urinary cAMP and phosphorus excretion. In patients with pseudohypoparathyroidism, the lack of response is diagnostic. The most common form of hypoparathyroidism is type Ia, Albright’s hereditary osteodystrophy, also associated with short stature, round faces, obesity, brachydactly and other defects27.
Tissue consumption of calcium
Hypocalcemia may result from precipitation into extra-skeletal tissue such as in pancreatitis. In addition, excess bone formation in some malignancies with blastic bone metastases, may lead the bone to acutely take up excess calcium. Following parathyroidectomy, there is an acute drop in calcium and phosphorus due to the hungry bone syndrome where calcium and phosphorus are rapidly taken up due to the sudden drop in PTH. In acute hyperphosphatemia due to rhabdomyolysis or tumor lysis syndrome, the phosphorus binds to calcium leading to a drop in ionized calcium. Similarly, the infusion of citrate, a preservative in blood and plasma transfusions, can similarly reduce ionized calcium. Sepsis is also associated with hypocalcemia, although the mechanism is not clear.
Treatment of hypocalcemia
Intravenous calcium infusions are only indicated in the setting of symptomatic hypocalcemia, and should not be given with severe hyperphosphatemia due to the risk of precipitation. Intravenous calcium comes in two forms, calcium gluconate (10 ml vial = 94 mg elemental calcium) or calcium chloride (10 ml vial = 273 mg elemental calcium). For obvious reasons, care must taken to order the correct formulation of calcium. A continuous infusion of 50 ml calcium gluconate in 450 ml D5W can also be used starting at 10 ml/min, titrating to desired ionized calcium level. If patients are not symptomatic, they should be repleted with oral calcium usually in the form of calcium carbonate. The amount of calcium absorbed will be increased if calcitriol is given with the calcium. In addition, hypomagnesemia should be treated concomitantly. If appropriate, patients may also be changed from loop diuretics to thiazide diuretics to decrease urinary calcium excretion. Most importantly, the underlying cause should be treated.
PHOSPHORUS
HYPERPHOSPHATEMIA
Hyperphosphatemia can occur from increased intestinal absorption, cellular release or rapid intracellular to extracellular shifts, or decreased renal excretion. Persistent hyperphosphatemia for more than 12 hours occurs almost exclusively in the setting of acute or chronic kidney disease. Increased intestinal absorption is generally due to large oral intake of phosphate containing laxatives or enemas (which are also absorbed when given rectally) and in vitamin D overdoses. Increased tissue release of phosphorus is commonly seen in acute tumor lysis syndrome, rhabdomyolysis, hemolysis, hyperthermia, profound catabolic stress, acute leukemia; these disorders can also lead to acute renal failure further exacerbating the hyperphosphatemia. Rarely thyrotoxicosis and acromegaly will lead to hyperphosphatemia. Acute hyperphosphatemia generally does not cause symptoms unless there is precipitation of phosphorus with calcium leading to symptoms of hypocalcemia. In addition, there have been multiple reports of acute renal failure with the administration of sodium phosphate containing laxatives for preparation for colonoscopy. In many cases these patients present with non proteinuric kidney disease months after a colonoscopy. Renal biopsy shows calcification due to calcium-phosphate precipitates in the interstitium and tubules29. It is likely that the majority of these cases did not adequately hydrate during the colonoscopy prep. The use of phosphate enemas is contraindicated in the setting of any impairment in kidney function.
In chronic kidney disease, patients should have their serum phosphorus checked at least at least annually for CKD stage 423. When patients are hyperphosphatemic, dietary phosphate restriction and oral phosphate binders should be given. There are several choices of phosphate binders, but in stage 3 and 4 CKD, calcium carbonate, calcium acetate, or sevelamer carbonate can be initiated with the largest meal13.
The treatment of acute hyperphosphatemia includes volume expansion, dialysis, and oral phosphate binders, although in the setting of normal, or even mild to moderate kidney disease it is usually self resolving due to the continued ability of the kidney to excrete a phosphorus load.
HYPOPHOSPHATEMIA
Hypophosphatemia can occur when there is decreased phosphorus intake (decreased intestinal absorption or increased gastrointestinal losses), or excess renal wasting from renal tubular defects or hyperparathyroidism. In addition, low serum phosphorus levels may also occur in the setting of extracellular to intracellular shifts. In the case of cellular shifts, total body phosphorus may not be depleted. By convention, hypophosphatemia is often graded as mild (< 3.5 mg/dl), moderate (< 2.5 mg/dl) and severe (< 1.0 mg/dl). Moderate and severe hypophosphatemia will generally only occur when there are multiple problems. The causes of hypophosphatemia are shown in Box 2.
Box 2
Etiology of Hypophosphatemia
Decreased intestinal absorption
Antacid abuse, malabsorption and chronic diarrhea, Vitamin D deficiency, starvation or anorexia, alcoholism
Increased urinary losses
Primary hyperparathyroidism, post renal transplant, extracellular fluid volume expansion
Glucosuria (after treating DKA)
Post obstructive or resolving ATN diuresis
Acetazolamide
Fanconi’s syndrome
X-linked and Vitamin D dependent rickets
Oncogenic osteomalacia
Redistribution
Respiratory alkalosis
Alcohol withdrawal
Severe burns,
Post feeding syndrome)
Leukemic blast crisis
Clinical manifestations of hypophosphatemia
Hypophosphatemia is a common finding observed in 3% of all hospitalized patients, 10% of hospitalized alcoholic patients, and 70% of ventilated ICU patients25. Symptoms of hypophosphatemia are usually only seen in patients with moderate or severe hypophosphatemia and include muscle weakness (and difficulty weaning from ventilator), hemolysis, impaired platelet and WBC function, rhabdomyolysis, and in rare cases neurologic disorders. Hypophosphatemia is probably over-treated in the ICU, where the “difficult to wean” patient is given phosphorus when the low levels are actually due to cellular shifts from respiratory alkalosis. A careful review of the trend in serum phosphorus with arterial blood pH can help discern which patients need to be treated.
Differential Diagnosis of hypophosphatemia
The differential diagnosis, and treatment approach will be based on the cause and site of phosphate loss (list 2). Usually the cause is clinically apparent, but if not, the simplest test is to measure a 24 hr urine phosphorus. In the setting of hypophosphatemia, the kidney should be retaining (reabsorbing) all phosphorus. If the urinary excretion of phosphorus is < 100 mg/24 hrs, then there are gastrointestinal losses or extracellular to intracellular shifts.
Redistribution
Approximately 15% of the extra-skeletal phosphorus is intracellular, and thus hypophosphatemia may result from a shift to intracellular stores. In most situations this shift is not clinically detected. However, if there is some underlying phosphorus depletion, more profound hypophosphatemia can be observed. The most common clinical causes of this form of hypophosphatemia is with hyperglycemia due to diabetic ketoacidosis or nonketotic hyperglycemia. The glucose induced osmotic urinary diuresis results in renal losses, and glucose further causes a shift of the extracellular phosphorus into cells. This is usually a transient hypophosphatemia and should not generally be treated. In patients who are malnourished, sudden ‘re-feeding’ may also shift phosphorus into the cell. Respiratory, but not metabolic, alkalosis also increases the intracellular flux of phosphorus6. Even in normal subjects, severe hyperventilation (to pCO2 <20 mm Hg) may lower serum phosphate concentrations to below 1.0 mg/dL. Therefore, in ventilated patients, arterial blood gases may be helpful in differentiating shifts from true phosphorus depletion. Lastly, in hungry bone syndrome after a parathyroidectomy there is increased bone uptake of phosphorus and resultant hypophosphatemia.
Decreased oral intake
All proteins and dairy products contain phosphorus, and there is additional phosphorus used as a preservative in processed foods. The average American diet contains nearly 2 times the needed phosphorus content. Thus, decreased intake of phosphorus is usually only seen with poor oral intake, gastrointestinal losses with diarrhea and malabsorption, and in alcoholics. Occasionally patients will abuse antacids, which will lower phosphorus absorption by acting as phosphate binders.
Increased urinary losses
Phosphorus clearance in the kidney is primarily determined by the phosphorus concentration, urinary flow, parathyroid hormone and FGF23 and other phosphatonins. Both genetic and acquired Fanconi’s syndrome will result in increased urinary phosphorus excretion from defects in the proximal tubule together with, renal glucosuria, hypouricemia, aminoaciduria, and type 2 renal tubular acidosis. The acquired from can be seen in multiple myeloma and from some chemotherapy drugs (Cisplatin, ifosfamide, and 6-mercaptopuri) and the anti-retroviral agent tenofovir. Patients with glucosuria and post obstructive diuresis will have increased urinary flow and losses. Patients with primary hyperparathyroidism, or tertiary hyperparathyroidism post renal transplant will have increased PTH mediated urinary phosphorus excretion.
Rickets and Osteomalacia
Hypophosphatemia can lead to impaired bone mineralization. There are several genetic disorders that present with hypophosphatemia, inducing rickets in children5. Tumor induced osteomalacia shares similarities with these genetic disorders in that these tumors of mesenchymal origin secrete a phosphatonin, upregulating the renal sodium phosphate co-transporter with resultant renal phosphate wasting. To date, these tumors have been found to secrete FGF23, MEPE, and FRP-445.
Treatment of hypophoshatemia
Acute management of hypophosphatemia is usually only necessary in patients with moderate to severe hypophosphatemia. Oral intake is preferable, as the acute intravenous administration of phosphate can complex calcium and lead to extra-skeletal calcification. Oral supplementation can be given with skim milk (1000mg/quart), whole milk (850 mg/quart), Neutraphosph K capsules® (250 mg/capsule; max dose is 3 tabs q 6 hrs), or Neutraphosph® solution (128 mg/ml solution). Milk is much better tolerated (and cheaper!) and the concomitant administration of vitamin D in the milk or as a supplement will enhance its absorption. Intravenously, phosphorus can be replaced as Kphosphate (3 mmol/ml of phosph, 4.4 meq/ml of K) or Na phosphate (3 mmol/ml of phosph, 4.0 meq/ml of Na).
HYPERMAGNESEMIA
Hypermagnesemia is rare due to the ability of the kidney to rapidly respond to eleveated serum levels. The main symptoms include lethargy and confusion, arrythmias, and muscle weakness. In pregnant patients treated with magnesium infusion to prevent eclampsia, serum levels of 4– 6 mg/dl are not usually associated with clinically significant symptoms suggesting that clinical toxicity occurs with severe acute changes or chronic toxicity. The differential diagnosis of hypermagensemia includes increased intake, decreased renal excretion (due to altered function, volume depletion, or lithium which impairs renal handling), and rarely redistribution with acidosis. Magnesium is commonly found in over the counter antacids, many laxatives/enemas, and herbal supplements. Thus, serum levels should be checked in patients in whom non-specific excess symptoms remain unexplained. Hypermagnesemia is usually sufficiently treated by stopping the intake and making sure the patient is adequately volume repleted. In severe cases, especially in patients with kidney disease, dialysis may be needed. Importantly, in the setting of cardiac arrhythmias thought to be secondary to elevations in serum magnesium, infusion of calcium can stabilize the cardiac membrane.
HYPOMAGNESEMIA
In contrast to elevations in serum magnesium, hypomagnesemia is very common. In the hospital, one study found 7–12% of patients and 20% of ICU patients had hypomagnesemia44 The symptoms of hypomagensemia include apathy, depression, delirium, seizures and parasthesias, tremors, general muscle weakness, ventricular arrhythmias, and increased susceptibility to digoxin related arrythmias. In addition, hypomagensemia is commonly associated with other electrolyte abnormalities, including hypokalemia in up to 40% of patients, and hyponatremia, hypocalcemia, and hypophosphatemia44. Magnesium is not routinely analyzed on standard clinical tests, and thus diagnosis requires a high index of suspicion. A very low serum level (< 1 mg/dl) is always indicative of deficiency, whereas a normal value may still be associated with a decreased ionized, or free, magnesium concentration. A low urinary fractional excretion of magnesium (< 2%) may be helpful to diagnose hypomagnesemia in patients with borderline serum levels. Hypomagnesemia should be aggressively looked for in patients with refractory hypocalcemia and hypokalemia, and levels routinely evaluated in alcoholics, patients on chronic diuretic therapy (both loop and thiazide diuretics), and those receiving digoxin, aminoglycosides, amphotericin, and cisplatin22.
The differential diagnosis of hypomagensemia (Box 3) is decreased intake, decreased gastrointestinal absorption or diarrhea, and increased urinary losses. Severe deficiency usually requires a combination of these factors. Treatment of hypomagnesemia should be oral supplements unless there is severe gastrointestinal disorders/malabsorption or symptomatic deficiency. Several forms of magnesium supplements are available, including magnesium oxide and magnesium lactate; the latter has more magnesium per dose.
Box 3
Differential Diagnosis of Hypomagnesemia
-
Reduced intake
Starvation, alcoholism, prolonged postoperative state
-
Redistribution from extracellular to intracellular fluids
Insulin administration post therapy of diabetic ketoacidosis, hungry bone syndrome post parathyroidectomy, catecholamine excess states such as ETOH withdrawal syndrome, acute pancreatitis, excessive lactation
-
Reduced absorption
Specific GI magnesium malabsorption, generalized malabsorption syndrome, post extensive bowel resections, diffuse bowel disease or injury, chronic diarrhea, laxative abuse
-
Extra-renal factors that increase magnesuria
Drug induced losses: Diuretics, aminoglycosides, digoxin, cisplatinum and cyclosporine.
Hormone induced magnesuria: aldosteronism, hypoparathyroidism, hyperthyroidism.
Ion or nutrient induced tubular losses: hypercalcemia, extracellular fluid volume expansion.
Miscellaneous causes: phosphate depletion syndrome, alcohol ingestion
Recently there has been an increased interest in the role of magnesium deficiency in chronic kidney diseases. Magnesium intake is inversely associated with the incidence of type 2 diabetes26, but interventional studies are inconsistent to date1. The use of magnesium in acute asthma and hypertension has also been reported, but definitive trials are lacking2,10. In contrast, the use of magnesium as adjuvant therapy for ventricular arrhythmias28, and atrial fibrillation19 may be beneficial although long term benefit remains unclear.
SUMMARY/SYNOPSIS
Disorders of mineral metabolism are common in both the office and hospital setting. The diagnosis can be simplified by remembering the target organs involved—intestine, kidney and bone—and assessing the presence of kidney disease and levels of parathyroid hormone and vitamin D status. Although the list of possible causes for these derangements is long, most patients with hypercalcemia have hyperparathyroidism or malignancy; those with hypocalcemia, hypophsophatemia and hypomagnesemia have gastrointestinal malabsorption, and those with hyperphosphatemia and hypermagnesemia have increased intake in the setting of kidney disease.
Acknowledgments
This work is supported by NIH DK and Veterans Administration Merit Award
Financial Disclosure: Dr. Moe is a consultant for Genzyme, Amgen, and Ineos. She has grant support from the NIH, Veterans Administration, Genzyme and Amgen.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Barbagallo M, Dominguez LJ. Magnesium metabolism in type 2 diabetes mellitus, metabolic syndrome and insulin resistance. Arch Biochem Biophys. 2007;458:40. doi: 10.1016/j.abb.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 2.Beasley R, Aldington S. Magnesium in the treatment of asthma. Curr Opin Allergy Clin Immunol. 2007;7:107. doi: 10.1097/ACI.0b013e328012ce4b. [DOI] [PubMed] [Google Scholar]
- 3.Bilezikian JP, Potts JT, Jr, Fuleihan Gel H, et al. Summary statement from a workshop on asymptomatic primary hyperparathyroidism: a perspective for the 21st century. J Bone Miner Res. 2002;17(Suppl 2):N2. [PubMed] [Google Scholar]
- 4.Bilezikian JP, Silverberg SJ. Clinical practice. Asymptomatic primary hyperparathyroidism. N Engl J Med. 2004;350:1746. doi: 10.1056/NEJMcp032200. [DOI] [PubMed] [Google Scholar]
- 5.Brame LA, White KE, Econs MJ. Renal phosphate wasting disorders: Clinical features and pathogenesis. Semin Nephrol. 2004;24:39. doi: 10.1053/j.semnephrol.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 6.Brautbar N, Leibovici H, Massry SG. On the mechanism of hypophosphatemia during acute hyperventilation: evidence for increased muscle glycolysis. Miner Electrolyte Metab. 1983;9:45. [PubMed] [Google Scholar]
- 7.Brown EM, Gamba G, Riccardi D, et al. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature. 1993;366:575. doi: 10.1038/366575a0. [DOI] [PubMed] [Google Scholar]
- 8.Brown EM, Pollak M, Hebert SC. Sensing of extracellular Ca2+ by parathyroid and kidney cells: cloning and characterization of an extracellular Ca(2+)-sensing receptor. American Journal of Kidney Diseases. 1995;25:506. doi: 10.1016/0272-6386(95)90118-3. [DOI] [PubMed] [Google Scholar]
- 9.Delmez JA, Slatopolsky E. Hyperphosphatemia: its consequences and treatment in patients with chronic renal disease. American Journal of Kidney Diseases. 1992;19:303. doi: 10.1016/s0272-6386(12)80446-x. [DOI] [PubMed] [Google Scholar]
- 10.Dickinson HO, Nicolson DJ, Campbell F, et al. Magnesium supplementation for the management of essential hypertension in adults. Cochrane Database Syst Rev. 2006;3:CD004640. doi: 10.1002/14651858.CD004640.pub2. [DOI] [PubMed] [Google Scholar]
- 11.Dusso AS, Brown AJ, Slatopolsky E. Vitamin D. Am J Physiol Renal Physiol. 2005;289:F8. doi: 10.1152/ajprenal.00336.2004. [DOI] [PubMed] [Google Scholar]
- 12.Endres DB, Villanueva R, Sharp CF, Jr, et al. Measurement of parathyroid hormone. Endocrinology & Metabolism Clinics of North America. 1989;18:611. [PubMed] [Google Scholar]
- 13.Fadem SZ, Moe SM. Management of chronic kidney disease mineral-bone disorder. Adv Chronic Kidney Dis. 2007;14:44. doi: 10.1053/j.ackd.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 14.Friedlander G. Regulation of renal phosphate handling: recent findings. Current Opinion in Nephrology & Hypertension. 1996;5:316. doi: 10.1097/00041552-199607000-00005. [DOI] [PubMed] [Google Scholar]
- 15.Fuss M, Pepersack T, Gillet C, et al. Calcium and vitamin D metabolism in granulomatous diseases. Clinical Rheumatology. 1992;11:28. doi: 10.1007/BF02207080. [DOI] [PubMed] [Google Scholar]
- 16.Goodman WG, Salusky IB, Juppner H. New lessons from old assays: parathyroid hormone (PTH), its receptors, and the potential biological relevance of PTH fragments. Nephrol Dial Transplant. 2002;17:1731. doi: 10.1093/ndt/17.10.1731. [DOI] [PubMed] [Google Scholar]
- 17.Hebert SC. Extracellular calcium-sensing receptor: implications for calcium and magnesium handling in the kidney. Kidney International. 1996;50:2129. doi: 10.1038/ki.1996.539. [DOI] [PubMed] [Google Scholar]
- 18.Hindie E, Urena P, Jeanguillaume C, et al. Preoperative imaging of parathyroid glands with technetium-99m-labelled sestamibi and iodine-123 subtraction scanning in secondary hyperparathyroidism. Lancet. 1999;353:2200. doi: 10.1016/S0140-6736(98)09089-8. [DOI] [PubMed] [Google Scholar]
- 19.Ho KM, Sheridan DJ, Paterson T. Use of intravenous magnesium to treat acute onset atrial fibrillation: a meta-analysis. Heart. 2007;93:1433. doi: 10.1136/hrt.2006.111492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofbauer LC, Heufelder AE. Role of receptor activator of nuclear factor-kappaB ligand and osteoprotegerin in bone cell biology. J Mol Med. 2001;79:243. doi: 10.1007/s001090100226. [DOI] [PubMed] [Google Scholar]
- 21.Holick MF. Vitamin D for health and in chronic kidney disease. Semin Dial. 2005;18:266. doi: 10.1111/j.1525-139X.2005.18402.x. [DOI] [PubMed] [Google Scholar]
- 22.Innerarity S. Hypomagnesemia in acute and chronic illness. Crit Care Nurs Q. 2000;23:1. doi: 10.1097/00002727-200008000-00002. [DOI] [PubMed] [Google Scholar]
- 23.K/DOQI NKF: Clinical Practice Guidelines for Bone Metabolism and Disease in Chronic Kidney Disease. American Journal of Kidney Diseases. 2003;42:S1. [PubMed] [Google Scholar]
- 24.Lambers TT, Bindels RJ, Hoenderop JG. Coordinated control of renal Ca2+ handling. Kidney Int. 2006;69:650. doi: 10.1038/sj.ki.5000169. [DOI] [PubMed] [Google Scholar]
- 25.Larsson L, Rebel K, Sorbo B. Severe hypophosphatemia--a hospital survey. Acta Med Scand. 1983;214:221. doi: 10.1111/j.0954-6820.1983.tb08598.x. [DOI] [PubMed] [Google Scholar]
- 26.Larsson SC, Wolk A. Magnesium intake and risk of type 2 diabetes: a meta-analysis. J Intern Med. 2007;262:208. doi: 10.1111/j.1365-2796.2007.01840.x. [DOI] [PubMed] [Google Scholar]
- 27.Levine MA. Pseudohypoparathyroidism: from bedside to bench and back. J Bone Miner Res. 1999;14:1255. doi: 10.1359/jbmr.1999.14.8.1255. [DOI] [PubMed] [Google Scholar]
- 28.Li J, Zhang Q, Zhang M, et al. Intravenous magnesium for acute myocardial infarction. Cochrane Database Syst Rev. 2007:CD002755. doi: 10.1002/14651858.CD002755.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Markowitz GS, Stokes MB, Radhakrishnan J, et al. Acute phosphate nephropathy following oral sodium phosphate bowel purgative: an underrecognized cause of chronic renal failure. J Am Soc Nephrol. 2005;16:3389. doi: 10.1681/ASN.2005050496. [DOI] [PubMed] [Google Scholar]
- 30.Marx SJ. Hyperparathyroid and hypoparathyroid disorders. N Engl J Med. 2000;343:1863. doi: 10.1056/NEJM200012213432508. [DOI] [PubMed] [Google Scholar]
- 31.Moe S. Calcific uremic arteriolopathy: a new look at an old disorder. NEPHSAP. 2004;3:77. [Google Scholar]
- 32.Moe SM. Calcium, Phosphorus, and Vitamin D Metabolism in Renal Disease and Chronic Renal Failure. In: Kopple JD, Massry SG, editors. Nutritional Management of Renal Disease. ed Second. Philadelphia: Lippincott Williams & Wilkins; 2004. p. 261. [Google Scholar]
- 33.Moe SM. The treatment of steroid-induced bone loss in transplantation. Current Opinion in Nephrology & Hypertension. 1997;6:544. doi: 10.1097/00041552-199711000-00008. [DOI] [PubMed] [Google Scholar]
- 34.Moe SM, Sprague S. Mineral Bone Disorders in Chronic Kidney Disease. In: Brenner B, editor. The Kidney. ed 8th. Vol 2. Philadelphia: Saunders; 2008. p. 1784. [Google Scholar]
- 35.Peacock M, Edmondson J. A Randomized, Double-Blind, Placebo-Controlled, Multi-Center, 6 Week Dose-Ranging Study to Assess the Safety, Pharmacokinetics, and Clinical Effect of an Oral Calcimimetic Agent (AMG 073) Primary Hyperparathyroidism. 1998 [Google Scholar]
- 36.Quarles LD. Extracellular calcium-sensing receptors in the parathyroid gland, kidney, and other tissues. Curr Opin Nephrol Hypertens. 2003;12:349. doi: 10.1097/00041552-200307000-00002. [DOI] [PubMed] [Google Scholar]
- 37.Schiavi SC. Fibroblast growth factor 23: the making of a hormone. Kidney Int. 2006;69:425. doi: 10.1038/sj.ki.5000168. [DOI] [PubMed] [Google Scholar]
- 38.Schiavi SC, Kumar R. The phosphatonin pathway: New insights in phosphate homeostasis. Kidney Int. 2004;65:1. doi: 10.1111/j.1523-1755.2004.00355.x. [DOI] [PubMed] [Google Scholar]
- 39.Stewart AF. Clinical practice. Hypercalcemia associated with cancer. N Engl J Med. 2005;352:373. doi: 10.1056/NEJMcp042806. [DOI] [PubMed] [Google Scholar]
- 40.Stewart AF. Hyperparathyroidism, humoral hypercalcemia of malignancy, and the anabolic actions of parathyroid hormone and parathyroid hormone-related protein on the skeleton. J Bone Miner Res. 2002;17:758. doi: 10.1359/jbmr.2002.17.5.758. [DOI] [PubMed] [Google Scholar]
- 41.Stewart AF, Adler M, Byers CM, et al. Calcium homeostasis in immobilization: an example of resorptive hypercalciuria. New England Journal of Medicine. 1982;306:1136. doi: 10.1056/NEJM198205133061903. [DOI] [PubMed] [Google Scholar]
- 42.Tenenhouse HS. Regulation of phosphorus homeostasis by the type IIa na/phosphate cotransporter. Annu Rev Nutr. 2005;25:197. doi: 10.1146/annurev.nutr.25.050304.092642. [DOI] [PubMed] [Google Scholar]
- 43.Wagner CA. Disorders of renal magnesium handling explain renal magnesium transport. J Nephrol. 2007;20:507. [PubMed] [Google Scholar]
- 44.Whang R, Oei TO, Watanabe A. Frequency of hypomagnesemia in hospitalized patients receiving digitalis. Archives of Internal Medicine. 1985;145:655. [PubMed] [Google Scholar]
- 45.White KE, Larsson TM, Econs MJ. The Roles of Specific Genes Implicated as Circulating Factors Involved in Normal and Disordered Phosphate Homeostasis: Frp-4, MEPE, and FGF23. Endocr Rev. 2006 doi: 10.1210/er.2005-0019. [DOI] [PubMed] [Google Scholar]
- 46.Zivin JR, Gooley T, Zager RA, et al. Hypocalcemia: a pervasive metabolic abnormality in the critically ill. Am J Kidney Dis. 2001;37:689. doi: 10.1016/s0272-6386(01)80116-5. [DOI] [PubMed] [Google Scholar]