

Abstract
After nitrogen, fluorine is probably the next most favorite hetero-atom for incorporation into small molecules in life science-oriented research. This review focuses on a particular fluorinated substituent, the trifluoromethoxy group, which is finding increased utility as a substituent in bioactives, but it is still perhaps the least well understood fluorine substituent in currency. The present review will give an overview of the synthesis, properties and reactivity of this important substituent.
Introduction
Nowadays, fluorine containing compounds are synthesized in pharmaceutical research on a routine basis and about 10% of all marketed pharmaceuticals contain a fluorine atom. There has been an enormous increase in the use of fluorine containing compounds for medicinal applications. For example, nine of the 31 new chemical entities approved in 2002 contain one or several fluorine atoms. According to the World Drug Index (WDI), there are 128 fluorinated compounds with US trade names. Even more fluorinated drugs are predicted to be developed in the near future, as fluoro-organic compounds continue to attract attention in the field of chemistry and biochemistry [1].
Fluorine as a substituent in active ingredients plays a significant and increasingly important role. Currently about 15% of the pesticides listed in the 13th edition of the Pesticide Manual contain at least one fluorine atom. The biggest group of fluorinated pesticides are the compounds containing a trifluoromethyl group (mainly at aromatic rings), followed by aromatic compounds containing an isolated fluorine atom (one and more). However, according to the 12th and 13th edition of the pesticide manual only five pesticides containing OCF3-groups are so far registered (see Figure 1). The proinsecticide Indoxacarb acting as a voltage-gated sodium channel (vgSCh) modulator, the insect growth regulant (IGR) Triflumuron, the plant growth regulator Flurprimidol, the inhibitor of the respiratory chain and succinate dehydrogenase (SD) Thifluzamide as well as the inhibitor of acetolactate synthase (ALS) Flucarbazone-sodium. It was estimated that the number of fluorinated compounds currently under development represent some 35–50% of the all active ingredients under development [2].
Figure 1.

OCF3-bearing pesticides.
One or several fluorine atoms as substituents at specific sites in an organic compound can dramatically alter its chemical and biological nature. In fact, the incorporation of fluorine into a bioactive compound allows a simultaneous change in the electronic, lipophilic and steric parameters, all of which can influence both the pharmacodynamic and pharmacokinetic properties of the candidate [3].
What is so particular about fluorine? Due to its comparable size, the fluorine atom (1.47 Å) can mimic a hydrogen atom (1.20 Å) or a hydroxy group (1.40 Å) in a bioactive compound with respect to steric requirements at receptor sites. Its high electronegativity (4.0 according to the Pauling scale) can have a pronounced influence on the reactivity pattern of a molecule. The most common reason for incorporating fluorine into a molecule is to reduce the rate of oxidative metabolism. However, the increased oxidative stability of fluorinated molecules has nothing to do with the greater strength of the carbon-fluorine bond relative to the carbon-hydrogen bond. In fact, biological oxidation does not involve the homolysis of C–H or C–F bonds. More relevant are the bond energies and heats of formation of H–O and C–O bonds relative to those of F–O bonds. As the latter are unfavorable all alternative mechanisms avoiding attack at fluorine always apply in biological systems [4].
Moreover, the presence of fluorine atoms in biologically active molecules can enhance their lipophilicity and thus their in vivo uptake and transport. In particular, the trifluoromethyl group (−CF3) confers increased stability and lipophilicity in addition to its high electronegativity [5–9]. However, another fluorinated substituent, the trifluoromethoxy group, is becoming more and more important in both agrochemical research and pharmaceutical chemistry [10–11].
The trifluoromethoxy group is perhaps the least well understood fluorine substituent. When asked to draw up a list of textbook substituents, hardly anyone would consider associating such an "exotic entity" like trifluoromethoxy to the lasting popularity of the carboxyl, acetyl, formyl, nitro, amino, hydroxyl and sulfo groups. Nevertheless, the occurrence of OCF3-substituted organics, the majority of which are aromatic compounds, has significantly increased in the recent years [12].
In the 1950s and 1960s the successful development of α-fluorinated ethers as volatile, non-toxic, non-explosive and fast-acting inhalation anesthetics was quickly followed by applications of anti-inflammatory agents. Investigations of the anesthetic properties of α-fluorinated ethers were undertaken on the rational basis that replacement of the hydrogen atom in already known "anesthetic molecules" by fluorine should result in derivatives having improved thermal stabilities relative to the inhalation anesthetics in common use at that time (cyclopropane and ether), like the halo ether anesthetic Fluoroxene (Fluoromar®, F3C-H2C-O-CH=CH2). Numerous analogues [13] were prepared and evaluated (Table 1). Meanwhile, cyclic analogues bearing the fluorinated 1,3-dioxolanes moiety [14] have largely replaced Fluoroxene in its clinical use. Many anesthetics currently used are powerful positive allosteric modulators of GABAA [15].
Table 1.
α-Fluorinated ethers used as Anesthetics.
| Entry | α-Fluorinated ethers | b.p. [°C] | Common names | Brand names |
| 1 | F2HC-O-CHFCF3 | 12.4 ± 25.0 | Desflurane | Suprane® |
| 2 | F2HC-O-CHClCF3 | 48.5 ± 0.0 | Isoflurane | Forane® |
| 3 | FH2C-O-CH(CF3)2 | 49.5 ± 25.0 | Sevoflurane | Sevofrane® |
| 4 | F2HC-O-CF2-CHFCl | 59.9 ± 25.0 | Enflurane | Ethrane® |
| 5 | F2HC-O-CHF-CF2-CHF2 | 60.9 ± 25.0 | BAX 3224 | Synthane® |
| 6 | H3C-O-CF2-CHFBr | 87.0 ± 25.0 | Roflurane | DA 893 |
| 7 | H3C-O-CF2-CHCl2 | 105.0 ± 0.0 | Methoxyflurane | Pentrane® |
Numerous new OCF3 containing compounds have been prepared, clinically evaluated and in many cases marketed as drugs with enhanced effectiveness, often coupled with diminished side-effects [10]. Between 2004 and 2007 the number of structures bearing an OCF3-substituent has more than doubled (from 30,000 to 74,514). They are documented in 18,000 literature references (SciFinder Scholar), most being patent applications (~11,000), but also in close to 7000 research articles. In contrast, trifluoromethoxy substituted heterocycles are relatively rare, although numerous structures are protected by patent applications.
Review
Preparation of Trifluoromethyl Ethers
Nucleophilic substitution
The first aryl trifluoromethylethers were prepared by L. Yagupol'skii in 1955 starting from substituted anisoles [16]. The displacement of chlorine by fluorine was realized with anhydrous hydrogen fluoride or with antimony trifluoride in the presence of antimony pentachloride (Scheme 1 and Table 2) [16–19].
Scheme 1.

Preparation of trifluoromethyl ethers via a chlorination/fluorination sequence.
Table 2.
Synthesis of ArOCF3 compounds starting from substituted anisoles.
| Anisole | ArOCCl3 | Yield (%) | ArOCF3 | Yield (%) |
| 4-ClC6H4OMe | 4-ClC6H4OCCl3 | 77 | 4-ClC6H4OCF3 | 80 |
| 2-ClC6H4OMe | 2-ClC6H4OCCl3 | 69 | 2-ClC6H4OCF3 | 40 |
| 4-FC6H4OMe | 4-FC6H4OCCl3 | 66 | 4-FC6H4OCF3 | 58 |
| 2,4-Cl2C6H3OMe | 2,4-Cl2C6H3OCCl3 | 70 | 2,4-Cl2C6H3OCF3 | 20 |
| 4-NCC6H4OMe | 4-NCC6H4OCCl3 | 50 | 4-NCC6H4OCF3 | 20 |
| 4-Cl(O)CC6H4OMe | 4-Cl(O)CC6H4OCCl3 | 83 | 4-F(O)CC6H4OCF3 | 69 |
The photochlorination which works well with electron-deficient anisoles cannot be applied to anisole itself. In fact, halogen attack on the phenyl ring proceeds more easily than radical chlorination of the methyl group. Louw and Franken could show that with elemental chlorine, photostimulated in refluxing tetrachloromethane, essentially trichloromethylanisole is obtained [20]. The fluorination of the trichloromethyl ether succeeds then easily as shown above. The chlorination/fluorination sequence described above can be simplified by producing the trichloromethyl aryl ethers without isolation and through in situ conversion into the final trifluoromethyl aryl ethers. As Feiring could show more recently, the phenol is heated together with tetrachloromethane, anhydrous hydrogen fluoride and catalytic amounts of boron trifluoride in a closed pressure vessel under autogeneous pressure up to 150 °C [21]. However, substrates containing ortho substituents capable of hydrogen bonding to the hydroxy group are not suitable starting materials. The stoechiometric use of tetrachloromethane lowers the yield and milder conditions afford essentially chlorodifluoromethoxy derivatives (Scheme 2 and Table 3).
Scheme 2.

Preparation of trifluoromethyl ethers via an in situ chlorination/fluorination sequence.
Table 3.
Synthesis of ArOCF3 compounds via an in situ Cl/F exchange.
| Phenol (mol) | CCl4 (mol) | HF (g) | Conditions | ArOCF3 | Yield (%) |
| C6H5OH | 100 °C/2 h | C6H5OCF3 | 10 | ||
| (0.05) | 0.15 | 30 | 150 °C/4 h | ||
| 4-O2NC6H4OH | 150 °C/8 h | 4-O2NC6H4OCF3 | 56 | ||
| (0.15) | 0.15 | 40 | |||
| 4-O2NC6H4OH | 100 °C/8 h | 4-O2NC6H4OCF2Cl | 45 | ||
| (0.06) | 0.15 | 40 | |||
| 4-ClC6H4OH | 150 °C/8 h | 2-ClC6H4OCF3 | 70 | ||
| (0.6) | 1.8 | 400 | |||
| 3-H2NC6H4OH | 140 °C/8 h | 3-H2NC6H4OCF3 | 26 | ||
| (0.6) | 1.8 | 400 | |||
| 2-FC6H3OH | 150 °C/8 h | 2-FC6H3OCF3 | 35 | ||
| (0.07) | 0.21 | 40 | |||
| 4-MeC6H4OH | 100 °C/2 h | 4-MeC6H4OCF3 | 20 | ||
| (0.05) | 0.12 | 30 | 150 °C/4 h | ||
Yarovenko and Vasil'eva developed an approach based on the readily accessible, although highly toxic aryl chlorothionoformates 1. They can be cleanly converted by chlorination into trichloromethyl aryl ethers [17]. This step is then followed by fluorination using antimony trifluoride and a catalytic amount of antimony pentachloride (Scheme 3). The latter compounds can be obtained directly when treated with molybdenum hexafluoride [22]. Unfortunately, the high percutaneous toxicity of the chlorothionoformates 1 prohibited any industrial exploitation so far.
Scheme 3.

Preparation of trifluoromethyl ethers via chlorothionoformates.
W. Sheppard described in 1964 the syntheses of aryl trifluoromethylethers [23] by reaction of SF4 with aryl fluoroformates. However, this approach implied the use of highly toxic reagents and the fluoroformates were rarely isolated (Scheme 4 and Table 4).
Scheme 4.

Preparation of trifluoromethyl ethers via fluoroformates.
Table 4.
Preparation of aryl trifluoromethyl ethers by two-step method from phenolsa.
| Phenol (mol) | COF2 (mol) | SF4 (mol) | ArOCF3 | Yield (%) |
| C6H5OH | C6H5OCF3 | 62 | ||
| (2.5) | 3.0 | 2.5 | ||
| 4-O2NC6H4OH | 4-O2NC6H4OCF3 | 81 | ||
| (1.0) | 1.5 | 1.5 | ||
| 4-ClC6H4OH | 2-ClC6H4OCF3 | 58 | ||
| (0.25) | 0.38 | 0.28 | ||
| 2-ClC6H4OH | 2-ClC6H4OCF3 | 17 | ||
| (0.25) | 0.38 | 0.28 | ||
| 4-FC6H4OH | 4-FC6H4OCF3 | 42 | ||
| (0.13) | 0.22 | 0.16 | ||
| 4-MeC6H4OH | 4-MeC6H4OCF3 | 9 | ||
| (1.0) | 1.35 | 1.2 | ||
aReactions run in "Haselloy-lined" pressure vessel of 140, 240, or 1000 mL. capacity at autogenous pressure. Normal heating pattern was 1 h at 100 °C followed by 2 to 3 h at 140 °C (or higher temperatures above phenol melting point) for the COF2 reaction; 2 h successively at 100, 140, or 150 °C, and 160 or 175 °C for the SF4 reaction.
Fluorodesulfurization methods
Recently, an elegant method towards trifluoromethyl ethers based on an oxidative desulfurization-fluorination has been disclosed by Hiyama [24–27]. When dithiocarbonates (2, xanthogenates) are exposed to a huge excess of hydrogen fluoride-pyridine and 1,3-dibromo-5,5-dimethylhydantoin, trifluoromethyl ethers form in moderate to excellent yields (Scheme 5 and Table 5).
Scheme 5.

Oxidative desulfurization-fluorination toward trifluoromethyl ethers.
Table 5.
Oxidative desulfurization-fluorination towards ROCF3 compounds.
| Xanthogenate 2 | Fluoride sourcea (mol) | N-halo imideb (mol) | ArOCF3c | Yield (%) |
| 4-n-Pr-C6H4- | 70% HF/Py (40) | DBH (3) | 4-n-Pr-C6H4-OCF3 | 81 |
| TBAH2F3 (5) | NBS (4) | 4-n-Pr-C6H4-OCF2SMe | 58 | |
| 4-n-Hex-C6H4- | 70% HF/Py (80) | DBH (3) | 4-n-Hex-C6H4-OCF3 | 50 |
| 4-PhCH2O-C6H4- | 70% HF/Py (80) | DBH (4) | 4-PhCH2O-C6H4-OCF3 | 56 |
| 4-Br-C6H4- | 70% HF/Py (80) | DBH (3) | 4-Br-C6H4-OCF3 | 62 |
| Ph-CH2CH2CH2- | 70% HF/Py (80) | DBH (3) | Ph-CH2CH2CH2-OCF3 | 75 |
| n-C16H33- | 70% HF/Py (80) | DBH (3) | n-C16H33-OCF3 | 95 |
aMol amounts of HF/Py (70%) or tetrabutylammonium dihydrogen trifluoride (TBAH2F3) for 1 mol of 2. bMol amounts of 1,3-dibromo-5,5-dimethylhydantoin (DBH) or N-bromosuccinimide (NBS). cIsolated yield.
What makes this procedure attractive is its applicability to the conversion of aliphatic alcohols into trifluoromethyl alkyl ethers provided that the alcohol is primary rather than benzylic, secondary or tertiary (in which case the reaction fails). The mechanism is based on the nucleophilic attack of the carbon-sulfur bond on a positively charged halogen which makes subsequently the nucleophilic substitution by a fluoride possible (Scheme 6). Under modified reaction conditions, for example by using TBAH2F3 instead of HF-pyridine, the transient monothioacetals 3 can be isolated [24].
Scheme 6.

Mechanism of the oxidative desulfurization-fluorination.
CF3-Transfer agents
Umemoto reported recently in detail on the preparation of O-(trifluoromethyl)dibenzofuranium salts 4 [28–31] and their use as CF3-transfer agents based on studies of Yagupol'skii [32]. The direct O- and N-trifluoromethylation of alcohols, phenols, amines, anilines and pyridines under mild conditions was described. However, the difficulty in the use of these reagents is the in situ preparation by photochemical decomposition of the corresponding 2-(trifluoromethoxy)biphenylyl-2'-diazonium salts at −100 °C (Scheme 7) [28]. The major drawback of this method is the necessity to work at very low temperature and on small scale.
Scheme 7.

Umemoto's O-(trifluoromethyl)dibenzofuranium salts 4 as CF3-transfer agents.
Togni managed very recently to circumvent these difficulties by using hypervalent iodine compounds such as 5 [33–35]. When 2,4,6-trimethylphenol was treated with the hypervalent iodine compound depicted below, the corresponding trifluoromethyl ether was obtained beside C-trifluoromethylation products (Scheme 8).
Scheme 8.

Togni's approach using hypervalent iodine compounds as CF3-transfer agents.
Alkyl trifluoromethyl ethers, still a rarity, have so far been prepared by the reaction of alkyl fluoroformates with sulfur tetrafluoride [36], the trifluoromethyl transfer from O-(trifluoromethyl)dibenzofuranium hexafluoroantimonate 4 [37] and the addition of trifluoromethyl hypofluorite (FOCF3) to olefins [38].
The introduction of the trifluoromethoxy substituent into carbohydrates was realized using tris(dimethylamino)sulfonium trifluoromethoxide (TASOCF3) as OCF3-transfer reagent [39]. This compound can be prepared by reaction of carbonyl fluoride with tris(dimethylamino)sulfonium difluorotrimethylsilicate in anhydrous THF at −75 °C (Scheme 9) [40]. The trifluoromethoxide anion is a relatively poor nucleophile. However, when reacted with primary triflate esters of carbohydrates, the anion displaced the triflate under mild conditions.
Scheme 9.

TAS OCF3 as a nucleophilic OCF3-transfer agent.
However, although aromatic trifluoromethyl ethers are well known and have many applications in pharmaceutical and agricultural domains, aliphatic trifluoromethyl ethers are still rare and difficult to make [41]. Methyl (trifluoromethoxy)acetate for example has been prepared [42] using the carbonyl fluoride/sulfur tetrafluoride method cited above [36]. Recent advances in the fluorodesulfurization reaction [24,41,43–45] enabled the preparation of some aliphatic trifluoromethyl ethers under mild conditions.
Properties
What makes the introduction of OCF3 into pharmaceutically relevant compounds particularly intriguing is their unique electron distribution. The geminal combination of an alkoxy or aryloxy group with a fluorine atom offers the possibility of bonding/non-bonding resonance which can be formally expressed by the superposition of a covalent and an ionic limiting structure (Figure 2).
Figure 2.
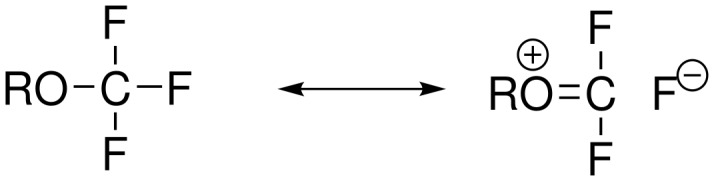
Mesomeric structures of the OCF3-group.
Structure
The anomeric effect reveals itself by a lengthening of the acceptor bond and a shortening of the donor bond. The differences are nevertheless small at least as far as the carbon-fluorine bond is concerned which in general is elongated by just 0.02 Å. In contrast, anomerically active carbon-oxygen bonds may shrink by almost 0.1 Å. The stable tris(dimethylamino)sulfonium (TAS) trifluoromethoxide (see Scheme 9 for structure) [40] represents an extreme case. The three carbon-fluorine bonds are stretched by approximately 0.07 Å and the carbon-oxygen bond contracted by 0.09 Å relative to trifluoromethanol [40] and by 0.21 Å relative to methanol [46].
The geminally 1,1-difluorinated 2,3,4,6-tetra-O-acetyl-1-deoxy-D-glucopyranose (6, Figure 3) [47] exhibits unequivocally non-identical C-F bond lengths, according to crystallography. The difference of 1.5 hundredth of an Å falls in the expected range.
Figure 3.

Structures of 6 and 7.
2,2,3,3-Tetrafluoro-2,3-dihydro-1,4-benzodioxine (7, Figure 3) which occupies a half-chair conformation, shows clearly differences between quasi-axial and quasi-equatorial fluorine atoms. The average bond lengths of 1.355 and 1.330 Å differ significantly [48].
The effect of replacing a methyl by a trifluoromethyl moiety on bond length is dependent upon the electronegativity of the atom to which the substituent is attached [49] and reflects the "anomeric effect" shown above [50]. The lengthening of the acceptor bond and the shortening of the donor bond are small, as far as the carbon fluorine bond are concerned. However the carbon-oxygen bond may decrease by almost one tenth of an Å (Table 6).
Table 6.
Effect of substituting a trifluoromethyl group for methyl on different heteroatoms.
| Atom/group Y-CX3 (X = H, F) | Allred-Rochow Electronegativity | C-Y bond length in Å | Δr = r(CF3)–r(CH3) in Å | |
| X = H | X = F | |||
| P-(CX3) | 2.06 | 1.844 | 1.904 | +0.060 |
| H-(CX3) | 2.20 | 1.099 | 1.102 | +0.003 |
| I-(CX3) | 2.21 | 2.139 | 2.138 | −0.001 |
| S-(CX3) | 2.44 | 1.805 | 1.819 | +0.014 |
| Se-(CX3) | 2.48 | 1.945 | 1.980 | +0.035 |
| Br-(CX3) | 2.74 | 1.939 | 1.923 | −0.016 |
| Cl-(CX3) | 2.83 | 1.781 | 1.752 | −0.029 |
| N-(CX3) | 3.07 | 1.458 | 1.426 | −0.032 |
| O-(CX3) | 3.50 | 1.416 | 1.369 | −0.047 |
| F-(CX3) | 4.10 | 1.385 | 1.319 | −0.066 |
A fluorine substituent can lead to a change in the preferred molecular conformation. For example, methoxybenzenes without ortho substituents favor a planar conformation. However, Roche researchers by searching trifluoromethoxybenzenes without ortho substituents in the Cambridge Structural Database, found that none of the entries has the –OCF3 group in the plane of the phenyl ring (Figure 4). From six compounds, five entries have a dihedral angle C-C-O-C of around 90° and one compound showed a skew conformation (dihedral angle C=C/OCF3 : 36°) [51].
Figure 4.

Conformational preference of the trifluoromethoxy group on aryl rings.
Lipophilicity
On the basis of its electronic properties, which are close to those of a chlorine or a fluorine atom [52], the trifluoromethoxy group has been referred to as a super- [53] or a pseudo-halogen [54]. The advantage of incorporating a trifluoromethoxy group into a molecule can be described in terms of its properties. The trifluoromethoxy group is both more electron withdrawing and lipophilic than its methoxy analogue.
The fluorinated carbon adjacent to an oxygen atom increases lipophilicity as shown by the high value of the OCF3 hydrophobic substituent parameter. While both trifluoromethyl and trifluoromethoxy substituents invariably boost the lipophilicity (Table 7), single fluorine atoms may alter this parameter in either direction. If the halogen occupies a vicinal or homovicinal position with respect to a hydroxy, alkoxy or carbonyl oxygen atom, it enhances the solvation energy in water more than in organic solvents (such as 1-octanol or chloroform) and hence lowers the lipophilicity [51]. It appears that the OCF3 substituent is far more lipophilic (π = +1.04) than the halogens and lies between a CF3 (π = +0.88) and a SCF3 (π = +1.44) group. It may thus replace advantageously a fluorine atom (π = +0.14) in most molecules with the benefit of increased lipid solubility.
Table 7.
Electronegativities and Hydrophobic Parameters for various substituents.
Acidity of trifluoromethyl ethers
As described previously, the trifluoromethoxy group is at the same time a strong electron-withdrawing substituent due to the three fluorine atoms and a π-donating substituent due to the oxygen lone pairs. Yagupol'skii [57–58] and Sheppard [53,59] provided detailed data on the pKa-values of benzoic acids and phenols which reveal that the trifluoromethoxy group is a moderately electron-withdrawing moiety which resembles a chlorine atom. The pKa values are lowered by the trifluoromethoxy group by 0.5 – 1.0 units [60–62].
Reactivity
The OCF3 group is thermally and chemically resistant to attack by acids, bases, organometallic reagents and oxidizing/reducing agents [23,36]. When substituted on an aromatic ring, the trifluoromethoxy group exhibits similar electron withdrawing behavior to the alkoxy group but also acts to deactivate the aromatic ring system [53].
Electrophilic Aromatic Substitution
Trifluoromethoxybenzene, for example, undergoes nitration considerably (up to 5 times) more slowly than benzene. The electrophilic substitution occurs selectively at the ortho and para position. This means the inductive electron-withdrawing effect compromises the attack of the electrophile, but is counterbalanced, to some extent, by the capacity of the ether oxygen to act through resonance as an electron donor. This antagonistic behavior is well known for chloro and bromo substituents. The trifluoromethoxy substituent has a pronounced preference for the para substitution. Unless the para position is occupied, ortho isomers are formed only in small amounts (≤ 10%) without any trace amounts of the meta isomers [52,63–64].
When nitration is carried out under standard conditions, the ortho/para ratio changes with the number of fluorine atoms as depicted in Scheme 10 [52,65–66]. At temperatures in the range of 25 – 50 °C, double nitration can be achieved. The resulting 2,4-dinitrophenyl ethers are isolated in moderate to excellent yield [29,66–67].
Scheme 10.

Nitration of trifluoromethoxy benzene.
The para-directing effect of a trifluoromethoxy group surpasses even that of an amide function. N-Acetyl-3-(trifluoromethoxy)aniline is nitrated mainly at the 6-position and to a minor extent (10%) at the 4-position (Scheme 11) [19]. N-Acetyl-4-(trifluoromethoxy)aniline reacts at the 3-position (again meta with respect to the nitrogen function and ortho to the trifluoromethoxy group!).
Scheme 11.

Synthesis and Nitration of N-Acetyl-(trifluoromethoxy)anilines.
The pronounced preference for para substitution of (trifluoromethoxy)benzene [52,63–64] holds for most electrophilic aromatic substitutions, in particular sulfonation [64], bromination [52], chloromethylation [68] and acylation [52,64]. Attack at the meta position has so far been observed only with the isopropylation and ethylation of (trifluoromethoxy)benzene (to the extent of 9 and 31%, respectively) [52].
Organometallic Reactions
Some very versatile methodology functionalizing trifluoromethoxy substituted aromatics is based on the synthesis-oriented organometallic chemistry. The metal is introduced into a substrate in general by either one of two favorite methods, the permutational interconversion of halogen against metal or hydrogen against metal and subsequently replaced by an electrophile [69–70].
The three isomeric bromo(trifluoromethoxy)benzenes react easily with butyllithium in diethyl ether at −75 °C to generate the corresponding aryllithium (Scheme 12) species which can be trapped by a variety of electrophiles furnishing a diversity of new products (Table 8) [71–72].
Scheme 12.

Bromine/lithium exchange of bromo(trifluoromethoxy)benzenes.
Table 8.
Reaction of 2-, 3- and 4-(trifluoromethoxy)phenyllithiums and electrophiles.
| El | 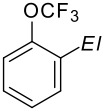 |
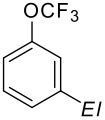 |
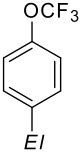 |
| B(OH)2 | 89% | 72% | 84% |
| OH | 88% | ||
| Br | 71% | ||
| I | 81% | 74% | 86% |
| CH3 | 52% | 77% | 73% |
| CH2CH2OH | <5% | 78% | 70% |
| CHO | 93% | 90% | 95% |
| COCOOC2H5 | 87% | 63% | 61% |
| COCH2COOC2H5 | 52% | 32% | 26% |
| COOH | 80% | 85% | 95% |
| CN | 49% | 21% | |
Trifluoromethoxybenzene reacts with sec-butyllithium in the presence of N,N,N',N'-tetramethylethylenediamine ("TMEDA") smoothly under hydrogen/metal permutation ("metalation") as shown in Scheme 13 [72].
Scheme 13.

Metalation of (trifluoromethoxy)benzene.
4-Trifluoromethoxybiphenyl can be metalated using the Schlosser superbase LIC-KOR made by combining butyllithium (LIC) with potassium tert-butoxide (KOR) in tetrahydrofuran at −100 °C. Upon trapping with molecular iodine, 3-iodo-4-trifluoromethoxybiphenyl was isolated in 90% yield [73]. Under the same conditions as employed with trifluoromethoxybenzene, 1- and 2-trifluoromethoxynaphthalene undergo selective lithiation at the 2- and 3-position, respectively (Scheme 14) [74].
Scheme 14.

Metalation of (trifluoromethoxy)naphthalenes.
Both the trifluoromethyl and the trifluoromethoxy group are strongly electron-withdrawing groups and both have a far-reaching activating effect [74]. In an intramolecular competition on 1-trifluoromethoxy-4-(trifluoromethyl)benzene it has been shown, that lithiation next to the OCF3 substituent is favoured, probably due to steric reasons. In fact, 1-trifluoromethoxy-4-(trifluoromethyl)benzene (Scheme 15) affords 2-trifluoromethoxy-5-(trifluoromethyl)benzoic acid after lithiation and carboxylation [75].
Scheme 15.

Competition between -CF3- and -OCF3 in Metalation reactions.
When the OCF3 substituent is in competition with fluorine, as in fluoro(trifluoromethoxy)benzenes, the fluorine-adjacent positions are always metalated (Scheme 16) [75].
Scheme 16.

Competition between -F- and -OCF3 in Metalation reactions.
The OCF3 group reveals a powerful π-polarization as it acidifies not only the ortho but also the meta and para positions strongly. Therefore, metalation of 2- and 4-(trifluoromethoxy)anisole occurs preferentially or exclusively at the methoxy-neighboring position. However, proton abstraction at the trifluoromethoxy-adjacent sites becomes dominant when sec-butyllithium in the presence of N,N,N',N'',N''-pentamethyldiethylenetriamine (PMDTA) is employed. 3-Trifluoromethoxyanisole undergoes deprotonation always at the doubly activated 2-position (Scheme 17). The trifluoromethoxy group enhances the kinetic acidity of anisole by a factor of 3 if in the ortho position, 300 if in the para position and almost 2000 if in the meta position [76].
Scheme 17.

Metalation of trifluoromethoxyanisoles.
The long-range effect of the trifluoromethoxy group was rationalized by Schlosser et al. by a synergy between two kinds of electronic perturbations. The electronegativity of nitrogen, oxygen, or a halogen atom pulls electrons in all σ-bonds towards the heteroelement. This σ-polarization diminishes with the distance. On the other hand, the substituent affects the π-electron cloud by attracting the whole sextet as one toward itself if it is both tetravalent and electrondeficient, e.g. trifluoromethyl or trimethylammonio groups. Alternatively, the π-cloud will remain, as in chlorobenzene, or even be pushed away from lone-pair carrying substituents (with progressively increasing strength from fluorine to alkoxy to dialkylamino). In this way, π-electron density can accumulate at the meta and para positions, where it counterbalances the σ-polarization. The trifluoromethoxy group has a slightly smaller σ-inductive effect than fluorine or a trifluoromethyl substituent. Its π-donating capacity is inferior to the one of the methoxy group, and even inferior to that of a fluorine atom. As a result, these two effects confer its electronic individuality to the trifluoromethoxy group. While acidifying ortho-positions only moderately, its anion-stabilizing effect is important at meta- and para-positions (Figure 5) [76].
Figure 5.

Direction of π-polarization depending on the substituent as described by Schlosser et al. [57].
By contrast, bromo(trifluoromethoxy)benzenes are metalated at −100 °C by bases such as LDA at a position next to the oxygen substituent (Scheme 18) [74].
Scheme 18.

Metalation of Bromo(trifluoromethoxy)benzenes.
At temperatures above −75 °C, lithium bromide elimination generates didehydro(trifluoromethoxy)benzenes ("arynes"). These short-lived species can be trapped with furan to form the corresponding Diels-Alder cycloadducts (Scheme 19) [74].
Scheme 19.

Aryne formation from bromo(trifluoromethoxy)phenyllithiums and subsequent Diels-Alder cycloaddition with furan.
Trifluoromethoxy substituted anilines require protection of the amino function. The BOC-protected ortho and para isomer gives the 3- and 4-(trifluoromethoxy)anthranilic acid after metalation with tert-butyllithium, followed by carboxylation (Scheme 20) [71]. When the amino function is protected instead of a BOC group by a silyl group, 3-trifluoromethoxy-N-(trimethylsilyl)aniline is metalated in position 2. However, 3- and 4-trifluoromethoxy-N,N-bis(trimethylsilyl)aniline are metalated at the oxygen-adjacent position [71].
Scheme 20.

Metalation of (trifluoromethoxy)anilines.
Conclusion
In the life science field, single fluorine atoms, trifluoromethyl or trifluoromethoxy groups are used to tailor pKa values, to facilitate cell membrane penetration and to increase the metabolic stability of compounds. These features of fluorine contribute to the critical "bioavailability" of therapeutically active compounds. The growing interest and utility of the trifluoromethoxy-substitutent in drugs and agrochemical products, presents challenging synthetic strategies which are increasing being tackled in industrial and academic research programmes.
Acknowledgments
We thank the Ministère de la Recherche of France and are grateful to the CNRS and Bayer CropScience for a PhD grant to BM.
References
- 1.Thayer A M. Chem Eng News. 2006;84(23):15–24. [Google Scholar]
- 2.Tomlin C D S, editor. The Pesticide Manual. 13th ed. Farnham: British Crop Protection Council; 2003. [Google Scholar]
- 3.Elliott A J. In: Chemistry of Organic Fluorine Compounds II: A Critical Review. Hudlicky M, Pavlath A E, editors. Washington, DC: American Chemical Society; 1995. pp. 1119–1125. ((ACS Monograph 187)). [Google Scholar]
- 4.Edwards P N. Uses of Fluorine in Chemotherapy. In: Banks R E, Smart B E, Tatlow J C, editors. Organofluorine Chemistry: Principles and Commercial Applications. New York: Plenum Press; 1994. [Google Scholar]
- 5.Filler R, Kobayashi Y, Yagupolskii L M, editors. Organofluorine Compounds in Medicinal Chemistry and Biomedical Applications. Amsterdam: Elsevier; 1993. [Google Scholar]
- 6.Filler R, Kobayashi Y, editors. Biomedicinal Aspects of Fluorine Chemistry. Tokyo: Kodansha Ltd.; 1982. [Google Scholar]
- 7.Ojima I, McCarthy J R, Welch J T, editors. Biomedical Frontiers of Fluorine Chemistry. Washington, DC: American Chemical Society; 1996. ((ACS Symposium Series 639)). [Google Scholar]
- 8.Welch J T, Eswarakrishnan S. Fluorine in Bioorganic Chemistry. New York: Wiley; 1991. [Google Scholar]
- 9.Banks R E, Smart B E, Tatlow J C, editors. Organofluorine Chemistry: Principles and Commercial Applications. New York: Plenum Press; 1994. [Google Scholar]
- 10.Jeschke P, Baston E, Leroux F R. Mini-Rev Med Chem. 2007;7:1027–1034. doi: 10.2174/138955707782110150. [DOI] [PubMed] [Google Scholar]
- 11.Jeschke P. ChemBioChem. 2004;5:570–589. doi: 10.1002/cbic.200300833. [DOI] [PubMed] [Google Scholar]
- 12.Leroux F, Jeschke P, Schlosser M. Chem Rev. 2005;105:827–856. doi: 10.1021/cr040075b. [DOI] [PubMed] [Google Scholar]
- 13.Larsen E R. Fluorine Chem Rev. 1969;3:1–44. [Google Scholar]
- 14.Bagnall R D, Bell W, Pearson K. J Fluorine Chem. 1977;9:359–375. doi: 10.1016/S0022-1139(00)82169-7. [DOI] [Google Scholar]
- 15.Franks N P, Lieb W R. Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- 16.Yagupol'skii L M. Dokl Akad Nauk SSSR. 1955;105:100–102. [Google Scholar]
- 17.Yarovenko N N, Vasil'eva A S. Zh Obshch Khim. 1958;28:2502–2504. [Google Scholar]
- 18.Yagupol'skii L M, Orda V V. Zh Obshch Khim. 1964;34:1979–1984. [Google Scholar]
- 19.Yagupol'skii L M, Troitskaya V I. Zh Obshch Khim. 1961;31:915–924. [Google Scholar]
- 20.Louw R, Franken P W. Chem Ind. 1977:127–128. [Google Scholar]
- 21.Feiring A E. J Org Chem. 1979;44:2907–2910. doi: 10.1021/jo01330a017. [DOI] [Google Scholar]
- 22.Mathey F, Bensoam J. Tetrahedron Lett. 1973;14:2253–2256. doi: 10.1016/S0040-4039(01)87608-5. [DOI] [Google Scholar]
- 23.Sheppard W A. J Org Chem. 1964;29:1–11. doi: 10.1021/jo01024a001. [DOI] [Google Scholar]
- 24.Kanie K, Tanaka Y, Suzuki K, Kuroboshi M, Hiyama T. Bull Chem Soc Jpn. 2000;73:471–484. doi: 10.1246/bcsj.73.471. [DOI] [Google Scholar]
- 25.Kuroboshi M, Kanie K, Hiyama T. Adv Synth Catal. 2001;343:235–250. doi: 10.1002/1615-4169(20010330)343:3<235::AID-ADSC235>3.0.CO;2-0. [DOI] [Google Scholar]
- 26.Kuroboshi M, Suzuki K, Hiyama T. Tetrahedron Lett. 1992;33:4173–4176. doi: 10.1016/S0040-4039(00)74681-8. [DOI] [Google Scholar]
- 27.Shimizu M, Hiyama T. Angew Chem, Int Ed. 2005;44:214–231. doi: 10.1002/anie.200460441. [DOI] [PubMed] [Google Scholar]
- 28.Umemoto T, Adachi K, Ishihara S. J Org Chem. 2007;72:6905–6917. doi: 10.1021/jo070896r. [DOI] [PubMed] [Google Scholar]
- 29.Umemoto T, Ishihara S. Tetrahedron Lett. 1990;31:3579–3582. doi: 10.1016/S0040-4039(00)94447-2. [DOI] [Google Scholar]
- 30.Umemoto T, Ishihara S. J Am Chem Soc. 1993;115:2156–2164. doi: 10.1021/ja00059a009. [DOI] [Google Scholar]
- 31.Umemoto T, Adachi K. J Org Chem. 1994;59:5692–5699. doi: 10.1021/jo00098a030. [DOI] [Google Scholar]
- 32.Yagupol'skii L M, Kondratenko N V, Timofeeva G N. Zh Org Khim. 1984;20:115–118. [Google Scholar]
- 33.Kieltsch I, Eisenberger P, Togni A. Angew Chem, Int Ed. 2007;46:754–757. doi: 10.1002/anie.200603497. [DOI] [PubMed] [Google Scholar]
- 34.Eisenberger P, Gischig S, Togni A. Chem–Eur J. 2006;12:2579–2586. doi: 10.1002/chem.200501052. [DOI] [PubMed] [Google Scholar]
- 35.Stanek, K.; Togni, A. Poster presentation, 15th European Symposium on Fluorine Chemistry, Prague, July 15–20, 2007
- 36.Aldrich P E, Sheppard W A. J Org Chem. 1964;29:11–15. doi: 10.1021/jo01024a002. [DOI] [Google Scholar]
- 37.Umemoto T. Chem Rev. 1996;96:1757–1777. doi: 10.1021/cr941149u. [DOI] [PubMed] [Google Scholar]
- 38.Rozen S. Chem Rev. 1996;96:1717–1736. doi: 10.1021/cr941147+. [DOI] [PubMed] [Google Scholar]
- 39.Trainor G L. J Carbohydr Chem. 1985;4:545–563. doi: 10.1080/07328308508082676. [DOI] [Google Scholar]
- 40.Farnham W B, Smart B E, Middleton W J, Calabrese J C, Dixon D A. J Am Chem Soc. 1985;107:4565–4567. doi: 10.1021/ja00301a043. [DOI] [Google Scholar]
- 41.Ben-David I, Rechavi D, Mishani E, Rozen S. J Fluorine Chem. 1999;97:75–78. doi: 10.1016/S0022-1139(99)00031-7. [DOI] [Google Scholar]
- 42.Yagupol'skii L M, Alekseenko A N, Il'chenko A Y. Ukr Khim Zh (Russ Ed) 1978;44:1057–1059. [Google Scholar]
- 43.Kollonitsch J, Marburg S, Perkins L M. J Org Chem. 1976;41:3107–3111. doi: 10.1021/jo00881a007. [DOI] [Google Scholar]
- 44.Sondej S C, Katzenellenbogen J A. J Org Chem. 1986;51:3508–3513. doi: 10.1021/jo00368a022. [DOI] [Google Scholar]
- 45.Blazejewski J-C, Anselmi E, Wakselman C. J Org Chem. 2001;66:1061–1063. doi: 10.1021/jo005701t. [DOI] [PubMed] [Google Scholar]
- 46.Kimura K, Kubo M. J Chem Phys. 1959;30:151–158. doi: 10.1063/1.1729867. [DOI] [Google Scholar]
- 47.Praly J-P, Brard L, Descotes G, Toupet L. Tetrahedron. 1989;45:4141–4152. doi: 10.1016/S0040-4020(01)81310-7. [DOI] [Google Scholar]
- 48.Praly, J.-P.; Scopelliti, R.; Gorecka, J.; Schlosser, M. Unpublished results
- 49.Oberhammer H. J Fluorine Chem. 1983;23:147–162. doi: 10.1016/S0022-1139(00)84976-3. [DOI] [Google Scholar]
- 50.McClinton M A, McClinton D A. Tetrahedron. 1992;48:6555–6666. doi: 10.1016/S0040-4020(01)80011-9. [DOI] [Google Scholar]
- 51.Böhm H-J, Banner D, Bendels S, Kansy M, Kuhn B, Müller K, Obst-Sander U, Stahl M. ChemBioChem. 2004;5:637–643. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- 52.Olah G A, Yamato T, Hashimoto T, Shih J G, Trivedi N, Singh B P, Piteau M, Olah J A. J Am Chem Soc. 1987;109:3708–3713. doi: 10.1021/ja00246a030. [DOI] [Google Scholar]
- 53.Sheppard W A. J Am Chem Soc. 1963;85:1314–1318. doi: 10.1021/ja00892a021. [DOI] [Google Scholar]
- 54.Haas A. Adv Inorg Chem Radiochem. 1984;28:167–202. [Google Scholar]
- 55.Hansch C, Leo A. Substituent Constants for Correlation Analysis in Chemistry and Biology. New York: John Wiley and Sons; 1979. [DOI] [PubMed] [Google Scholar]
- 56.Leo A, Jow P Y C, Silipo C, Hansch C. J Med Chem. 1975;18:865–868. doi: 10.1021/jm00243a001. [DOI] [PubMed] [Google Scholar]
- 57.Yagupol'skii L M, Yagupol'skaya L N. Dokl Akad Nauk SSSR. 1960;134:1381–1383. [Google Scholar]
- 58.Yagupol'skii L M, Bystrov V F, Stepanyants A U, Fialkov Y A. Zh Obshch Khim. 1964;34:3682–3690. [Google Scholar]
- 59.Sheppard W A. J Am Chem Soc. 1962;84:3072–3076. doi: 10.1021/ja00875a007. [DOI] [Google Scholar]
- 60.Kortüm G, Vogel W, Andrussow K. Dissociation Constants of Organic Acids in Aqueous Solution. London: Butterworths & Co.; 1961. [Google Scholar]
- 61.Kortüm G, Vogel W, Andrussow K. Pure Appl Chem. 1961;1:187–536. [Google Scholar]
- 62.Hansch C, Leo A, Taft R W. Chem Rev. 1991;91:165–195. doi: 10.1021/cr00002a004. [DOI] [Google Scholar]
- 63.Ansink H R W, Cerfontain H. Recl Trav Chim Pays-Bas. 1992;111:215–221. [Google Scholar]
- 64.Desbois M. Bull Soc Chim Fr. 1986:885–890. [Google Scholar]
- 65.Bunton C A, Minkoff G J, Reed R I. J Chem Soc. 1947:1416–1418. doi: 10.1039/JR9470001416. [DOI] [Google Scholar]
- 66.Shelyazhenko S V, Fialkov Y A, Yagupol'skii L M. Zh Org Khim. 1992;28:1652–1659. [Google Scholar]
- 67.Meldola R, Woolcott G H, Wray E. J Chem Soc, Trans. 1896;69:1321–1334. doi: 10.1039/CT8966901321. [DOI] [Google Scholar]
- 68.Fujii, K.; Nakamoto, Y.;Umeyama, H. (Ube Industries, Japan). Jpn. Kokai Tokkyo Koho, filed on December 15, 1999; Chem. Abstr. 2001, 135, 45998.
- 69.Leroux F, Schlosser M, Zohar E, Marek I. The Preparation of Organolithium Reagents and Intermediates. In: Rappoport Z, Marek I, editors. The Chemistry of Organolithium Compounds. Chichester: John Wiley & Sons Ltd.; 2004. pp. 435–493. [DOI] [Google Scholar]
- 70.Schlosser M. Angew Chem, Int Ed. 2005;44:376–393. doi: 10.1002/anie.200300645. [DOI] [PubMed] [Google Scholar]
- 71.Leroux F, Castagnetti E, Schlosser M. J Org Chem. 2003;68:4693–4699. doi: 10.1021/jo0267798. [DOI] [PubMed] [Google Scholar]
- 72.Castagnetti E, Schlosser M. Eur J Org Chem. 2001:691–695. doi: 10.1002/1099-0690(200102)2001:4<691::AID-EJOC691>3.0.CO;2-A. [DOI] [Google Scholar]
- 73.Pauluth D, Haas H, inventors. (Merck Patent GmbH, Darmstadt, Germany) Verfahren zur Herstellung von substituiertem Trifluormethoxybenzol. 4219281C1. German Patent DE. 1993 Dec 23;
- 74.Schlosser M, Castagnetti E. Eur J Org Chem. 2001:3991–3997. doi: 10.1002/1099-0690(200111)2001:21<3991::AID-EJOC3991>3.0.CO;2-J. [DOI] [Google Scholar]
- 75.Schlosser, M.; Castagnetti, E.; Cottet, F. Unpublished results. See: Castagnetti, E. Ph.D. Thesis, Université de Lausanne, 2001
- 76.Castagnetti E, Schlosser M. Chem–Eur J. 2002;8:799–804. doi: 10.1002/1521-3765(20020215)8:4<799::AID-CHEM799>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]