

Abstract
Both caspase-1- and caspase-3-like activities are required for Fas-mediated apoptosis. However, the role of caspase-1 and caspase-3 in mediating Fas-induced cell death is not clear. We assessed the contributions of these caspases to Fas signaling in hepatocyte cell death in vitro. Although wild-type, caspase-1−/−, and caspase-3−/− hepatocytes were killed at a similar rate when cocultured with FasL expressing NIH 3T3 cells, caspase-3−/− hepatocytes displayed drastically different morphological changes as well as significantly delayed DNA fragmentation. For both wild-type and caspase-1−/− apoptotic hepatocytes, typical apoptotic features such as cytoplasmic blebbing and nuclear fragmentation were seen within 6 hr, but neither event was observed for caspase-3−/− hepatocytes. We extended these studies to thymocytes and found that apoptotic caspase-3−/− thymocytes exhibited similar “abnormal” morphological changes and delayed DNA fragmentation observed in hepatocytes. Furthermore, the cleavage of various caspase substrates implicated in mediating apoptotic events, including gelsolin, fodrin, laminB, and DFF45/ICAD, was delayed or absent. The altered cleavage of these key substrates is likely responsible for the aberrant apoptosis observed in both hepatocytes and thymocytes deficient in caspase-3.
Apoptosis originally was used to describe a unique type of cell death that exhibits a distinct set of morphological and biological changes, including cytoplasmic membrane blebbing, nuclear condensation, and fragmentation of chromosomal DNA. It now is recognized as a crucial biological process for development and homeostasis of all multicellular organisms (1). Although apoptosis can be triggered by diverse stimuli ranging from intracellular stress to extracellular receptor signaling, emerging evidence in recent years suggests that the central execution machinery of apoptosis is evolutionarily conserved (2). As in Caenorhabditis elegans where the cysteine protease ced-3 is absolutely required for programmed cell death, the mammalian homologs of ced-3, the caspase family proteases, now are considered the central players in all apoptotic events in mammals (3). Like many other cellular proteases, all caspases first are synthesized as inactive proenzymes that can be activated upon apoptotic stimulation. It is believed that apoptosis is a result of the proteolysis of various cellular components initiated by activated caspases. Because more than 10 caspases have been identified in mammals, the precise contribution of individual caspases in this process and how they functionally relate to each other in vivo becomes a key question. Although the tissue-specific phenotypes in caspase-1, -2, -3, and -11-deficient mice seem to indicate a tissue/cell type-specific function of these proteases (4–8), biochemical studies in Fas signaling and caspase substrate specificities strongly suggested a cascade activation model for caspases (9, 10).
A member of the tumor necrosis factor receptor family, Fas (CD95, APO-1) mediates apoptotic signals upon FasL engagement (11). In recent years, Fas-FasL interaction has been shown to play crucial roles in maintaining the homeostasis of the immune system, immunological privilege observed in the eye and testis, and the pathogenesis of autoimmune diseases such as type I diabetes (12–14). Although the potential physiological function of constitutive Fas expression in several other tissues, including heart, kidney and thymus, remains to be established, several lines of evidence suggested that Fas-FasL interaction may be involved in the destruction of hepatocytes that occurs in both acute and chronic liver diseases (15). It therefore would be of potential clinical importance to understand the molecular basis of Fas-mediated apoptosis in hepatocytes.
During apoptosis, many cellular proteins undergo caspase-dependent degradation. Although the relevance of cleavage of structural proteins, like gelsolin, fodrins, actins, and lamins, is easily conceivable, the functional importance of these and other cleavages, such as those of signaling molecules, including D4-GDI and MEKK1, is not yet clear (16). It is, however, widely assumed that the caspase-specific cleavage of these proteins is responsible for the various hallmarks of apoptosis such as nuclear fragmentation, cytoplasmic membrane blebbing, and DNA fragmentation. This hypothesis is greatly supported by the demonstrations that cleavage of gelsolin is important for nuclear and DNA fragmentation and cleavage of the protein kinase PAK2 and fodrin is involved in the formation of cytoplasmic blebs (17–19). More recently, factors that mediate the classic apoptotic diagnostic DNA laddering also have been identified, and it was further shown that activation of this DNase is mediated by the caspase-specific cleavage of its associated inhibitor (20–22).
In this study, our original goal was to assess the contribution of caspase-1 and caspase-3 in mediating Fas-mediated hepatocyte apoptosis by using an in vitro coculture system (23). Although our results indicate that neither caspase is necessary for Fas-mediated hepatocyte death, they revealed a crucial role of caspase-3 in mediating the various morphological changes during apoptosis through the cleavage of key substrates.
MATERIALS AND METHODS
Animals and Cell Lines.
All mice were between 3 and 5 weeks old and housed in facilities at Yale. Wild-type littermates were used as controls for caspase-1−/− and caspase-3−/− mice. CD-1 mice were used for caspase inhibitor experiments. FasL-expressing NIH 3T3 fibroblasts were generated as described (24).
Reagents and Antibodies.
Caspase inhibitor zVAD.fmk was purchased from Bachem. Polyclonal chicken antibody against LaminB was kindly provided by Scott Kauffman (Mayo Clinics, Rochester, MN). Rabbit polyclonal antibodies against DFF45 and gelsolin were kindly provided by Xiaodong Wang (Texas Southwestern Medical Center, Dallas) and David Kwiatkowski (Harvard Medical School, Boston), respectively. Anti-Fodrin antibody was purchased from Chemicon.
Coculture Experiments.
Mouse hepatocytes were prepared at the Isolation Core of Yale Liver Center according to a modified protocol (31, 32). Isolated hepatocytes then were cultured overnight at a density of 5 × 104 cells/cm2. The next day fibroblasts with or without FasL cultured to confluency were trypsinized and resuspended in Waymouth MB medium. The coculture was initiated by addition of these fibroblasts at a density of 7.5 × 104 cells/cm2 to the overnight hepatocyte cultures.
Determination of Hepatocyte Viability and Nuclear Morphology.
Hepatocyte viability and nuclear morphology were assessed by propidium iodide (PI) (3 μM, 10 min) and 4′,6′-diamidino-2-phenylindole (2 μM, 10 min) staining, respectively, and a total of 150–180 cells/coverslip were examined by randomly selecting six visual fields/coverslip. The following cell populations were distinguished: for viability, PI-positive dead cells, PI-negative apoptotic cells with cytoplasmic bleb formation, and PI-negative viable cells with normal morphology; for nuclear morphology, cells with round nuclei and organized chromatin structure, cells with no detectable nuclear material, cells with condensed nuclei, and cells with fragmented nuclei.
For thymocytes, cells were spun onto glass slides by using cytospin at 600 rpm for 4 min. Cells then were fixed and stained by using Leukostat solutions (Sigma) and visualized under a light microscope.
DNA Fragmentation Assay and Terminal Deoxynucleotidyltransferase-Mediated dUTP Nick-End Labeling (TUNEL).
Hepatocytes or thymocytes were harvested at indicated time points after treatment and digested in lysis buffer (10 mM EDTA/50 mM Tris/0.5% N-Laurosacosine, pH 8.0/200 μg/ml of proteinase K) at 55°C for 1 hr followed by 100 μg/ml of RNaseA treatment (37°C, 1 hr). Genomic DNAs were isolated by phenol/chloroform extraction and ethanol precipitation and were dissolved in Tris-EDTA buffer (pH 7.5). DNA ladders were separated on 1% agarose gel. TUNEL was performed by using a kit (Boehringer Mannheim).
Western Blot Analysis.
At various time points after treatment, wild-type or caspase-3−/− thymocytes were lysed in whole-cell lysing buffer (400 mM NaCl/25 mM Tris, pH 7.5/1% SDS) and were analyzed by 6% (fodrin), 10% (laminB and gelsolin), or 14% (DFF45) SDS/PAGE. Proteins were transferred by using a semidry method and immunoblotted with various primary antibodies followed by appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies. For laminB, the primary chicken anti-lamin antibody and HRP-conjugated anti-rabbit Ig antibody were sandwiched with a rabbit anti-chicken Ig to increase the signal/noise ratio. All immunoblots were visualized by ECL.
Statistics and Data Analysis.
Where appropriate, values were given as mean ± SD. Statistical comparisons were made by using Student’s t test or ANOVA, and P values < 0.05 were taken as significant.
RESULTS
Membrane-Bound FasL Induces Mouse Hepatocyte Apoptosis Through Caspase Activation.
When cocultured with FasL-expressing fibroblasts, virtually all wild-type hepatocytes were dead within 24 hr, whereas only a minor reduction in survival was observed in hepatocytes cocultured with control fibroblasts (Figs. 1 and 2). Furthermore, hepatocytes cocultured with FasL-expressing fibroblasts displayed characteristic transient bleb formation (Fig. 1 b and c), and nuclear and DNA fragmentation (Fig. 1e). As in many other experimental systems, addition of a pan-caspase inhibitor zVAD.fmk (10 μM) completely prevented hepatocyte death, as well as cytoplasmic bleb formation and nuclear fragmentation (Fig. 2). These results clearly showed that FasL-expressing fibroblasts efficiently induce apoptosis of cocultured hepatocytes and that zVAD.fmk-inhibitable caspases are required for the induction of apoptosis and its associated morphological changes.
Figure 1.

(a) Light microscopic view (×600) of 24-hr coculture of wild-type hepatocytes and control fibroblasts. Arrows indicate normal hepatocytes. ∗ indicates fibroblast monolayer. (b) 24-hr coculture of wild-type hepatocytes with FasL-expressing fibroblasts (×600). Arrows point to dying hepatocytes. (c) Light microscopic view of blebbing hepatocytes with granular cytoplasm on the surface of monolayer of FasL-expressing fibroblasts (original magnification ×600, digitally enlarged). (d) DAPI (4′,6′-diamidino-2-phenylindole) staining illustrating hepatocyte nuclear condensation and (e) 24-hr coculture of wild-type hepatocytes with FasL-expressing fibroblasts (×320).
Figure 2.

Time course of (a) cell death (PI negative cells), (b) blebbing, and (c) nuclear fragmentation of wild-type, caspase-1−/−, and caspase-3−/− hepatocytes after coculture with NIH 3T3 fibroblasts (○) or FasL-expressing NIH 3T3 fibroblasts with (▵) or without (□) caspase inhibitor z-VAD.fmk (10 μM).
Wild-Type, Caspase-1−/−, and Caspase-3−/− Hepatocytes Are Equally Susceptible to FasL-Induced Apoptosis.
To examine the involvement of caspase-1 and caspase-3 in mediating FasL-induced hepatocyte apoptosis, hepatocytes from caspase-1−/− or caspase-3−/− mice were isolated and cocultured with FasL-expressing fibroblasts. Fas expression levels on these hepatocytes were analyzed by FACS, and no difference was observed among wild-type, caspase-1−/−, or caspase-3−/− hepatocytes (data not shown).
After 6 and 24 hr of coculture with FasL-expressing fibroblasts, the viability of hepatocytes was determined by their ability to take up PI. Wild-type, caspase-1−/−, and caspase-3−/− hepatocytes all became PI positive after 24 hr with no significant difference in the kinetics of PI uptake (Fig. 2a). Deficiency in either caspase-1 or caspase-3 expression therefore did not prevent hepatocytes from undergoing apoptosis. Thus, both caspase-1 and caspase-3 are dispensable for the induction of Fas-mediated apoptosis in hepatocytes.
Caspase-3−/− Hepatocytes Undergo an Aberrant Form of Apoptosis.
Although no difference in the kinetics of PI uptake was observed, further examination revealed that caspase-3−/− but not caspase-1−/− hepatocytes exhibited altered morphological changes during Fas-mediated apoptosis. With both wild-type and caspase-1−/− hepatocytes, transient cytoplasmic bleb formation was found in a high percentage of cells after 6 hr and was not detectable after 24 hr (Fig. 2b). Nuclear fragmentation was clearly visible in more than 25% of the hepatocytes after 6 and 24 hr (Fig. 2c). These results indicate that lack of caspase-1 has no discernible impact on FasL-induced hepatocyte apoptosis in vitro.
Drastically different morphological features, however, were observed in apoptotic caspase-3−/− hepatocytes. After 6 hr of coculture, no bleb formation could be detected in caspase-3−/− hepatocytes, and this deficiency was not caused by a delay in kinetics as no blebbing caspase-3−/− hepatocytes were found up to 24 hr (data not shown). Furthermore there was virtually no detectable nuclear fragmentation in caspase-3−/− hepatocytes at any time point, compared with nuclear fragmentation that could be clearly observed in up to 25% of the wild-type and caspase-1−/− cells (Fig. 2c). In fact, although wild-type hepatocytes displayed a typical nuclear fragmentation profile with widely distributed small nuclear fragments that are difficult to visualize in one focal plane (Fig. 3a), the nuclei of apoptotic caspase-3−/− hepatocytes exhibited irregular clumping of chromatin without radical change in its distribution (Fig. 3b), a phenomenon often associated with necrosis. Together, these results demonstrated that although absence of caspase-3 did not render hepatocytes resistant to FasL-induced apoptosis in vitro, caspase-3 was required to mediate cytoplasmic and nuclear events typically associated with apoptosis.
Figure 3.

DAPI (4′,6′-diamidino-2-phenylindole) staining illustrating typical small nuclear fragments in wild-type hepatocytes after coculture with FasL-expressing fibroblasts (a) and altered staining pattern in apoptotic caspase-3−/− hepatocytes (b). (c and d) Different TUNEL staining pattern in wild-type and caspase-3−/− hepatocytes 6 hr post-coculture. (e) Delayed DNA fragmentation in caspase-3−/− hepatocytes after coculture with FasL-expressing fibroblasts.
Besides these altered morphological changes, the DNA laddering assay revealed much slower kinetics of DNA breakdown in caspase-3−/− hepatocytes. Although a 24-hr coculture with FasL-expressing fibroblasts clearly resulted in internucleosomal cleavage of chromosomal DNA in hepatocytes from both wild-type and caspase-3−/− mice, very little DNA cleavage was observed in caspase-3−/− hepatocytes at 6 hr when DNA laddering was already evident in wild-type hepatocytes (Fig. 3e). The TUNEL method, a more sensitive detection of DNA double-strand breaks, confirmed the results seen with the DNA laddering assay. Although a large number of wild-type hepatocytes possessed TUNEL positive fluorescent signals after 6 hr of coculture (Fig. 3c), the signal was much weaker in caspase-3−/− hepatocytes and could be observed in only a few cells (Fig. 3d). More interestingly, the positive TUNEL signal patterns observed in wild-type and caspase-3−/− hepatocytes were also different. In wild-type apoptotic hepatocytes, a typical pattern with small, fragmented TUNEL positive nuclear fragments could be easily observed. In contrast, the TUNEL signal seen in caspase-3−/− hepatocytes seemed to be from clumped, but nonfragmented, chromatin DNAs, an observation that is confirmed by direct examination of nuclear morphological changes during apoptosis (Fig. 3 a and b).
Caspase-3 Mediates Morphological Changes and DNA Fragmentation in Thymocytes Through Cleavage of Various Substrates.
To address whether the above observations are hepatocyte specific, we examined anti-Fas antibody-induced apoptosis in thymocytes by using various criteria. As we previously have reported, caspase-3−/− thymocytes were equally susceptible to Fas-mediated apoptosis when measured by Annexin-V staining as no difference in the kinetics of phosphotidylserine exposure was observed (Fig. 4A). When we further examined the morphological changes, caspase-3−/− thymocytes exhibited similar unusual features as the hepatocytes. Although both nuclear and DNA fragmentation were clearly observed in wild-type thymocytes upon anti-Fas antibody crosslinking, caspase-3−/− thymocytes exhibited no nuclear fragmentation and significantly delayed DNA fragmentation as measured by the DNA ladder assay (Fig. 4 B and C). Instead, irregular clumping of the nuclear material was the main feature of apoptotic caspase-3−/− thymocytes.
Figure 4.
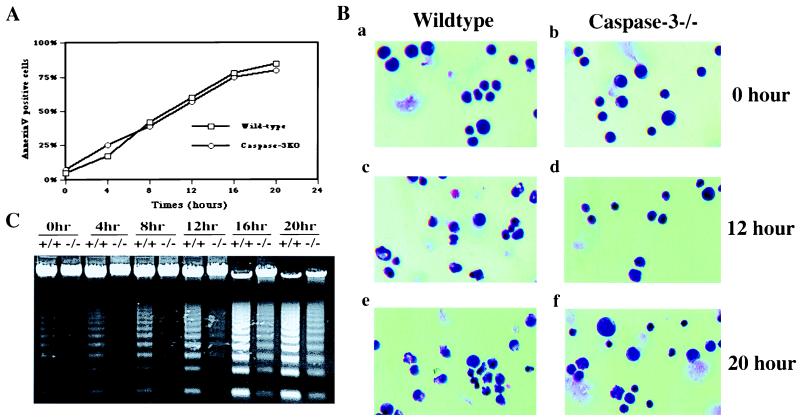
Kinetics of phosphatidylserine (PS) flip (A), nuclear morphology (B), and DNA fragmentation (C) in apoptotic wild-type and caspase-3−/− thymocytes. Thymocytes from either wild-type or caspase-3−/− mice were cultured in RPMI medium (10% fetal bovine serum) at 37°C in the presence of coated anti-Fas antibody Jo2. At each indicated time point, cells were harvested and subjected to morphological analysis, PS flipping, and DNA fragmentation assay as described in Materials and Methods.
To address the molecular basis of these observations, we examined the cleavage of various caspase substrates that have been implicated in mediating these morphological changes. Because recent studies have suggested that the cleavage of fodrin and gelsolins has profound effects on the cytoplasmic and nuclear morphology, respectively, in cells undergoing apoptosis (17, 19), we examined their cleavage in caspase-3−/− thymocytes. Our results showed that although the cleavage of gelsolin was greatly reduced, caspase-dependent cleavage of fodrin was essentially abrogated in caspase-3−/− thymocytes (Fig. 5). Previously studies also have suggested that the nuclear membrane breakdown could be mediated through the cleavage of nuclear membrane structural proteins such as lamins (25). We therefore examined the cleavage of laminB, the only lamin expressed in thymocytes, in both wild-type and caspase-3−/− thymocytes. Our result indicated that laminB was cleaved in caspase-3−/− thymocytes upon Fas signaling, albeit at a slower rate.
Figure 5.

Comparison of caspase substrate cleavage in wild-type and caspase-3−/− thymocytes during apoptosis. At each indicated time point after treatment (same as in Fig. 4), cells were lysed, and cleavage of various proteins were analyzed as described in Materials and Methods.
Finally, because delayed kinetics of cleavage of internucleosomal chromosomal DNA also was observed in caspase-3−/− hepatocytes and thymocytes, we analyzed the cleavage of DFF45/ICAD in both wild-type and caspase-3−/− thymocytes undergoing Fas-mediated apoptosis. As expected, although DFF45/ICAD was cleaved in a time-dependent fashion in wild-type apoptotic thymocytes, its cleavage in caspase-3−/− thymocytes was greatly delayed, though not completely abrogated (Fig. 5).
DISCUSSION
Previous studies have shown that upon Fas signaling, caspase-1-like activity is rapidly increased and is followed by a slower and gradual increase of caspase-3-like activity (9). Our results, however, clearly showed that deficiency in either caspase-1 or caspase-3 failed to protect hepatocytes from FasL-induced apoptosis. Given that zVAD.fmk, an inhibitor with broad specificity, was able to completely prevent the apoptosis of FasL-induced hepatocyte apoptosis, it is almost certain that the presumed caspase-1-like and caspase-3-like activities are provided by other caspases. For example, the caspase-1-like activity is most likely caused by the activation of caspase-8, a YVAD.CHO-sensitive caspase and apparently the most upstream caspase in the Fas signaling pathway as it is physically associated with Fas itself via the adapter protein FADD (26, 27). Although caspase-3 itself indeed is activated after Fas signaling, caspase-3-like activity conceivably also can be provided by caspase-7, a close homologue of caspase-3 (28–30). It is therefore likely that in hepatocytes Fas-FasL engagement leads to rapid activation of caspase-8, which in turn activates either caspase-3 and/or caspase-7 proteolytically. Such a scenario also is supported by recent biochemical studies on the substrate specificities of all the caspases, which suggested that group III caspases (caspase-6, -8, and -9) possessed the highest efficiency in activating group II caspases (caspase-2, -3, and -7) (10). The relevance of this grouping system was proven recently when caspase-9 was demonstrated to be the activating caspase for caspase-3 and caspase-7 in cytochrome c-mediated apoptosis in Hela cell extracts (31) and mice in vivo (32).
The observation that caspase-3−/− hepatocytes and thymocytes displayed morphologically distinct features during apoptosis was unexpected and significant. Although it has been suggested that caspases might be responsible for the distinct morphological features that are associated with apoptosis (33, 34), the notion that a single caspase controls multiple events ranging from the formation of cytoplasmic blebs to chromatin condensation and nuclear breakdown, is striking. These results not only demonstrate that the morphological features occurring during apoptosis depend on specific caspase activities, but also distinguish cell death from the associated morphological changes.
Cytoplasmic membrane changes associated with apoptosis include the formation of blebs and loss of phosphatidylserine (PS) membrane asymmetry. It was proposed that the blebbing resulted from the cleavage of several important cytoskeletal structure proteins such as fodrin, actin, and Gas2 (19, 35, 36) and signaling molecules like PAK2 and cdc42. The failure of Ac-DEVD.CHO to inhibit fodrin cleavage previously led to the proposal that fodrin is not cleaved by caspase-3 (37). However, our results clearly showed that although caspase-3 apparently is not involved in regulating PS flipping during apoptosis, it is indeed required for the cleavage of fodrinα, as well as the formation of membrane blebs.
Nuclear events are other hallmarks of apoptosis. Because neither the peripheral condensation of chromatin or fragmentation of the nucleus was observed in caspase-3−/− cells, it is almost certain that the factor(s) controlling both processes are caspase-3 dependent. Previously, it was proposed that cleavage of nuclear envelope protein lamins and actin-severing proteins gelsolins play a crucial role in mediating these events. Our results, however, showed that both laminB and gelsolin were cleaved, albeit at a slower rate, in apoptotic caspase-3−/− thymocytes without leading to nuclear breakdown. It is therefore possible that another caspase such as caspase-7 also may process these molecules, and that nuclear breakdown requires caspase-3-dependent cleavage of other molecule(s).
The significantly delayed internucleosomal cleavage of genomic DNA in both caspase-3−/− hepatocytes and thymocytes is intriguing. According to one model (21), the enzymatic activity of CAD normally is inhibited by its direct association with its inhibitor DFF45/ICAD. Upon receipt of apoptotic stimuli, activation of caspases leads to the specific cleavage of DFF45/ICAD by caspases and the loss of its CAD inhibitory activity. Based on our observation that cleavage of DFF45/ICAD in caspase-3−/− cells is greatly delayed, it is tempting to postulate that in caspase-3-deficient cells, the delayed cleavage of DFF45/ICAD resulted in the delayed DNA cleavage by CAD. However, careful examination of the data suggests a more complicated scenario. Although the DNA cleavage in wild-type thymocytes at 12 hr posttreatment is comparable to that of caspase-3−/− thymocytes at 24 hr posttreatment, the corresponding cleavage of DFF45/ICAD is significantly more evident in wild-type thymocytes (Figs. 4C and 5). This observation indicates that the cleavage of DFF45/ICAD does not fully correlate with the internucleosomal cleavage of DNA and therefore suggests that, for example, there might well be more than one CAD protein, a possibility strengthened by the recent cloning of other DFF45/ICAD homologs (38). It is conceivable that other DFF45/ICAD and CAD complexes also exist and can be activated in the absence of caspase-3.
After this paper was submitted, two other groups reported altered nuclear events in apoptotic cells lacking caspase-3 by using either MEF cells derived from caspase-3−/− mice (39) or the human breast carcinoma cell line MCF-7, which is defective in caspase-3 expression (40). Although their reports support our observations that caspase-3-deficient cells undergo apoptosis without nuclear breakdown, those groups observed no DNA fragmentation in those mutant cells undergoing apoptosis. In our studies, however, we were always able to detect some residual level of DNA fragmentation in both caspase-3−/− thymocytes and hepatocytes. It is likely that this apparent discrepancy reflects differences in experimental systems such as cell types, apoptotic stimuli, and the sensitivity of the assays used.
Apoptosis originally was proposed to describe a type of death that was distinct from necrosis based on morphological criteria. To date, cell morphology continues to be the most convincing operational definition of apoptosis. Our observation that caspase-3−/− hepatocytes and thymocytes can be induced to undergo cell death without exhibiting many of the classical morphological features raises an important philosophical question: what is the molecular basis that distinguishes apoptosis from necrosis? The answer might be whether there is activation of key caspases such as caspase-3. In fact, the cell death phenomenon observed in both caspase-3-deficient hepatocytes and thymocytes after FasL-Fas induction is reminiscent of many of the features associated with necrosis rather than apoptosis, e.g., lack of membrane blebbing, irregular chromatin clumping, and late DNA fragmentation. Thus, whether a cell commits to apoptosis or necrosis might not only depend on its intracellular energy level, as previously proposed, but also the type of caspases that are being activated. This hypothesis also might explain the puzzling phenomenon that certain cell death stimuli, notably tumor necrosis factor α, can induce either apoptosis and/or necrosis, depending on the cell type and experimental conditions (41).
Acknowledgments
We thank M. Pate for expert help with isolation of hepatocytes and F. Manzo for help with manuscript preparation. This work was supported by grants from the Deutsche Forschungsgemeinschaft SCHL-403/1-1 (S.F.S.) and the National Institutes of Health DK 25636 (J.L.B.), AI 37554 (I.N.C.), and AI36529 (R.A.F.). R.A.F. is an Investigator of the Howard Hughes Medical Institute. Support from the Yale Liver Center morphology and cell isolation core also is acknowledged (DK 34989).
ABBREVIATIONS
- PI
propidium iodide
- TUNEL
terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling
References
- 1.Thompson C B. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 2.Steller H. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 3.Cohen G M. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuida K, Lippke J A, Ku G, Harding M W, Livingston D J, Su M S, Flavell R A. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 5.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, et al. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 6.Kuida K, Zheng T S, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell R A. Nature (London) 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 7.Bergeron L, Perez G I, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham K E, Flaws J A, Salter J C M, et al. Genes Dev. 1998;12:1304–1314. doi: 10.1101/gad.12.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang S, Miura M, Jung Y-K, Zhu H, Li E, Yuan J. Cell. 1998;92:501–509. doi: 10.1016/s0092-8674(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 9.Enari M, Talanian R V, Wong W W, Nagata S. Nature (London) 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- 10.Thornberry N A, Rano T A, Peterson E P, Rapser D M, Timkey T, Garcia-Calvo M, Houtzager V M, Nordstrom P A, Roy S, Vaillancourt J P, et al. J Biol Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 11.Nagata S, Golstein P. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 12.Chervonsky A V, Wang Y, Wong F S, Visintin I, Flavell R A, Janeway C A, Jr, Matis L A. Cell. 1997;89:17–24. doi: 10.1016/s0092-8674(00)80178-6. [DOI] [PubMed] [Google Scholar]
- 13.Griffith T S, Brunner T, Fletcher S M, Green D R, Ferguson T A. Science. 1995;270:1189–1192. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- 14.Bellgrau D, Gold D, Selawry H, Moore J, Franzusoff A, Duke R C. Nature (London) 1995;377:630–632. doi: 10.1038/377630a0. [DOI] [PubMed] [Google Scholar]
- 15.Feldmann G. J Hepatol. 1997;26:1–11. doi: 10.1016/s0168-8278(97)80491-6. [DOI] [PubMed] [Google Scholar]
- 16.Cryns V, Yuan J. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- 17.Kothakota S, Azuma T, Reinhard C, Klippel A, Tang J, Chu K, McGarry T J, Kirschner M W, Koths K, Kwiatkowski D J, Williams L T. Science. 1997;278:294–298. doi: 10.1126/science.278.5336.294. [DOI] [PubMed] [Google Scholar]
- 18.Rudel T, Bokoch G M. Science. 1997;276:1571–1574. doi: 10.1126/science.276.5318.1571. [DOI] [PubMed] [Google Scholar]
- 19.Martin S J, O’Brien G A, Nihioka W K, McGahon A J, Mahboubi A, Saido T C, Green D R. J Biol Chem. 1995;270:6425–6428. doi: 10.1074/jbc.270.12.6425. [DOI] [PubMed] [Google Scholar]
- 20.Liu X, Zou H, Slaughter C, Wang X. Cell. 1997;89:175–184. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- 21.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. Nature (London) 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 22.Sakahira H, Enari M, Nagata S. Nature (London) 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- 23.Dao T, Huleatt J W, Hingorami R, Crispe I N. J Immunol. 1997;159:4261–4267. [PubMed] [Google Scholar]
- 24.Hughes D P, Crispe I N. J Exp Med. 1995;182:1395–1401. doi: 10.1084/jem.182.5.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lazebnik J A, Takahashi A, Moir R D, Goldman R D, Poirier G G, Kaufmann S H, Earnshaw W C. Proc Natl Acad Sci USA. 1995;92:9042–9046. doi: 10.1073/pnas.92.20.9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muzio M, Chinnaiyan A M, Kischkel F C, O’Rourke K, Shevchenko A, Ni J, Schffidi C, Bretz J D, Zhang M, Gentz R, et al. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 27.Boldin M P, Goncharov T M, Goltev Y V, Wallach D. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 28.Fernandes-Alnemri T, Takahashi A, Armstrong R, Krebs J, Fritz L, Tomaselli K J, Wang L, Yu Z, Croce C M, Salveson G. Cancer Res. 1995;55:6045–6052. [PubMed] [Google Scholar]
- 29.Duan H, Chinnaiyan A M, Hudson P L, Wing J P, He W, Dixit V M. J Biol Chem. 1996;271:1621–1625. doi: 10.1074/jbc.271.3.1621. [DOI] [PubMed] [Google Scholar]
- 30.Lippke J A, Gu Y, Sarnecki C, Caron P R, Su M S-S. J Biol Chem. 1996;271:1825–1828. doi: 10.1074/jbc.271.4.1825. [DOI] [PubMed] [Google Scholar]
- 31.Li P, Nijhawan D, Budihardjo I, Srinivasula S M, Ahmad M, Alnemri E S, Wang X. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 32.Kuida K, Haydar T, Kuan C-Y, Gu Y, Taya C, Karasuyama H, Su M S-S, Rakic P, Flavell R A. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 33.Martin S J, Green D R. Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 34.Fraser A, Evan G. Cell. 1996;85:781–784. doi: 10.1016/s0092-8674(00)81005-3. [DOI] [PubMed] [Google Scholar]
- 35.Kayalar C, Ord T, Testa M P, Zhong L-T, Bredesen D E. Proc Natl Acad Sci USA. 1996;93:2234–2238. doi: 10.1073/pnas.93.5.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brancolini C, Benedetti M, Schneider C. EMBO, J. 1995;14:5179–5190. doi: 10.1002/j.1460-2075.1995.tb00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cryns V L, Bergeron L, Zhu H, Li H, Yuan J. J Biol Chem. 1996;271:21277–31282. doi: 10.1074/jbc.271.49.31277. [DOI] [PubMed] [Google Scholar]
- 38.Inohara N, Koseki T, Chen S, Wu X, Nüñez G. EMBO J. 1998;17:2526–2533. doi: 10.1093/emboj/17.9.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woo M, Hakem R, Soengas M S, Duncan G S, Shahinian A, Kagi S, Hakem A, McCurrach M, Khoo W, Kaufman S, et al. Genes Dev. 1998;12:806–819. doi: 10.1101/gad.12.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janicke R U, Sprengart M L, Wati M R, Porter A G. J Biol Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- 41.Laster S M, Wood J G, Gooding L R. J Immunol. 1988;141:2629–2634. [PubMed] [Google Scholar]