

Abstract
The TNFR family members OX40 (CD134) and 4-1BB (CD137) have been found to play major roles as costimulatory receptors for both CD4 and CD8 T cells. In particular, in many situations, they can control proliferation, survival, and cytokine production, and hence are thought to dictate accumulation of protective T cells during anti-viral and anti-tumor responses and pathogenic T cells during autoimmune reactions. As opposed to simply controlling the activity of naïve, effector, and memory T cells, recent data have suggested that both molecules are also instrumental in controlling the generation and activity of so-called regulatory or suppressor T cells (Treg), perhaps in both positive and negative manners. Part of the action on Treg might function to further promote protective or pathogenic T cells, but alternate activities of OX40 and 4-1BB on Treg are also being described that suggest there might be control by these molecules at multiple levels that will alter the biological outcome when these receptors are ligated. This review specifically focuses on recent studies of regulatory T cells, and regulatory or suppressive activity, that are modulated by OX40 or 4-1BB.
Keywords: OX40, 4-1BB, Treg, T cells
1. Introduction
OX40 (CD134) and 4-1BB (CD137) are typical members of the TNFR superfamily whose genes are clustered on human chromosome 1 together with those encoding GITR, CD30, HVEM, TNFRII, and DR3. Strong published data, as well as still emerging evidence, shows that all of these molecules have some similarities in regulating T cell biology and playing important roles in immune disease that are controlled by T cells. The regulation of T cells and immunity by OX40 and 4-1BB have been reviewed extensively elsewhere [1-7], with much of the early focus on these molecules concentrating around the idea that they primarily regulate effector-like CD4 or CD8 T cells that could be protective or pathogenic depending on the situation in which they arise. Both OX40 and 4-1BB signal through TRAF adaptor molecules that are shared as well as distinct, and inflammatory cascades can be triggered through these receptors such as activating PI-3-kinase and Akt/PKB, and triggering both canonical and non-canonical NF-κB pathways. Similarly, certain common target genes and functional activities have been described for both molecules, including upregulating anti-apoptotic members of the Bcl-2 family, promoting CD4 and CD8 T cell proliferation and survival, and enhancing cytokine secretion and CTL differentiation. However, recent data has suggested that at least functionally there may be a significant divergence in activities, which might be explained either by alternate expression on cell types other than T cells, and the subject of this review which is the notion that OX40 and 4-1BB are also strong controllers of immuno-suppressive or immuno-modulatory cells that can include a number of recognized regulatory T cell subsets (Treg).
Whereas the overall biology of OX40 still appears to be fairly straightforward, that of 4-1BB is apparently more complicated. Studies of OX40, with knockout animals, blocking reagents that suppress OX40 binding to its one known ligand, OX40L, or with agonist reagents that stimulate the OX40 receptor, all support the idea that OX40 controls the development of effector and memory T cells that in the setting of cancer or infectious disease are protective, and in the setting of autoimmunity or allergy are pathogenic. As described below, these actions most likely manifest as two interrelated activities in both allowing effector T cells to develop and concomitantly preventing the generation and/or activity of Treg whose primary function is arguably to suppress the effector T cells. In contrast, blocking the ligand for 4-1BB (4-1BBL), or studies of 4-1BBL knockout animals, has not revealed extensive phenotypes in models of autoimmunity, which contrasts strongly with suppressed autoimmunity in the absence of OX40 or OX40L. Furthermore, although initial and continuing reports show that stimulatory antibodies to 4-1BB can enhance CTL generation and promote anti-tumor as well as anti-viral immunity, it has now been found in most of the recognized mouse models of autoimmunity that an agonist antibody to 4-1BB can result in suppressed disease symptoms, a finding opposite to what would have been predicted several years ago. Although an understanding of the latter is not complete, and alternate modes of activity might manifest in different disease situations, many of the results from stimulating 4-1BB are potentially due to promoting as opposed to suppressing Treg development and/or activity. This review will specifically focus on recent studies of both OX40 and 4-1BB that relate directly or indirectly to control of Treg and suppressive mechanisms.
2. The Biology of Treg cells
Recent advances in identification and classification of regulatory T cells highlight their crucial function in immunity and the importance of the balance between effector/memory T cells and Treg. There are many types of Treg that have been described (Fig. 1), such as natural CD4+CD25+Foxp3+ Treg (nTreg), adaptive/inducible CD4+Foxp3+ Treg (iTreg) or CD4+Foxp3- IL-10-secreting Treg (Tr1) that both derive in the periphery from a naïve T cell, as well as CD8+ regulatory cells that can be Foxp3+ or Foxp3- [8, 9].
Figure 1.
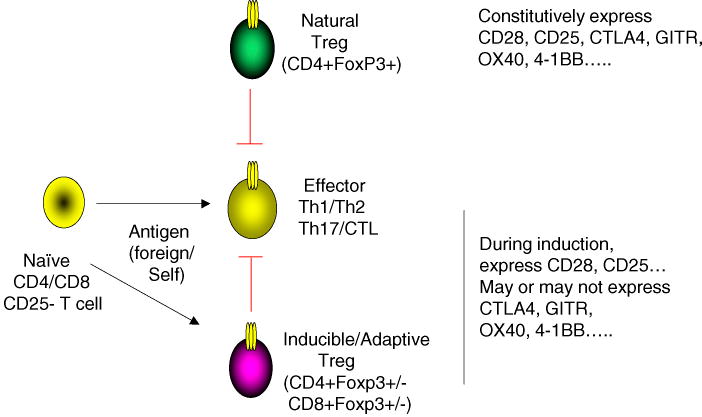
Subsets of Treg Control Effector CD4 and CD8 cells. Regulatory T cells can exist as either natural Treg (nTreg) that derive in the thymus and express CD4 and Foxp3, or as inducible or adaptive Treg (iTreg) that derive in the periphery from naïve CD4+ or CD8+ T cells in response to factors such as TGF-β or IL-10. The latter can be Foxp3+ or Foxp3- and may make suppressive cytokines such as TGF-β or IL-10. Emerging evidence suggests that either nTreg or iTreg limit inflammation and autoimmunity by directly or indirectly suppressing the activity of effector CD4 or CD8 T cells (Th1/Th2/Th17/CTL). Both effector T cells and Treg express surface receptors, either constitutively or inducibly, that can modulate their activity, such as CD28 and CD25, and TNFR superfamily molecules such as OX40, 4-1BB, and GITR.
CD4 T cell subsets expressing CD25 and Foxp3 (forkhead box P3) have been characterized extensively because of the demonstration of their definitive roles in immune homeostasis. Foxp3 is a master regulator of Treg lineage commitment and gain of suppressive function. CD25+Foxp3+ Treg develop either in the thymus or in the periphery. The former are referred to as natural Treg (nTreg) and the latter adaptive or inducible Treg (iTreg). Critical signals for induction and/or maintenance of Foxp3 in developing T cells have been shown to be antigen, IL-2, TGF-β, and retinoic acid. Disruption of LAT-PLC-γ1 interaction by introducing a single amino acid mutation caused the disappearance of Foxp3+ Treg in both the thymus and peripheral organs [10], suggesting that TCR engagement is primarily important for the development of Foxp3+ Treg. For induction of iTreg from naïve CD4 T cells extrathymically, a requirement for stimulation by limiting amounts of antigen and costimulation have been demonstrated [11, 12]. Costimulatory signals mediated by CD28 engagement of CD80 or CD86 are important in shaping the repertoire and size of the Treg cell compartment both in the thymus and peripheral lymphoid organs [13, 14]. However, the primary requirement for CD28 may reflect its strong activity in promoting IL-2. STAT5 activation through IL-2Rβ/γ (CD122/CD132) was found to be critical for Foxp3 induction and the viability of CD25+Foxp3+ nTreg both in the thymus and in the periphery. IL-2- or CD25 (IL-2Rα)-deficient mice had markedly reduced numbers of nTreg [15], and a severe defect in Foxp3 expression was observed in thymocytes or peripheral T cells in IL-2-/- × IL-15-/-, IL-2Rβ-/-, IL-2Rγ-/-, and Stat5-/-mice [16]. Furthermore, Stat5 activation in IL-2Rβ-/- mice could restore Foxp3+ Treg development, and Stat5 was found to bind to the promoter of the Foxp3 gene [16].
TGF-βR signals are also required for the maintenance of Foxp3+ nTreg in the periphery and perhaps as importantly for the induction of Foxp3+ iTreg differentiated by antigen from naïve CD4 T cells in the secondary lymphoid organs [12, 17-19]. Retinoic acid made by gut-associated dendritic cells can further enhance the TGF-β-dependent conversion of naïve CD4 T cells into Foxp3+ iTreg [20, 21]. Additional iTreg subsets include those that make IL-10, inducible by exposure to several factors including IL-10 itself [22] and Vitamin D3 and Dexamethasone [23]. So-called Tr1 cells make IL-10 but do not express Foxp3, whereas other Foxp3-expressing CD4 T cells can be generated, particularly in the gut environment, that also make IL-10 [24]. Furthermore, CD8 T cells differentiated from naïve cells can also display suppressive function and in turn these may be Foxp3+ or Foxp3-, with Foxp3 induction also driven by TGF-β [25, 26].
Of significance to the current discussion, it is now recognized that as well as being expressed on activated and responding effector T cells, many TNFR family members, including OX40 and 4-1BB, are expressed on CD4+CD25+Foxp3+ nTreg, as well as being constitutive or inducible on adaptive CD4 and CD8 T cells that turn into iTreg or display regulatory activity (Fig. 1). It was reported that reduced numbers of CD4+CD25+ T cells (nTreg) are present in OX40-/- mice both in the thymus and in the spleen early in life. However, by 12-13 weeks of age Treg numbers in OX40-/- mice reverted to normal [27]. Also, no defects in peripheral Foxp3+ Treg cells were found in Foxp3-GFP knock-in mice on the OX40-/- background [28], indicating that OX40 has no major role in homeostasis of nTreg in vivo. Similarly, numbers of nTreg are unaltered in 4-1BB-/- mice [29, 30](S.W.L, unpublished), also suggesting that this molecule does not control their homeostasis. However, studies described below suggest that ligating both OX40 and 4-1BB on Treg can strongly influence their responsiveness to self or non-self antigen, both in positive and negative manners.
3. OX40 Antagonizes Induction of Foxp3 in Naïve CD4 T cells
CD25-Foxp3- naïve CD4 T cells can acquire Foxp3 driven by TGF-βR and IL-2R signals leading to differentiation into an iTreg. Two studies recently found that costimulatory signals from OX40 are antagonistic for Foxp3 induction in antigen-responding naïve CD4 T cells and suppress the development of high numbers of CD25+Foxp3+ iTreg [28, 31]. Lower doses of antigen favored induction of Foxp3 in naïve CD4 T cells in a TGF-β-dependent manner and an anti-OX40 agonist antibody and OX40 interaction with its natural ligand, OX40L, strongly suppressed this antigen- and TGF-β-driven Foxp3 expression [31]. As described above, CD28 has been found to play a role in the generation of nTreg, and correspondingly, naïve CD4 T cells deficient in CD28 were impaired in upregulating Foxp3 in response to antigen and TGF-β, contrasting with enhanced induction of Foxp3 in OX40-deficient CD4 cells [31]. The requirement for CD28 to promote Foxp3 in peripheral naïve CD4 cells also largely reflects poor production of IL-2 and feedback through IL-2R [31] rather than a specific role for CD28 signaling that would have implied OX40 and CD28 engage fundamentally different signaling intermediates to explain divergent roles on Foxp3. Data with agonistic anti-CD28, or with CD80 and CD86, have shown that ligation of CD28 can also suppress Foxp3 in CD4 T cells if IL-2 is added exogenously [32, 33]. It is therefore likely that OX40 and CD28 share a common signaling pathway that can lead to inhibition of Foxp3 expression, but CD28 plays an alternate enhancing rather than suppressing role as its critical activity in promoting IL-2 overshadows any other activity. OX40 can promote IL-2, but arguably is not a primary regulator of IL-2, and hence suppression of Foxp3 predominates.
It is presently unknown how OX40 (or CD28) signals might interfere with the induction of Foxp3 in antigen-responding naïve T cells, but the same signals that promote effector/memory formation might concomitantly lead to suppression of Foxp3. OX40 signaling activates multiple signaling pathways, such as those involving PI3K/Akt, AP-1, and NF-κB, which it shares in common with CD28, which promote cell division, survival, and cytokine production [2, 34]. OX40 might act at several levels and negatively regulate induction and/or stability of Foxp3 that is induced by TGF-β. Foxp3 mRNA was initially promoted by TGF-β even when OX40 was engaged, but sustained and maximal levels of Foxp3 mRNA were affected [31]. It also should be noted that OX40 can downregulate Foxp3 in cells recently differentiated to become Foxp3+ (T.S. unpublished data, and [28]), further supporting the idea that signals that sustain Foxp3 expression are primary targets for inhibition by OX40. Several groups have found that persistent or periodic TGF-βR signaling is necessary to maintain intracellular Foxp3 [17, 35], likely due to the requirement for DNA methylation and epigenetic modification, suggesting that upstream or downstream signals from the TGF-βR might be the primary targets of suppression by OX40. What these could be is not clear, but phosphorylation of Smad proteins, such as Smad3, that control many TGF-β-mediated events, is an obvious target. Studies in non-lymphoid cells have found that Akt might directly bind Smad3 and antagonize phosphorylation induced by TGF-β [36], leading to the notion that OX40-activated Akt could be a primary suppressive signal. Recently, TGF-βR signaling was found to promote demethylation of the Foxp3 gene locus, which enhances Foxp3 expression, through an unknown mechanism potentially involving suppression of a DNA methyltransferase [37]. This also suggests another alternate target for OX40 antagonism of Foxp3.
Results showing OX40 can concomitantly promote effector T cell generation while antagonizing the differentiation of iTregs [2, 31, 38] additionally indicate that cytokines produced from OX40-triggered T cells may be important for suppressing Foxp3, essentially providing an indirect route for antagonizing Treg generation. Th1, Th2, Th17, and iTreg populations are all derived from naïve CD4 T cells and promoted in a cytokine-dependent manner through specific lineage-specific transcription factors, T-bet, GATA-3, RORγt, and Foxp3, respectively. Interestingly, recent data found that lineage specific cytokines (IFN-γ and IL-12 for Th1; IL-4 for Th2) and more specifically the transcription factors they elicit (T-bet for Th1; GATA-3 for Th2), strongly inhibited differentiation into Foxp3+ iTreg [39]. Thus, it is possible that OX40 antagonizes continued transcription of Foxp3 in response to TGF-β through directly targeting T-bet or GATA-3, or indirectly promoting IFN-γ or IL-4 that feedback in an autocrine fashion to promote these transcription factors. This type of action could still involve signaling pathways such as through Akt or NF-κB, but without the need to directly suppress signals delivered through the TGF-βR.
Further studies will be required to understand how the Foxp3 gene is regulated, and then how costimulatory signals from molecules such as OX40 might antagonize its expression. No reports as yet have shown that 4-1BB signals can suppress the induction of Foxp3 in naïve CD4 T cells. However, given some overlap in signaling characteristics between OX40 and 4-1BB, as well as other members of the TNFR family such as GITR, it is likely that if these molecules are expressed and ligated before or shortly after TGF-βR initiates Foxp3 transcription, they will display the same antagonistic ability as OX40 and ultimately skew the response towards an effector CD4 T cell and away from a CD4+ iTreg. Whether the same type of regulation by OX40 (or 4-1BB) might apply to a naïve CD8 T cell that is exposed to TGF-β and can differentiate into a Foxp3+ CD8 Treg has not been investigated, but again this likely would depend on the timing of when OX40 or 4-1BB were expressed and ligated and when TGF-βR signaling might be initiated.
4. OX40 and 4-1BB Support Division and Survival of CD25+Foxp3+ CD4 T cells
It has been well characterized for OX40, and slightly less so for 4-1BB, that signals from these molecules can promote proliferation and survival of effector CD4 and CD8 T cells through enhancing expression or activity of molecules such as survivin that regulate cell cycle progression, and anti-apoptotic molecules such as Bcl-xL and Bfl-1 that regulate survival, while also perhaps suppressing expression of pro-apoptotic molecules such as Bad and Bim [3, 6, 7]. Interestingly, several reports have now found that OX40 and 4-1BB signals can also promote proliferation and/or survival of CD4+CD25+(Foxp3+) nTreg in vitro, especially in the presence of IL-2 [27, 40, 41]. This suggests that an already existing CD25+Foxp3+ Treg subset might utilize these receptor/ligand systems as a means to survive and persist as a population, an action somewhat at odds with the idea that OX40 (and perhaps 4-1BB) can prevent the development of similar iTreg in the periphery. However, as described below, it is not clear whether this type of apparent activity in reversing the anergic state of Treg (i.e. promoting their proliferation) correlates with loss or retention of their suppressive activity. Also it has not been well characterized whether OX40 or 4-1BB signaling to a Treg that already expresses intracellular Foxp3 will affect expression of Foxp3. One report claimed that OX40 ligation on a Foxp3+ nTreg did induce a reduction in Foxp3 mRNA [28], whereas other data has not shown any difference in Foxp3 expression in nTreg or highly differentiated iTreg when OX40 is triggered and cell division is enhanced (T.S, unpublished). It is therefore possible that these discrepancies relate to the differentiation state of Foxp3+ Treg. In recently differentiated Foxp3+ cells where extensive epigenetic modifications have not yet occurred and Foxp3 is unstable, OX40 and 4-1BB might concomitantly promote proliferation and also downregulate Foxp3 or at least suppress further or continued Foxp3 transcription. The result here would again be to tip the balance in favor of effector activity (protective or pathogenic) and away from immunoregulation. In contrast, in highly differentiated nTreg or iTreg, where Foxp3 is now stable due to chromatin changes, OX40 and 4-1BB may not alter Foxp3 expression and the result would be outgrowth or enhanced survival of these cells. How this would affect the overall immune response in vivo is not clear. As both molecules could equally promote outgrowth or survival of effector T cells as well as stable Foxp3+ Treg, the effector/Treg balance would likely not change. However, as described below, another factor that potentially could affect the overall nature of the immune response, is whether an OX40- or 4-1BB-triggered Foxp3+ Treg that does not downregulate Foxp3 will still retain short or long-term suppressive function.
5. OX40 and 4-1BB Antagonize the Suppressive Function of CD4+Foxp3+ Treg
Several studies have in part addressed how engagement of OX40 and 4-1BB on CD25+Foxp3+ Treg can influence suppressor/regulatory activity in typical in vitro assays for Treg where proliferation or IL-2 production by a naïve/effector CD4+CD25- responder T cell is measured [27, 28, 30, 42]. These studies have shown that ligation of either OX40 or 4-1BB can reduce suppressor activity. In vivo experiments in a mouse model of inflammatory bowel disease [27], and in models of rejection of allogenic bone marrow [42] and skin transplants [28] and GVHD [30] support these conclusions. Two types of mechanism for resistance against Treg-mediated suppression have been demonstrated. One is by direct engagement of receptors on the Treg, which inhibits suppressive function. Another is by triggering signals on the naïve/effector/memory responder T cell population, which renders the responder cells resistant to Treg suppression. It has been concluded that both OX40 and 4-1BB use the former mechanism to reverse suppression and can directly act on Treg to inhibit their activity [27, 28, 30], although more studies are needed on both molecules before definitive conclusions can be drawn.
It is therefore possible that OX40 and 4-1BB may induce unique negative signals into nTreg, or stably differentiated iTreg, that counteract suppressive mechanisms. However, in all cases examined to date, the suppressive mechanism was not delineated and hence the target(s) of action of OX40 or 4-1BB are not yet known. Furthermore, it is unclear what types of intracellular signals might be responsible for attenuation of suppressive function in Treg. Interestingly, a defect in activation of the Akt pathway has been suggested to be necessary for suppressive function, and transduction of an active form of Akt into nTreg was recently shown to render them less suppressive [43]. As data have shown that both OX40 and 4-1BB are capable of activating the PI-3-kinase/Akt pathway [34, 44], this axis may play a critical role for attenuating suppressive function. As discussed above, one target could be Foxp3 itself, and downregulation of Foxp3 most likely would alter regulatory activity as Foxp3 transcribes many key genes that organize suppressive function and determine the identity as suppressor cells [45, 46]. However, it is unlikely that this is the primary target of action of OX40 or 4-1BB in a mature nTreg or iTreg and expression or production of suppressive cytokines or membrane molecules are more likely targets. The complication is that many Treg-related suppressive molecules have been proposed in the literature, such as CTLA4, membrane TGF-β, IL-10, IL-35, adenosine, CD73, granzyme B, and several galectins, and a single mechanism of suppression and hence target of OX40 or 4-1BB might not exist.
6. OX40 Blocks the Generation of IL-10-producing CD4+ Tr1 cells
In addition to the reported activity of OX40 on Foxp3+ CD4 Treg, OX40 also has been found to suppress the generation of CD4 cells producing IL-10 that do not express Foxp3 (Tr1-like). Stimulation of naïve and memory human CD4 T cells via OX40L interacting with OX40, under conditions that favor IL-10-producing cells (dexamethasone/vitamin D3, ICOS signaling), suppressed the development of this population of regulatory cells [47]. This implies that OX40 might antagonize the differentiation of several Treg subsets from naïve CD4 (or memory CD4) T cells in response to a variety of immunomodulatory stimuli, again reinforcing the concept that OX40-OX40L interactions strongly favor the development of protective or pathogenic effector T cells, minimizing potential limits on effector T cell reactivity by Treg. In further support of this, OX40 engagement was also found to decrease IL-10 production from already differentiated Tr1-like cells [47], implying that if IL-10 secretion is part of a suppressive mechanism, OX40 could prevent regulatory activity by targeting production or release of the active cytokine. In these studies, antagonism for Tr1 cell generation was specific for OX40 in that neither signals from 4-1BB nor GITR suppressed development of IL-10-producing cells from naïve CD4 T cells. This might suggest a unique function of OX40 in inhibiting the generation of IL-10-producing CD4 regulatory T cells, although further studies will be needed to confirm such a novel role. How OX40 might control IL-10 is not yet known.
7. CD8+ Treg and Novel Immune Regulation from 4-1BB
Although, as described above, 4-1BB signals might promote the proliferation or survival of Foxp3+ Treg as well as potentially inhibiting regulatory activity of these cells, other data have implied that 4-1BB might have additional and opposing activities. Targeting 4-1BB with agonist antibodies has enhanced the generation of antigen-specific effector T cells that can mediate anti-viral and anti-tumor immunity [2, 3], in line with a possible concomitant action in preventing the induction of Foxp3+ Treg or their regulatory activity. In contrast, the same agonistic 4-1BB antibody has been found to ameliorate many autoimmune and inflammatory diseases. Anti-4-1BB was shown to enhance the number of CD4+CD25+Foxp3+ Treg in NOD mice [48] and in mice undergoing colitis [49], both situations in which the antibody suppressed these diseases, correlating with a potential proliferation/survival activity on nTreg discussed before. However, it is likely that regulatory activity brought about by stimulating 4-1BB might not always be controlled by these mechanisms, and in some cases it has been suggested to be due to the development of novel regulatory CD8+ T cell populations that do not express Foxp3.
Anti-4-1BB was first shown to ameliorate both the incidence and severity of experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis (MS) [50], and to inhibit the relapse that occurs in EAE that is characteristic of the human disease. It was suggested that the antibody treatment inhibited antigen (MOG peptide) specific CD4 T cell responses by increasing activation-induced cell death (AICD). A similar action of anti-4-1BB was reported in mouse models of graft-versus-host disease (GVHD) in which treatment resulted in death of donor CD4 T cells [51, 52]. Whether any of these activities were due to a direct effect of signaling to the CD4 T cells is not known. Suppression of CD4 T cell reactivity by anti-4-1BB was also shown in mouse models of allergic asthma [53, 54] accompanied by prevention of the development of disease as well as reversal of established disease. The protective function in this case was associated with a marked reduction of Th2 cytokines but augmented IFN-γ. In one study, CD4 T cells from anti-4-1BB-treated mice did not proliferate to antigen in vitro unless IL-2 was provided, leading to the suggestion that targeting 4-1BB induced an anergic state in CD4 T cells [53].
It is possible however that all of these results are explained by a single common mechanism involving expansion of CD8 cells. In several cases, anti-4-1BB treatment has been found to specifically induce regulatory cells in the CD8 lineage. In one study, CD8 T cells generated by priming with antigen, anti-4-1BB, and TLR ligands were found to possess a type of Treg suppressive activity against other T cells in vitro [55, 56], a function dependent on IFN-γ priming and mediated through TGF-β. Agonistic anti-4-1BB also inhibited the development of collagen-induced arthritis (CIA) in mice, a model reminiscent of human rheumatoid arthritis [57]. Here, anti-4-1BB again suppressed antigen-specific CD4 T cell responses as well as the production of antibody to collagen, and data again supported a role for IFN-γ in this regulation, as well as indoleamine-2,3-dioxygenase (IDO), an enzyme that regulates tryptophan metabolism and suppresses T cell proliferation. In this case, it was noted that anti-4-1BB expanded a novel population of cells T cells expressing CD8 and CD11c and these cells were proposed to directly or indirectly mediate the suppression. Through secretion of IFN-γ this regulatory population was proposed to induce IDO production from monocytes and DC, ultimately leading to suppression of CD4 T cell proliferation. Interestingly, this anti-4-1BB-induced CD8+CD11c+ population was also found to arise in situations where anti-4-1BB proved efficacious in enhancing anti-tumor [58] and anti-viral [59] activity. This obviously raises the question of whether the CD8 T cells elicited by targeting 4-1BB in each individual situation were really different, or whether they were functionally identical CD8 populations that can be both regulatory or protective depending on diverse patho-physiological situations. The CD8 populations that were characterized to be regulatory/suppressive might have represented alternate subsets of cells, since one population expressed CD11c and did not produce TGF-β [57], whereas another did not express CD11c but did produce TGF-β [56]. Despite these apparent differences, both populations exerted suppressive activity that was dependent on IFN-γ, a feature that was shared with CD8 T cells elicited by anti-4-1BB that protected against tumor growth and viral replication.
A common trait in studies targeting 4-1BB with agonist antibody is the down-regulation of immunoglobulin responses and/or depletion of B cells. Anti-4-1BB ameliorated acute and established lupus-like symptoms in MRL/lpr [60] and NZB × NZW F1 [61] mice that spontaneously develop a disease resembling human systemic lupus erythematosus (SLE). In MRL/lpr mice, anti-4-1BB treatment resulted in a loss of auto- reactive B cells in the periphery that was dependent on IFN-γ. B cell loss was not seen in the NZB × NZW F1 strain, but, germinal center (GC) formation, which is central to the development of functional class-switched B cells, was abolished. In accordance with the latter, a recent report also showed that anti-4-1BB inhibited GC formation apparently by diminishing follicular dendritic cell (FDC) networks in B cell follicles [62]. Whether this activity was direct or indirect via another cell type is not clear, but it is possible that again suppression of B cell immunity was accompanied by enhanced CD8 responses. One possible mechanism for negative regulation of B cells is through down-regulation of CD4 responses that help B cell responses, however, it is more likely that a positive effect on augmenting a T cell population could explain most suppressive activities. Anti-4-1BB dramatically reduces the number of B cells in the spleen and bone marrow of lethally irradiated mice reconstituted with bone marrow, through a T cell and IFN-γ-dependent mechanism (S-W.L. unpublished). In line with the studies suggesting a regulatory role for 4-1BB-activated CD8 T cells, one can argue that 4-1BB signaling elicits strong T cell activation, particular in CD8 cells, leading to secretion of unusually high amounts of IFN-γ that block the development of B cells in the bone marrow, ultimately reducing the output of immature B cells into the periphery.
Further studies will be required to understand whether 4-1BB signaling can differentially induce alternate subsets of T cells, and if so what will dictate whether they become regulatory or non-regulatory, and in particular whether 4-1BB preferentially elicits CD8+ Treg. At present, no studies have described that OX40 might be able to promote or modulate CD8+ Treg.
8. Conclusions and Future Directions
Emerging evidence now suggests that costimulatory signals from multiple cell surface receptors will modulate the development and activity of regulatory T cells. OX40 and 4-1BB are two such molecules that have already been shown to have the potential to control Treg as well as control pathogenic or protective effector T cells (Fig. 2). How much of any immune response is ultimately dictated by a direct action on Treg as opposed to a direct action on developing effector T cells is not clear, but an argument can be made that all are linked with the final balance of activities being dictated by positive or negative signals that are delivered to both of these divergent T cell subsets. At present, OX40 and 4-1BB have several potential activities in controlling Treg that might or might not be common to both receptors, and include (i) suppressing the induction and/or maintenance of Foxp3 and IL-10 in CD4 T cells; (ii) promoting the proliferation and/or survival of CD4+Foxp3+ Treg subsets as well as CD8+Foxp3- Treg; and (iii) antagonizing the suppressive function of CD4+Foxp3+ Treg. How many of these activities are directly shared between OX40 and 4-1BB is not clear. Although both molecules are potentially similar in their signaling capabilities, and common activities and signaling cascades have been described between them, it is clear from in vivo experiments that their overall role in some immune responses is not the same. Ultimately, this might be dictated by their expression characteristics, when and for how long they are expressed, and on which cell types, and furthermore the availability of their ligands. In particular, dramatic suppressive activities have been shown with administration of agonist anti-4-1BB in vivo, which appear in part to be explained by enhancing the development or outgrowth of CD8+ Treg or some other regulatory mechanism that does not involve a particular Treg subset. Parallel activities have not been seen with agonist anti-OX40, although in reality many of the same studies have not been carried out targeting both molecules. However, this divergent suppressive mechanism may simply be explained by enhanced or more persistent expression of 4-1BB on CD8 T cells compared to OX40, or alternatively an unknown signaling capacity not shared between the two molecules.
Figure 2.
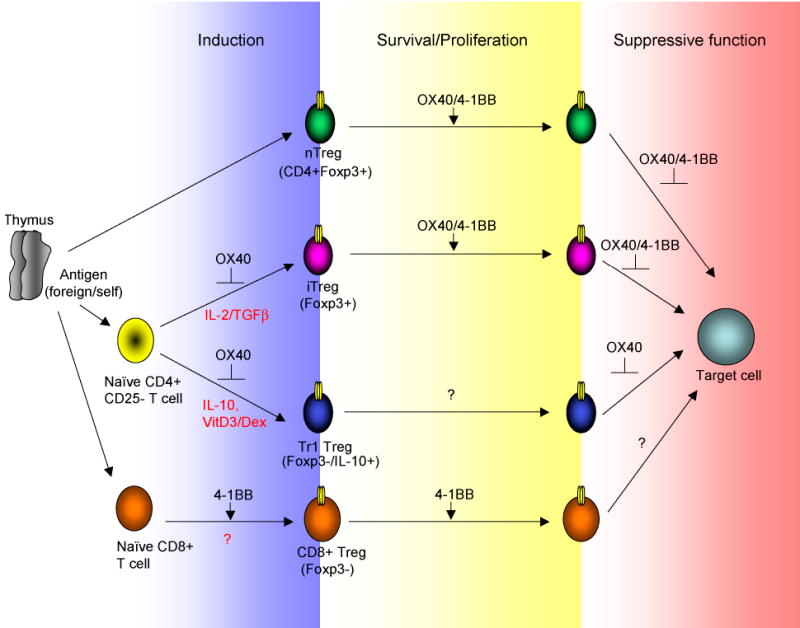
OX40 and 4-1BB May Exert Multiple Activities in Controlling Treg Generation or Function. OX40 and 4-1BB potentially can modulate Treg activity at several stages. OX40 inhibits development of Foxp3+ iTreg and Foxp3- IL-10-producing Tr1 Treg that are generated from naïve CD4 T cells. Whether 4-1BB possesses this activity is not known. Both OX40 and 4-1BB can promote proliferation and/or survival of already existing Foxp3+ iTreg and nTreg, and also can inhibit the regulatory/suppressive activity of these cells. Whether promotion of proliferation/survival are directly linked to loss of suppressive capacity is not known. OX40 can also downregulate IL-10 secretion, likely linked to loss of suppressive activity by Tr1 Treg. 4-1BB on the other hand can promote the development or expansion of CD8+ Treg that are Foxp3- and may in some cases express CD11c. It is not known if OX40 has the latter capacity.
What is important in terms of Treg biology is how signals from costimulatory molecules control Foxp3 expression, and then proliferation, survival, and regulatory function of these Foxp3+ Treg as well as other subsets of Treg, and what are the nature of those signals. Once the latter are known, this might explain overlapping or divergent activity of a number of receptors. Of critical importance is also how Treg suppression is mediated and then what are the signals that control this activity. Both OX40 and 4-1BB appear to share the ability to downregulate suppression mediated by CD4+Foxp3+ Treg but it is not clear at what level this may operate. Several potential mechanisms for neutralizing regulatory T cell activity might be suggested based on the literature, such as decreasing the production of suppressive cytokines, inhibiting cell-to-cell contact events that could engage co-inhibitory receptors, preventing IDO and cAMP production, and antagonizing the ability to make or release target cell killing enzymes, and so on. Without first understanding how various subsets of Treg suppress, it will not be possible to fully understand how OX40 or 4-1BB engagement controls such differentiated Treg. Further issues that need to be resolved include whether there are fundamental differences between Foxp3+ nTreg and iTreg, including questions regarding the stability of Foxp3, the regulatory elements for each cell, and also whether the latter are absolutely linked to Foxp3 expression. The majority of data to date have been directed to nTreg, but emerging evidence suggests that regulation of the induction of Foxp3 and generation and activity of inducible antigen-reactive iTreg might represent an area for crucial control of many inflammatory diseases by molecules such as OX40 and 4-1BB. As more studies are carried out on these latter cells, and further evidence is provided regarding the importance of the Treg to effector T cell balance in disease situations, it will be increasingly important to understand the Treg phenotypes, the receptors that are constitutive or inducible on their surface, and how ligation of these receptors alters Treg persistence and function. OX40 and 4-1BB are likely to be central to some of these issues, although their integration and co-operation or antagonism with other membrane receptors is likely to be just as crucial to overall Treg activity.
Acknowledgments
M.C. is supported by NIH grants AI49453, AI42944, CA91837, and AI070535.
abbreviations used
- Treg
regulatory T cells
- TRAF
TNFR associated factor
- PKB
protein kinase B
- Foxp3
forkhead box P3
- TGF
transforming growth factor
- IDO
indoleamine-2,3-dioxygenase
Biographies

Michael Croft is a Professor and Member in the Molecular Immunology Division of the La Jolla Institute for Allergy and Immunology. Dr. Croft received his PhD in Immunology from Sussex University in the United Kingdom, and completed postdoctoral training in the Biology Department of the University of California San Diego. The focus of research in Dr. Croft’s laboratory is the cellular regulation of T cell immunity and tolerance, and how membrane bound costimulatory molecules, particularly in the TNFR-TNF superfamily, control T cell function. Dr. Croft has been the recipient of many research awards from the National Institutes of Health, and has served as a member of several National Institutes of Health grant review study sections, as well as being an Associate Editor for the Journal of Immunology.

Dr. Takanori So is a biochemist, graduated from Faculty of Pharmaceutical Sciences, Kyushu University, Japan in 1993. He obtained his Ph.D. degree in Pharmaceutical Sciences at the same institution in 1999. Between 1997 and 2003, he held a position as assistant professor at the same institution. His work in Japan was conducted basic immunological studies for regulation of protein immunogenicity and tolerogenicity by introducing amino acid mutations and chemical modifications into protein antigens, which is important for development of protein pharmaceutical agents. In 2002, he moved to the La Jolla Institute for Allergy and Immunology as a postdoctoral fellow. In Dr. Michael Croft’s Lab, his research interest relates to T cell activation/tolerance and the lineage commitment mechanism of peripheral CD4+ T cells. His current research focuses on identification of downstream signaling targets of OX40 which are critical for T helper cell differentiation and reversal of immunological tolerance state.

Dr. Seung-Woo Lee is an immunologist, graduated from department of Life Science, Pohang University of Science and Technology, Korea in 1994. He obtained his Ph.D. degree in Viral Immunology at the same institution in 2000. His work in Korea focused on vaccine development with DNA immunization against Hepatitis C Virus and basic immunological studies about CpG DNA and cytokine IL-12. In 2003, he joined Dr. Michael Croft Lab in La Jolla Institute for Allergy and Immunology as a postdoctoral fellow and has studied the role of 4-1BB, a member of TNFR superfamily, in the biology of many types of immune cells. His current research focuses on the role of 4-1BB/4-1BBL in hematopoiesis, T cell activation/tolerance, and the generation of autoimmunity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Croft M. Costimulation of T cells by OX40, 4-1BB, and CD27. Cytokine Growth Factor Rev. 2003;14:265–73. doi: 10.1016/s1359-6101(03)00025-x. [DOI] [PubMed] [Google Scholar]
- 2.Croft M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nature reviews. 2003;3:609–20. doi: 10.1038/nri1148. [DOI] [PubMed] [Google Scholar]
- 3.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 4.Vinay DS, Kwon BS. Immunotherapy targeting 4-1BB and its ligand. International journal of hematology. 2006;83:23–8. doi: 10.1532/IJH97.05125. [DOI] [PubMed] [Google Scholar]
- 5.Vinay DS, Cha K, Kwon BS. Dual immunoregulatory pathways of 4-1BB signaling. J Mol Med. 2006;84:726–36. doi: 10.1007/s00109-006-0072-2. [DOI] [PubMed] [Google Scholar]
- 6.Salek-Ardakani S, Song A, Humphreys IR, Croft M. OX40:OX40L Axis: Emerging targets for immunotherapy of human disease. Curr Immunol Rev. 2006;2:37–53. [Google Scholar]
- 7.Lee SW, Croft M. 4-1BB as a therapeutic target for human disease. Therapeutic Targets of the TNFR Superfamily. 2007 doi: 10.1007/978-0-387-89520-8_8. Landes bioscience: http://eurekah.com/chapter/3496. [DOI] [PubMed]
- 8.Shevach EM. From vanilla to 28 flavors: multiple varieties of T regulatory cells. Immunity. 2006;25:195–201. doi: 10.1016/j.immuni.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 9.Campbell DJ, Ziegler SF. FOXP3 modifies the phenotypic and functional properties of regulatory T cells. Nature reviews. 2007;7:305–10. doi: 10.1038/nri2061. [DOI] [PubMed] [Google Scholar]
- 10.Koonpaew S, Shen S, Flowers L, Zhang W. LAT-mediated signaling in CD4+CD25+ regulatory T cell development. J Exp Med. 2006;203:119–29. doi: 10.1084/jem.20050903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Apostolou I, von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J Exp Med. 2004;199:1401–8. doi: 10.1084/jem.20040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nature immunology. 2005;6:1219–27. doi: 10.1038/ni1265. [DOI] [PubMed] [Google Scholar]
- 13.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–40. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 14.Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nature immunology. 2005;6:152–62. doi: 10.1038/ni1160. [DOI] [PubMed] [Google Scholar]
- 15.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nature immunology. 2005;6:1142–51. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 16.Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178:280–90. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- 17.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–7. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25:441–54. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 19.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–71. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 20.Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–85. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–64. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vieira PL, Christensen JR, Minaee S, O’Neill EJ, Barrat FJ, Boonstra A, et al. IL-10-secreting regulatory T cells do not express Foxp3 but have comparable regulatory function to naturally occurring CD4+CD25+ regulatory T cells. J Immunol. 2004;172:5986–93. doi: 10.4049/jimmunol.172.10.5986. [DOI] [PubMed] [Google Scholar]
- 23.Barrat FJ, Cua DJ, Boonstra A, Richards DF, Crain C, Savelkoul HF, et al. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med. 2002;195:603–16. doi: 10.1084/jem.20011629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY, et al. Regulatory T cells expressing interleukin 10 develop from Foxp3(+) and Foxp3(-) precursor cells in the absence of interleukin 10. Nature immunology. 2007;8:931–41. doi: 10.1038/ni1504. [DOI] [PubMed] [Google Scholar]
- 25.Faunce DE, Terajewicz A, Stein-Streilein J. Cutting edge: in vitro-generated tolerogenic APC induce CD8+ T regulatory cells that can suppress ongoing experimental autoimmune encephalomyelitis. J Immunol. 2004;172:1991–5. doi: 10.4049/jimmunol.172.4.1991. [DOI] [PubMed] [Google Scholar]
- 26.Kapp JA, Honjo K, Kapp LM, Xu X, Cozier A, Bucy RP. TCR transgenic CD8+ T cells activated in the presence of TGFbeta express FoxP3 and mediate linked suppression of primary immune responses and cardiac allograft rejection. International immunology. 2006;18:1549–62. doi: 10.1093/intimm/dxl088. [DOI] [PubMed] [Google Scholar]
- 27.Takeda I, Ine S, Killeen N, Ndhlovu LC, Murata K, Satomi S, et al. Distinct roles for the OX40-OX40 ligand interaction in regulatory and nonregulatory T cells. J Immunol. 2004;172:3580–9. doi: 10.4049/jimmunol.172.6.3580. [DOI] [PubMed] [Google Scholar]
- 28.Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, et al. OX40 costimulation turns off Foxp3+ Tregs. Blood. 2007;110:2501–10. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwon BS, Hurtado JC, Lee ZH, Kwack KB, Seo SK, Choi BK, et al. Immune responses in 4-1BB (CD137)-deficient mice. J Immunol. 2002;168:5483–90. doi: 10.4049/jimmunol.168.11.5483. [DOI] [PubMed] [Google Scholar]
- 30.Choi BK, Bae JS, Choi EM, Kang WJ, Sakaguchi S, Vinay DS, et al. 4-1BB-dependent inhibition of immunosuppression by activated CD4+CD25+ T cells. J Leukoc Biol. 2004;75:785–91. doi: 10.1189/jlb.1003491. [DOI] [PubMed] [Google Scholar]
- 31.So T, Croft M. Cutting edge: OX40 inhibits TGF-beta- and antigen-driven conversion of naive CD4 T cells into CD25+Foxp3+ T cells. J Immunol. 2007;179:1427–30. doi: 10.4049/jimmunol.179.3.1427. [DOI] [PubMed] [Google Scholar]
- 32.Fu S, Zhang N, Yopp AC, Chen D, Mao M, Chen D, et al. TGF-beta induces Foxp3 + T-regulatory cells from CD4 + CD25 - precursors. Am J Transplant. 2004;4:1614–27. doi: 10.1111/j.1600-6143.2004.00566.x. [DOI] [PubMed] [Google Scholar]
- 33.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204:1765–74. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song J, Salek-Ardakani S, Rogers PR, Cheng M, Van Parijs L, Croft M. The costimulation-regulated duration of PKB activation controls T cell longevity. Nature immunology. 2004;5:150–8. doi: 10.1038/ni1030. [DOI] [PubMed] [Google Scholar]
- 35.Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Remy I, Montmarquette A, Michnick SW. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat Cell Biol. 2004;6:358–65. doi: 10.1038/ncb1113. [DOI] [PubMed] [Google Scholar]
- 37.Kim HP, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med. 2007;204:1543–51. doi: 10.1084/jem.20070109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.So T, Song J, Sugie K, Altman A, Croft M. Signals from OX40 regulate nuclear factor of activated T cells c1 and T cell helper 2 lineage commitment. Proc Natl Acad Sci U S A. 2006;103:3740–5. doi: 10.1073/pnas.0600205103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei J, Duramad O, Perng OA, Reiner SL, Liu YJ, Qin FX. Antagonistic nature of T helper 1/2 developmental programs in opposing peripheral induction of Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2007;104:18169–74. doi: 10.1073/pnas.0703642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng G, Wang B, Chen A. The 4-1BB costimulation augments the proliferation of CD4+CD25+ regulatory T cells. J Immunol. 2004;173:2428–34. doi: 10.4049/jimmunol.173.4.2428. [DOI] [PubMed] [Google Scholar]
- 41.Elpek KG, Yolcu ES, Franke DD, Lacelle C, Schabowsky RH, Shirwan H. Ex Vivo Expansion of CD4+CD25+FoxP3+ T Regulatory Cells Based on Synergy between IL-2 and 4-1BB Signaling. J Immunol. 2007;179:7295–304. doi: 10.4049/jimmunol.179.11.7295. [DOI] [PubMed] [Google Scholar]
- 42.Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood. 2005;105:2845–51. doi: 10.1182/blood-2004-07-2959. [DOI] [PubMed] [Google Scholar]
- 43.Crellin NK, Garcia RV, Levings MK. Altered activation of AKT is required for the suppressive function of human CD4+CD25+ T regulatory cells. Blood. 2007;109:2014–22. doi: 10.1182/blood-2006-07-035279. [DOI] [PubMed] [Google Scholar]
- 44.Starck L, Scholz C, Dorken B, Daniel PT. Costimulation by CD137/4-1BB inhibits T cell apoptosis and induces Bcl-xL and c-FLIP(short) via phosphatidylinositol 3-kinase and AKT/protein kinase. B Eur J Immunol. 2005;35:1257–66. doi: 10.1002/eji.200425686. [DOI] [PubMed] [Google Scholar]
- 45.Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445:931–5. doi: 10.1038/nature05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445:936–40. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
- 47.Ito T, Wang YH, Duramad O, Hanabuchi S, Perng OA, Gilliet M, et al. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc Natl Acad Sci U S A. 2006;103:13138–43. doi: 10.1073/pnas.0603107103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Irie J, Wu Y, Kachapati K, Mittler RS, Ridgway WM. Modulating protective and pathogenic CD4+ subsets via CD137 in type 1 diabetes. Diabetes. 2007;56:186–96. doi: 10.2337/db06-0793. [DOI] [PubMed] [Google Scholar]
- 49.Lee J, Lee EN, Kim EY, Park HJ, Chang CY, Jung DY, et al. Administration of agonistic anti-4-1BB monoclonal antibody leads to the amelioration of inflammatory bowel disease. Immunol Lett. 2005;101:210–6. doi: 10.1016/j.imlet.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 50.Sun Y, Lin X, Chen HM, Wu Q, Subudhi SK, Chen L, et al. Administration of agonistic anti-4-1BB monoclonal antibody leads to the amelioration of experimental autoimmune encephalomyelitis. J Immunol. 2002;168:1457–65. doi: 10.4049/jimmunol.168.3.1457. [DOI] [PubMed] [Google Scholar]
- 51.Kim J, Kim HJ, Park K, Kim J, Choi HJ, Yagita H, et al. Costimulatory molecule-targeted immunotherapy of cutaneous graft-versus-host disease. Blood. 2007;110:776–82. doi: 10.1182/blood-2006-08-043612. [DOI] [PubMed] [Google Scholar]
- 52.Kim J, Choi WS, La S, Suh JH, Kim BS, Cho HR, et al. Stimulation with 4-1BB (CD137) inhibits chronic graft-versus-host disease by inducing activation-induced cell death of donor CD4+ T cells. Blood. 2005;105:2206–13. doi: 10.1182/blood-2004-06-2080. [DOI] [PubMed] [Google Scholar]
- 53.Polte T, Foell J, Werner C, Hoymann HG, Braun A, Burdach S, et al. CD137- mediated immunotherapy for allergic asthma. J Clin Invest. 2006;116:1025–36. doi: 10.1172/JCI23792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun Y, Blink SE, Liu W, Lee Y, Chen B, Solway J, et al. Inhibition of Th2-mediated allergic airway inflammatory disease by CD137 costimulation. J Immunol. 2006;177:814–21. doi: 10.4049/jimmunol.177.2.814. [DOI] [PubMed] [Google Scholar]
- 55.Myers L, Takahashi C, Mittler RS, Rossi RJ, Vella AT. Effector CD8 T cells possess suppressor function after 4-1BB and Toll-like receptor triggering. Proc Natl Acad Sci U S A. 2003;100:5348–53. doi: 10.1073/pnas.0837611100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Myers L, Croft M, Kwon BS, Mittler RS, Vella AT. Peptide-specific CD8 T regulatory cells use IFN-gamma to elaborate TGF-beta-based suppression. J Immunol. 2005;174:7625–32. doi: 10.4049/jimmunol.174.12.7625. [DOI] [PubMed] [Google Scholar]
- 57.Seo SK, Choi JH, Kim YH, Kang WJ, Park HY, Suh JH, et al. 4-1BB-mediated immunotherapy of rheumatoid arthritis. Nat Med. 2004;10:1088–94. doi: 10.1038/nm1107. [DOI] [PubMed] [Google Scholar]
- 58.Ju SA, Park SM, Lee SC, Kwon BS, Kim BS. Marked expansion of CD11c+CD8+ T-cells in melanoma-bearing mice induced by anti-4-1BB monoclonal antibody. Mol Cells. 2007;24:132–8. [PubMed] [Google Scholar]
- 59.Kim YH, Seo SK, Choi BK, Kang WJ, Kim CH, Lee SK, et al. 4-1BB costimulation enhances HSV-1-specific CD8+ T cell responses by the induction of CD11c+CD8+ T cells. Cell Immunol. 2005;238:76–86. doi: 10.1016/j.cellimm.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 60.Sun Y, Chen HM, Subudhi SK, Chen J, Koka R, Chen L, et al. Costimulatory molecule-targeted antibody therapy of a spontaneous autoimmune disease. Nat Med. 2002;8:1405–13. doi: 10.1038/nm1202-796. [DOI] [PubMed] [Google Scholar]
- 61.Foell J, Strahotin S, O’Neil SP, McCausland MM, Suwyn C, Haber M, et al. CD137 costimulatory T cell receptor engagement reverses acute disease in lupus-prone NZB × NZW F1 mice. J Clin Invest. 2003;111:1505–18. doi: 10.1172/JCI17662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun Y, Blink SE, Chen JH, Fu YX. Regulation of follicular dendritic cell networks by activated T cells: the role of CD137 signaling. J Immunol. 2005;175:884–90. doi: 10.4049/jimmunol.175.2.884. [DOI] [PubMed] [Google Scholar]