

Abstract
Ageing, or increased mortality with time, coupled with physiologic decline, is a nearly universal yet poorly understood biological phenomenon. Studies in model organisms suggest that two conserved pathways modulate longevity: DNA damage repair and insulin/Igf1-like signaling. In addition, homologs of yeast Sir2 – the sirtuins – regulate lifespan in diverse organisms. Here, we focus on one particular sirtuin, SIRT6. Mice lacking SIRT6 develop a degenerative disorder that in some respects mimics models of accelerated ageing [1]. We discuss how sirtuins in general and SIRT6 specifically relate to other evolutionarily conserved pathways affecting ageing, and how SIRT6 might function to ensure organismal homeostasis and normal lifespan.
Keywords: Ageing, DNA Damage, Metabolism
INTRODUCTION
Although most eukaryotes exhibit a limited lifespan, a molecular understanding of natural ageing has remained elusive. However, recent studies have begun to shed some light into this process. In this regard, several mechanisms influence lifespan in a conserved fashion even among distantly related species. Specifically, the maintenance of genomic stability, Insulin/Igf1-like signaling (IIS), and homologs of the yeast Sir2 protein (sirtuins), all represent conserved regulators of lifespan in diverse organisms.
Sirtuins and mammalian longevity: lessons from mouse knockouts
Overexpression or hyperactivity of sirtuins in many organisms – including yeast, worms, flies, and potentially fish and mammals – promotes longevity [2]. Mammals possess at least seven sirtuins, termed SIRT1–SIRT7 [3, 4]. Sirtuins exert their effects via NAD+-dependent enzymatic modification of other proteins: yeast Sir2 (the founding member of this family) and its mammalian ortholog SIRT1 possess both robust protein deacetylase [5–8] and ADP-ribosyltransferase activity [4, 9]. Some sirtuins, such as SIRT4 and SIRT6, possess primarily ADP-ribosyltransferase activity with little or no deacetylase activity on standard substrates such as core histones or histone peptides [1, 10–12]. Sir2 apparently only modifies histones. However other sirtuins have evolved to modify a large array of proteins; for example SIRT1 binds to and/or deacetylates a wide variety of factors impacting on numerous cellular processes [13].
To better define sirtuin function in mammals, our group has generated germline knockouts of the seven mammalian sirtuins, SIRT1–SIRT7. Among these, SIRT1- [14], and SIRT6-deficient mice [1] demonstrate strong phenotypes, whereas the remaining mouse strains show no grossly evident defects ([11] and unpublished). We and others have found that SIRT1-deficient mice die perinatally on a pure 129-strain background, exhibiting heart and retinal developmental defects [14, 15]. In addition to its developmental functions, SIRT1 deacetylates and inactivates the tumor suppressor p53 in vitro [16–18] and in vivo [14]; consequently SIRT1-deficient thymocytes show increased levels of apoptosis in response to genomic insult [14]. A detailed analysis of SIRT4-deficient mice has uncovered a role for this factor in ADP-ribosylating the enzyme glutamate dehydrogenase (GDH); this modification reduces GDH activity and in turn suppresses insulin secretion in response to amino acids [11]. During periods of prolonged fasting such as calorie restriction (see below), inhibition of GDH by SIRT4 appears to be relieved via an unknown mechanism, allowing regulation of insulin secretion by amino acids rather than glucose in the setting of overall reduced carbohydrate metabolism [11]. We have recently found that SIRT3 plays a major role in deacetylating numerous mitochondrial proteins in mice in vivo (D.L., B.S., R.M., F.W.A., et al., in preparation). For the remaining knockouts (SIRT2, SIRT5, and SIRT7), in-depth characterization will very likely uncover specific phenotypes. Such studies, along with generation and characterization of sirtuin overexpressors, will also be required to address whether and how these factors may affect mammalian lifespan.
SIRT6 deficiency causes a degenerative syndrome with progeroid features
From the standpoint of ageing research, SIRT6 deficiency causes the most striking phenotype among all the sirtuin knockouts. At the cellular level, SIRT6 deficiency leads to slow growth and increased sensitivity to certain forms of genotoxic damage. In addition, SIRT6-deficient cells show increased spontaneous genomic instability, characterized by numerous non-clonal chromosomal aberrations [1]. These findings suggest a defect in the ability of SIRT6-deficient cells to cope with DNA damage. Indeed, these cells exhibit increased sensitivity to alkylating and oxidizing agents, such as methyl-methane sulfonate (MMS) and hydrogen peroxide (H2O2), but normal response to UV damage and normal repair of DNA double-strand breaks.
In the whole organism, metabolic effects dominate the manifestations of SIRT6 deficiency. SIRT6 knockout animals are born at a Mendelian ratio, and, though somewhat smaller than wild-type littermates, appear relatively normal until approximately two weeks of age. At this point, serum glucose begins a precipitous decline, eventually reaching levels inconsistent with life. On a pure 129-strain background, SIRT6 deficiency is lethal with 100% penetrance prior to one month of age. SIRT6-deficient mice possess extremely low levels of the hormone Igf1 (see below), and, as Igf1 is critical for proper bone development, these mice show thin bones and spinal curvature (termed lordokyphosis) often observed in so-called “prematurely ageing” mouse models. Although SIRT6-deficient mice initially show a normal lymphocyte compartment, these cells are lost in a wave of apoptosis at around three weeks of age due to systemic (i.e., non-cell-autonomous) causes. Overall, SIRT6 deficiency is associated with a complex phenotype resembling some aspects of models of “premature ageing” [1].
For the remainder of this review, we will discuss how loss of SIRT6 might lead to these pleiotropic effects. The manifestations of SIRT6 deficiency are particularly intriguing in light of the fact that the pathways affected – genomic stability and IIS – both affect lifespan in multiple organisms. We first consider issues in DNA damage repair and IIS that are relevant for longevity in general and sirtuins in particular. We then focus on SIRT6; we discuss how SIRT6 might modulate DNA repair and IIS to affect lifespan.
DNA REPAIR PATHWAYS AND LIFESPAN
Defective DNA repair can result in ageing-like phenotypes
The eukaryotic cell is faced with more than 1×105 DNA lesions per day that require repair in order to avert chromosomal instability, mutations and ultimately, cell death or dysfunction [19]. Therefore, eukaryotic organisms have evolved five major DNA repair pathways, each responsible for repairing different (but in some cases overlapping) types of lesions. Specifically, double strand breaks (DSBs) are repaired through homologous recombination (HR), non-homologous end joining (NHEJ) [20], and in some cases by an alternative end-joining pathway (Yan et al., in press). Single-strand lesions are repaired through base excision repair (BER), nucleotide excision repair (NER) and mismatch repair (MMR) [20, 21]. Despite these multiple repair pathways, DNA mutations accumulate with age [22]. A great deal of data exists supporting the notion that unrepaired nuclear DNA damage is a significant causative factor in the manifestations of ageing [20]. One important observation supporting this hypothesis is that certain DNA repair defects in mice and humans manifest phenotypically as “segmental progerias” [23] – that is, they recapitulate some (but not all) features of ageing in an accelerated fashion. Defects in NHEJ, HR, and NER have been associated with such progeroid (ageing-like) phenotypes [20]. For purposes of this review, we will first describe DNA lesions that are most relevant to ageing and then summarize BER and NER, the two main pathways related to the topics discussed herein.
Reactive oxygen species and ageing
Reactive oxygen species (ROS), produced as a consequence of endogenous metabolism, likely produce much of the DNA damage leading to the manifestations of ageing [24]. Three well-characterized types of ROS are superoxide anion, hydroxyl radical, and hydrogen peroxide. Although these and related molecules can damage protein, lipids, and RNA as well as DNA, these other cellular constituents can be replaced, whereas damaged DNA must instead be repaired. It is clear that increased mutations in mitochondrial DNA can cause a progeroid phenotype [25, 26], apparently via chronically elevated rates of apoptosis in many tissues [25]; the relative contribution of mitochondrial versus nuclear DNA damage in ageing has not been resolved. Here we focus exclusively on ROS-related nuclear DNA damage.
In lower organisms, the connection between ROS and lifespan has been well-established by a large body of experimental work [27]. In mammals, overexpression of catalase artificially targeted to mitochondria, or of p53 and Arf together, extends murine lifespan along with lower levels of oxidative damage [28, 29]. Mutation of the p66shc gene or the adenylyl cyclase AC5 is also associated with increased oxidative stress resistance and extended lifespan [30, 31]. However, mice heterozygous for mitochondrial superoxide dismutase (SOD2), a critical detoxifier of mitochondrial ROS, suffer increased nuclear DNA damage and an elevated incidence of cancer but do not show evidence of premature ageing [32]. Mice lacking Hif-2α, a regulator of many antioxidant genes, show elevated levels of oxidative stress and complex pathologies in mitochondria-rich tissues, as well as metabolic disturbances including hypoglycemia and a short lifespan [33]. However the phenotype of this mutant does not resemble natural mouse ageing. Complicating the picture still further, ROS, along with their deleterious effects on cellular macromolecules, play critical roles in many signaling pathways, at least in part by inhibiting activity of tyrosine phosphatases [34]. Overall, although unanswered questions remain regarding the role of ROS in mammalian ageing, ROS represent an important determinant of lifespan in lower organisms and likely affect lifespan in mammals as well.
BER and ageing
The main sentinel against ROS-generated damage is the BER pathway. In general, BER is responsible for repair of small chemical alterations in DNA [35] such as methylation, alkylation and oxidative damage generated during endogenous cellular metabolism. Although these lesions may not impede transcription, they frequently miscode; therefore BER is particularly relevant for preventing mutagenesis. In its core reaction, a set of DNA glycosylases, each recognizing a specific lesion, cleaves the damaged base, generating an abasic site, which is then processed by the APE1 endonuclease to generate a nick (Figure 1). In short-patch repair, DNA polymerase β (polβ) performs a one-nucleotide gap-filling reaction and removes the 5′ baseless sugar residue via its dRP-lyase activity. The XRCC1-LigaseIII complex completes the repair process (Figure 1). Additional factors, such as the chromatin factor PARP-1, a poly-ADP-ribosyltransferase, are known to participate in the reaction, although their precise roles remain unclear [35]. In the alternative pathway (long-patch repair), 2–10 nucleotides are removed and synthetized de novo, in a reaction that involves additional repair factors, such as Polδ/ε, the cofactor PCNA, the FEN1 endonuclease and Ligase I [35] (Figure 1). Although BER represents the main pathway to repair oxidative lesions, BER defects do not produce ageing-like manifestations [35, 36], perhaps due to redundancy among glycosylases, whereas core BER factors are required for viability [36]. It is noteworthy that even though deficiencies in core BER factors do not lead to progeroid syndromes, some BER factors prevent ageing-associated diseases, namely cancer and neurodegeneration [35]. More work is needed to clarify the potential relationship between ageing and BER, including the use of mice bearing conditionally inactive and hypomorphic alleles of the core factors.
Figure 1. Schematic of base excision repair.
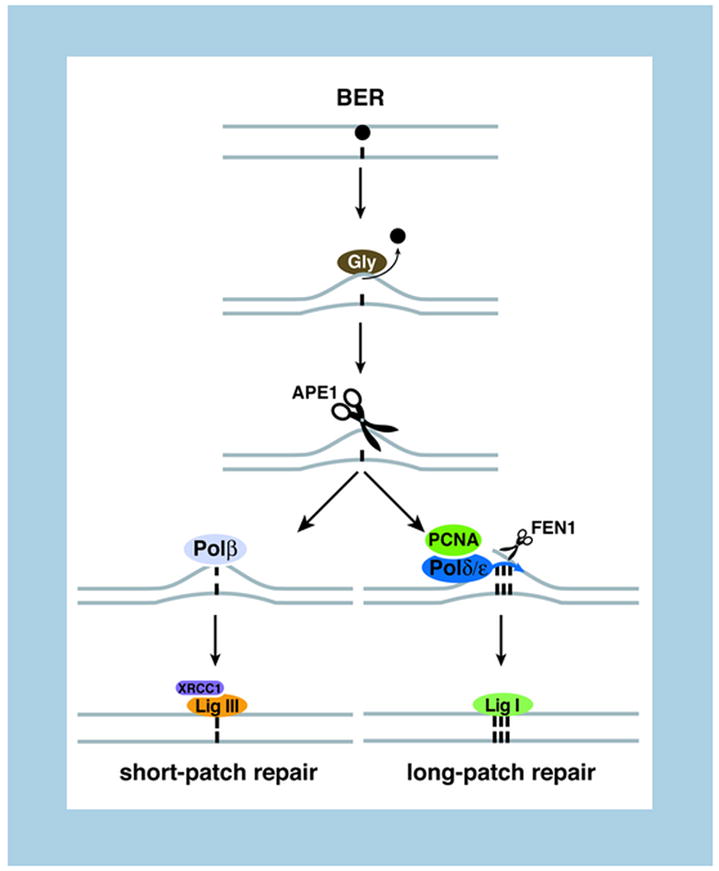
For details see text.
NER and ageing
In contrast with BER, perturbations in NER lead to progeroid syndromes. NER deals with a diverse array of helix-distorting lesions that interfere with base pairing and obstruct transcription and replication. This pathway is the most versatile in terms of lesion recognition and can be subdivided into global genome NER (GG-NER), which surveys the entire genome for lesions, and transcription-coupled repair (TC-NER) which targets damage that blocks transcription [37] (Figure 2). The major difference between these two pathways lies in the mechanism of lesion recognition. For GG-NER, a specific complex (XPC-hHR23B) recognizes disrupted base pairing (Figure 2). In the case of TC-NER, it is the ability of the lesion to block the RNA polymerase (RNAP II) that activates the pathway; two specific proteins are involved in this process, CSA and CSB (Figure 2). The subsequent stages are identical and involve helicases (XPB-XPD), endonucleases (XPG-ERCC1/XPF), auxiliary factors (such as the single-strand binding protein RPA) and the DNA replication machinery to fill the gap (Figure 2).
Figure 2. Nucleotide excision repair pathways.
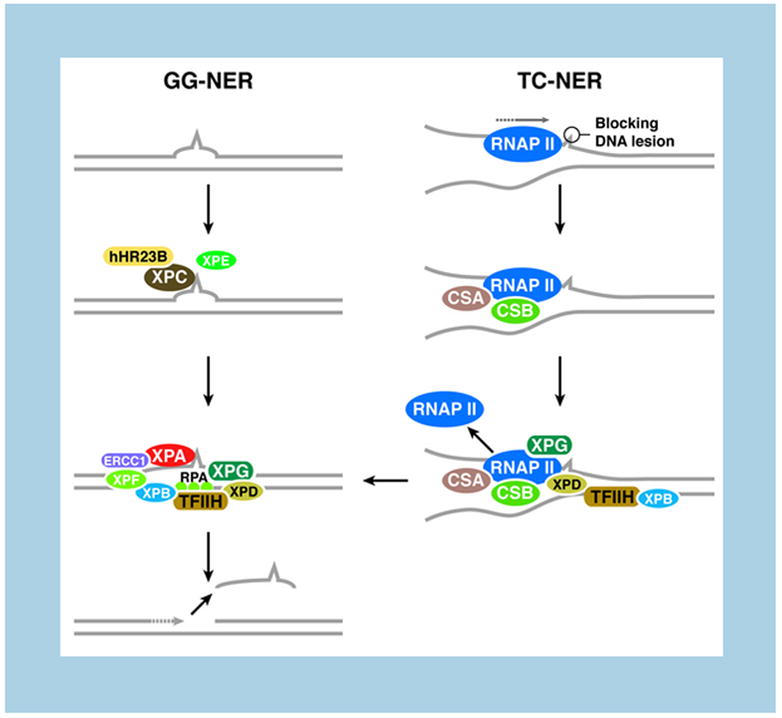
Global genome NER (GG-NER) scans the entire genome for lesions. The XPC-hHR23B complex detects disrupted base pairing and subsequently attracts other repair factors. Transcription-coupled repair (TC-NER) is activated when elongating RNA polymerase II (RNAP II) is blocked by DNA damage on the actively transcribed strand. Arrested RNAP II recruits CSA, CSB, and XPG, followed by TFIIH, XPD, XPB and auxiliary factors and is later released in an ATP-dependent reaction. The subsequent steps of GG-NER and TC-NER are identical. Gap filling by the DNA replication machinery completes the repair process.
Several human patients and mouse strains with defects in NER factors have phenotypes resembling premature ageing [37]. Defects in the TC-NER factors CSA or CSB lead to Cockayne syndrome (CS) in humans, a complex disorder with developmental delay, neurodegeneration and progeroid features. In addition, patients with defects in XPD suffer from trichothiodystrophy (TTD), a disease characterized by brittle hair, skin defects and shortened lifespan [38]. Notably, mice with a mutation in XPD mimicking the one observed in TTD develop similar ageing associated changes, including wasting, osteoporosis and melanocyte loss [39].
Mice deficient in CSB/XPA or XPD/XPA rapidly degenerate and die within a few weeks of birth, a result of a severe NER defect [40, 41]. More recently, a human patient with a dramatic progeroid syndrome was found with mutations in XPF [42]. Mice deficient for XPF or its binding partner ERCC1 also exhibit severe acute degeneration, dying prior to one month of age with osteopenia, skin and bone marrow abnormalities, kyphosis, and, at the cellular level, sensitivity to oxidative stress [42, 43, 44]. ERCC1/XPF play roles in other DNA repair pathways besides canonical NER – cross-link repair, HR, and telomere maintenance [20]. Recently, the mRNA expression profile of progeroid NER-defective mice was shown to resemble that of old mice and genotoxin-treated mice [42, 45], demonstrating directly that DNA repair defects, specifically in NER, can lead to changes resembling natural ageing in mammals.
THE INSULIN/IGF1-LIKE SIGNALING (IIS) PATHWAY AND LIFESPAN
The IIS pathway
IIS represents a conserved pathway regulating metabolism and, in many organisms, lifespan. Mammals possess two distinct hormones that provoke IIS to signal different outcomes in the postnatal state: insulin, secreted by the beta cells of the pancreas, directs anabolic metabolism, whereas Igf1, secreted mostly by the liver in response to pituitary growth hormone (GH), predominantly directs somatic growth and differentiation [46]. GH release is in turn driven by growth hormone releasing hormone (GHRH) secreted by the hypothalamus. This system (GHRH-GH-Igf1) is termed the somatotrophic axis, due to its role in driving growth. IIS itself is initiated when insulin or related peptide hormones bind to their cognate heterotetrameric transmembrane receptors, leading to receptor autophosphorylation [47] (Figure 3). This in turn allows tyrosine phosphorylation of insulin receptor substrate (IRS) proteins as well as other factors, allowing recruitment of other proteins and initiating multiple signaling cascades. One such cascade involves phosphatidylinositol-3-OH kinase (PI3K). Activation of PI3K leads to activation of PDK1 kinase, which in turn participates in activation of AKT/PKB kinase (Figure 3); activated AKT phosphorylates proteins impacting multiple processes: metabolism, stress resistance, cell cycle, survival, and growth [48]. The FOXO family of Forkhead transcription factors represents a critical target of AKT (and of the related kinase SGK); activated AKT/SGK phosphorylates nuclear FOXO proteins leading to their cytoplasmic sequestration in association with 14-3-3 chaperone proteins (Figure 3). Reduced IIS allows translocation of FOXO proteins to the nucleus, where they activate or repress transcription at various promoters [49] (Figure 3). Nuclear FOXOs promote cell death, stress resistance, cell cycle arrest, and/or metabolic changes in a cell-type specific manner [49].
Figure 3. Insulin/Igf1-like signaling pathway.
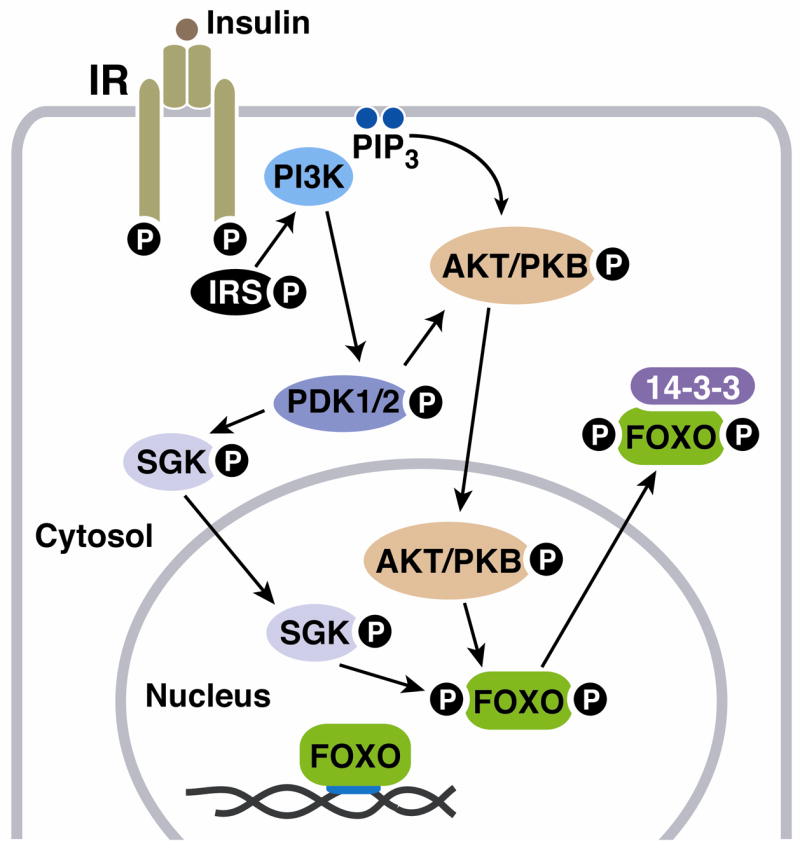
For details see text.
Decreased IIS is associated with increased lifespan
Reduction in IIS is associated with longevity in many model organisms. Deletion of the AKT homolog sch9 promotes stress resistance and increased lifespan in yeast in both chronological and replicative ageing assays [50, 51]. In C. elegans and Drosophila, many different naturally occurring or engineered mutations in IIS confer extended longevity [52]. Perturbations in the mTOR pathway, also impacting on nutrient sensing and IIS, increase lifespan in lower organisms as well [51, 53–55]. In mammals, reduced IIS promotes extended longevity in mice [46]. Deletion of Irs2 specifically in the brain also leads to increased longevity [56]. By contrast, organism-wide reductions in insulin sensitivity or insulin secretion produce diabetes and shorten lifespan in mice and humans. Deletion of the insulin receptor specifically in adipose tissue does extend longevity; however this effect may stem from reduced adiposity in these animals rather than altered IIS per se [57]. In humans, reduced IIS has not been clearly linked with longevity; indeed low levels of Igf1 have been linked to cardiovascular disease and diabetes, whereas high Igf1 levels may confer susceptibility to cancer [46]. Overall, convincing evidence links reduction of IIS to increased lifespan in diverse organisms, including mammals; however there are no firm data as yet in support of this connection in humans.
SIRTUINS AFFECT LIFESPAN VIA MULTIPLE PATHWAYS
How do sirtuins modulate longevity? In yeast, Sir2 functions in transcriptional silencing at the silent mating-type loci, the rDNA array, and telomeres [2, 58]. S. cerevisiae Sir2 exhibits at least two activities relevant to lifespan [2]. First, Sir2 mediates genomic stability. Sir2 suppresses recombination at the rDNA array, and thus the formation of extrachromosomal ribosomal circles (ERCs), which replicate autonomously and accumulate preferentially in yeast mother cells, eventually reaching toxic levels [59]. Modest overexpression of Sir2 increases yeast lifespan in association with reduced rDNA recombination – higher levels of Sir2 expression are toxic [60], whereas loss of Sir2 reduces lifespan and increases rDNA instability [61, 62]. Second, Sir2 directs segregation of oxidatively damaged proteins to the yeast mother cell [63], preventing inheritance of these proteins by daughter cells. The beneficial effect of Sir2 on lifespan does not occur under all conditions in yeast, however: it is strain specific [64], and moreover, Sir2 can actually shorten yeast lifespan in culture-based assay [65].
ERC formation is apparently a yeast-specific ageing mechanism. In higher organisms such as worms and flies, sirtuins may exert lifespan-extending effects via modulation of factors involved in IIS. This connection will be discussed in more detail below. In mammals, indirect evidence points to potential roles for sirtuins in regulating longevity; most studies have focused on SIRT1. SIRT1 possesses multiple metabolic functions that might indicate a beneficial role in longevity: these include increasing cellular stress resistance and suppressing apoptosis via deacetylation of FOXO proteins, p53, and E2F1 [16–18, 49, 66–69]; and decreasing white adipose tissue formation [70]. SIRT1 also plays a protective role in model systems of neurodegeneration [71] (reviewed in [72]).
Other data are potentially inconsistent with the simple hypothesis that hyperactivity of sirtuins will extend lifespan in mammals. In this context, sirtuin dosage appears to be an important factor in mammals, as it is in yeast; while modest overexpression of SIRT1 protects cardiac muscle from age-related decline in mice, higher levels of SIRT1 actually promote degenerative changes in this tissue [67]. SIRT1 can play a role in promoting tumorigenesis, thus potentially negatively affecting lifespan [73, 74]. As described more fully below, SIRT1 appears to promote insulin secretion and Igf1 signaling, activities that might be predicted to reduce longevity. Furthermore, our lab has shown that SIRT1 promotes fibroblast senescence via regulation of the tumor suppressor p19 [75]. Senescence likely represents an important tumor suppressor mechanism [76]; if SIRT1 promotes senescence in vivo in critical cell populations (for example, stem cells), it might be expected to suppress tumor formation but promote tissue decline via stem cell depletion. Some of the data supporting a potential beneficial effect of SIRT1 on mammalian lifespan comes from studies using resveratrol, a polyphenol reported to stimulate Sir2/SIRT1 activity, which increases longevity in several organisms, including notably mice fed a high-fat diet [77–81]. However, resveratrol affects several other cellular signaling pathways potentially relevant to longevity [82–84], and may increase lifespan in C. elegans by actually inhibiting Sir2.1 activity [85]. Altogether, these results highlight the complexity of SIRT1 function; future studies will hopefully uncover potential therapeutic effects of SIRT1 activators/inhibitors in regulating lifespan and/or ageing-associated diseases.
LINKS BETWEEN PATHWAYS REGULATING AGEING: SIRTUINS AND DNA REPAIR
Sirtuins modulate DNA repair
The genomic instability and genotoxin sensitivity of SIRT6-deficient cells suggest that SIRT6 might function in DNA repair although there are other potential interpretations of this finding (see below). Other sirtuins play roles in modulating DNA repair, potentially providing insight into SIRT6 function. S. cerevisiae Sir2 enhances DNA repair via an indirect mechanism [86–90]. In mammals, SIRT1 deacetylates NBS1, a protein critical in the checkpoint response to DNA damage and in DSB repair, to promote cell survival [91]. Several sirtuins affect DNA repair via chromatin alterations. The yeast sirtuins Hst3 and Hst4 deacetylate K56 of histone H3; lack of this activity results in DNA damage sensitivity, genomic instability, and silencing defects [92–94]. How this modification promotes genomic stability mechanistically is unclear. In Trypanosoma brucei, the sirtuin TbSIR2RP1 deacetylates and ADP-ribosylates histones and promotes survival in reponse to DNA damage. TbSIR2RP1 increases chromatin accessibility to micrococcal nuclease following genotoxin treatment; thus one plausible model of TbSIR2RP1 function is that this sirtuin modifies chromatin following genomic insult to allow greater access to DNA repair enzymes [95]. In summary, several sirtuin proteins, potentially including SIRT6, modulate DNA repair.
SIRT6 and BER
The spectrum of sensitivities exhibited by SIRT6-deficient cells suggested a role for SIRT6 in BER; in support of this notion, these sensitivities were rescued by expression of the dRP-lyase domain of DNA polymerase β (polβ) [1]. Other repair pathways appear intact in SIRT6-deficient cells, as they are not sensitive to UV – arguing against a defect in NER – and show unimpaired DSB repair [1]. How might SIRT6 influence BER? One possibility is that SIRT6 modifies a BER factor directly, via deacetylation or ADP-ribosylation. In this regard, SIRT6 is able to deacetylate polβ weakly in vitro [1]; deacetylation of this factor is required for dRP-lyase activity [96]. However, whether SIRT6 can catalyze this modification in vivo in a manner that is functionally significant is unclear. Alternatively, given its tight association with chromatin, SIRT6 might affect the chromatin environment to allow access of BER factors to sites of damage. Notably, the activity of several BER factors is strongly inhibited on chromatinized templates [97, 98]. In this regard, extracts from SIRT6-deficient cells performed as efficiently as wild-type extracts in an in vitro BER assay with naked non-chromatinized DNA templates [1]. As noted above, the genotoxin sensitivity of SIRT6-deficient cells can be rescued by introduction of the 8-kDa dRP-lyase fragment of polβ. As this fragment is much smaller than the full-length enzyme, it might bypass a putative requirement for SIRT6 in altering chromatin structure to permit effective BER. A third possibility is that SIRT6 does not influence BER directly, but rather influences accumulation of DNA damage via modulation of metabolism and ROS levels which negatively impact on BER; this possibility is discussed in more detail below.
LINKS BETWEEN PATHWAYS REGULATING AGEING: SIRTUINS AND IIS
Sirtuins modulate IIS
SIRT6 also might affect metabolism directly, instead of or in addition to playing a direct role in DNA repair. Specifically, the hypoglycemia of SIRT6-deficient mice suggests that SIRT6 might play a role in IIS. In lower organisms and in mammals, sirtuins affect IIS in multiple ways. In mammals, sirtuins influence insulin secretion at several levels. Mice overexpressing SIRT1 specifically in the beta cells of the pancreas produce higher levels of insulin [99], whereas SIRT1-deficient mice secrete lower levels of this hormone [100], effects occurring via regulation of uncoupling protein 2 levels by SIRT1. SIRT1-deficient animals also possess elevated levels of Igf1 binding protein (IGFBP1), an effect likely to suppress Igf1 signaling via sequestration of circulating Igf1; it has been suggested that many of the effects of SIRT1 deficiency – in particular, small size, failure to thrive, and sterility – stem from reduced Igf1 signaling [101]. These results must be interpreted with some caution, given that analysis of adult SIRT1-deficient mice necessarily occurs using a highly preselected group of rare survivors in a pure strain background [14, 15]. As described above, SIRT4 inhibits insulin secretion in response to amino acids via modulation of GDH activity [11]. With regard to intracellular signaling, in C. elegans, the sirtuin Sir2.1 acts through the FOXO protein DAF-16 to extend lifespan [102]. Similarly, in mammals, SIRT1 deacetylates FOXO proteins, allowing them to activate a subset of target genes, including those involved in stress resistance [49]. Recently, SIRT2 has also been shown to deacetylate a FOXO protein, causing decreased ROS levels under conditions of mild stress but promoting cell death under severe stress [103].
One context in which sirtuins may modulate IIS is calorie restriction (CR), an intervention capable of extending lifespan in a wide variety of eukaryotic organisms [104]. CR leads to multiple endocrine changes, including alterations in IIS: reduction in Igf1, insulin, and blood glucose levels, as well as increased insulin sensitivity [46, 105]. Based on work in yeast and other invertebrates, studies using resveratrol, and indirect data in mammals, sirtuins have been proposed to mediate the effects of CR [11, 66, 78, 106]; however this connection is currently controversial, particularly in yeast [2]. There are conflicting data as to whether CR in worms and flies acts via sirtuins [107–109]. In mammals, levels of SIRT1, SIRT2, and SIRT3 proteins rise in tissues of animals subject to CR [103, 110–112]. In response to nutrient deprivation, SIRT1 deacetylates and activates the metabolic transcriptional regulator PGC-1α, potentially permitting metabolic compensation to lowered calorie intake [113, 114]. SIRT1 may mediate behavioral aspects of the CR response [66]. Thus some of the lifespan-extending effects of CR could be mediated by alterations in IIS, which may in turn be regulated by sirtuins.
SIRT6 affects IIS
As described above, SIRT6-deficient mice show prominent metabolic defects, in particular, hypoglycemia, low Igf1 [1], and low insulin levels (unpublished results). These defects indicate potential perturbations in IIS in these animals, and suggest that SIRT6 might directly down-modulate this pathway to promote normoglycemia. Indeed, recent studies in several cell types demonstrate that SIRT6 down-regulates AKT phosphorylation, allowing nuclear relocalization of FOXO proteins (D.L., R.M., B.S., and F.W.A., unpublished results). In this regard, SIRT6-deficient mice exhibit increased AKT phosphorylation in some tissues, despite low levels of circulating insulin and Igf1, suggesting that these mice might possess increased insulin sensitivity. These results could explain the hypoglycemia and the compensatory low insulin and Igf1 levels observed in these mice. Since AKT responds to a variety of stimuli, however, more work is needed to define a potential mechanistic role for SIRT6 in IIS.
SIRT6 IMPACTS ON TWO CONSERVED AGEING PATHWAYS
SIRT6 deficiency impacts on fundamental aspects of cellular function, genomic stability and metabolic homeostasis. What is the connection between these roles of SIRT6? One possibility is that SIRT6 affects both DNA repair and IIS independently of one another, presumably via distinct substrates. As outlined above, SIRT1 modifies numerous proteins affecting a plethora of cellular processes [72], and we have obtained evidence that SIRT3 deacetylates several mitochondrial proteins as well (unpublished data). However, another possibility is that SIRT6 primarily affects a single cellular process. Indeed, as discussed below, the DNA damage response can be accompanied by changes in the IIS pathway, suggesting that in the context of SIRT6 deficiency, a primary DNA repair defect might trigger a metabolic response. Alternatively, SIRT6 might play a direct role only in regulating metabolism, influencing genomic stability indirectly, a possibility also considered below.
DNA repair defects can provoke metabolic phenotypes
Mutations in several DNA repair factors cause metabolic phenotypes along with ageing-like manifestations. This finding may imply the existence of cellular compensatory mechanisms for DNA repair defects, working to minimize genomic damage by adjusting metabolic parameters. For example, defects in DSB repair can lead to ageing-like phenotypes and perturbed IIS. In this regard, certain mutations in checkpoint factors, such as the ATM kinase and the p53 tumor suppressor protein, as well as the HR factor BRCA1, are accompanied by progeroid syndromes. In each of these cases, changes in IIS are also observed [115–121]. In the context of chromatin and DNA repair, mice deficient in the chromatin factor HMG1 exhibit severe hypoglycemia [122]; HMG1-deficient cells display pronounced genomic instability and increased sensitivity to genotoxic agents [123]. Loss of HMG1 is thought to promote higher levels of damage via a more susceptible chromatin state, rather than defective repair per se [123]; it will be of interest to test whether SIRT6 acts in concert with HMG1 to promote genomic stability and metabolic homeostasis.
The most informative models regarding how DNA repair defects might lead to metabolic defects – and the mutants with the greatest similarity to the SIRT6 knockout –are mice defective in NER. As discussed, several NER mutants show progeroid phenotypes. Additionally, inhibition of the somatotrophic axis is observed in ERCC1-deficient mice, as well as CSB/XPA and XPD/XPA mutant mice [41, 42, 45]. In these models, it has been suggested that unrepaired DNA damage triggers a metabolic response mediated via downregulation of the somatotrophic axis to shift energetic investment from growth to somatic maintenance and to avert further DNA damage via decreased ROS levels. Indeed, like SIRT6-deficient animals, these mice show low insulin and low Igf1 levels. This response does not represent a universal reaction to all forms of DNA damage, as mice deficient in the NHEJ factor Ku80 do not show this metabolic effect [41]. One important question concerning this potential DNA damage-metabolism connection is whether low levels of insulin and Igf1 in these models might occur secondarily to insulin hypersensitivity itself; indeed, hypoglycemia in the NER mutants and SIRT6 knockout mice argues for increased, not decreased overall IIS. In this case, increased insulin sensitivity, potentially with increased metabolic rate and ROS production, might directly contribute to the observed DNA damage, which might synergize with the putative primary DNA repair defect. Overall, mechanistic insights into exactly how unrepaired DNA damage might modulate IIS in this setting are clearly required.
Metabolism can affect DNA damage accumulation and repair
A second model for SIRT6 function is that SIRT6 modulates metabolism directly via regulation of IIS, indirectly influencing genomic stability. In this regard, metabolic perturbations, or lesions in factors implicated in IIS can impact upon DNA damage accumulation and repair [124]. For example, in mammals, CR is associated with lower levels of ROS, and concomitantly, a lower frequency of ROS-induced mutations [125, 126]. Cells deficient in Pten, a phosphatase that negatively regulates IIS, show genomic instability as a consequence of hyperactivity of Akt, which phosphorylates Chk1 leading to its degradation, and concomitantly defective checkpoint responses [127, 128]. Pten also functions to maintain genomic stability via regulation of the HR protein Rad51 and centromeric stability [129]. FOXO proteins negatively regulate ROS levels, and thus potentially levels of DNA damage, through regulation of the expression of factors that modulate oxidant levels such as SOD2 and catalase [49]. FOXOs also directly induce cell cycle arrest and DNA repair via induction of Gadd45 [49]. Indeed, deletion of three FOXO factors (FOXO1, FOXO3 and FOXO4) in mice decreases hematopoietic stem cell renewal capacity, a phenotype rescued with antioxidant treatment [130, 131].
Thus, alterations in IIS can impact on genomic instability via multiple mechanisms. A metabolic defect conferred by SIRT6 deficiency – for example, hyperactive IIS – could promote DNA damage accumulation via increased overall levels of ROS due to elevated metabolic activity and decreased levels of antioxidant enzymes as a consequence of a reduction in FOXO activity. Increased ROS levels could in turn overwhelm DNA repair mechanisms and promote accumulation of DNA lesions, mimicking a BER defect. A similar outcome might occur even if SIRT6 regulates metabolic processes apart from IIS itself. As noted above, mutations in Hif-2α lead to decreased levels of antioxidant proteins and consequently elevated ROS, systemic degeneration, and hypoglycemia. Thus a putative mitochondrial defect conferred by SIRT6 deficiency might produce many of these same phenotypes as well as increased levels of oxidative DNA damage.
PERSPECTIVE
Studies in model organisms have revealed common lifespan-regulatory mechanisms that influence lifespan in many diverse species: DNA damage repair, IIS, and sirtuins, which interact with and influence one another in complex ways. Some mouse mutants with defective DNA repair show progeroid phenotypes, implicating effective DNA repair as an important longevity assurance mechanism. Among these, some – like certain NER mutants – show disordered metabolism as well. SIRT6 could in principle affect DNA repair in several ways: by affecting expression of or directly modifying a factor involved in BER, by modifying chromatin to facilitate access for repair factors, or by modulating metabolic processes to minimize ROS production and DNA damage. Conversely, metabolic defects conferred by SIRT6 deficiency could conceivably lead to higher ROS production, thus increasing DNA damage. More generally, sirtuins can potentially modulate both DNA damage repair and IIS and thus are likely to interact with these responses in complex ways. In this regard, SIRT6 might play a direct role in reducing IIS in response to genotoxic stress, thereby decreasing ROS levels and retarding accumulation of DNA damage. Overall, elucidation of the mechanistic interplay between these pathways will no doubt yield important new insights into ageing, that may in the long term result in new therapeutic approaches to treat and prevent age-related ailments.
Acknowledgments
We thank members of the Alt laboratory for helpful discussions. This work was supported by an Ellison Medical Foundation Senior Scholar Award to F.W.A. D.B.L. is supported by a K08 award from NIA/NIH. R.M. is supported by a Senior Postdoctoral Fellowship from The Leukemia and Lymphoma Society. B.S. is the recipient of a UCSF Sandler Postdoctoral Research Fellowship Award. F.W.A. is an Investigator of the Howard Hughes Medical Institute. F.W.A is a member of the scientific advisory board of Sirtris Pharmaceuticals.
Footnotes
Conflict of interest statement
No conflict of interest was declared.
References
- 1.Mostoslavsky R, Chua KF, Lombard DB, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–29. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 2.Longo VD, Kennedy BK. Sirtuins in aging and age-related disease. Cell. 2006;126:257–68. doi: 10.1016/j.cell.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000;273:793–8. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 4.Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun. 1999;260:273–9. doi: 10.1006/bbrc.1999.0897. [DOI] [PubMed] [Google Scholar]
- 5.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD- dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 6.Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci U S A. 2000;97:5807–11. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith JS, Brachmann CB, Celic I, et al. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci U S A. 2000;97:6658–63. doi: 10.1073/pnas.97.12.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanner KG, Landry J, Sternglanz R, Denu JM. Silent information regulator 2 family of NAD- dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc Natl Acad Sci U S A. 2000;97:14178–82. doi: 10.1073/pnas.250422697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanny JC, Dowd GJ, Huang J, Hilz H, Moazed D. An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell. 1999;99:735–45. doi: 10.1016/s0092-8674(00)81671-2. [DOI] [PubMed] [Google Scholar]
- 10.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The Human Sir2 Ortholog, SIRT2, Is an NAD(+)-Dependent Tubulin Deacetylase. Molecular cell. 2003;11:437–44. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 11.Haigis MC, Mostoslavsky R, Haigis KM, et al. SIRT4 Regulates Glutamate Dehydrogenase and Insulin Secretion in Pancreatic Beta Cells. Cell. 2006;126:941–54. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 12.Liszt G, Ford E, Kurtev M, Guarente L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. The Journal of biological chemistry. 2005;280:21313–20. doi: 10.1074/jbc.M413296200. [DOI] [PubMed] [Google Scholar]
- 13.Sauve AA, Wolberger C, Schramm VL, Boeke JD. The Biochemistry of Sirtuins. Annu Rev Biochem. 2006;75:435–65. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 14.Cheng H, Mostoslavsky R, Saito Si, et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci USA. 2003;100:10794–9. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McBurney MW, Yang X, Jardine K, et al. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. 2003;23:38–54. doi: 10.1128/MCB.23.1.38-54.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo J, Nikolaev AY, Imai S, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–48. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 17.Vaziri H, Dessain SK, Ng Eaton E, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–59. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 18.Langley E, Pearson M, Faretta M, et al. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. Embo J. 2002;21:2383–96. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol. 2000;65:127–33. doi: 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- 20.Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005;120:497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 21.Hsieh P. Molecular mechanisms of DNA mismatch repair. Mutation research. 2001;486:71–87. doi: 10.1016/s0921-8777(01)00088-x. [DOI] [PubMed] [Google Scholar]
- 22.Vijg J. Somatic mutations and aging: a re-evaluation. Mutation research. 2000;447:117–35. doi: 10.1016/s0027-5107(99)00202-x. [DOI] [PubMed] [Google Scholar]
- 23.Martin GM. Genetic modulation of senescent phenotypes in Homo sapiens. Cell. 2005;120:523–32. doi: 10.1016/j.cell.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 24.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Kujoth GC, Hiona A, Pugh TD, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–4. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 26.Trifunovic A, Wredenberg A, Falkenberg M, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–23. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 27.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–47. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 28.Schriner SE, Linford NJ, Martin GM, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–11. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 29.Matheu A, Maraver A, Klatt P, et al. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448:375–9. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- 30.Migliaccio E, Giorgio M, Mele S, et al. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–13. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- 31.Yan L, Vatner DE, O’Connor JP, et al. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell. 2007;130:247–58. doi: 10.1016/j.cell.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 32.Van Remmen H, Ikeno Y, Hamilton M, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics. 2003;16:29–37. doi: 10.1152/physiolgenomics.00122.2003. [DOI] [PubMed] [Google Scholar]
- 33.Scortegagna M, Ding K, Oktay Y, et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nature genetics. 2003;35:331–40. doi: 10.1038/ng1266. [DOI] [PubMed] [Google Scholar]
- 34.Finkel T. Oxidant signals and oxidative stress. Current opinion in cell biology. 2003;15:247–54. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 35.Wilson DM, 3rd, Bohr VA. The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair (Amst) 2007;6:544–59. doi: 10.1016/j.dnarep.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 36.Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J. Aging and genome maintenance: lessons from the mouse? Science. 2003;299:1355–9. doi: 10.1126/science.1079161. [DOI] [PubMed] [Google Scholar]
- 37.Andressoo JO, Hoeijmakers JH, Mitchell JR. Nucleotide excision repair disorders and the balance between cancer and aging. Cell Cycle. 2006;5:2886–8. doi: 10.4161/cc.5.24.3565. [DOI] [PubMed] [Google Scholar]
- 38.Bergmann E, Egly JM. Trichothiodystrophy, a transcription syndrome. Trends Genet. 2001;17:279–86. doi: 10.1016/s0168-9525(01)02280-6. [DOI] [PubMed] [Google Scholar]
- 39.de Boer J, Andressoo JO, de Wit J, et al. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296:1276–9. doi: 10.1126/science.1070174. [DOI] [PubMed] [Google Scholar]
- 40.Murai M, Enokido Y, Inamura N, et al. Early postnatal ataxia and abnormal cerebellar development in mice lacking Xeroderma pigmentosum Group A and Cockayne syndrome Group B DNA repair genes. Proc Natl Acad Sci U S A. 2001;98:13379–84. doi: 10.1073/pnas.231329598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van de Ven M, Andressoo JO, Holcomb VB, et al. Adaptive Stress Response in Segmental Progeria Resembles Long-Lived Dwarfism and Calorie Restriction in Mice. PLoS Genet. 2006;2:e192. doi: 10.1371/journal.pgen.0020192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niedernhofer LJ, Garinis GA, Raams A, et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–43. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- 43.Tian M, Shinkura R, Shinkura N, Alt FW. Growth retardation, early death, and DNA repair defects in mice deficient for the nucleotide excision repair enzyme XPF. Mol Cell Biol. 2004;24:1200–5. doi: 10.1128/MCB.24.3.1200-1205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McWhir J, Selfridge J, Harrison DJ, Squires S, Melton DW. Mice with DNA repair gene (ERCC-1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nature genetics. 1993;5:217–24. doi: 10.1038/ng1193-217. [DOI] [PubMed] [Google Scholar]
- 45.van der Pluijm I, Garinis GA, Brandt RM, et al. Impaired genome maintenance suppresses the growth hormone--insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biol. 2006;5:e2. doi: 10.1371/journal.pbio.0050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rincon M, Rudin E, Barzilai N. The insulin/IGF-1 signaling in mammals and its relevance to human longevity. Exp Gerontol. 2005;40:873–7. doi: 10.1016/j.exger.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 47.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 48.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 49.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–25. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 50.Fabrizio P, Pozza F, Pletcher SD, Gendron CM, Longo VD. Regulation of longevity and stress resistance by Sch9 in yeast. Science. 2001;292:288–90. doi: 10.1126/science.1059497. [DOI] [PubMed] [Google Scholar]
- 51.Kaeberlein M, Powers RW, 3rd, Steffen KK, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–6. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- 52.Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–60. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 53.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–90. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Current opinion in cell biology. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 55.Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- 56.Taguchi A, Wartschow LM, White MF. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science. 2007;317:369–72. doi: 10.1126/science.1142179. [DOI] [PubMed] [Google Scholar]
- 57.Bluher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572–4. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- 58.Moazed D. Enzymatic activities of Sir2 and chromatin silencing. Current opinion in cell biology. 2001;13:232–8. doi: 10.1016/s0955-0674(00)00202-7. [DOI] [PubMed] [Google Scholar]
- 59.Sinclair DA, Guarente L. Extrachromosomal rDNA circles--a cause of aging in yeast. Cell. 1997;91:1033–42. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 60.Holmes SG, Rose AB, Steuerle K, Saez E, Sayegh S, Lee YM, Broach JR. Hyperactivation of the silencing proteins, Sir2p and Sir3p, causes chromosome loss. Genetics. 1997;145:605–14. doi: 10.1093/genetics/145.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–80. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weitao T, Budd M, Campbell JL. Evidence that yeast SGS1, DNA2, SRS2, and FOB1 interact to maintain rDNA stability. Mutation research. 2003;532:157–72. doi: 10.1016/j.mrfmmm.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 63.Aguilaniu H, Gustafsson L, Rigoulet M, Nystrom T. Asymmetric Inheritance of Oxidatively Damaged Proteins During Cytokinesis. Science. 2003;299:1751. doi: 10.1126/science.1080418. [DOI] [PubMed] [Google Scholar]
- 64.Kaeberlein M, Andalis AA, Liszt GB, Fink GR, Guarente L. Saccharomyces cerevisiae SSD1-V confers longevity by a Sir2p-independent mechanism. Genetics. 2004;166:1661–72. doi: 10.1534/genetics.166.4.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fabrizio P, Gattazzo C, Battistella L, Wei M, Cheng C, McGrew K, Longo VD. Sir2 blocks extreme life-span extension. Cell. 2005;123:655–67. doi: 10.1016/j.cell.2005.08.042. [DOI] [PubMed] [Google Scholar]
- 66.Chen D, Steele AD, Lindquist S, Guarente L. Increase in activity during calorie restriction requires Sirt1. Science. 2005;310:1641. doi: 10.1126/science.1118357. [DOI] [PubMed] [Google Scholar]
- 67.Alcendor RR, Gao S, Zhai P, et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512–21. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 68.Alcendor RR, Kirshenbaum LA, Imai S, Vatner SF, Sadoshima J. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ Res. 2004;95:971–80. doi: 10.1161/01.RES.0000147557.75257.ff. [DOI] [PubMed] [Google Scholar]
- 69.Wang C, Chen L, Hou X, et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat Cell Biol. 2006;8:1025–31. doi: 10.1038/ncb1468. [DOI] [PubMed] [Google Scholar]
- 70.Picard F, Kurtev M, Chung N, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–6. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim D, Nguyen MD, Dobbin MM, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. Embo J. 2007;26:3169–79. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–21. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 73.Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437–48. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 74.Pruitt K, Zinn RL, Ohm JE, et al. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2006;2:e40. doi: 10.1371/journal.pgen.0020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chua KF, Mostoslavsky R, Lombard DB, et al. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab. 2005;2:67–76. doi: 10.1016/j.cmet.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 76.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–22. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 77.Valenzano DR, Terzibasi E, Genade T, Cattaneo A, Domenici L, Cellerino A. Resveratrol prolongs lifespan and retards the onset of age-related markers in a short-lived vertebrate. Curr Biol. 2006;16:296–300. doi: 10.1016/j.cub.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 78.Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–9. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 79.Baur JA, Pearson KJ, Price NL, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–42. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Howitz KT, Bitterman KJ, Cohen HY, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–6. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 81.Lagouge M, Argmann C, Gerhart-Hines Z, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–22. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 82.Zhang J. Resveratrol inhibits insulin responses in a SirT1-independent pathway. Biochem J. 2006;397:519–27. doi: 10.1042/BJ20050977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Frojdo S, Cozzone D, Vidal H, Pirola L. Resveratrol is a class IA phosphoinositide 3-kinase inhibitor. Biochem J. 2007 doi: 10.1042/BJ20070236. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A. 2007;104:7217–22. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Viswanathan M, Kim SK, Berdichevsky A, Guarente L. A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span. Developmental cell. 2005;9:605–15. doi: 10.1016/j.devcel.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 86.Tsukamoto Y, Kato J, Ikeda H. Silencing factors participate in DNA repair and recombination in Saccharomyces cerevisiae. Nature. 1997;388:900–3. doi: 10.1038/42288. [DOI] [PubMed] [Google Scholar]
- 87.Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell. 1999;97:621–33. doi: 10.1016/s0092-8674(00)80773-4. [DOI] [PubMed] [Google Scholar]
- 88.Mills KD, Sinclair DA, Guarente L. MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell. 1999;97:609–20. doi: 10.1016/s0092-8674(00)80772-2. [DOI] [PubMed] [Google Scholar]
- 89.Astrom SU, Okamura SM, Rine J. Yeast cell-type regulation of DNA repair. Nature. 1999;397:310. doi: 10.1038/16833. [DOI] [PubMed] [Google Scholar]
- 90.Lee SE, Paques F, Sylvan J, Haber JE. Role of yeast SIR genes and mating type in directing DNA double-strand breaks to homologous and non-homologous repair paths. Curr Biol. 1999;9:767–70. doi: 10.1016/s0960-9822(99)80339-x. [DOI] [PubMed] [Google Scholar]
- 91.Yuan Z, Zhang X, Sengupta N, Lane WS, Seto E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Molecular cell. 2007;27:149–62. doi: 10.1016/j.molcel.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maas NL, Miller KM, DeFazio LG, Toczyski DP. Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Molecular cell. 2006;23:109–19. doi: 10.1016/j.molcel.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 93.Celic I, Masumoto H, Griffith WP, Meluh P, Cotter RJ, Boeke JD, Verreault A. The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr Biol. 2006;16:1280–9. doi: 10.1016/j.cub.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 94.Brachmann CB, Sherman JM, Devine SE, Cameron EE, Pillus L, Boeke JD. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 1995;9:2888–902. doi: 10.1101/gad.9.23.2888. [DOI] [PubMed] [Google Scholar]
- 95.Garcia-Salcedo JA, Gijon P, Nolan DP, Tebabi P, Pays E. A chromosomal SIR2 homologue with both histone NAD-dependent ADP-ribosyltransferase and deacetylase activities is involved in DNA repair in Trypanosoma brucei. Embo J. 2003;22:5851–62. doi: 10.1093/emboj/cdg553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hasan S, El-Andaloussi N, Hardeland U, et al. Acetylation regulates the DNA end-trimming activity of DNA polymerase beta. Molecular cell. 2002;10:1213–22. doi: 10.1016/s1097-2765(02)00745-1. [DOI] [PubMed] [Google Scholar]
- 97.Beard BC, Wilson SH, Smerdon MJ. Suppressed catalytic activity of base excision repair enzymes on rotationally positioned uracil in nucleosomes. Proc Natl Acad Sci U S A. 2003;100:7465–70. doi: 10.1073/pnas.1330328100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nilsen H, Lindahl T, Verreault A. DNA base excision repair of uracil residues in reconstituted nucleosome core particles. Embo J. 2002;21:5943–52. doi: 10.1093/emboj/cdf581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moynihan KA, Grimm AA, Plueger MM, et al. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2:105–17. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 100.Bordone L, Motta MC, Picard F, et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006;4:e31. doi: 10.1371/journal.pbio.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lemieux ME, Yang X, Jardine K, et al. The Sirt1 deacetylase modulates the insulin-like growth factor signaling pathway in mammals. Mech Ageing Dev. 2005;126:1097–105. doi: 10.1016/j.mad.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 102.Tissenbaum HA, Guarente L. Increased dosage of a sir–2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–30. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 103.Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6:505–14. doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- 104.Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Barja G. Free radicals and aging. Trends Neurosci. 2004;27:595–600. doi: 10.1016/j.tins.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 106.Lamming DW, Latorre-Esteves M, Medvedik O, et al. HST2 mediates SIR2-independent life-span extension by calorie restriction. Science. 2005;309:1861–4. doi: 10.1126/science.1113611. [DOI] [PubMed] [Google Scholar]
- 107.Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A. 2004;101:15998–6003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang Y, Tissenbaum HA. Overlapping and distinct functions for a Caenorhabditis elegans SIR2 and DAF-16/FOXO. Mech Ageing Dev. 2006;127:48–56. doi: 10.1016/j.mad.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 109.Tsuchiya M, Dang N, Kerr EO, et al. Sirtuin-independent effects of nicotinamide on lifespan extension from calorie restriction in yeast. Aging Cell. 2006;5:505–14. doi: 10.1111/j.1474-9726.2006.00240.x. [DOI] [PubMed] [Google Scholar]
- 110.Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. The Journal of biological chemistry. 2005;280:13560–7. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- 111.Cohen HY, Miller C, Bitterman KJ, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–2. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 112.Al-Regaiey KA, Masternak MM, Bonkowski M, Sun L, Bartke A. Long-lived growth hormone receptor knockout mice: interaction of reduced insulin-like growth factor i/insulin signaling and caloric restriction. Endocrinology. 2005;146:851–60. doi: 10.1210/en.2004-1120. [DOI] [PubMed] [Google Scholar]
- 113.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–8. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 114.Gerhart-Hines Z, Rodgers JT, Bare O, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. Embo J. 2007;26:1913–23. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Viniegra JG, Martinez N, Modirassari P, et al. Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. The Journal of biological chemistry. 2005;280:4029–36. doi: 10.1074/jbc.M410344200. [DOI] [PubMed] [Google Scholar]
- 116.Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. The Journal of biological chemistry. 2004;279:41189–96. doi: 10.1074/jbc.M406731200. [DOI] [PubMed] [Google Scholar]
- 117.Peretz S, Jensen R, Baserga R, Glazer PM. ATM-dependent expression of the insulin-like growth factor-I receptor in a pathway regulating radiation response. Proc Natl Acad Sci U S A. 2001;98:1676–81. doi: 10.1073/pnas.041416598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shahrabani-Gargir L, Pandita TK, Werner H. Ataxia-telangiectasia mutated gene controls insulin-like growth factor I receptor gene expression in a deoxyribonucleic acid damage response pathway via mechanisms involving zinc-finger transcription factors Sp1 and WT1. Endocrinology. 2004;145:5679–87. doi: 10.1210/en.2004-0613. [DOI] [PubMed] [Google Scholar]
- 119.Schneider JG, Finck BN, Ren J, et al. ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab. 2006;4:377–89. doi: 10.1016/j.cmet.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 120.Shukla V, Coumoul X, Cao L, et al. Absence of the full-length breast cancer-associated gene-1 leads to increased expression of insulin-like growth factor signaling axis members. Cancer research. 2006;66:7151–7. doi: 10.1158/0008-5472.CAN-05-4570. [DOI] [PubMed] [Google Scholar]
- 121.Maier B, Gluba W, Bernier B, et al. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004;18:306–19. doi: 10.1101/gad.1162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Calogero S, Grassi F, Aguzzi A, Voigtlander T, Ferrier P, Ferrari S, Bianchi ME. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nature genetics. 1999;22:276–80. doi: 10.1038/10338. [DOI] [PubMed] [Google Scholar]
- 123.Giavara S, Kosmidou E, Hande MP, Bianchi ME, Morgan A, d’Adda di Fagagna F, Jackson SP. Yeast Nhp6A/B and mammalian Hmgb1 facilitate the maintenance of genome stability. Curr Biol. 2005;15:68–72. doi: 10.1016/j.cub.2004.12.065. [DOI] [PubMed] [Google Scholar]
- 124.Levine AJ, Feng Z, Mak TW, You H, Jin S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 2006;20:267–75. doi: 10.1101/gad.1363206. [DOI] [PubMed] [Google Scholar]
- 125.Dempsey JL, Pfeiffer M, Morley AA. Effect of dietary restriction on in vivo somatic mutation in mice. Mutation research. 1993;291:141–5. doi: 10.1016/0165-1161(93)90153-q. [DOI] [PubMed] [Google Scholar]
- 126.Aidoo A, Mittelstaedt RA, Bishop ME, Lyn-Cook LE, Chen YJ, Duffy P, Heflich RH. Effect of caloric restriction on Hprt lymphocyte mutation in aging rats. Mutation research. 2003;527:57–66. doi: 10.1016/s0027-5107(03)00072-1. [DOI] [PubMed] [Google Scholar]
- 127.Puc J, Keniry M, Li HS, et al. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell. 2005;7:193–204. doi: 10.1016/j.ccr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 128.Puc J, Parsons R. PTEN loss inhibits CHK1 to cause double stranded-DNA breaks in cells. Cell Cycle. 2005;4:927–9. doi: 10.4161/cc.4.7.1795. [DOI] [PubMed] [Google Scholar]
- 129.Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–70. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 130.Tothova Z, Kollipara R, Huntly BJ, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–39. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 131.van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nature reviews. 2007;8:440–50. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]