

Abstract
Pituitary gonadotropins follicle-stimulating hormone (FSH) and luteinizing hormone stimulate the gonads by regulating germ cell proliferation and differentiation. FSH receptors (FSH-Rs) are localized to testicular Sertoli cells and ovarian granulosa cells and are coupled to activation of the adenylyl cyclase and other signaling pathways. Activation of FSH-Rs is considered essential for folliculogenesis in the female and spermatogenesis in the male. We have generated mice lacking FSH-R by homologous recombination. FSH-R-deficient males are fertile but display small testes and partial spermatogenic failure. Thus, although FSH signaling is not essential for initiating spermatogenesis, it appears to be required for adequate viability and motility of the sperms. FSH-R-deficient females display thin uteri and small ovaries and are sterile because of a block in folliculogenesis before antral follicle formation. Although the expression of marker genes is only moderately altered in FSH-R −/− mice, drastic sex-specific changes are observed in the levels of various hormones. The anterior lobe of the pituitary gland in females is enlarged and reveals a larger number of FSH- and thyroid-stimulating hormone (TSH)-positive cells. The phenotype of FSH-R −/− mice is reminiscent of human hypergonadotropic ovarian dysgenesis and infertility.
The pituitary glycoprotein hormone follicle-stimulating hormone (FSH) plays an essential role in mammalian reproduction (1) through interaction with a specific receptor (2). The FSH receptor (FSH-R) is a G-protein-coupled, seven-transmembrane receptor linked to the adenylyl cyclase or other pathways (3–5). FSH-R activation initiates a cascade of intracellular events leading to modification of cell response (6–8) and receptor desensitization (3).
In the male, the FSH-R is expressed exclusively in Sertoli cells (9, 10). FSH signaling is considered essential for pubertal initiation of spermatogenesis and maintenance of normal sperm production in the adult (3). This function is thought to be exerted via intimate contacts between Sertoli cells and differentiating spermatogonia (11). Upon FSH and testosterone stimulation, Sertoli cells provide a critical support required for germ cell differentiation (12).
In the female, the FSH-R is expressed in granulosa cells and is thought to tightly regulate the various phases of follicle maturation in response to periodic pituitary FSH release (3, 10, 13). During the reproductive life only a fraction of follicles undergo differentiation, whereas more than 99% enter a degenerative process called atresia. The onset of FSH stimulation at puberty reduces apoptosis, enhances proliferation, and induces follicular maturation leading to ovulation (3, 13).
The critical role played by FSH signaling is illustrated by the phenotype of mice carrying a targeted mutation in the FSHβ subunit gene (14) and by the effect of FSH-R mutations in humans (15, 16). An inactivating mutation (Ala-189–Val) found in females with pure ovarian dysgenesis leads to a disease characterized by normal karyotype, high gonadotropins, and streaky gonads associated with primary amenorrhea (13, 17). This mutation lies in the extracellular domain and is thought to modify protein folding (17). Importantly, males with the same mutation display various degrees of spermatogenic failure, lack of azoospermia, or absolute infertility (18).
Thus, the same inactivating mutation differentially influences reproduction in males or females. One activating mutation has been found in a male hypophysectomized patient who, under testosterone therapy, was unexpectedly fertile in spite of undetectable gonadotropin levels (16). This heterozygous Asp-567–Gly substitution in the third intracytoplasmic loop leads to constitutively increased cAMP levels independently of FSH stimulation (16).
We have generated mutant mice for the FSH-R by homologous recombination. The conserved organization of the human, mouse, and rat FSH-R genes has been established (19–22). Mutant males display small testes, partial spermatogenic failure, and reduced fertility. FSH signaling is not essential for initiating spermatogenesis, but is required to sustain adequate viability and motility of the sperms. The phenotype of mutant females is much more severe. They display thin uteri and small ovaries and are sterile because of a block in folliculogenesis before antral follicle formation. Drastic changes in hormone levels prompted us to analyze the pituitary anatomy. There is a moderate, but significant, enlargement in the anterior lobe accompanied by a drastic increase of FSH-positive cells. These animals constitute a model to study the physiological link between gonads and pituitary, hypergonadotropic ovarian dysgenesis, and infertility.
MATERIALS AND METHODS
Generation of FSH-R −/− Mice.
An 11-kb genomic fragment covering exon I and a large part of intron I of the FSH-R gene was isolated from a 129SV phage library. A 7-kb XbaI–SacI fragment was subcloned for subsequent insertions. A EcoRV fragment (−420 to +228) was replaced by a PGK-Neo cassette with an internal EcoRI site. A GTI-GTII promoter-driven herpes simplex virus thymidine kinase cassette was inserted at the 5′ SalI site. Embryonic stem cells (107) were electroporated with targeting plasmid at 400 V and 125 μF. Two mutant clones were injected in C57BL/6 blastocysts. Ten male chimeras were bred with C57BL/6 females, and germ-line transmission was obtained with three of them.
Histological Analysis.
Sections fixed in Bouin solution were prepared and stained (hematoxylin/eosin) by using standard procedures. Analysis of pituitary sections was performed as described (23). For the analysis of the estrous cycle vaginal smears were stained with hematoxylin/eosin as described (23).
FSH Binding and Hormone Assays.
Total homogenates and crude membranes from testes were assayed for FSH binding. Aliquots of 400 μg were incubated at 23°C with 5 ng/ml of 125I-FSH (NEN/DuPont) in 50 mM Tris⋅HCl, 0.5% BSA. Nonspecific binding was assessed in the presence of 50 μg/ml of cold ovine FSH (Sigma). Bound hormone was separated by centrifugation at 3,000 × g.
Serum FSH, luteinizing hormone (LH), inhibin, and testosterone levels were determined by RIA using Amersham and Peninsula assay systems according to the manufacturers’ procedures. Intracellular testosterone levels were measured in testis homogenates after ether extraction.
RNA and Protein Analyses.
RNA preparation, Northern, reverse transcription–PCR, in situ, and RNase-protection analyses were according to standard methods (24). Poly(A)+ RNA was prepared by using the Pharmacia purification kit. Equal loading and RNA quality were confirmed with a G6PD probe.
Tissues were homogenized in Laemmli buffer. For Western analysis the following antibodies were as indicated by the manufacturer: a polyclonal antiprolactin (National Institutes of Health); a mouse monoclonal antiprolactin receptor (Interchim, Montluçon, France); a rat monoclonal anticyclin D2 (Santa Cruz Biotechnology); and a rat polyclonal raised against the regulatory subunit RIIβ of the cAMP-dependent protein kinase (gift of K. Tasken, University of Olso, Norway).
RESULTS
Homologous Recombination of the Mouse FSH-R Gene.
The single gene encoding the FSH-R generates a variety of alternatively spliced forms displaying different structural motifs (3, 20). We constructed a targeting vector deleting all FSH-R isoform transcripts (Fig. 1 A and B). Testis and ovaries RNA analysis from FSH-R +/− and FSH-R −/− mutant mice showed reduced or no expression, respectively (Fig. 1 C and D). Ligand binding assays with 125I-FSH demonstrate the lack of functional FSH-R in membranes from testis of mutant mice (Fig. 1E). Mutant mice were macroscopically indistinguishable from wild-type littermates. No changes in body weight and growth or apparent differences in the structure of internal organs were detected. Heterozygous males and females were fertile and viable, although with reduced fertility, as the litter size was reduced by 35–50% as compared with wild-type mice.
Figure 1.

Generation of FSH-R gene-deficient mice. (A) Strategy for generating the FSH-R null mutation. The EcoRV fragment containing exon 1 of the FSH-R gene was replaced by a PGK-Neo cassette. (B) DNA extracted from tail biopsies of wild-type (+/+), heterozygous (+/−), and homozygous (−/−) mice were digested with EcoRI; hybridization with probe 1 yielded 11-kb (wild type) and 9.5-kb (mutant) bands. (C) Northern analysis of poly(A)+ RNA extracted from mouse testis. (D) Reverse transcription–PCR from testis and ovary total RNA; primers used were selected within the sequence of exon 2 (sense primer: 5′-GTGCTCACCAAGCTTCGA-3′) and exon 7 (antisense primer: 5′-GAATCCCATTCTTATTCAGC-3′); all isoforms of the FSH-R are apparent. (E) FSH binding to crude membrane preparation obtained from testes of wild-type, heterozygous, and homozygous mice.
FSH-R Mutant Males Are Fertile, But Display Partial Spermatogenic Failure.
Beginning at 6–7 weeks FSH-R −/− males display reduced fertility and a drastic decrease in testis size (Fig. 2A). Accessory glands (seminal vesicles, prostate, and epididymis) appear normal. Sperm analysis indicates a significant decrease in cell number and motility and a percent increase of aberrant spermatozoa (Table 1). Affected sperm had abnormal and bent tails with cytoplasmic droplets. As FSH signaling influences germ cell function via the Sertoli cells (11), we performed histological analysis of the seminiferous epithelium. The decreased testes size is paralleled by a 20–30% volume reduction of the seminiferous tubuli, which revealed a decreased thickness of the epithelium and a smaller lumen (Fig. 2 C–E). The spermatogenic wave and the respective cellular associations appeared normal, as the shape and the numbers of somatic Sertoli and Leydig cells. The FSH-R −/− tubules had an unusual consistency, being transparent and sticky. To understand this altered consistency, we analyzed the tunica propria by electron microscopy. The epithelium anchors on a basement membrane adjacent to which type I collagen fibrils can be seen (11). Mutant mice testes show an altered organization of the collagen fibrils (Fig. 2 G and H), explaining the modified texture of the tubules. Thus, lack of FSH-R may influence the structural and functional organization of the basement membrane, leading to disarray in spermatogenesis.
Figure 2.
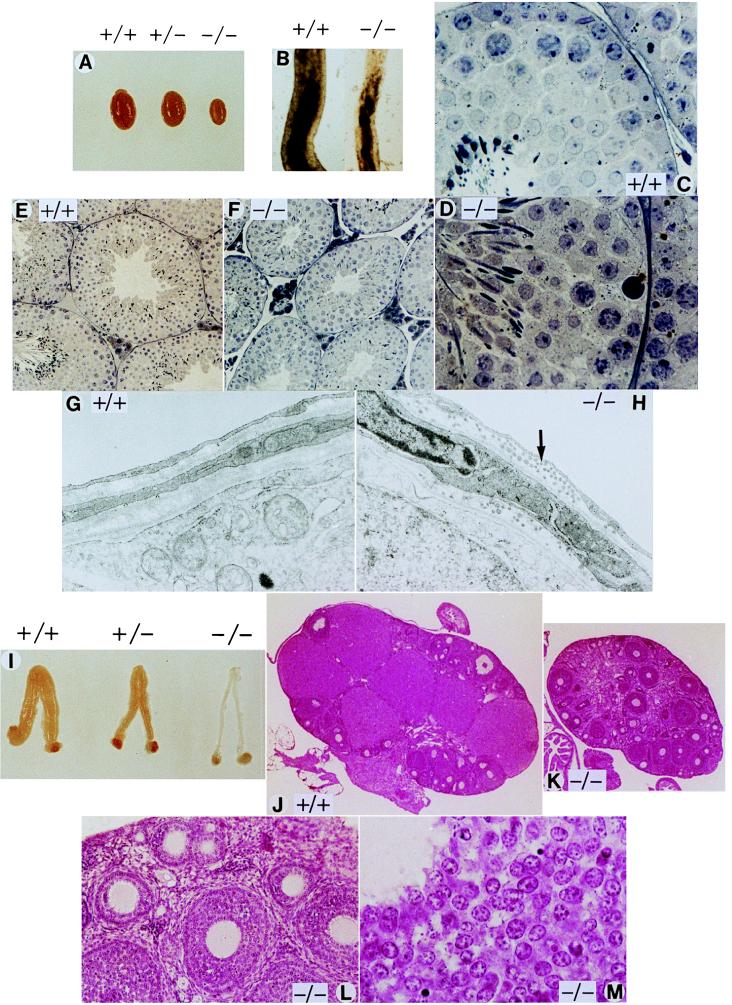
Analysis of testis and ovary from FSH-R mutant mice. (A) Testes comparison from 8-week-old wild-type, heterozygous, and homozygous littermates. (B) Transilluminated seminiferous tubules (stage VII–VIII of the seminiferous epithelium cycle) dissected from wild-type and homozygous mice; note the smaller tubule diameter in the mutant as compared with the wild type. Mutant mice conserve a normal spermatogenic wave. Histological sections at lower (E) and higher (C) magnification of seminiferous tubuli from a wild-type and mutant (D and F) mouse. (G and H) Electron micrographs of the basal membrane; note the altered organization of the collagen fibrils in mutant mouse tubules (arrow). (I) Reproductive tracts from 8-week-old littermates; ovaries and uteri from FSH-R mutant mice are smaller and thinner compared with the wild type; a reduction in size is also evident in the heterozygous. (J–M) Histological sections of the ovary from wild-type (J) and homozygous (K) mice. The decreased ovary size in the FSH-R −/− females is evident. There are no Graafian follicles nor corpora lutea. (L and M) Higher magnification of detailed structures in the FSH-R −/− ovary. Primary and secondary follicles appear normal (L), as well as the granulosa cells composition and organization (M). Magnifications: C and D ×370; E and F ×111; G and H ×18,000; J and K ×10; L ×96; M ×370.
Table 1.
Sperm count and motility
| +/+ | −/− | |
|---|---|---|
| Spermatozoa/mouse | 5.6 × 106 | 3.6 × 106 |
| Motility, % moving spermatozoa | 62 | 47 |
| Aberrant structure, % | 18 | 47 |
Sperm cells were collected from the epididymis and counted with an hemocytometer. Aberrant sperm cells had abnormal and bent tails with cytoplasmic droplets. Sperm motility was assessed by using a microscope connected to a video camera.
FSH-R −/− Females Are Sterile.
In contrast to the males, the mutant females are infertile. Ovaries and uteri from mutant mice appeared strikingly smaller and thinner as compared with wild-type littermates (Fig. 2I). FSH-R +/− females displayed an intermediate phenotype. In contrast to the normal stages of folliculogenesis and the presence of corpora lutea, ovaries from FSH-R −/− females lacked corpora lutea and mature Graafian follicles (Fig. 2 J–M). Mutant mice demonstrate only primordial, primary, and secondary follicles and an apparently normal number and location of granulosa and thecal cells at these stages. Thus, the decreased size of the ovary in the FSH-R −/− females is caused by the lack of large Graafian follicles and by the absence of corpora lutea caused by ovulatory failure. Hence, the infertility of mutant females arises from a block in follicular maturation.
Effects on Gene Expression.
The coordinated expression of several genes at defined stages of sperm and follicular development has been described (13, 25). Surprisingly, the expression of most of these genes is only marginally altered in both male and female mutants as compared with wild-type littermates (Fig. 3). Relevant examples are represented by cAMP-responsive element modulator (CREM) in males and cyclin D2 in females. CREM expression is under FSH regulation during spermatogenesis (26), while targeted disruption of the gene blocks spermiogenesis and increases germ cells apoptosis (24). Cyclin D2 expression is FSH responsive, and mutation of the gene blocks granulosa cell proliferation, causing sterility (27). The reasons for the unaltered expression of these genes in the FSH-R mutants are unclear. It is plausible that alternative signaling routes exist that could insure the function of some key genes as a compensatory mechanism.
Figure 3.

Analysis of gene expression. Gonadal-specific gene expression evaluated by reverse transcription–PCR using 1-μg total RNA aliquots from 8-week-old wild-type, heterozygous, and homozygous mice. The identity of the amplified fragments was confirmed by Southern analysis hybridizing with internal probes.
Drastic Alterations in Hormonal Levels.
Regulation of the hypothalamic-pituitary axis is controlled by a feedback loop system operating from the gonads to the pituitary (3). To study how this control may be altered in the absence of FSH signaling, we measured FSH and LH levels in males and females. FSH levels increased by 3-fold in FSH-R −/− adult males, whereas there is a drastic increase of at least 15-fold in mutant females. Interestingly, although heterozygous males show an increase with respect to wild-type controls, this increase is not observed in the females (Fig. 4A). LH and LH-receptor levels appeared unchanged in male and female mutants (not shown). The robust change in FSH levels suggested that the feedback control exerted by inhibin might be altered. Regulated α-inhibin is responsible for gonadal cell proliferation and differentiation (27). Although α-inhibin rates drop to about 50% of the wild type in the mutant males, no significant changes are detected in the females (Fig. 4B). As the most important increase in FSH levels is observed in mutant females, it appears that inhibin production from other sites is possible in the absence of FSH signaling and that inhibin-mediated retroinhibition is not essential for FSH-modulated synthesis.
Figure 4.

Alteration in hormone levels. Serum from 8-week-old wild-type, heterozygous, and homozygous littermates was assayed for FSH, testosterone, and inhibin by RIA. Intracellular testosterone changes paralleled the variations in serum (not shown). (B) RNase protection assay of α-inhibin is shown.
Testosterone levels in mutant males decrease to about 35% of the wild type, with intermediate values in the heterozygous (Fig. 4C). Comparative analyses of the estrous cycle of normal and mutant mice (ref. 23; not shown) showed that a rhythmic periodicity with a recognizable estrous every 4 days is present in both populations.
Analysis of the Pituitary Phenotype.
The dramatic increase of FSH levels observed in the female mutants and the direct link between gonads and pituitary function prompted us to study the pituitary gland anatomy of the FSH-R mutants. We analyzed pituitary glands from mice of both sexes and of various ages (1 week to 1 year). No anatomical abnormalities are present in the males. On the contrary, in all adult females starting from 4 months of age there is a moderate, but significant, enlargement of the anterior lobe (Fig. 5). Histological analysis (23) revealed a reproducible 20% increase in the size of the anterior lobe in the mutants, whereas the intermediate and posterior lobes were unaltered. This enlargement appears related to an increase in the number of cells and not of their size.
Figure 5.

Analysis of pituitary phenotype. Pituitary glands from 7-month-old wild-type and FSH-R mutant mice were fixed in OCT compound, and 10-μm cryostatic sections were prepared. The expression of the indicated gene was analyzed. Note the increased signal for FSH and TSH in mutant mice. GH, growth hormone; PRL, prolactin; POMC, proopiomelanocortin.
Expression analysis by in situ hybridization of pituitary hormones genes reveal a drastic increase in FSH-positive cells in the mutants and a moderate increase in thyroid-stimulating hormone (TSH)-positive cells (Fig. 5). Expression levels and distribution of other hormones appear normal. Analysis of other genes involved in pituitary physiology revealed no major differences (Fig. 6).
Figure 6.

Expression analysis in the pituitary. Pituitary RNA (10 μg) was analyzed by Northern with GSUα and prolactin probes (23). Pituitary extracts were analyzed with a prolactin antibody (PRL); ovary extracts were analyzed with RIIβ, cyclin D2, and PRL-R antibodies.
DISCUSSION
Targeted disruption of the FSH-R gene brings critical information to the study of the role played by FSH signaling in maintaining spermatogenesis and fertility. Mutation of the FSHβ subunit gene leads to a similar, but slightly less severe, phenotype (14, 28). Mutation of the FSHβ subunit gene may not fully interfere with FSH-R-dependent signaling because of the existence of possible compensatory mechanisms (1). Interesting is the complete block of gametogenesis in both males and females mice lacking the glycoprotein hormone α-subunit (29, 30). We have selectively perturbed signaling in Sertoli and granulosa cells, yet allowed evaluation of the peripheral hormonal response.
FSH and testosterone levels change in the FSH-R mutants. The magnitude of these changes is sex specific, as FSH levels increase 15-fold in the female and only 3- to 4-fold in the male. The drop in testosterone in the males is remarkable (Fig. 4C), but only moderately affects fertility. This result is interesting for several reasons. First, it shows that lower testosterone levels are sufficient to sustain sex accessories. Second, it points to a link between FSH signaling and testosterone production. This link could involve an intercellular communication pathway that appears compromised, despite normal LH levels, in the mutants.
The sex-specific increase in FSH levels and the pituitary anterior lobe enlargement (Figs. 4 and 5) may suggest a role for FSH in gonadotroph proliferation. Interestingly, we also observe an increase in the number of TSH-positive cells. This finding is somewhat surprising as TSH-positive cells belong to a different ontogenic pathway than gonadotrophs (31). TSH-positive cells are thought to originate from an α subunit of glycoprotein hormone (GSUα)-positive precursor by two alternative lineages, which are either Pit1 dependent or Pit1 independent (31). Pit1 is a transcription factor required for the establishment of cell types expressing prolactin, growth hormone, and TSH (32). GSUα-positive precursors also yield the gonadotrophs, but via a distinct lineage that is likely to involve cAMP-responsive transcription factors (32). Similarly, it has been shown that thyrotrope- and gonadotrope-specific expression of the α-subunit gene involves distinct sets of transcriptional elements (33, 34). Thus, the FSH-R mutant mice will become essential for more detailed studies of pituitary cell lineages.
The role played by FSH in the differentiation of germ cells in various species has been debated considerably (21, 35–37). Description of humans homozygous for a FSH-R inactivating mutation revealed differential effects in men and women (15, 18). The abnormalities in sperm morphology and motility in the FSH-R −/− mice are reminiscent of changes observed under conditions of sustained blockade of FSH-R function in adult monkeys after immunization (36), suggesting application of the mutant mice in further studies. FSH-R mutant females demonstrate the crucial role of FSH signaling in ovarian follicle maturation and fertility. They provide a model to study hypergonadotropic-hypogonadism, which is a hereditary syndrome characterized by failure of follicular development, lack of ovarian response, and elevated levels of circulating gonadotropins (15). Thus, our genetically altered mice mimic menopausal conditions in women, arguing for their potential utility in human health.
Acknowledgments
We thank P. Chambon, E. Borrelli, E. Lalli, V.S.R. Subbarayan, and M. Chrétien for interest and support; E. Borrelli, P. Mellon, and K. Tasken for reagents; and E. Blondelle, J. M. Garnier, and E. Heitz for technical help. M.R.S. was supported by a visiting scientist award of the Fond de la Recherche Scientifique du Québec-Institut National de la Santé et de la Recherche Médicale exchange program while on leave from Institut de Recherches Cliniques de Montréal. L.M. was supported by Fondation pour la Recherche Médicale. G.M.F. is supported by the European Community. This work was funded by the Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, Centre Hospitalier Universitaire Régional, Fondation pour la Recherche Médicale, Medical Research Council Canada, and Association pour la Recherche sur le Cancer.
ABBREVIATIONS
- FSH
follicle-stimulating hormone
- FSH-R
FSH receptor
- LH
luteinizing hormone
- TSH
thyroid-stimulating hormone
References
- 1.Ulloa-Aguirre A, Midgley A R, Beitins I Z, Padmanabhan V. Endocr Rev. 1995;16:765–787. doi: 10.1210/edrv-16-6-765. [DOI] [PubMed] [Google Scholar]
- 2.Sprengel R, Braun T, Nikolics K, Segaloff D, Seeburg P H. Mol Endocrinol. 1990;4:525–530. doi: 10.1210/mend-4-4-525. [DOI] [PubMed] [Google Scholar]
- 3.Simoni M, Gromoll J, Nieschlag E. Endocr Rev. 1997;18:739–773. doi: 10.1210/edrv.18.6.0320. [DOI] [PubMed] [Google Scholar]
- 4.Grasso P, Reichert L E. Endocrinology. 1990;128:949–956. doi: 10.1210/endo-127-2-949. [DOI] [PubMed] [Google Scholar]
- 5.Sairam M R, Jiang L G, Yarney T A, Khan H. Biochem Biophys Res Comm. 1996;226:717–722. doi: 10.1006/bbrc.1996.1419. [DOI] [PubMed] [Google Scholar]
- 6.Blok L J, Mecknebach P, Trapman J, Themmen A P N, Brinkmann A O, Grootegoed J A. Mol Cell Endocrinol. 1989;63:269–271. doi: 10.1016/0303-7207(89)90104-4. [DOI] [PubMed] [Google Scholar]
- 7.Monaco L, Foulkes N S, Sassone-Corsi P. Proc Natl Acad Sci USA. 1995;92:10673–10677. doi: 10.1073/pnas.92.23.10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stegtenhorst-Eegdeman K, Post M, Baarends M, Themmen A, Grootegoed J A. Mol Cell Endocrinol. 1995;108:115–124. doi: 10.1016/0303-7207(94)03468-9. [DOI] [PubMed] [Google Scholar]
- 9.Kangasniemi M, Kaipia A, Toppari J, Perheentupa A, Huhtaniemi I, Parvinen M. J Androl. 1990;11:336–343. [PubMed] [Google Scholar]
- 10.Ranniki A S, Zhang F P, Huhtaniemi I. Mol Cell Endocrinol. 1995;107:199–208. doi: 10.1016/0303-7207(94)03444-x. [DOI] [PubMed] [Google Scholar]
- 11.Jégou B. Int Rev Cytol. 1993;147:25–96. [PubMed] [Google Scholar]
- 12.Parvinen M. Endocr Rev. 1982;3:404–417. doi: 10.1210/edrv-3-4-404. [DOI] [PubMed] [Google Scholar]
- 13.Richards J S. Endocr Rev. 1994;15:725–751. doi: 10.1210/edrv-15-6-725. [DOI] [PubMed] [Google Scholar]
- 14.Rajendrakumar T, Wang Y, Lu N, Matzuk M M. Nat Genet. 1997;15:201–204. doi: 10.1038/ng0297-201. [DOI] [PubMed] [Google Scholar]
- 15.Aittomäki K, Herva R, Stenman U, Juntunen K, Ylöstalo P, Hovata O, de la Chapelle A. J Clin Endocrinol Metab. 1996;81:3722–3726. doi: 10.1210/jcem.81.10.8855829. [DOI] [PubMed] [Google Scholar]
- 16.Gromoll J, Simoni M, Nieschlag E. J Clin Endocrinol Metab. 1996;81:1367–1370. doi: 10.1210/jcem.81.4.8636335. [DOI] [PubMed] [Google Scholar]
- 17.Aittomäki K, Dieguez Lucena J L, Pakarinen P, Sistonen P, Tapanainen J, Lehväslaiho H, Reyes Engel A, Nieschlag E, Huhtaniemi I, de la Chapelle A. Cell. 1995;82:959–968. doi: 10.1016/0092-8674(95)90275-9. [DOI] [PubMed] [Google Scholar]
- 18.Tapanainen J S, Aittomaki K, Min J, Vaskivuo T, Huhtaniemi I L. Nat Genet. 1997;15:205–206. doi: 10.1038/ng0297-205. [DOI] [PubMed] [Google Scholar]
- 19.Heckert L L, Daley I J, Griswold M D. Mol Endocrinol. 1992;6:70–80. doi: 10.1210/mend.6.1.1738373. [DOI] [PubMed] [Google Scholar]
- 20.O’Shaughnessy P, Marsh P, Dudley K. Mol Cell Endocrinol. 1994;101:197–201. doi: 10.1016/0303-7207(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 21.Gromoll J, Pekel E, Nieschlag E. Genomics. 1996;35:308–311. doi: 10.1006/geno.1996.0361. [DOI] [PubMed] [Google Scholar]
- 22.Huhtaniemi I T, Eskola V, Pakarinen P, Matikainen T, Sprengel R. Mol Cell Endocrinol. 1992;88:55–66. doi: 10.1016/0303-7207(92)90009-u. [DOI] [PubMed] [Google Scholar]
- 23.Saiardi A, Bozzi Y, Baik J H, Borrelli E. Neuron. 1997;19:114–127. doi: 10.1016/s0896-6273(00)80352-9. [DOI] [PubMed] [Google Scholar]
- 24.Nantel F, Monaco L, Foulkes N S, Masquilier D, LeMeur M, Henriksen K, Dierich A, Parvinen M, Sassone-Corsi P. Nature (London) 1996;380:159–162. doi: 10.1038/380159a0. [DOI] [PubMed] [Google Scholar]
- 25.Sassone-Corsi P. Cell. 1997;88:163–166. doi: 10.1016/s0092-8674(00)81834-6. [DOI] [PubMed] [Google Scholar]
- 26.Foulkes N S, Schlotter F, Pévet P, Sassone-Corsi P. Nature (London) 1993;362:264–267. doi: 10.1038/362264a0. [DOI] [PubMed] [Google Scholar]
- 27.Sicinsky P, Donaher J, Geng Y, Parker S, Gardner H, Park M Y, Robker R L, Richards J S, McGinnis L, Biggers J, et al. Nature (London) 1996;384:470–474. doi: 10.1038/384470a0. [DOI] [PubMed] [Google Scholar]
- 28.Philip M, Arbelle J E, Seger Y, Parvari R. N Engl J Med. 1998;338:1729–1732. doi: 10.1056/NEJM199806113382404. [DOI] [PubMed] [Google Scholar]
- 29.Kendall S K, Samuleson L C, Saunders T L, Wood R I, Camper S A. Genes Dev. 1995;9:2007–2019. doi: 10.1101/gad.9.16.2007. [DOI] [PubMed] [Google Scholar]
- 30.Markkula M, Huhtaniemi I. Rev Reprod. 1996;1:97–106. doi: 10.1530/ror.0.0010097. [DOI] [PubMed] [Google Scholar]
- 31.Borrelli E. Trends Genet. 1994;10:222–224. doi: 10.1016/0168-9525(94)90154-6. [DOI] [PubMed] [Google Scholar]
- 32.Voss J W, Rosenfeld M G. Cell. 1992;70:527–530. doi: 10.1016/0092-8674(92)90422-9. [DOI] [PubMed] [Google Scholar]
- 33.Hammernik D, Keri R, Clay C, Clay J, Sherman G, Sawyer R, Nett T, Nilson J. Mol Endocrinol. 1992;6:1745–1755. doi: 10.1210/mend.6.10.1280329. [DOI] [PubMed] [Google Scholar]
- 34.Kendall S K, Gordon D F, Birkmeier T S, Petrey D, Sarapura V D, O’Shea K S, Wood W M, Lloyd R V, Ridgway E C, Camper S A. Mol Endocrinol. 1994;8:1420–1433. doi: 10.1210/mend.8.10.7531821. [DOI] [PubMed] [Google Scholar]
- 35.Lerchl A, Sotiviadon S, Behre H, Pierce J, Weinbauer G, Nieschlag E. Biol Reprod. 1993;49:1108–1117. doi: 10.1095/biolreprod49.5.1108. [DOI] [PubMed] [Google Scholar]
- 36.Moudgal N R, Sairam M R, Krishnamurthy H N, Sridhar S, Krishnamurthy M, Khan H. Endocrinology. 1997;138:3065–3068. doi: 10.1210/endo.138.7.5381. [DOI] [PubMed] [Google Scholar]
- 37.Mougdal N R, Sairam M R. Hum Reprod. 1998;13:916–919. doi: 10.1093/humrep/13.4.916. [DOI] [PubMed] [Google Scholar]