

Summary
The solution structure of Alg13, the glycosyl-donor binding domain of an important bipartite glycosyltransferase in the yeast S. cerevisiae, is presented. This glycosyl transferase is unusual in that it is only active in the presence of a binding partner, Alg14. Alg13 is found to adopt a unique topology amongst glycosyltransferases. Rather than the conventional Rossmann fold found in all GT-B type enzymes, the N-terminal half of the protein is a Rossmann-like fold with a mixed parallel and anti-parallel β sheet. The Rossmann fold of the C-terminal half of Alg13 is conserved. However, while conventional GT-B type enzymes usually possess three helices at the C-terminus, only two helices are present in Alg13. Titration of Alg13 with both UDP-GlcNAc, the native glycosyl donor, and a paramagnetic mimic, UDP-TEMPO, shows that the interaction of Alg13 with the sugar donor is primarily through the residues in the C-terminal half of the protein.
Introduction
Glycosyltransferases (GTs), enzymes that form new glycosidic bonds in glycoconjugates by transferring sugars from sugar nucleotide donors to various acceptors, are vital players in cellular functions that range from quality control in protein synthesis, to intra-cellular signaling, metabolism and inter-cellular communication (Varki et al., 1999). Their ability to modulate disease states in higher organisms has also been documented (Fuster and Esko, 2005; Ohtsubo and Marth, 2006). In humans, the cell-surface products of GTs have been connected with viral & bacterial infection, cancer, diabetes, arthritis and a host of other diseases.
In eukaryotes an important subset of glycosyltransferases is involved in the synthesis of N-linked glycosides, a process that begins with construction of a tetradecasaccharide on the lipid carrier, dolichol. The second step in the dolichol pathway involves the transfer of N-acetylglucosamine (GlcNAc) from the UDP-GlcNAc donor to the Dol-PP-GlcNAc acceptor. In the yeast S. cerevisiae this transformation is catalyzed by a heterodimeric enzyme Alg13/Alg14 (Chantret et al., 2005). Here we present the NMR structure for Alg13, the donor subunit of this heterodimeric enzyme.
Despite their importance and prevalence (it is estimated that nearly 1 % of genes in every genome code for enzymes involved in glycoside bond synthesis (Coutinho et al., 2003)), the structures of GTs are severely under represented in the protein data bank PDB (Breton et al., 2006; Schuman et al., 2007). By the latest tabulation of the GLYCO3D database (http://www.cermav.cnrs.fr/glyco3d), structures of only 50 distinct GTs have been solved. Interestingly, of the structures solved, the majority belong to one of only two structural folds, classified as GT-A and GT-B type folds (Breton et al., 2006; Coutinho et al., 2003; Qasba et al., 2005). This is surprising considering the diversity in the GT sugar acceptor specificity and further emphasizes the need to understand the intricacy of the GT substrate selection process.
To date, NMR has played a minor role in the study of GTs as the size of most GTs puts GT structure determination at the limit of solution NMR methods. In the future however, contributions from NMR may gain prominence. Improvements in technology are pushing back size limitations, and NMR-based methods can provide a route to structures of proteins that have traditionally been difficult to crystallize, namely proteins that are glycosylated and proteins that have substantial unstructured segments. Both factors decrease the possibility of crystal formation needed for X-ray crystallography (Breton et al., 2006). Many GTs of eukaryotic origin are glycosylated and many are known to have disordered loops. At least in GT-A type GTs, the these loops are implicated in aspects of catalysis (Breton et al., 2006; Qasba et al., 2005; Schuman et al., 2007). Studying the dynamics of these loops and their interactions with ligands by NMR may also help to further understand GT reactions and their specificity.
Alg13, a protein that is highly conserved in most eukaryotic organisms, does not require glycosylation for activity in yeast, thus making the expression of the active protein in prokaryotic expression systems feasible. It also presents a unique opportunity to make a step toward application of NMR to the investigation of more general problems in GT structure and function. In most GT reactions, a single protein is responsible for catalyzing the sugar transfer reaction. However, as shown by Chantret et al. (2005) both Alg13 and Alg14, are required to catalyze the transfer of GlcNAc to Dol-PP-GlcNAc (Burda and Aebi, 1999; Kelleher and Gilmore, 2006; Weerapana and Imperiali, 2006). Sequence alignment showed that Alg13 is a member of the protein family PF03033, which also includes C-terminal sugar donor binding domains from several GT-B type GTs including the E. coli protein MurG, which catalyzes a similar reaction to Alg 13/14 in the biosynthesis of peptidoglycan. The other gene product, Alg14, belongs to the same protein family as N-terminal sugar acceptor binding domains of GT-B type GTs. Subsequent genetic and biochemical studies proved that Alg13 forms a complex with Alg14 on the surface of ER, where the catalytic reaction is carried out (Gao et al., 2005).
Since the Alg13 and Alg14 subunits represent only half the molecular weight of a typical intact GT, the unique natural partition of the two-domain enzyme provides the opportunity to study the GT-B type GTs using NMR. Here we present the NMR structure of Alg13, along with data showing regions of significant flexibility and regions of interaction with the sugar nucleotide donor, UDP-GlcNAc. The structure of Alg13 retains typical GT-B structural characteristics in its C-terminal half, but has significant deviations in its N-terminal half that may be important for its interaction with Alg14.
Results
Oligomer formation of Alg13
Fully protonated Alg13 produced well-dispersed 15N-HSQC but gave little signal in the three-dimensional experiments normally used for NMR resonance assignment and structure determination. Gel-filtration chromatography suggested that at high concentrations the protein existed as a dimer, or higher order oligomer. This was supported when the total correlation time of the protein, as measured by relaxation interference (Lee et al., 2006), was determined to be 18 ns (this is typical of a protein with molecular weight ~45 kDa). Further characterization of Alg13 was therefore done through sedimentation equilibrium experiments. At the concentrations used (7 to 54 μM), little dimerization was detected, suggesting that the dimer dissociation constant must be greater than 400 μM, implying that less than 50 % of the protein would be in the dimer form at the concentrations used for NMR experiments. Although this fraction of dimer may be enough to explain the enhanced rotational correlation time and loss of signal intensities through chemical exchange, the relatively weak association is not likely to lead to many inter-subunit NOEs and resultant ambiguities in the assignments of NOE cross peaks used in structure determination. Therefore, data were analyzed in terms of a monomeric structure. However, the loss of signal sensitivity due to the weak dimerization meant that deuteration of Alg13 and subsequent specific protonation of methyl groups and Phe residues were crucial to overcoming data collection difficulties associated with the enhanced correlation times.
Assignment
Backbone assignment of the deuterated Alg13 was carried out using conventional backbone directed experiments. Of the 193 residues identifiable in the 15N-HSQC, 174 are assigned. Figure 2 shows the 1H,15N-HSQC of deuterated Alg13 annotated with the residue number and type of each peak. The only significant missing segment is composed of residues 74 to 79 (all residue numbering in the paper refers to the recombinant His-tagged Alg13, see Experimental Procedures for details).
Figure 2.
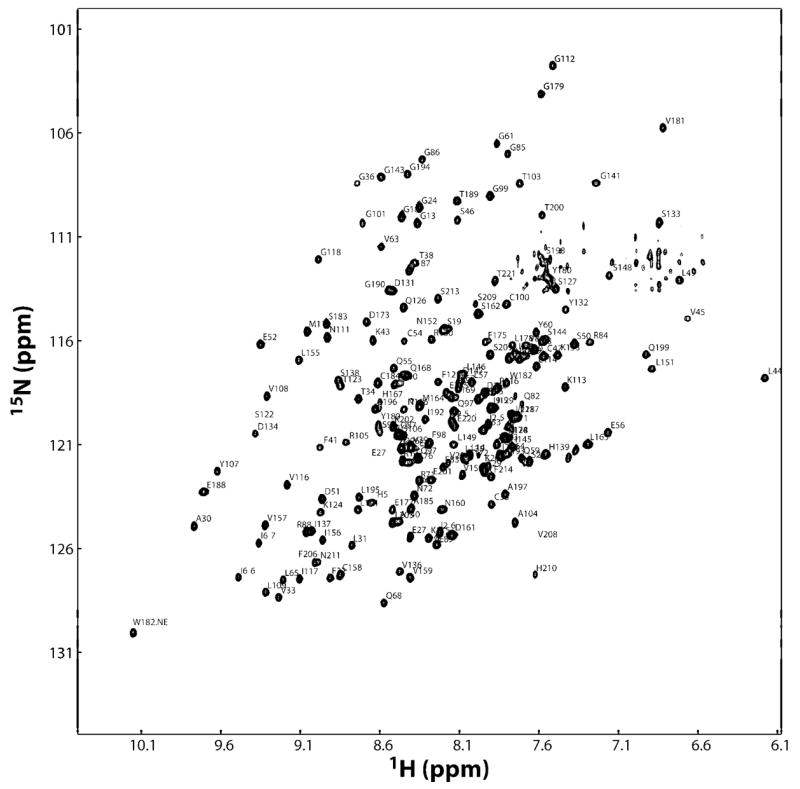
Annotated HSQC spectrum of deuterated Alg13. Each assigned peak is labeled with the residue number and one letter residue name.
To assign the methyl groups in the side chains of specifically methyl-protonated Alg13, a newly coded CCH-TOCSY pulse sequence was used. In this experiment, 13C magnetizations from Cα and Cβ were frequency labeled and transferred to other 13C atoms via TOCSY. To compensate for the loss in signal intensity due to the lack of initial sensitivity enhancement from a 1H-13C INEPT segment, the signal is detected exclusively on methyl groups by transferring only the methyl 13C magnetization to the protons. Utilizing CCH-TOCSY, chemical shifts of all Leu, Val and Ile Cδ1 methyl groups were assigned. We were also able to make full or partial assignment of the chemical shifts of aromatic protons for ten out of twelve Phe residues by correlating Phe amide proton resonances with Hβ cross peaks in the 15N-NOESY-HSQC experiment and then by correlating Phe Hβ resonances with aromatic proton cross peaks in 2D homonuclear NOESY spectra of specifically Phe-protonated Alg13 samples. In all, 72% of 13C atoms were assigned, 89% of 15N atoms were assigned and 38% of all 1H atoms were assigned. Although the percentage of 1H assignments is low, this would not lead to mis-assigned NOEs as all NOESY data were collected with specifically protonated samples in which only signals between assigned protons were visible.
Based solely on the chemical shifts of backbone resonances, a crude assessment of the accuracy of threading models can be made. The structure produced by the mGenTHREADER algorithm (Jones, 1999) shows a typical GT-B structure with two central Rossmann folds flanked by extra helices at both ends as well as a short extra βstrand at the N-terminus. Chemical shift indexing analysis of the assigned backbone chemical shifts showed the secondary structural element distribution of Alg13 largely agrees with the prediction made by the mGenTHREADER algorithm. The only exceptions are residues 95 to 100, which were predicted to form a β-strand but are in fact an α-turn and residues 105 to 110, which form a β-strand despite being predicted to be a α-helix (Figure 1). The latter transformation is especially significant as the replacement of a helix with β-strand disrupted the alternating helix/strand secondary structure arrangement typical of a Rossmann fold, thus creating an irregular mixed α/β domain rather than the predicted N-terminal Rossmann motif.
Figure 1.
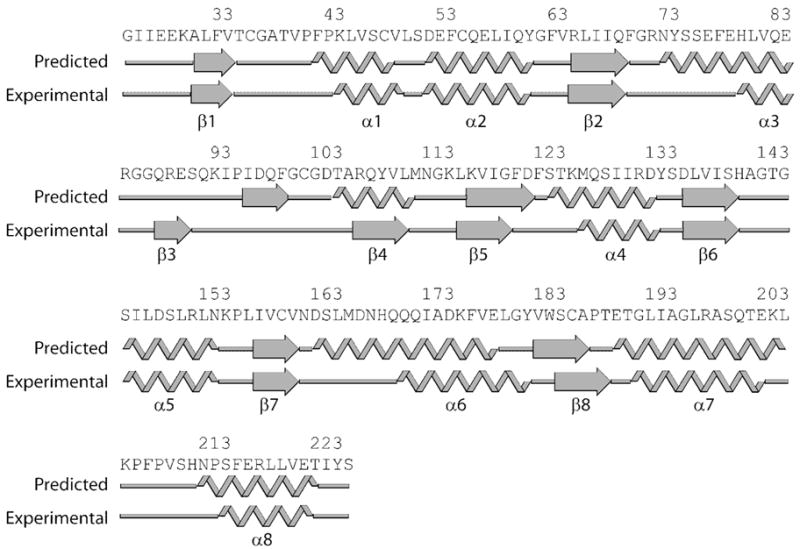
Predicted secondary structural elements and experimentally determined secondary structural elements. Prediction is done using secondary prediction function of mGenthreader (Jones, 1999). Experimental secondary structure is determined using back bone dihedral angles predicted using TALOS and chemical shifts.
Structure determination from NOEs
Data from 15N-edited and 13C-edited NOESY-HSQC of methyl protonated or methyl and Phe protonated Alg13 were first manually assigned, producing 174, 155 and 53 assignments respectively in each spectrum. Our original expectations of obtaining long range methyl-methyl distance restraints from methyl protonated Alg13 was not met owing to lack of proton chemical shift dispersion and broad line widths in the 13C-edited NOESY-HSQC. This necessitated the use of methyl and Phe protonated samples in which the large difference in aromatic proton and methyl proton chemical shifts allow resolution of many more cross peaks. The use of this sample gave us an extra 63 distance restraints, 31 of which were long range distance restraints involving Phe aromatic protons. The manually assigned peak lists were then supplemented with automatically assigned cross peaks using CYANA. This produced a total of 897 distance restraints. Combining the distance restraints with 122 phi and 122 psi angle restraints generated using TALOS, ensembles of the Alg13 structure with typical RMSDs of 1.9 Å for ordered residues were generated with CYANA. The structures were further refined in XPLOR-NIH using the same distance restraints and backbone dihedral angles as well as a set of NH residual dipolar couplings (RDCs) collected in a compressed gel. Figure 3A shows the ensemble of 10 lowest energy structures from a total of 50 structures calculated. The backbone RMSD for the ordered residues fell to 1.4 Å after refinement (RMSD is 1.2 Å if the last helix is excluded). Although the backbone RMSD is higher than the typical RMSD for smaller proteins, it is sufficient to provide a structure of good quality for subsequent studies. A second set of NH RDCs was also collected in an alkylated polyethylene glycol medium, but was not used in structure refinement as this medium caused significant protein precipitation and chemical shift perturbation. However, 89 selected RDCs from cross peaks that were not perturbed by the medium were used to cross validate the refined structures. The cross validation produced an average Cornilescu Q factor of 0.58 for the final set of structures, which, in our experience, is a typical value obtained for NMR structures not refined using the validating RDCs. Table 1 lists some of the statistics for the structure.
Figure 3.
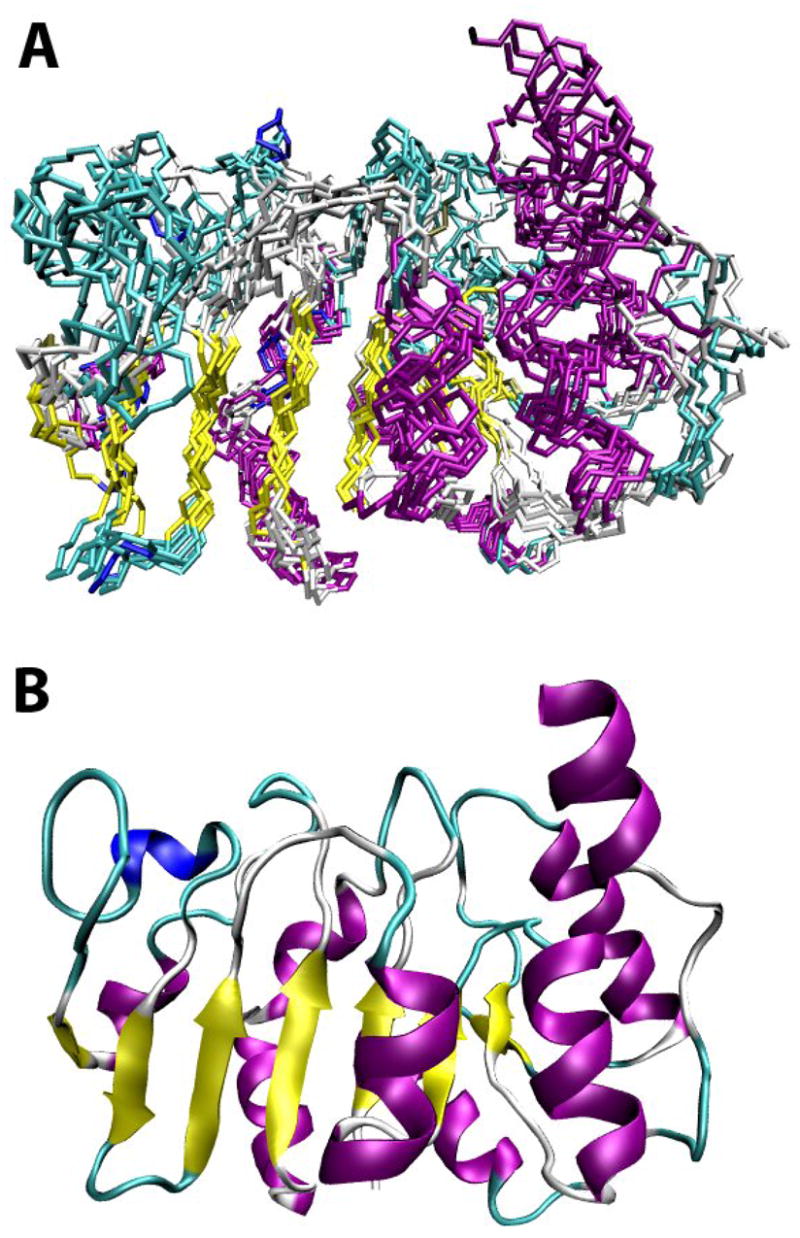
A) Backbone ensemble of 10 lowest energy Alg13 structures from 50 structures calculated using XPLOR-NIH. Residues 30 to 220 are shown. The back bone is colored based on secondary structure. B) Schematic ribbon diagram of the Alg13 structure.
Table 1.
NMR experiments parameters.
| Experiment | Nucleia | 1H freq. (MHz) | t1 points | t2 points | t3 points | F1 SW (Hz) | F2 SW (Hz) | F3 SW (Hz) | Mixing time (ms) |
|---|---|---|---|---|---|---|---|---|---|
| HNCACB | 13C,15N,1H | 600 | 64 | 32 | 512 | 12062 | 2200 | 8000 | |
| HN(CA)CB | 13C,15N,1H | 600 | 64 | 32 | 1024 | 12062 | 2200 | 8000 | |
| HN(CO)CA | 13C,15N,1H | 600 | 50 | 32 | 819 | 4525 | 2200 | 8000 | |
| HN(COCA)CB | 13C,15N,1H | 600 | 68 | 32 | 512 | 12062 | 2200 | 8000 | |
| HNCO | 13C,15N,1H | 600 | 36 | 32 | 512 | 3770 | 2200 | 8000 | |
| HN(CA)CO | 13C,15N,1H | 600 | 36 | 32 | 512 | 3770 | 2200 | 8000 | |
| 15N-edited NOESY-HSQC | 1H,15N,1H | 600 | 80 | 32 | 976 | 8000 | 8000 | 2200 | 150–200 |
| 13C-edited NOESY HSQC | 1H,13C,1H | 900 | 64 | 34 | 890 | 10799 | 11313 | 12626 | 100 |
| CCH-TOCSY | 13C,13C,1H | 800 | 63 | 32 | 512 | 12001 | 6033 | 8000 | 90 |
| 2D 1H-1H NOESY | 1H,13C,1H | 600 | 310 | 2048 | 8000 | 8000 | 150 | ||
| gNHSQC (T2) | 15N,1H | 600 | 128 | 976 | 2200 | 8000 | |||
| 1H, 15N-S3-TROSY | 15N,1H | 800 | 256 | 1024 | 2836 | 11204 |
The nucleus acquired during each evolution time, e. g. 15N,1H indicates 15N is acquired during t1 and 1H is acquired during t2.
During the analysis of amide proton to amide proton contacts from 15N-edited NOESY, a data set that is very diagnostic of β strand pairing, it became obvious that the Alg13 structure adopts a unique topology amongst GT domains. In particular, the N-terminal β-strand (β1, refer to Figure 1 for the numbering of the structural elements) in Alg13, which threads to the last strand in the sugar acceptor binding domain of MurG, forms a parallel β-sheet with residues 64 to 68 (β2) and residues 135 to 138 (β6). It also became clear that the adjacent β strands of residues 105 to 109 (β4) and residues 114 to 118 (β5) form a strong anti-parallel β sheet with residues 114 to 118 also participating in the formation of a parallel β sheet with residues 64 to 68 (β2). Residues 87 to 90, which were predicted to form an unstructured loop, form an extra short β strand next to the anti-parallel β-strand pair. Figure 4 illustrates the difference between the predicted topology of Alg13 and its actual topology. The structural consequence of these changes is the formation of an 8-stranded mixed parallel/anti-parallel β-sheet with the β-strand order of 34521678 (the underlined number indicates anti-parallel orientation). The existence of this extended and mixed β-sheet is not predicted in threading models. The lack of chemical shift assignments for residues 74 to 79 could easily be the result of dynamics in an unstructured loop in this region. Taken together, these changes in the N-terminal half of Alg13 mean that instead of having a Rossmann fold, the N-terminal half is only a Rossmann-like fold. Examination of all known GT structures showed that this fold is unique amongst GTs.
Figure 4.
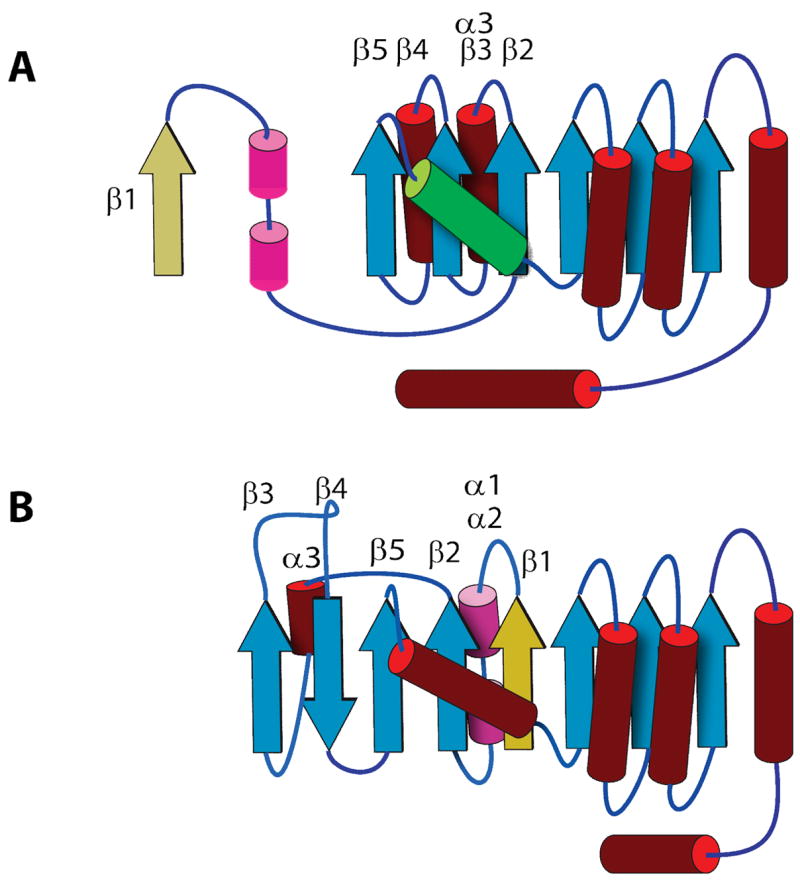
A) Schematic illustration of the predicted topology of Alg13, The numbering of the element is according to the scheme from Figure 1. Note that the predicted helix after β2 gave rise to both α3 and β3. B) Schematic illustration of the experimentally determined topology of Alg13.
The second half of the protein adopts the traditional Rossmann fold, with a three-strand parallel β-sheet and two alpha helices connecting the strands clustered on the same side, as predicted by the threading model. However, sugar donor binding domains of GT-B enzymes typically end with three helices, with the last helix stretching back to and interacting with the sugar acceptor binding domain, perhaps acting as a tether that ensures close contacts between the separate domains. In Alg13, the protein possesses only two helices at its C-terminus. Since no long range distance restraints between the last helix and the rest of the protein exist, the orientation of the last helix is difficult to define.
A possible reason for the lack of long range NOEs connecting the last helix to the rest of the protein was uncovered in data from residue specific rotational correlation time measurements of Alg13. These were carried out using a recently devised constant time relaxation interference experiment (Liu & Prestegard, In press). The measurements showed that C-terminal helix residues possess significantly shorter correlation times than the rest of the protein (Figure 5). This indicates the presence of internal motion relative to other parts of the protein. The motion is apparently fast (ns time scale) as no significant motion at the μs and ms time scale was observed in a relaxation-compensated CPMG experiments (Loria et al., 1999). Some experimental information regarding the possible placement of the last helix will, however, come from titration of Alg13 with UDP-TEMPO as described below. Using these data, the most frequently populated position of the last helix appears to be between helices 5 and 6.
Figure 5.
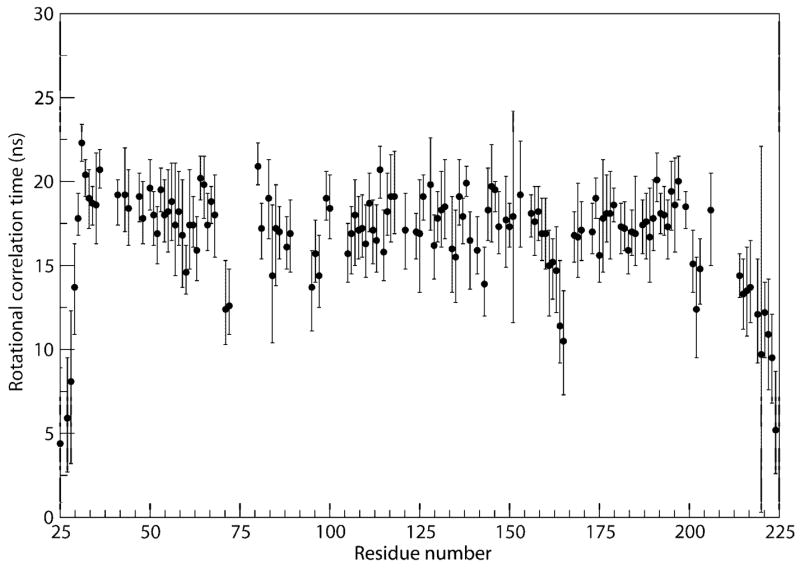
Rotational correlation time measurements of Alg13. The C-terminus is considerably more dynamic than the rest of the protein.
It is interesting to note that besides the residues in the C-terminal helix, residues 164 and 165 also have shorter correlation times, and the cross peak intensities of residues 166 and 167 in the 1H,15N-HSQC were weaker than residues surrounding it in the sequence. This confirms that the segment from residues 164 to 167, which is close to the proposed sugar binding site, may be undergoing conformational exchange. However, a relaxation-compensated CPMG experiment showed that the dynamics of these residues did not change in the presence of the ligand, thus the role of these motions in recognizing sugar is unclear.
Interaction of Alg13 with UDP-GlcNAc and UDP-TEMPO
To probe the interaction of Alg13 with the glycosyl donor, UDP-GlcNAc, we first titrated Alg13 with the native donor, and monitored chemical shift changes in the 15N-HSQC cross peaks of the protein. Figure 6A shows the normalized changes in HSQC cross peak chemical shifts at a UDP-GlcNAc concentration of 2.6 mM. The regions that are most disturbed by the ligand include residues 35, 126, 143 to 145, 147, 148, 150, 151, 158 to 160 and 166. Mapping these residues on the structures of Alg13 (Figure 6B) showed the residues are all in the same part of the molecule with those in helix 5 being most perturbed. Although no precise characterization of the Kd was carried out, it was observed that it took a ligand-protein ratio of 10:1 to saturate the protein sample. Based on this observation, the Kd should be near 0.5 mM. This is similar to sugar donor dissociation constants observed in other GTs (Macnaughtan et al., 2007).
Figure 6.
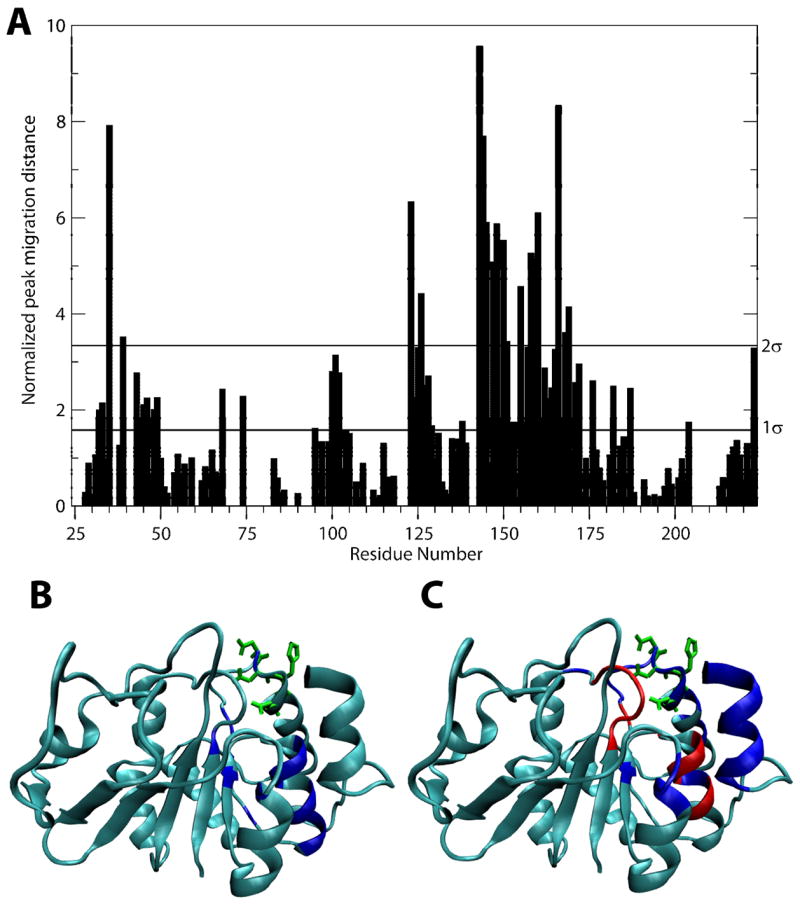
A) Chemical shift changes produced by 2.6 mM UDP-GlcNAc. The two horizontal lines on the graph represent standard deviations in chemical shift changes. B) Mapping of UDP-GlcNAc-generated chemical shift changes on Alg13 structure. Residues with perturbation bigger than two standard deviations are colored red. This includes residues 35, 39, 123, 126, 143, 144, 145, 147, 148, 150, 155, 158, 159, 160, 166, 168 and 169. Side chains of possible carbohydrate-interacting residues (residues 165 to 158) are shown in green. C) Mapping of UDP-TEMPO-generated signal dispersion on Alg13 structure. Residues 41, 124 to 126, 139, 146, 150, 161 to 169, 171, 174, 175 and 213 to 220 are peaks that have disappeared in the presence of TEMPO and reappeared after reduction and are colored in blue. Residues 35 to 39, 141, 143, 144, 145, 147, 148 and 160 are peaks that have disappeared in the presence of TEMPO but failed to reappear after reduction of TEMPO and are colored in red. Side chains of possible carbohydrate-interacting residues (residues 165 to 158) are shown in green.
To derive further structural information as well as unambiguously map the ligand binding site, we also titrated the protein with UDP-TEMPO and monitored the intensity loss of HSQC cross peaks. TEMPO contains a nitroxide radical, and loss of intensity is a good measure of amide proton T2 reduction from the presence of this paramagnetic site. The loss can be related to a nitroxide to proton distance in an approximate 1/r6 manner, particularly when proton spin diffusion effects are minimized by deuteration of other sites in the protein (Battiste and Wagner, 2000; Jain et al., 2001). Addition of just half an equivalent of UDP-TEMPO (0.2 mM) caused peaks from residues 35, 36, 38, 39, 141, 143 to 148, 158, 160, 163 to 169 and 213 to disappear. Further additions of UDP-TEMPO led to the disappearance of peaks from residues 41, 124 to 129, 150, 151, 155, 161, 162, 171, 174,175, 214 to 218, 220 and 221. Reduction of TEMPO with ascorbic acid, which should reverse the effects of paramagnetic interactions, leads to reappearance of all peaks except those from residues 35, 36, 38, 39, 127, 141, 143 to 145, 147, 148, 158 and 160. The failure of these peaks to reappear can be due to exchange broadening. Figure 6C shows the most strongly perturbed (red) and second most strongly perturbed set (blue). They overlap well with residues showing chemical shift perturbation in the UDP-GlcNAc titration except TEMPO influences a greater number of residues in the area as well as residues in the C-terminus of the protein, which showed only moderate chemical shift perturbation in the UDP-GlcNAc titration. Paramagnetic relaxation enhancement is usually a more reliable indicator of interactions than chemical shift perturbation as chemical shifts of remote groups can be affected by allosteric effects. It is interesting to note that the C-terminal helix is perturbed by TEMPO while helix 7, which immediately precedes the last helix, shows no change. This implies that although the position of the last helix is dynamic, as implied by the rotational correlation time measurements, the position of the helix is close to the ligand binding site for a significant fraction of time. Based on these data, the last helix should either be above and perpendicular to the β strands, or parallel to the strands and close to helices 5 and 6. Thus, the linker between helix 7 and the last helix is most likely not fully extended. We also tried to quantify the distances between the paramagnetic center on TEMPO and amide protons of perturbed residues. However, most cross peaks from residues perturbed by TEMPO disappeared after the first addition, thus making quantitation impossible. Some peaks from residues in the C-terminal helix did remain even at a TEMPO concentration of 2.5 mM and using the method specified by Jain et al. (Jain et al., 2001), the distance from this helix to the paramagnetic center is estimated to be from 10 to 13 Å.
Discussion
The Alg13/Alg14 pair has been classified as a member of the GT-1 family by the CAZy database (www.cazy.org) and its structure, as described above, does have many of the characteristics predicted based on the preferred threading of its sequence to GT-B type GTs, namely two Rossmann-like folds, and the presence of a sugar-nucleotide binding site between helices of the second Rossmann fold. In fact, the second Rossmann fold is very well preserved; it is only the helices following the fold that deviate in structure from the threading model. Aligning the sequence of Alg13 with that of MurG, for which a crystal structure with UDP-GlcNAc exists, showed that the MurG residues that made contact with UDP-GlcNAc in the complex structure (residues 258 to 260, 263 282,283 and 286) are aligned with Alg13 residues that were also perturbed in the UDP-GlcNAc and UDP-TEMPO titrations (residues 143,144, 147, 167,168 and 171). This indicates that the location of the UDP-GlcNAc binding site is equivalent in the two proteins. It is also interesting to note that residues 143, 144 and 147, which are aligned with MurG residues contacting the nucleotide, were perturbed far more than the residues 167, 168 and 171 which are aligned with MurG residues contacting the sugar. A previous STD NMR study has shown that GnTV, a GT catalyzing branching of mammalian N-glycans, binds nucleotide sugar primarily by interacting with the nucleotide portion of the sugar donor rather than the sugar portion of the donor (Macnaughtan et al., 2007), thus it is reasonable to observe more perturbation in residues contacting the nucleotide of UDP-GlcNAc. Some additional information regarding possible sugar interacting residues in Alg13 can also be derived by using the structure of MurG as a guide and taking into consideration the polarity of the amino acids. This type of analysis shows that residues 165 to 168 are most likely the residues involved in contacting the sugar as they are aligned with MurG residues that contacts the sugar. They are also perturbed by UDP-TEMPO (Figure 6C), and they exhibit both slow and fast time scale dynamics.
Despite the similarity of the sugar donor binding domain to other GT-B type GTs and the high threading scores (p-value < 0.001) to other GT-B enzymes, the sequence identity between Alg13 and other GT-B sugar binding domains is quite low (<13.5 %) and the structure of Alg13 shows several significant differences compared to other known GT-B structures, particularly in the N-terminal half and the extreme C-terminus. The first half of Alg13 is no longer a Rossmann fold but an irregular 3 layer α/β/α sandwich that has no equivalent topology in existing GT structures. The central β-sheet is an eight-stranded mixed anti-parallel and parallel (34521678) sheet that has one more parallel strand and an inserted anti-parallel β-strand that are absent in the GT-B threading model. The distinct topology of Alg13 is partly the result of the presence of the N-terminal strand and helix, which thread to parts of the sugar acceptor binding domain of GT-B enzymes rather than the sugar donor binding domain. At least in the absence of the sugar acceptor domain, the strand and helix are incorporated into the main sheet rather than forming a flexible linker, as predicted by the threading algorithm. The helix-for-strand substitution at residues 105 to 110 also led to deviations from the Rossmann fold by creating an anti-parallel strand. These unique features of Alg13 may warrant the reclassification of Alg13/Alg14 bipartite enzyme as a new class of GTs and make it interesting to explore more broadly the relationship of the structure to other proteins found in the PDB.
It is noteworthy that GT-A type GTs possesses some similarity to what is observed in Alg13. GT-A type GTs have 7 strands as opposed to 8, but they do have an intervening anti-parallel strand (position 5 instead of 2). Searching more broadly, using the DALI structural homology search tool (Holm and Sander, 1995) on only the N-terminal half of Alg13, revealed a number of other proteins containing a similar topology. Many of them are molybdenum cofactor binding proteins. In fact, the three structures deemed most similar to the N-terminal of Alg13 (backbone RMSDs between 3.5 and 4 Å) are all molybdenum cofactor biosynthetic enzymes. There is no reason that there should be a functional relationship between Alg13 and these proteins, but it is reassuring that the structural utility of this type of fold is documented.
The last two helices in the enzyme also clearly depart from positions in models built by threading to GT-B GTs. The last helix unfortunately shows no long range NOE-based distance restraints to the rest of the enzyme. Yet the TEMPO perturbation data clearly show the helix to be associated with the protein. It is unlikely to be directly involved in glycosyl-donor binding as the cross peaks associated with this helix did not move appreciably during UDP-GlcNAc titration. Our hypothesis is that the last helix is involved in interaction with Alg14. It may be that, in the presence of Alg14, the helix will move away and adopt a new orientation, and that contacts with other parts of Alg13 would be at a minimum in the absence of Alg14. Experimental evidence from mutagenesis studies also indicates the C-terminal region plays a crucial role in modulating Alg13/Alg14 interactions (Gao et al., 2007).
It is tempting to consider the possibility that the tendency of Alg13 to dimerize is related to the normal tendency of Alg13 and Alg14 to interact. The predicted folds of Alg13 and Alg14 are, in fact, similar, and a second Alg13 in the dimer might substitute for Alg14. Examination of conventional GT-B structures shows that, if Alg13 dimerization is in a manner similar to the interactions between sugar acceptor and sugar donor binding domains of GT-B type enzymes, the C-terminal helix, as well as helix 5 (Figure 1) should be part of the dimer interface. However, no concentration-dependent changes in the chemical shifts of these residues were observed and the C-terminal helix remained dynamic relative to the rest of the protein even at the highest concentration of protein possible. Therefore, the dimerization interface for Alg13 is most likely different from its interaction interface with Alg14.
The topology of Alg13 is different from all the GT structures known to date. Of the 50 unique glycosyltransferases structures, 47 are either of GT-A type or GT-B type GTs. The three previous exceptions are bacterial sialyltransferases Cst I & II, whose fold is similar to the GT-A fold (Breton et al., 2006; Chiu et al., 2007; Chiu et al., 2004), and the GT domain of the bifunctional peptidylglycan cross-linking enzyme, which is a mostly αhelical domain (Lovering et al., 2007). In fact, none of the proteins similar to the N-terminus of Alg13 contains a Rossmann fold at its C-terminus, thus it is possible that not only is the Alg13 fold unique amongst GTs, it might be unique amongst all known protein structures.
It is tempting to speculate on the evolutionary connection between Alg13 and other glycosyltransferases. Alg13 and Alg14 most likely derived from different domains of a GT-B enzyme, with Alg13 evolving from the C terminus of a GT-B enzyme. Within this C-terminal comparison, the N-terminal half of Alg13 is not involved in binding ligands. Hence it may have been allowed more structural plasticity than the C-terminal half of Alg13. This evolutionary deviation must have occurred during the development of the first eukaryotic organisms as Alg13 and Alg14 are well conserved proteins in all eukaryotic organisms. The recent discovery of protozoan Alg13/Alg14 homologs (Averbeck et al., 2007) lends weight to this hypothesis. In protozoa, Alg13 and Alg14 once again exist as a single polypeptide chain. However, in two protozoa the order of Alg13 and Alg14 on the chain is inverted. The existence of the swapped domain order brings to light the possibility that after separation, the two domains might have rejoined again in protozoa. These examples demonstrate the possible flexibility available in combining GT substrate domains. It is also worth mentioning that MurG may not be the bacterial protein from which Alg13/14 are derived. The discovery of protein N-glycosylation system in the Gram-negative bacteria Campylobacter jejuni (for review see (Weerapana and Imperiali, 2006)) shows that there is precedence for the existence of N-glycosylation system in prokaryotes. These systems could have served as the basis for the eukaryotic N-glycosylation system. Indeed, the Campylobacter jejuni protein PglA, which catalyzes an analogous reaction in the Campylobacter jejuni N-glycoslyation pathway, is similar in size as MurG and threads to the MurG structure with a p-value much less than 0.0001.
Why the sugar-donor binding domain and the acceptor-binding domain exist separately in Alg13 and Alg14 in some organisms prompts some interesting speculation. Potentially, bipartitioning can offer many advantages such as flexibility in regulation and ability to recruit substrates in both the cytosol and in the membrane. Alg13 might also be a general UDP-GlcNAc carrier, regulating the delivery of UDP-GlcNAc to a variety of membrane-associated proteins involved in complex carbohydrate synthesis. However, a search through the yeast protein-protein interaction database hosted at the Munich Information Center for Protein Sequences (Guldener et al., 2006) showed that other than Alg14, no proteins are known to interact with Alg13. Nevertheless, the unusual structure of the N-terminal half of Alg13 and its structural similarity to molybdenum cofactor binding proteins may provide some additional clues to its function.
Another item of most interest is what the structure of Alg13 might be in the presence of Alg14. Alg14 is a membrane-associated protein, having a single trans-membrane helix and cytosolic domain much like GT-B GTs. It may well be that the C-terminal domain of Alg14 folds independently and its structure, as well as the structure of its complex with Alg13 can be pursued by methods similar to those described above.
Experimental Procedure
Protein expression and purification
The Alg13 ORF, cloned directly from yeast genomic DNA, was inserted into the pET15b vector (Stratagene), which added a His tag and a thrombin cleavage site (a total of 22 residues) to the N-terminus of the protein. All residue numbering in the manuscript refers to this recombinant His-tagged Alg13; thus, the actual Alg13 sequence consists of G24 to S224. D2O conditioned E. coli strain BL21(DE3) was used to express the protein in D2O M9 minimal media in all subsequent sample preparations. The only exception was the unlabeled Alg13 sample used in the sedimentation equilibrium experiment, which was expressed in LB with unconditioned BL21(DE3). To produce deuterated Alg13, D2O-conditioned BL21(DE3), transformed with the aforementioned vector, was grown over night in 50ml of D2O M9 media containing 10g/L uniformly 13C, 2H-labeled glucose and 1g/L 15N-NH4Cl at 37°C. The overnight culture was used to seed a 500ml D2O M9 media containing 2g/L uniformly 13C, 2H-labeled glucose and 1g/L 15NH4Cl. This was grown at 37°C to O.D.600 of 1.0, upon which time protein expression was induced with the addition of 0.5mM IPTG. The culture was harvested after growing for 16 hours at 18°C.
Selective protonation of Leu, Val and Ile.δ1 methyl groups in Alg13 was achieved by following a protocol outlined in Goto et al. (Goto et al., 1999). In particular, the culture was grown according to the procedure above except one hour before induction, uniformly 13C labeled 3,3-2H2-α-ketobutyrate and uniformly 13C labeled 3-2H-α-ketoisovalerate (Cambridge Isotope Laboratories) were added to the large scale culture to concentrations of 100mg/L. Specific protonation of Phe was accomplished by adding protonated L-Phe to the large scale culture one hour before the induction to a concentration of 100mg/L. To make samples with Leu, Val, and Ile.δ1 methyl prottonation, as well as Phe protonation, isotopically labeled α-ketobutyrate, α-ketoisovalerate, and unlabeled L-Phe were added to the media to concentrations of 100mg/L one hour before induction.
Induced cells were lysed using a French press and the cleared supernatant was applied to a HIS-select HF Ni affinity column (Sigma, St. Louis, MI). Pure His-tagged Alg13 was eluted with 300mM imidazole after extensive washing of the column with 100mM NaCl, 20mM phosphate, pH 8.0 buffer. The protein eluted was homogenous according to SDS-PAGE. The typical protein yield was approximately 10mg/L. To fully protonate amide protons, deuterated protein was treated under mildly denaturing conditions (2 M urea, 100 mM NaCl, 20 mM phosphate, pH 6.7) for 16 hours at room temperature. Pure protein was buffer exchanged into 100mM NaCl, 10mM DTT, 20mM phosphate, pH 6.7 buffer using Centricon concentrators (10×4 fold). Typical concentrations of the protein samples were approximately 400 μM.
Characterization of Alg13 Oligomerization
To examine the Alg13′s potential for oligomerization, gel filtration chromatography was carried out using a Superdex 75 gel filtration column (GE Health Care). 200 μL of 0.3 mM Alg13 in 150mM NaCl, 1 mM DTT and 20 mM phosphate, pH 7 buffer was loaded onto a Superdex 75 16/60 column and eluted with the same buffer at a flow rate of 1 ml/min. To more accurately gauge the fraction of Alg13 dimer that might be in the NMR sample, an attempt to measure the dimerization constant of Alg13 was made using sedimentation equilibrium measurements. The sedimentation equilibrium experiment was performed using a Beckman-Coulter optima XL-A analytical ultracentrifuge with a four-position AN-60-Ti rotor and cells with six-sector epon center pieces. Unlabeled Alg13 in 100 mM NaCl, 1 mM DTT, 20 mM phosphate, pH 6.7 was prepared with concentrations of 6.9, 16.9 and 27.0 μM and spun at 20 °C and at five speeds ranging from 20,000 to 42,000 rpm. Scans were collected in a radial increment mode with 0.001 increments and 4 point averaging at 280 nm. The samples were allowed to equilibrate for 24 hours prior to recording the scan. The data were fit using the Ultrascan 7.0 software (www.ultrascan.uthscsa.edu). To explore a more extended range of Kds, the experiment was also carried out with protein concentrations of 14, 34 and 54 μM and absorbance data were collected at 291 nm.
Weak alignment of Alg13
For the measurement of residual dipolar couplings, Alg13 was aligned in both compressed positively charged 7% polyacrylamide gels (Cierpicki and Bushweller, 2004) and 4% C12E5 polyethylene glycol (PEG) bicelles. In particular, positively charged 7% acrylamide produced by mixing equal proportions of (3-acrylamidopropyl)-trimethylammonium chloride (APTMAC, Sigma-Aldrich, Inc.) and normal acrylamide was formed in 3.2 mm inner diameter plastic tubes. The gel was then washed extensively with deionized water and desiccated. The dry gel was swelled using 500 μl of protein solution inside a 5mm Shigemi tube and compressed to a length of 14 mm using the upper plunger. 4% C12E5 PEG alignment media was prepared according to Ruckert & Otting (Ruckert and Otting, 2000). Specifically, 8% C12E5 PEG in 100mM NaCl, 10 mM DTT, 20 mM phosphate, pH 6.7 buffer was converted to a liquid crystalline state by additions of successive 4-μl aliquots of hexanol with rigorous vortexing until the solution was clear. The stock PEG alignment media was diluted to 4% with the addition of equal volume of protein solution. The extent of alignment was examined using the splitting of the signal from 2H1HO in the buffer. In the case of the compressed gel, the deuterium splitting was around 5 Hz where as in the 4% PEG medium it was usually around 15 Hz.
NMR data collection
Varian Inova 900, and 600 MHz spectrometers equipped with cold probes were used to collect data for Alg13 samples. Pulse sequences for all experiments were from the Biopack distributed by Varian Inc unless stated otherwise. For backbone assignment purposes, HNCO, HN(CA)CO, HN(COCA)CB (modified from the Biopack version to correct an offset error in an off resonance CO inversion pulse), HN(CO)CA and two HNCACB experiments with 13C evolution time of 3.5 and 7 ms respectively were collected. To assign the specifically protonated methyl groups in a deuterated background, a CCH-TOCSY sequence was written in house. The pulse sequence selectively applies a 90° pulse to Cα and then transfers the magnetization throughout the carbon on the side chains using TOCSY. The magnetization on the methyl group is finally transferred to protons for detection. Since Cα, Cβ chemical shifts for each residue are known, the correlation between Cα, Cβ and methyl groups allowed each methyl to be assigned to a specific residue. NOE information was derived from 15N-edited NOESY-HSQC spectra collected on specific methyl protonated Alg13 and 13C-edited NOESY-HSQC spectra collected on specific methyl protonated Alg13 as well as on specific Phe and methyl protonated Alg13.
HN residual dipolar coupling constants (RDCs) were measured from differences in peak positions in 1H,15N-S3-TROSY (Permi and Annila, 2000) and HSQC spectra on aligned and isotropic samples. Although additional N-C′ and HN-C′ RDCs would have been valuable, these are in general small and difficult to measure in larger proteins.
The overall correlation time of the Alg13 was measured using a 15N-filtered one dimensional relaxation interference difference experiment according to Lee et al. (Lee et al., 2006) and the residue specific correlation times were measures with recently devised constant time relaxation interference experiment (Liu & Prestegard, In press). Table 2 lists detailed experimental parameters.
Table 2.
Summary of structural statistics of the ensemble of 10 (out of total 50 calculated) Alg13 solution structures
| NOE-derived distance restraints | ||
| Short range (|i-j|<2) | 317 | |
| Medium range (2≤|i-j|<5) | 246 | |
| Long range (|i-j|>5) | 334 | |
| Total | 897 | |
| Backbone dihedral angle restraints | ||
| φ | 122 | |
| ψ | 122 | |
| RDC | ||
| NH | 117 | |
| Axial component | 9.2 ± 0.7 Hz | |
| Rhombicity | 0.6 ± 0.1 | |
| RMSD | All | Well-defined regionsa |
| Backbone | 2.1 Å | 1.4 Å |
| All heavy atoms | 2.7 Å | 2.0 Å |
| Distance restraint violation > 0.5 Å | 0 | |
| Dihedral angle violation > 10 ° | 0 | |
| Backbone dihedral angle distribution | ||
| % in most favored regions | 90.1 | |
| % in additionally allowed regions | 9.9 | |
| % in other regions | 0 | |
Well ordered residues are defined as residues with sum of phi and psi order parameters > 1.8. This includes residues 29 – 34, 40 –61, 63 –67, 72 –73, 80 –84, 87 –88, 98 –99, 105 –109, 112 –117, 120 –121, 125 –132, 136 –138, 143 –159, 167 –178, 183 –199, 213 – 221.
Structure calculation
A threading model of the Alg13 structure was obtained using the mGenTHREADER algorithm (Jones, 1999) via the online server at http://bioinf.cs.ucl.ac.uk/psipred/. Backbone dihedral angle restraints for secondary structure determination and subsequent 3D structure determination were generated from Alg13 backbone chemical shifts using TALOS (Cornilescu et al., 1999). Approximately 382 unambiguous cross peaks in 15N and 13C-edited NOESY-HSQC spectra were manually assigned based on chemical shift correlations in the HNCACB, HN(CO)CACB and CCH-TOCSY experiments as well as on the basis of secondary structural information. Partially assigned peak lists were then further assigned using the automatic NOE assignment and structure calculation algorithm implemented in CYANA (Guntert et al., 1997; Herrmann et al., 2002). In total, 50 structures were calculated and the top 20 most energetically favorable structures were kept. CYANA calculated structures were further refined with compressed gel RDCs and CYANA defined distance restraints using XPLOR-NIH (Schwieters et al., 2003). The refinement protocol used in XPLOR-NIH followed closely the procedure outlined in the example script refine.py from the GB1 RDC refinement example in the XPLOR-NIH distribution. However, the number of steps during torsion angle high temperature dynamics has been increased from 5000 to 7500 and the number of steps at each temperature during cooling has been increased from 100 to 150. The weighting factor for the van der waals potential during cooling has been changed from a range of 0.9 to 0.8 to a range of 0.94 to 0.84. The radius-of-gyration potential has also been used for residues 30 to 220 as the protein is globular (Kuszewski et al., 1999). The dihedral angle database potential for side chain and back bone dihedral angles has also been activated (Clore and Kuszewski, 2002; Kuszewski and Clore, 2000). For RDC refinement, the peptide plane ω angles were fixed using the IVM_groupRigidBackbone function to eliminate any NH bond vector rotation. A histogram of NH RDCs was used to obtain initial values of 8.6 Hz and 0.8 for the Da and Rh parameters for the alignment tensor (Clore et al., 1998). The values of Da and Rh were allowed to vary during the high temperature step and the final average values of Da and Rh were 10.9 ± 0.8 Hz and 0.6 ± 0.1 for the ten lowest energy structures (Clore and Schwieters, 2004). Torsion angle dynamics has been used exclusively for refinement at high temperature as the use of Cartesian dynamics will distort peptide plan ω angles to satisfy NH RDC potentials. Again 50 total structures were calculated and the top 10 structures were used to form the final ensemble.
Titration of Alg13 with UDP-GlcNAc & UDP-TEMPO
To probe the interaction of Alg13 with its ligands, Alg13 was titrated with both UDP-GlcNAc, the native sugar donor, and UDP-TEMPO (Jain et al., 2001) a radical-containing UDP-sugar mimetic. In the titration of Alg13 with UDP-GlcNAc, 15N-HSQCs of 0.35mM [15N, 2H]-Alg13 sample containing 0.2, 0.4, 0.7, 1.1, 1.5, 2.0 and 2.6 mM UDP-GlcNAc were acquired. In the titration of Alg13 with UDP-TEMPO, 15N-HSQCs of 0.35mM [15N, 2H]-Alg13 sample containing 0.2, 0.3, 0.5, 0.7, 0.9, 1.1, 1.5, 2.0 and 2.5 mM UDP-TEMPO were acquired. The spectra were overlayed and changes in chemical shifts tabulated. As a control to ensure that peaks only disappeared because of paramagnetic broadening, 5 mM ascorbic acid was added to the UDP-TEMPO titrated sample. Ascorbic acid specifically reduces the radical, therefore peaks that disappeared owing to paramagnetic broadening should reappear whereas peaks broadened by conformational exchange would remain undetectable. To eliminate the possibility of broadening due to adventitious binding of the hydrophobic TEMPO moiety HSQCs of Alg13 with and without 3 mM unligated TEMPO were also run.
Chemical shift assignments of Alg13 have been deposited in BMRB with the accession number 15617. Coordinates of the Alg13 structure have been deposited in RCSB with the PDB ID of 2jzc. The Alg13 structure is solved as part of the community outreach effort of the Northeast Structural Genomics Consortium and Alg13 bears the NESG target number of YG1.
Acknowledgments
This work is supported by NIH NIGMS grant number GM033225 (JHP) and GM068692 (BI) as well as a grant from the NIH in support of the Northeast Structural Genomics Consortium (U54-GM074958, G. Montelione, PI). XW is supported by Fellowships from Alberta Heritage Foundation for Medical Research and Canadian Institutes for Health Research. We also want to thank Dr. Fang Tian of University of Georgia for contributing the CCH-TOCSY pulse sequence and numerous insightful discussions, Dr. Jeffery Urbauer of University of Georgia for performing the sedimentation equilibrium experiment as well as assistance in analyzing the data, and Dr. Geert-Jan Boons and Dr. Andre Venot for the synthesis of UDP-TEMPO.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Averbeck N, Keppler-Ross S, Dean N. Membrane topology of the Alg14 endoplasmic reticulum UDP-GlcNAc transferase subunit. J Biol Chem. 2007;282:29081–29088. doi: 10.1074/jbc.M704410200. [DOI] [PubMed] [Google Scholar]
- Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- Breton C, Snajdrova L, Jeanneau C, Koca J, Imberty A. Structures and mechanisms of glycosyltransferases. Glycobiology. 2006;16:29r–37r. doi: 10.1093/glycob/cwj016. [DOI] [PubMed] [Google Scholar]
- Burda P, Aebi M. The dolichol pathway of N-linked glycosylation. Bba-Gen Subjects. 1999;1426:239–257. doi: 10.1016/s0304-4165(98)00127-5. [DOI] [PubMed] [Google Scholar]
- Chantret I, Dancourt J, Barbat A, Moore SEH. Two proteins homologous to the N- and C-terminal domains of the bacterial glycosyltransferase Murg are required for the second step of dolichyl-linked oligosaccharide synthesis in Saccharomyces cerevisiae (vol 280, pg 9236, 2005) J Biol Chem. 2005;280:18551–18552. doi: 10.1074/jbc.M413941200. [DOI] [PubMed] [Google Scholar]
- Chiu CPC, Lairson LL, Gilbert M, Wakarchuk WW, Withers SG, Strynadka NCJ. Structural analysis of the alpha-2,3-sialyltransferase cst-i from Campylobacter jejuni in apo and substrate-analogue bound forms. Biochemistry. 2007;46:7196–7204. doi: 10.1021/bi602543d. [DOI] [PubMed] [Google Scholar]
- Chiu CPC, Watts AG, Lairson LL, Gilbert M, Lim D, Wakarchuk WW, Withers SG, Strynadka NCJ. Structural analysis of the sialyltransferase CstII from Campylobacter jejuni in complex with a substrate analog. Nature Structural & Molecular Biology. 2004;11:163–170. doi: 10.1038/nsmb720. [DOI] [PubMed] [Google Scholar]
- Cierpicki T, Bushweller JH. Charged gels as orienting media for measurement of residual dipolar couplings in soluble and integral membrane proteins. J Am Chem Soc. 2004;126:16259–16266. doi: 10.1021/ja046054g. [DOI] [PubMed] [Google Scholar]
- Clore GM, Gronenborn AM, Szabo A, Tjandra N. Determining the magnitude of the fully asymmetric diffusion tensor from heteronuclear relaxation data in the absence of structural information. J Am Chem Soc. 1998;120:4889–4890. [Google Scholar]
- Clore GM, Kuszewski J. chi(1) Rotamer populations and angles of mobile surface side chains are accurately predicted by a torsion angle database potential of mean force. J Am Chem Soc. 2002;124:2866–2867. doi: 10.1021/ja017712p. [DOI] [PubMed] [Google Scholar]
- Clore GM, Schwieters CD. How much backbone motion in ubiquitin is required to account for dipolar coupling data measured in multiple alignment media as assessed by independent cross-validation? J Am Chem Soc. 2004;126:2923–2938. doi: 10.1021/ja0386804. [DOI] [PubMed] [Google Scholar]
- Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol Nmr. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- Coutinho PM, Deleury E, Davies GJ, Henrissat B. An evolving hierarchical family classification for glycosyltransferases. J Mol Biol. 2003;328:307–317. doi: 10.1016/s0022-2836(03)00307-3. [DOI] [PubMed] [Google Scholar]
- Fuster MM, Esko JD. The sweet and sour of cancer: Glycans as novel therapeutic targets. Nat Rev Cancer. 2005;5:526–542. doi: 10.1038/nrc1649. [DOI] [PubMed] [Google Scholar]
- Gao XD, Moriyama S, Miura N, Nishimura SI. C-termini of both alg13 and alg14 proteins are required for formation of the alg13/alg14 complex. Glycobiology. 2007;17:1277. [Google Scholar]
- Gao XD, Tachikawa H, Sato T, Jigami Y, Dean N. Alg14 recruits alg13 to the cytoplasmic face of the endoplasmic reticulum to form a novel bipartite UDP-N-acetylglucosamine transferase required for the second step of N-linked glycosylation. J Biol Chem. 2005;280:36254–36262. doi: 10.1074/jbc.M507569200. [DOI] [PubMed] [Google Scholar]
- Goto NK, Gardner KH, Mueller GA, Willis RC, Kay LE. A robust and cost-effective method for the production of Val, Leu, Ile (delta 1) methyl-protonated N-15-, C-13-, H-2-labeled proteins. J Biomol Nmr. 1999;13:369–374. doi: 10.1023/a:1008393201236. [DOI] [PubMed] [Google Scholar]
- Guldener U, Munsterkotter M, Oesterheld M, Pagel P, Ruepp A, Mewes HW, Stumpflen V. MPact: the MIPS protein interaction resource on yeast. Nucleic Acids Research. 2006;34:D436–D441. doi: 10.1093/nar/gkj003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntert P, Mumenthaler C, Wuthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Dali - a Network Tool for Protein-Structure Comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- Jain NU, Venot A, Umemoto K, Leffler H, Prestegard JH. Distance mapping of protein-binding sites using spin-labeled oligosaccharide ligands. Protein Science. 2001;10:2393–2400. doi: 10.1110/ps.17401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT. GenTHREADER: An efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol. 1999;287:797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- Kelleher DJ, Gilmore R. An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology. 2006;16:47r–62r. doi: 10.1093/glycob/cwj066. [DOI] [PubMed] [Google Scholar]
- Kuszewski J, Clore GM. Sources of and solutions to problems in the refinement of protein NMR structures against torsion angle potentials of mean force. J Magn Reson. 2000;146:249–254. doi: 10.1006/jmre.2000.2142. [DOI] [PubMed] [Google Scholar]
- Kuszewski J, Gronenborn AM, Clore GM. Improving the packing and accuracy of NMR structures with a pseudopotential for the radius of gyration. J Am Chem Soc. 1999;121:2337–2338. [Google Scholar]
- Lee D, Hilty C, Wider G, Wuthrich K. Effective rotational correlation times of proteins from NMR relaxation interference. J Magn Reson. 2006;178:72–76. doi: 10.1016/j.jmr.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Loria JP, Rance M, Palmer AG. A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J Am Chem Soc. 1999;121:2331–2332. [Google Scholar]
- Lovering AL, de Castro LH, Lim D, Strynadka NCJ. Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science. 2007;315:1402–1405. doi: 10.1126/science.1136611. [DOI] [PubMed] [Google Scholar]
- Macnaughtan MA, Kamar M, Alvarez-Manilla G, Venot A, Glushka J, Pierce JM, Prestegard JH. NMR structural characterization of substrates bound to N-acetylglucosaminyltransferase V. J Mol Biol. 2007;366:1266–1281. doi: 10.1016/j.jmb.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Permi P, Annila A. Transverse relaxation optimised spin-state selective NMR experiments for measurement of residual dipolar couplings. J Biomol Nmr. 2000;16:221–227. doi: 10.1023/a:1008362211560. [DOI] [PubMed] [Google Scholar]
- Qasba PK, Ramakrishnan B, Boeggeman E. Substrate-induced conformational changes in glycosyltransferases. Trends Biochem Sci. 2005;30:53–62. doi: 10.1016/j.tibs.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Ruckert M, Otting G. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J Am Chem Soc. 2000;122:7793–7797. [Google Scholar]
- Schuman B, Alfaro JA, Evans SV. Glycosyltransferase structure and function. Bioactive Conformation I. 2007:217–257. [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- Varki A, Cummings R, Esko JD, Freeze H, Hart G, Marth JD, editors. Essentials of Glycobiology. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1999. [PubMed] [Google Scholar]
- Weerapana E, Imperiali B. Asparagine-linked protein glycosylation: from eukaryotic to prokaryotic systems. Glycobiology. 2006;16:91R–101R. doi: 10.1093/glycob/cwj099. [DOI] [PubMed] [Google Scholar]