

Abstract
Early embryonic development in the pig requires DNA methylation remodeling of the maternal and paternal genomes. Aberrant remodeling, which can be exasperated by in vitro technologies, is detrimental to development and can result in physiological and anatomic abnormalities in the developing fetus and offspring. Here, we developed and validated a microarray based approach to characterize on a global scale the CpG methylation profiles of porcine gametes and blastocyst stage embryos. The relative methylation in the gamete and blastocyst samples showed that 18.5% (921/4992) of the DNA clones were found to be significantly different (P<0.01) in at least one of the samples. Furthermore, for the different blastocyst groups, the methylation profile of the in vitro-produced blastocysts was less similar to the in vivo-produced blastocysts as compared to the parthenogenetic- and somatic cell nuclear transfer (SCNT)-produced blastocysts. The microarray results were validated by using bisulfite sequencing for 12 of the genomic regions in liver, sperm, and in vivo-produced blastocysts. These results suggest that a generalized change in global methylation is not responsible for the low developmental potential of blastocysts produced by using in vitro techniques. Instead, the appropriate methylation of a relatively small number of genomic regions in the early embryo may enable early development to occur.
Introduction
Epigenetic remodeling of the paternal and maternal genomes is necessary for the development of the early embryo and occurs immediately after fertilization and continues through the blastocyst stage. In some species, the male pronucleus undergoes active demethylation within 4 hours after fertilization (Mayer et al. 2000a; Mayer et al. 2000b; Santos et al. 2002), but not in sheep (Wilmut et al. 2002) or rabbits (Beaujean et al. 2004; Shi et al. 2004). The maternal genome undergoes passive demethylation until the blastocyst stage (Rougier et al. 1998). Remethylation or remodeling of the genome appears to occur around the time of implantation and is maintained in somatic tissues. De novo remethylation is differentially applied in the cells of the inner cell mass (ICM) and trophectoderm (TE) (Dean et al. 2001; Santos et al. 2002); with the TE being hypomethylated relative to the ICM.
In vitro techniques of embryo production, such as in vitro fertilization and nuclear transfer, are very inefficient and sometimes result in offspring with severe abnormalities (Carter et al. 2002). The precise causes of the phenotypic abnormalities observed in cloned animals are thought, in part, to be related to epigenetic defects because the offspring of cloned animals appear to be normal (Conway 1996; Tamashiro et al. 2002; Zhang et al. 2004). For somatic cell nuclear transfer to produce viable offspring requires that DNA remodeling and transcriptional reprogramming approximate that of the in vivo-produced embryo. The inefficiency of the epigenetic reprogramming is demonstrated by the analysis of transcription and methylation status of imprinted genes in the preimplantation stage mouse cloned embryo where only 4% of the SCNT derived embryos reproduced the expression of the imprinted genes H19, Meg3, Igf2r, Ascl2, and Snrpn relative to in vivo derived blastocysts (Mann et al. 2003). The cloned embryos were also found to have substantial loss of allele-specific DNA methylation at the imprinting control regions of the Snrpn and H19 genes.
The dynamics of methylation remodeling in the early pig embryo and cloned offspring have been studied for several sequences. IVF and SCNT blastocysts show gradual demethylation of centromeric satellite and Pre-1 in the development to the blastocyst (Kang 2001). The demethylation of these sequences was also shown to be similar to the in vivo produced blastocysts. Pre-1 and centromeric satellite sequences were found to have similar methylation levels in healthy cloned pigs and control pigs (Archer et al. 2003). However, even normal appearing pigs can have aberrant expression of imprinted genes (Jiang et al. 2007).
In this study we used a global microarray-based approach, Porcine Differential Methylation Hybridization (PDMH), based on a similar tool that has been developed for humans (Huang et al. 1999), to analyze the CpG methylation in porcine gametes, and embryos. The specific question addressed was to determine if there is a difference in the global methylation profiles in the parthenogenetic-, SCNT-, in vitro, and in vivo-produced blastocysts that explain the developmental differences of these samples. Our hypothesis was that the blastocysts with the highest development rates (in vitro- and in vivo-produced blastocysts) would be more similar to each other, and the blastocysts with the lowest development rates (parthenogenetic- and SCNT-produced blastocysts) would be least similar to in vivo-produced blastocysts.
Materials and Methods
Oocyte acquisition and in vitro maturation
Cumulus-oocyte-complexes (COCs) were aspirated from ovaries of prepubertal gilts collected from a local abattoir. Germinal vesicle (GV) stage oocytes were collected for PDMH analysis or matured in vitro prior to in vitro fertilization (Abeydeera 2002). For PDMH analysis, COCs with numerous layers of intact cumulus cells were vortexed in 0.1% (w/v) hyaluronidase in Hepes-buffered saline for at least 5 minutes to remove the cumulus cells. Denuded GV-stage oocytes were rinsed three times in phosphate buffered saline containing 3 mg/ml Bovine Serum Albumin (BSA) (Fraction V) before the removal of the zona pellucida by incubation in 5 mg/ml pronase. The zona free GV-stage oocytes were rinsed 3 times in DEPC-treated phosphate buffered saline (PBS) and flash frozen in liquid nitrogen prior to storage at -80°C. For in vitro fertilization (IVF) the COCs were incubated in Tissue Culture Medium 199 (TCM199) (Gibco BRL, Grand Islands, NY) containing 0.14% (w/v) PVA, 10 ng/ml (w/v) Epidermal Growth Factor (EGF), 0.57 mM (w/v) cysteine, 0.5 μg/ml (w/v) porcine follicle stimulating hormone and 0.5 μg/ml (w/v) porcine lutenizing hormone (Abeydeera 1998). The maturation media was pre-equilibrated in 5% CO2 in air at 39°C in a humidified atmosphere overnight. COCs were matured for 40-44 hours in 5% CO2 in air at 39°C prior to the removal of the cumulus cells by vortexing for three minutes in Hepes-buffered medium with 0.1% (w/v) hyaluronidase. Denuded oocytes were washed and held in modified Tris-buffered medium (mTBM) prior to fertilization (Abeydeera and Day 1997).
In vitro fertilization and embryo culture
Thirty-five oocytes were delivered to 50 μl of mTBM under oil and held in 5% CO2 in air at 39°C. Freshly collected semen for IVF was diluted 3:1 in Androhep Enduraguard semen extender (Minitub, Verona, WI). The extended semen was washed in Dubelcco's PBS (Gibco BRL) with PVA/TL-Hepes/0.1% (w/v) BSA and centrifuged for 4 minutes at 1900 ×g. This wash was repeated two more times and the sperm pellet was resuspended in 100 μl of mTBM. Fresh, extended sperm were diluted to 4 ×104 per ml. The diluted sperm were preincubated for 2 hours in 5% CO2 in air at 39°C. Fifty μl of the diluted sperm was added to the oocytes and incubated for 5 hours in 5% CO2 in air at 39°C. The presumptive zygotes were then washed three times and incubated in PZM3 embryo culture medium (Yoshioka et al. 2003) in 5% CO2 at 39°C. Blastocysts were removed after 6 days of culture and the zonae pellucidae of the blastocysts were removed as described. The zona free blastocysts were rinsed 3 times in DEPC-treated PBS and flash frozen in liquid nitrogen prior to storage at -80°C.
Parthenogenetic embryo production
Cumulus cells were removed from in vitro matured (IVM) oocytes by vortexing for three minutes in Hepes-buffered medium with 0.1% (w/v) hyaluronidase, and the oocytes were equilibrated in activation medium (0.3 M mannitol, 0.5 mM Hepes, 0.01% (w/v) BSA, 0.1 mM CaCl2, and 0.1 mM MgCl2) for 5 minutes and placed in an activation chamber with electrodes 1 mm apart containing activation medium. Two 30-μsec electrical pulses of 1.2 kV/cm were delivered (BTX Electro-cell manipulator: BTX San Diego, CA). The activated oocytes were cultured in PZM3 in 5% CO2 in air at 39°C (Lai and Prather 2003).
SCNT
Reconstructed embryos were produced by using SCNT techniques as previously described (Lai and Prather 2003). Porcine fetal fibroblast-like (PFF) cultures were established from a day 30 porcine conceptus. PFFs were cultured in Dubelcco's Modified Eagle Medium containing 15% (v/v) fetal calf serum (FCS), 100 IU/ml penicillin and 100 μg/ml streptomycin in 5% CO2 in air at 39°C. PFFs were harvested with the addition of Hank's Balanced Salt Solution with 0.1% (w/v) trypsin and 0.02% (w/v) EDTA.
In vivo embryo collection
The use of animals was conducted in accordance with protocols approved by the University of Missouri Animal Care and Use Committee. Crossbred Landrace gilts were bred on Day 0 of estrus by using artificial insemination (AI). Blastocysts were flushed on Day 6 according to previously published procedures (Machaty et al. 1998). Zonae pellucidae were removed from the blastocysts as described and frozen in pools of 5-14 embryos.
Porcine Differential Methylation Hybridization
Differential methylation hybridization analysis was conducted based on a technique developed for global scanning of methylation changes in the human genome (Huang et al. 1999). Porcine CpG island clones from a Porcine CpG Island Library (United Kingdom Human Genome Mapping Project, Hinxton, Cambridge, United Kingdom) were cultured in 96-well plates. The cloned inserts were amplified by polymerase chain reaction (PCR) using the library specific primers 3558 (5′- CGG CCG CCT GCA GGT CGA CCT TAA) and 3559 (5′- AAC GCG TTG GGA GCT CTC CCT TAA). The PCR amplification was performed in a 10 μl reaction containing 1X Deep Vent DNA Polymerase Buffer, 10% DMSO, 400 pM of each primer, 100 pm each dATP, dTTP, dCTP, and dGTP, and 0.018 units Deep Vent Polymerase (New England Biolabs, Beverly, MA). The PCR program consisted of a denaturation step at 98°C for 4 minutes followed by 30 cycles of denaturation at 95°C for 30 seconds, annealing at 55°C for 30 seconds and extension at 72°C for 1 minute. A final extension at 72°C completed the program. PCR products were stored at -20°C until needed. Restriction digestion with Bstu I was performed using 1.5 μl of the PCR reaction in 1X NEB 2 and 0.4 units Bstu I at 60°C for at least 1 hr. The digested and undigested PCR products were run on a 1.5% 0.5X TBE agarose gel. Bstu I positive clones where the PCR product was cut, indicating the presence of a Bstu I site (CGCG) in the insert, were reracked and recultured in 96-well plates. Plates with all Bstu I positive clones were PCR amplified in a 50μl reaction and purified in Millipore 96-well PCR Purification plates in preparation for printing. The purified PCR products were dried and resuspended in 10 μl 50% DMSO (v/v) 1% CHAPS (w/v) (Rickman et al. 2003). The resuspended PCR products were printed on Gold Seal glass microscope slides (Fisher Scientific, Hampton, NH) that were coated with 0.02% (v/v) poly-L-lysine (Sigma, St. Louis, MO) in 0.5X PBS (Eisen 1999). The slides were stored for 3 weeks at room temperature under desiccation before printing with a pick and place robot. The printed slides were crosslinked at 120 mJ/cm2 for 20 s (Spectrolinker; Spectronics Corp., Westbury, NY) prior to blocking in 0.018% succinic anhydride (Sigma, St. Louis, MO) and 0.043 M sodium borate (Sigma, St. Louis, MO) in 1-methyl-2-pyrrolidinone (Sigma, St. Louis, MO) (Eisen and Brown 1999). Slides were stored under desiccation at room temperature until hybridization. Spotting buffer only (50% DMSO/1% CHAPS) and a whole CpG island library amplification (192 spots for each) served as the negative and positive controls, respectively. The whole CpG island library amplification was generated by PCR as a means of providing a general positive control on the microarray slides. A scraping of the frozen CpG island library was collected, amplified by using the PCR program shown above and purified by the methods described herein. A total of 384 control spots and 4,992 test spots were printed on the array.
DNA Isolation
DNA was isolated from three replicates of each of the sample types. DNA was isolated from the pooled blastocysts and GV stage oocytes by adding H2O to a final volume of 25 μl and incubating at 98°C for 15 minutes. DNA from motile sperm was isolated by gently layering the extended semen on a 60%/80% Percoll gradient and centrifuging for 600 × g for 10 minutes. The sperm pellet was removed and resuspended in PBS with 0.1% BSA. Contaminating somatic cells were eliminated by incubating the sperm pellet in PBS/ Triton X-100/SDS for 10 minutes at room temperature (RT). The sperm pellet was rinsed three times in 10 mM Tris (pH 8.0)/ 1 mM EDTA/100 mM NaCl (STE) and resuspended in 700 μl STE followed with the addition of 70 μl 20% (w/v) sodium dodecyl sulfate (SDS), 25 μl 1M dithiothreitol, and 5 μl 20 mg/ml proteinase K and incubated at 56°C overnight. The DNA was purified with phenol chloroform, precipitated with EtOH and ammonium acetate, and resuspended in 10 mM Tris/1 mM EDTA (TE).
Amplicon Generation, Labeling and Hybridization
Amplicons were produced by digesting DNA from liver (reference sample), GV stage oocyte, sperm, or blastocysts with the restriction enzyme Mse I (50 units) in 1X NEB 2, 1X BSA at 37°C overnight as recommended (New England Biolabs, Beverly, MA). The restricted DNA was ligated to PCR linkers produced by mixing oligomers (H-24, 5′- AGG CAA CTG TGC TAT CCG AGG GAT and H-12, 5′-TAA TCC CTC GGA), heating to 65°C, and cooling to room temperature. DNA was then digested with the methylation sensitive restriction enzyme BstuI (NEB) as recommended. The intact DNA fragments were amplified by PCR using H-24 as the linker specific primer. The PCR products were labeled with amino allyl-dUTP (Sigma, St. Louis, MO) by using the BioPrime labeling system with modifications. The PCR products were purified with a Qiagen PCR Purification Kit and resuspended in 29 μl H2O, mixed with 1X BioPrime buffer, dNTPs (2:3 dUTP:dTTP, dATP, dGTP, dCTP), 40 units Klenow, and incubated for 60 minutes at 37°C. Amino allyl-dUTP incorporated PCR products were purified with the Qiaquick columns using PB buffer, (phosphate washing buffer: 5 mM KPO4, 80% EtOH, pH 8.0), and phosphate elution buffer (4 mM KPO4, pH 8.5). The samples were dried and resuspended in 0.1 M sodium carbonate buffer (pH 9.0) and labeled with Cy3 for the oocyte, sperm and blastocyst samples or Cy5 for the liver reference samples. The samples were incubated for 60 minutes at room temperature in the dark. The labeling reactions were purified with Qiaquick columns by using PB buffer, PE buffer, and EB buffer. The labeling efficiency was then analyzed spectrophotometrically by using a Nanodrop ND-1000 (Nanodrop, Wilmington, DE). Comparable amounts of labeled test sample and liver reference sample were mixed together based on the incorporation of the Cy 3 and Cy5 dyes, respectively. The combined samples were dried and resuspended in 26 μl hybridization buffer (50% formamide, 5X SSC, 0.1% SDS). The samples were denatured at 95°C for 3 minutes and immediately transferred to ice before being applied to a microarray slide with a coverslip. The array was incubated at 42°C for 8-12 hours before removing the coverslip in Wash I (1X SSC/0.2% (v/v) SDS), and washing in Wash II (1X SSC/0.2% (w/v) SDS), Wash III (0.1X SSC/0.2% (w/v) SDS), Wash IV (0.1X SSC), and Wash V (H2O). The slides were immediately centrifuged for 5 minutes at 1500 ×g and scanned with an Axon 4000B scanner.
Microarray Analysis
Microarray images were analyzed with GenePix 4.0. Spots with intensities where at least 25% of the pixels were greater than 1 standard deviation higher than the background in either the Cy3 or Cy5 channel were further analyzed with Gene Spring version 7.2 (Silicon Genetics). The LOWESS normalized data was analyzed by Analysis of Variance (ANOVA) (P<0.01) using the Benjamini and Hochberg False Discovery Rate for multiple testing. Clones (n=106) were initially selected for sequencing based on the presence or absence of a significant difference between the in vivo-produced blastocysts and the other samples. The sequence of the selected clones was determined by sequencing the clone from the appropriate well of the BstuI positive clones 96-well plates after amplification by PCR using library specific 3558 and 3559 primers to amplify the insert. The PCR products were purified and sequenced by using the library specific primer 3558.
Sequencing was performed at the University of Missouri-Columbia DNA Core facility by using a 3730 96-capillary DNA Analyzer with Applied Biosystems Big Dye Terminator cycle sequencing chemistry. Sequence homology was determined by using the National Center for Biotechnology Information nucleotide-nucleotide Basic Local Alignment Search Tool. The clonal sequences were identified as homologous when a single gene was exclusively or predominantly identified (n= 42). The scores of these regions ranged from 58 to 910 with an average score (bits) of 178.
Bisulfite Sequencing
Clones were selected for bisulfite analysis because of potential biological significance to embryogenesis (e.g. transcription factors, WNT8B) and repeated detection of the same region in multiple spots in the microarray analysis (e.g. PPOX). In order to ascertain the analytical capacity of the microarray analysis procedures, additional spots were selected because microarray analysis showed that there was not a significant difference between the in vivo-produced blastocysts and the other biological samples or there was a significant difference between the in vivo-produced blastocysts and the other biological samples. This provided an independent method to verify the microarray data. Sperm and liver DNA (1 μg) were treated with sodium bisulfite by using the EZ DNA Methylation Kit (Zymo Research, Orange, CA) according to the vendors recommendations. DNA from 50 day 6 in vivo-produced blastocysts was treated with bisulfite by using the EZ DNA Methylation-Gold Kit (Zymo Research, Orange, CA) according to the vendor's recommendations. Primers were designed for bisulfite treated DNA by using the MethPrimer software (Li 2002). Primer sequences are shown in Table 1. PCR was performed with the following reagents (H2O 32.5 μl, DNTP 1.3 μl, 10X Buffer (TagGold) 5 μl, MgCl2 5 μl, Forward Primer (10 μM) 2 μl, Reverse Primer (10 μM) 2 μl, DNA (Bisulfite treated) 2 μl, Ampli Taq Gold (5 u/μl) 0.25 μl, for a total of 50 μl in the reaction. The PCR program consisted of a denaturation step at 98°C for 3 minutes followed by 50 cycles of denaturation at 95°C for 15 seconds, annealing at 55°C for 30 seconds and extension for 72°C for 30 seconds. A final extension of 72°C for 5 minutes completed the program.
Table 1.
Bisulfite Modification Specific Primers.
| CPG Clone | Position | 5′-------------------------------------------------------3′ |
|---|---|---|
| B G2 | LEFT | TTT TAT TAA TGG GAG GTA GAA TTA G |
| B G2 | RIGHT | TAA AAA CAA AAT TCT CCC AAC CTC |
| CC C1 | LEFT | TTT GAA ATT AGG GTT GTA AGG TAG GT |
| CC C1 | RIGHT | CCA CCC TCT AAC AAA AAA CTC TTA C |
| EE A11 | RIGHT | AAAAATAACTCTAACCAAAATAAAC |
| EE A11 | LEFT | TTTTAGTTAATAGGGAGGTAGTGTA |
| EEE D4 | LEFT | GGT ATT GTA GAA AGT GGG TTT GAG T |
| EEE D4 | RIGHT | AAAAATAATATAAAACCA AAA ATA ACA C |
| HH A7 | LEFT | GTT AAA GTT TGG AGT AAA AGG TG |
| HH A7 | RIGHT | AAT TTA AAA CCC CAT ATT AAA ACC |
| K D3 | LEFT | AAT AAT AAA GTT TTA GGA GGG ATT T |
| K D3 | RIGHT | ATA CTA CCC AAC CCA AAC AAA AAA |
| L E8 | LEFT | GGG TTT TAT TTT GTT TTT TTA AG |
| L E8 | RIGHT | TAT CAC TAA AAA TCA ATC CCC AAA A |
| O D10 | LEFT | GTA GAA GGT AGA TGA TTT TTT TT |
| O D10 | RIGHT | TAA AAC AAA TTT TTC AAA CCC AAA C |
| QQ E4 | RIGHT | ACAAAACTAAAACATCTCTTTACCTAAAAT |
| QQ E4 | LEFT | GTTTGGATTGGGTTTTTTGAT |
| S E3 | RIGHT | AAA AAA AAT AAC AAT TCC ACC ACC |
| S E3 | LEFT | GTT TAT GGG GAA GTT TAG GGT AGA G |
| WW G4 | LEFT | GGT TTT TTA GTT TTT TAT TTG TTT AG |
| WW G4 | RIGHT | AACTAAATCTTACCCTACTTTCTA TAA ATA |
| X G2 | RIGHT | TAA ACA CTA ACC CAA AAA AAC CTT C |
| X G2 | LEFT | GTT TGG TAG GGG AGT TTG TAG AGT |
The PCR reaction was purified by using the Qiaquick columns as described. The PCR products were cloned by using the pGEM T-Easy Kit (Promega, Madison, WI). The vectors were transformed into DH10B cells (Invitrogen) and grown on LB/IPTG/X-Gal/Ampicillin agar plates. Recombinant colonies were selected for sequencing based on the blue/white screening criteria. The cytosines of the CpG sites were identified as methylated or unmethylated if a C or T was present in the sequence, respectively. The percent methylation was calculated for the respective sequence and the methylation status of the microarrays and bisulfite sequencing were compared. A ratio of liver:donor cell methylation was calculated by using the following formula: Rm = (100-MS) / (100-ML). Where: MS is the % CpG methylation in the sample and ML is the % CpG methylation in the liver reference.
The use of this formula provided a means to calculate a ratio that indicateed the relative levels of methylation in a given sequence when one of the samples lacks methylated CpG dinucleotides. The ratios produced from the microarray and bisulfite analysis were classified as consistent when the bisulfite analysis-produced ratio indicated the sample was hypomethylated (>1) or hypermethylated (<1) and matched the hypermethylation status of the microarray-produced data. The threshold of hypermethylation and hypomethylation for the microarray results was identified as the smallest deviation from 1 for those regions which were validated by using bisulfite analysis. Specifically, the microarray results for CPG X G2 of the sperm sample was 1.251. Accordingly, microarray results were classified as hypomethylated when the microarray ratio was >1.25 and the sample was classified as hypermethylated when the microarray ratio was <0.75.
Results
Differential Methylation in the GV Oocyte, Sperm, and Blastocysts
The relative methylation in the gamete and blastocyst samples analyzed by using GeneSpring 7.2 revealed that 18.5% (921/4,992) of the clones were found to be significantly different (P<0.01) in at least one of the samples. A Self Organized map shows this relationship (Fig. 1). Of greatest interest are those clones in Figure 1C and 1D. In Figure 1C those clones that were hypermethylated in the in vivo-produced blastocysts tended to be relatively hypomethylated in the IVF embryos. Conversely, those clones that were hypomethylated in the in vivo-produced blastocysts were relatively hypermethylated in most of the other samples.
Figure 1.
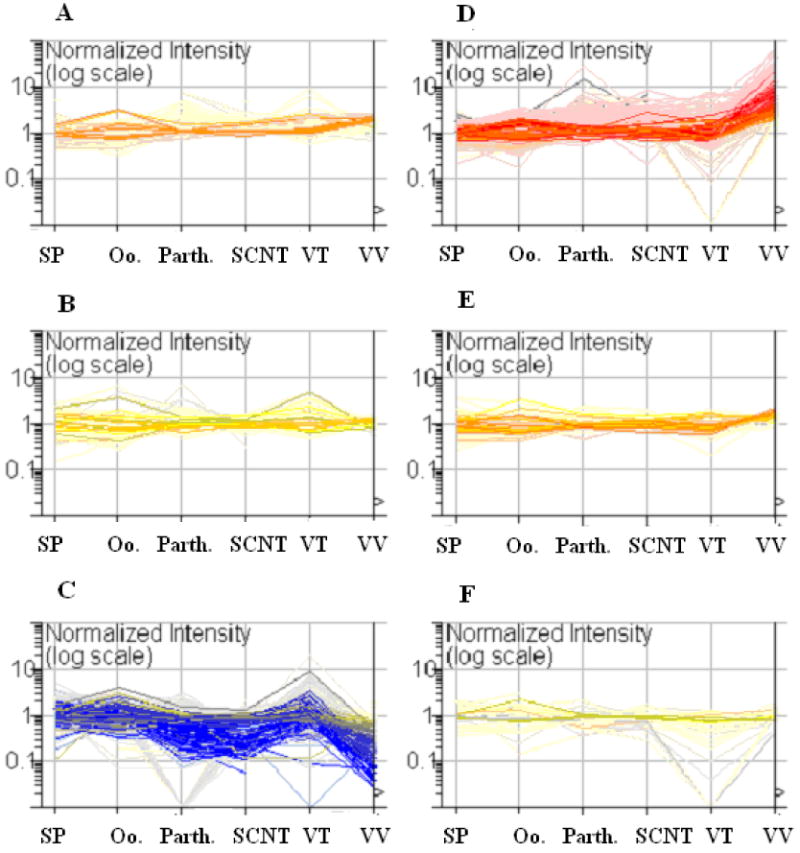
Self Organized Map analysis of methylation profiles of porcine sperm (Sp), germinal vesicle oocytes (Oo), parthenogenetic- (P), nuclear transfer- (N), in vitro- (VT), and in vivo-(VV) produced blastocysts generated by using PDMH analysis. This graph shows the Reference/Sample ratios for the 921 clones that were significantly different (P<0.01) in at least one of the sample groups. A Reference/Sample ratio greater than 1 indicates that the reference is hypermethylated relative to the sample and a Reference/Sample ratio less than one indicates that the reference is hypomethylated relative to the sample. Each line represents the methylation status of a single clone at the different stages listed on the X axis. The in vivo-produced blastocysts have more genes that are hypomethylated relative to the reference as compared to the other samples. Extensive hypermethylation is measured in the parthenogenetic-, SCNT-, and in vivo-produced blastocysts but not in the in vitro-produced blastocysts. The clones are colored by the Reference/Sample ratios in the in vivo-produced blastocyst sample.
Genomic clones were selected to be sequenced based the relationship between the ratios for the sperm and in vivo-produced blastocysts. Of the 104 clones that were successfully sequenced, relevant annotation was available for only 33 of the clones. One sequence was represented twice and another represented three times. Thus, there was little redundancy on the array and most (97.1%: 101/104) of the clones represented different genomic regions. These sequences were deposited in GenBank (DQ915200-DQ915250, EF185170-EF185205, and EF189709-EF189710).
Validation of the Microarray Analysis by using Bisulfite Sequencing
In order to validate the microarray data, clones (n=12) were selected when the ratios of the sperm and in vivo-produced blastocysts were similar and when the ratios were significantly different (Table 2). For the selected clones, the methylation status of the in vivo-produced blastocysts relative to the liver reference samples was identified as hypomethylated for 6/12 (50.0%), equivalent for 2/12 (16.7%), and hypermethylated for 4/12 (33.3%) of the samples. ANOVA was used to detect a significant difference between the in vivo-produced blastocysts and the other samples in 32/60 (53.3%) of the comparisons (Table 2).
Table 2.
Microarray reference/sample ratio ± standard error of microarray clones identifies the methylation status of the selected clones. A ratio greater than one indicates the region is hypomethylated in the test sample relative to the reference sample. A ratio less than one indicates the region is hypermethylated in the test sample relative to the reference sample. These clones were selected to validate the microarray results by using bisulfite sequencing. A significant difference was detected by using ANOVA between the in vivo-produced blastocysts and the other samples (a - P<0.05, b- P<0.01, c-P<0.001, d -P<0.0001). There was a significant difference between the in vivo-produced blastocysts versus the other samples in 32/60 (53.33%) of the samples. n.d.- no data.
| Sperm | Oocyte | Parth. blast. | SCNT blast. | In vitro blast. | In vivo blast. | |
|---|---|---|---|---|---|---|
| B G2 | 0.960±0.172 c | 1.043±0.181 c | 0.992±0.203 c | 1.748±0.241 b | 0.878±0.187 c | 21.520±11.047 |
| CC C1 | 2.055±0.545 b | 1.145±0.607 | 0.354±0.455 | 0.324±0.293 | n.d. | 0.375±0.565 |
| EE A11 | 1.262±0.208 c | 0.867±0.201 b | 0.560±0.243 b | 0.476±0.240 a | 1.232±0.199 c | 0.173±0.354 |
| EEE D4 | 1.074±0.191 c | 2.825±0.223 b | 3.381±0.484 a | 2.544±0.710 a | 1.463±0.235 b | 32.965±20.055 |
| HH A7 | 0.619±0.427 c | 2.033±0.773 a | 2.539±2.283 a | 0.354±0.677 | 4.885±1.886 b | 0.558±0.673 |
| K D3 | 0.593±0.214 c | 0.411±0.548 a | 1.674±0.501 | 1.432±0.291 | 3.364 | 2.806±1.259 |
| L E8 | 1.033±0.229 a | 0.804±0.253 | 0.485±0.242 | 0.552±0.242 | 0.721±0.446 | 0.464±0.450 |
| O D10 | 1.105±0.175 d | 1.068±0.188 c | 1.178±0.214 c | 1.546±0.239 b | 0.990±0.196 b | 6.897±0.966 |
| QQ E4 | 0.462±0.271 | 0.179±0.320 | 3.438±1.776 | 0.965±0.401 | n.d. | 1.135±0.716 |
| S E3 | 0.910±0.290 | 1.691±0.721 | 1.235±0.574 | 0.833±0.318 | n.d. | 0.859±0.578 |
| WW G4 | 1.506±0.413 b | 2.378±0.943 | 2.054±1.158 | 0.218±1.187 | 3.959±0.976 | 5.927±2.425 |
| X G2 | 1.251±0.203 b | 1.203±0.185 b | 1.240±0.328 | 2.129±0.313 | 0.938±0.322 b | 3.596±0.692 |
Bisulfite sequencing indicated high levels of methylation for the clones B G2, HH A7, WW G4, and X G2 (Fig. 2) in liver, sperm or in vitro-produced blastocysts. The percent methylation levels of cytosines in the CpG dinucleotides is shown in Table 3 for the clones B G2, HH A7, WW G4, and X G2 in the in vivo-produced blastocysts. The microarray values are LOWESS normalized Cy5/Cy3 ratios representing the methylation status of the specified clones in the liver (Cy5) and the sperm and in vivo-produced blastocyst samples (Cy3). The Bisulfite (Reference/Sample) values represent the relative methylation levels in the liver and in vivo-produced blastocyst at selected regions of the specified clones. The Bisulfite (Reference/Sample) values were calculated from the equation shown in the Materials and Methods section. The methylation status, determined by microarray analysis and bisulfite sequencing, of these four regions of the in vivo-produced blastocysts is graphically depicted in Figure 3. The methylation status, determined by microarray analysis and bisulfite sequencing, of these four regions of the sperm is shown in Figure 4. The reference/sample ratios were consistent with the microarray data in 87.5% (7/8) of the regions that were tested.
Figure 2.
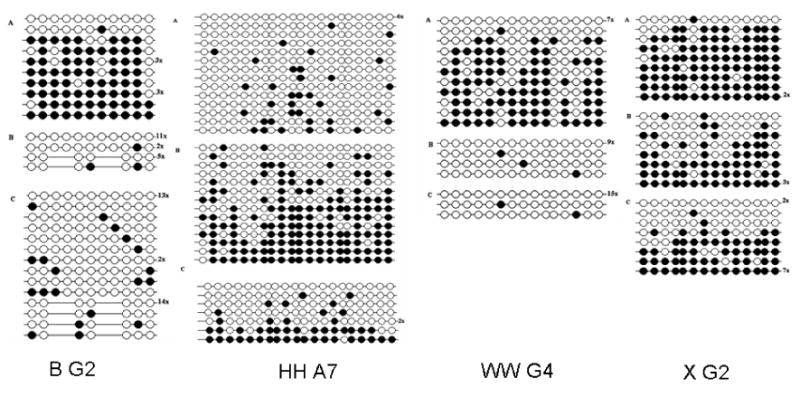
Methylation status of 4 clones in the liver (A), sperm (B) and in vivo-produced blastocyst (C) detected by using bisulfite sequencing. Closed circles identify methylated cytosines and open circles identify unmethylated cytosines in the sequenced clones. The number of clones with the same methylation pattern is shown to the right of the sequence. Also, internal regions of B G2 identify an alternate sequence in the sperm and in vivo-produced blastocyst. This alternate sequence may be a duplicated region that retained regions homologous to the primers but include two modified regions.
Table 3.
Methylation status of B G2, HH A7, WW G4, and X G2 for in vivo-produced blastocysts analyzed by using microarray and bisulfite sequencing analysis. A.Shown here is the percent cytosine methylation at all the CpG dinucleotides that were analyzed by using bisulfite sequencing in the liver DNA, sperm DNA, and in vivo-produced blastocyst DNA for the regions analyzed by using bisulfite sequencing. B) Bisulfite analysis data and the microarray analysis data are in agreement for 87.5% (7/8) of the samples.
| A | ||||
| Bisulfite Analysis | ||||
| CPG clone | Liver | Sperm | In vivo blast. | |
| B G2 | 0.692 | 0.024 | 0.053 | |
| HH A7 | 0.092 | 0.543 | 0.313 | |
| WW G4 | 0.341 | 0.018 | 0.008 | |
| X G2 | 0.807 | 0.657 | 0.667 | |
| B | ||||
| Sperm (Ref/Sample) | In vivo-produced Blast (Ref/Sample) | |||
| CPG clone | Bisulfite | Microarray | Bisulfite | Microarray |
| B G2 | (3.173) | (0.960) | 3.078 | 21.520 |
| HH A7 | 0.504 | 0.619 | 0.757 | 0.558 |
| WW G4 | 1.490 | 1.506 | 1.505 | 5.927 |
| X G2 | 1.778 | 1.251 | 1.728 | 3.596 |
Figure 3.

Methylation status of in vivo-produced blastocysts measured by using microarray and bisulfite analysis. Bisulfite analysis confirmed the microarray data at these four regions in the in vivo-produced blastocyst. Bisulfite analysis data and the microarray analysis data are in agreement for all 4 clones. The ratios produced from the microarray and bisulfite analysis were classified as consistent when the bisulfite analysis-produced ratio indicated the sample was hypomethylated (>1) or hypermethylated (<1) and matched the methylation status of the microarray-produced data. From the microarray-produced ratios, the samples were classified as hypermethylated when the ratio was < 0.75 and the sample was classified as hypomethylated when the ratio was >1.25. The microarray values are LOWESS normalized Cy5/Cy3 ratios representing the methylation status of the specified clones in the liver (Cy5) and in vivo-derived blastocyst (Cy3) samples. The Bisulfite ratios (Ref/Sample) were calculated from the equation shown in the Materials and Methods section.
Figure 4.

Methylation status of sperm measured by microarray and bisulfite sequencing analysis. The methylation status identified by using the PDMH microarrays was confirmed by bisulfite sequencing (as described in the legend for Figure 3) for all regions except for B G2).
The remaining 8 regions showed very little methylation (<10%) in the liver, sperm, and in vivo-produced blastocyst samples (Fig. 5). The low levels of methylated CpGs were consistent with the microarray data in 62.5% of the samples (10/16) (Table 4). Table 4A shows the percent methylation for the clones CC C1, EEE D4, EE A11, K D3, L E8, O D10, QQ E4, and S E3 in the liver (Reference), sperm, and in vivo-produced blastocyst samples. The microarray values are LOWESS normalized Cy5/Cy3 ratios representing the methylation status of the specified clones in the liver (Cy5) and in the sperm and in vivo-produced blastocyst samples (Cy3) (Table 4B). The Bisulfite (Reference /Sample) values represent the relative methylation levels in the liver and in vivo-produced blastocysts at selected regions of the specified clones.
Figure 5.
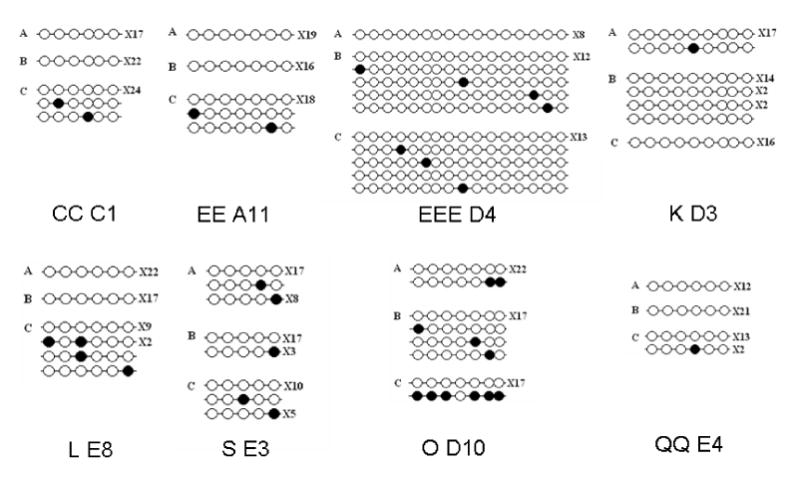
Methylation status of 8 clones in the liver (A), sperm (B) and in vivo-produced blastocyst (C) detected by using bisulfite sequencing. Closed circles identify methylated cytosines and open circles identify unmethylated cytosines in the sequenced clones. The number of clones with the same methylation pattern is shown to the right of the sequence
Table 4.
Minimal methylation was detected by using bisulfite sequencing in 8 of the 12 clones selected for bisulfite sequencing. A) Shown here is the percent cytosine methylation at the CpG dinucleotides in the liver DNA, sperm DNA, and in vivo-produced blastocyst DNA for the regions analyzed by using bisulfite sequencing in clones CC C1, EEE D4, EE A11, K D3, L E8, O D10, QQ E4, and S E3. B) Bisulfite analysis data and the microarray analysis data are in agreement for 37.5% (6/16) of the samples (shown in bold type). The ratios produced from the microarray and bisulfite analysis were classified as consistent when the bisulfite analysis-produced ratio indicated the sample was hypomethylated (>1) or hypermethylated (<1) and matched the methylation status of the microarray-produced data. From the microarray-produced ratios, the samples were classified as hypermethylated when the ratio was < 0.8 and the sample was classified as hypomethylated when the ratio was >1.2. The ratios produced from bisulfite analysis data and the microarray analysis data are not in agreement for 62.5% (10/16) of the samples (shown in parentheses). The microarray values are LOWESS normalized Cy5/Cy3 ratios representing the methylation status of the specified clones in the liver (Cy5) and in vivo-derived blastocyst (Cy3) samples. The Bisulfite ratios (Ref/Sample) were calculated from the equation shown in the Materials and Method section.
| A | |||
| Percent methylation | |||
| Liver | Sperm | IVP Blast | |
| CC C1 | 0.00 | 0.00 | 1.54 |
| EE A11 | 0.00 | 0.00 | 1.43 |
| EEE D4 | 0.00 | 1.56 | 1.10 |
| K D3 | 0.62 | 0.00 | 0.00 |
| L E8 | 0.00 | 0.00 | 7.14 |
| O D10 | 1.24 | 2.14 | 4.76 |
| QQ E4 | 0.00 | 0.00 | 2.22 |
| S E3 | 6.92 | 3.00 | 7.50 |
| B | |||
| Sperm (Ref/Sample) | IVP Blast (Ref/Sample) | ||
| Bisulfite | PDMH microarray | Bisulfite | PDMH microarray |
| (1.000) | (2.055) | (0.985) | (0.375) |
| (1.000) | (1.262) | (0.986) | (0.173) |
| 0.984 | 1.074 | (0.989) | (32.960) |
| (1.006) | (0.593) | (1.006) | (2.806) |
| 1.000 | 1.033 | (0.929) | (0.464) |
| 0.991 | 1.105 | (0.964) | (6.897) |
| (1.000) | (0.462) | 0.978 | 1.135 |
| 1.042 | 0.910 | 0.994 | 0.859 |
Figure 6 shows a typical arrangement of the location of a CpG island relative to the start of a gene. A differentially methylated CpG island was identified 184 bases upstream of the start of the myeloid leukemia factor 1 gene (WW G4). The methylation profile of WW G4 (myeloid leukemia factor 1 (MLF1) was measured by using PDMH microarrays. This region was hypomethylated in all samples relative to the reference sample except for the SCNT-produced blastocysts where the region was hypermethylated relative to the liver reference sample (Table 2).
Figure 6.
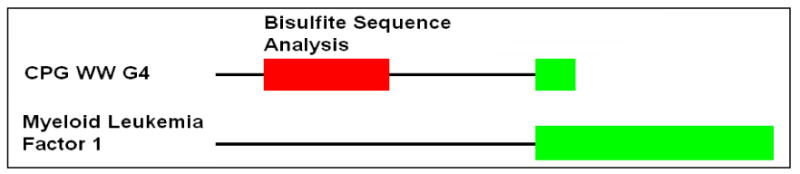
Bisulfite sequencing and BLAST analysis of WW G4. The region 184 bases upstream of the start site of myeloid leukemia factor 1 (MLF1) is hypomethylated in the sperm and egg relative to the liver.
Analysis Based on Methylation Profiles
Figure 1 shows the relative methylation status of the sperm, GV stage oocyte and blastocysts by using Self Organizing Map Analysis. BLAST analyses of clones that are hypomethylated relative to the liver in the in vivo-produced sequenced clones shown are shown in Table 5. ANOVA identified a significant difference between the in vivo-produced blastocysts versus the other samples with 134/185 (72.43%) of the samples. BLAST analyses of clones that are hypomethylated relative to the liver in the in vivo-produced sequenced clones shown are shown in Table 6. ANOVA identified a significant difference between the in vivo-produced blastocysts versus the other samples with 97/165 (58.78%) of the samples. The BLAST analysis for the clones shown in Figure 1 is on our web site (http://animalsciences.missouri.edu/faculty/prather/ ). Figure 7 shows a condition tree based on the similarity of the methylation profiles of the sperm, GV stage oocyte, and blastocysts for the clones where there was a significant difference (P>0.01) in the methylation status in at least one of the samples. The in vivo-produced blastocysts clustered with the SCNT- and parthenogenetic- produced blastocysts. Interestingly the sperm and GV stage oocytes clustered together and were more similar to each other than any of the blastocyst stage embryos. It should be noted that while the parthenogenetic blastocysts clustered with the SCNT- and in vivo-produced blastocysts the clustering is not strong as illustrated in the Supplemental Figures 1 and 2.
Table 5.
BLAST analysis of sequenced clones that were hypermethylated in the in vivo-produced blastocysts relative to the liver and their ratios. A significant difference was detected by using ANOVA between the in vivo-produced blastocysts and the other samples (a - P<0.05, b- P<0.01, c-P<0.001, d -P<0.0001). There was a significant difference between the in vivo-produced blastocysts versus the other samples with 129/180 (71.6%) of the samples. n.d.- no data.
| Clone ID | Annotation | Gene | IVP±SE | Oocyte±SE | Sperm±SE | Parth±SE | SCNT±SE | IVF±SE |
|---|---|---|---|---|---|---|---|---|
| AA A1 | NS | 12.490±1.608 | 3.084±0.695 b | 1.184±0.169 d | 1.753±0.370 b | 1.621±0.283 b | 0.730±0.198 c | |
| B G2 | NS | 21.521±11.047 | 1.043±0.181 c | 0.960±0.172 c | 0.992±0.203 c | 1.748±0.241 c | 0.878±0.187 b | |
| BBB A12 | Sus scrofa glutamate decarboxylase 2 (GAD2), mRNA | GAD2 | 15.517±1.937 | 1.259±0.210 d | 1.027±0.175 d | 1.333±0.224 d | 2.515±0.546 d | 2.478±0.340 b |
| BBB H7 | Multiple | 10.564±2.250 | 0.887±0.183 d | 0.887±0.183 d | 1.107±0.197 c | 1.240±0.248 c | 0.735±0.224 b | |
| CCC H7 | Multiple | 3.517±1.258 | 1.795±0.190 | 29.000±0.000 | 2.284±0.440 | 3.594 | 0.591±0.205 | |
| D C10 | NS | 2.676±0.722 | 0.727±0.231 b | 2.187±0.498 | 1.524±0.298 | 1.186±0.433 | 1.469±0.187 a | |
| D D10 | NS | 30.503±13.704 | 0.000±0.000 | 1.282±0.211 c | 2.061±0.245 b | 1.087±0.201 b | 1.484±0.289 c | |
| D D6 | Multiple | 8.786±1.431 | 0.795±0.211 c | 1.607±0.383 c | 1.938±0.391 b | 1.197±0.209 b | 2.543±0.868 c | |
| EEE B7 | Multiple | 58.478±25.479 | 0.818±0.213 c | 1.264±0.216 c | 3.582±0.764 b | 4.234±1.233 b | 0.761±0.207 b | |
| EEE B9 | Multiple | 20.484±31.499 | 0.789±0.169 b | 1.019±0.203 a | 1.287±0.238 a | 0.806±0.211 a | 1.468±0.209 b | |
| EEE E3 | only Bac matches | 15.919±2.731 | 1.255±0.176 d | 2.078±0.305 c | 2.919±0.359 b | 2.231±0.245 b | 0.934±0.180 c | |
| F E10 | Multiple | 24.933±27.214 | 1.082±0.173 b | 1.043±0.198 b | 1.425±0.251 a | 1.043±0.198 a | 1.125±0.187 b | |
| F F10 | NS | 4.001±0.781 | 1.287±0.188 b | 2.041±0.298 | 2.057±0.391 | 1.940±0.479 | 1.953±0.201 | |
| FF G1 | only Bac matches | 6.205±5.827 | 0.835±0.240 a | 1.986±0.314 | 2.356±0.373 | 2.604±1.120 | 0.862±0.296 | |
| G G10 | PREDICTED: Canis familiaris similar-DEAD (Asp-Glu-Ala-Asp) box | DDX10 | 1.745±1.198 | 0.836±0.245 a | 1.371±1.820 | 1.465±0.296 | 0.000±0.000 | 1.587±0.676 |
| GGG D4 | WNT8B gene | WNT8B | 0.750±0.759 | 1.735±1.364 | 4.797±2.769 a | 1.293±0.332 | 7.000±0.000 | 1.769±0.232 a |
| II H10 | Multiple | 5.443±1.633 | 1.248±0.193 b | 1.806±0.496 | 2.961±0.471 | 1.875±1.612 | 0.866±0.185 | |
| III D1 | Multiple | 27.565±10.203 | 0.793±0.173 d | 1.334±0.207 c | 1.809±0.390 b | 0.690±0.200 b | 0.793±0.237 d | |
| JJ B10 | Bos taurus similar-Homeobox Protein SIX6 (Sine oculis homeobox homolog 6) | SIX6 | 22.006±8.660 | 0.971±0.182 c | 0.995±0.198 c | 1.162±0.242 c | 0.729±0.217 c | 0.901±0.179 c |
| JJ D12 | H.sapiens CpG island DNA | 19.756±9.884 | 0.789±0.172 c | 1.250±0.202 b | 1.468±0.421 b | 0.852±0.185 b | 0.804±0.183 c | |
| JJ E10 | NS | 30.306±19.817 | 0.801±0.171 c | 1.158±0.231 b | 1.565±0.277 b | 0.917±0.223 b | 1.262±0.185 b | |
| K G10 | Human cyclic AMP transcriptional regulator binding protein (CRE-BP1) | ATF2 | 2.837±0.464 | 1.005±0.171 c | 1.034±0.211 b | 1.353±0.241 a | 0.604±0.509 a | 1.370±0.183 b |
| LL E4 | NS | 11.712±1.302 | 1.196±0.169 d | 1.187±0.221 d | 1.610±0.245 d | 1.176±0.190 d | 0.647±0.187 d | |
| NN G9 | NS | 2.848±0.427 | 1.709±0.204 a | 1.221±0.553 | 2.106±0.820 | 1.522±0.315 | 0.786±0.183 a | |
| O D12 | Multiple | 26.245±27.961 | 0.993±0.257 b | 1.191±0.201 b | 1.244±0.257 a | 1.152±0.181 a | 1.008±0.263 b | |
| P F6 | Bos taurus similar-protoporphyrinogen oxidase, Last enzyme of heme synthesis | PPOX | 2.328±0.674 | 0.873±0.347 a | 2.094±0.586 | 1.582±0.372 | 1.228±0.818 | 0.770±0.204 |
| P H5 | PREDICTED: Bos Taurussimilar-zinc finger, CSL domain | ZCSL2 | 34.769±19.815 | 2.245±0.778 b | 1.042±0.347 c | 1.124±0.371 b | 1.135±0.272 c | n.d. |
| QQ A6 | Multiple | 29.106±36.958 | 0.926±0.169 b | 1.133±0.205 b | 1.632±0.247 a | 0.943±0.205 a | 1.014±0.185 b | |
| RR G5 | H. sapiens genes for histones H2B.1 and H2A | HIST2H2BE | 12.783±5.020 | 1.007±0.198 c | 2.118±0.446 a | 2.115±0.370 a | 0.902±0.292 a | 1.526±0.381 b |
| UU C10 | NS | 1.458±0.862 | 0.697±0.258 | 2.993±0.859 | 1.066±0.325 | 0.010±0.000 | 0.882±0.182 b | |
| UU H3 | NS | 1.723±0.653 | 1.096±0.210 | 0.838±0.251 b | 1.767±0.350 | 1.412±0.466 | n.d. | |
| X F12 | Homo sapiens prostate antigen PARIS-1 mRNA, complete cds | TBC1D2 | 8.198±1.664 | 0.847±0.185 d | 1.431±0.325 b | 1.554±0.246 b | 0.723±0.204 b | 1.203±0.185 c |
| X G2 | NS | 3.596±0.692 | 1.251±0.203 b | 1.240±0.328 a | 2.129±0.313 | 0.938±0.322 | 1.042±0.210 b | |
| XX H10 | NS | 15.403±5.999 | 0.979±0.178 c | 1.450±0.245 b | 2.382±0.781 a | 0.961±0.213 a | 1.166±0.181 b | |
| XX H12 | Homo sapiens splicing factor 3a, subunit 3, 60kDa (SF3A3), mRNA | SF3A3 | 30.199±16.716 | 1.102±0.169 c | 1.089±0.213 b | 1.642±0.405 b | 0.775±0.194 b | 1.016±0.187 c |
| Z D3 | NS | 22.118±9.730 | 0.877±0.189 c | 1.226±0.205 c | 1.485±0.253 b | 0.812±0.255 b | n.d. |
Table 6.
BLAST analysis of sequenced clones that were hypermethylated in the in vivo-produced blastocysts relative to the liver and their ratios. A significant difference was detected by using ANOVA between the in vivo-produced blastocysts and the other samples (a - P<0.05, b- P<0.01, c-P<0.001, d -P<0.0001). There was a significant difference between the in vivo-produced blastocysts versus the other samples with 97/160 (58.78%) of the samples. n.d.- no data.
| Clone ID | Annotation | Gene | IVP blast±SE | Oocyte±SE | Sperm±SE | Parth blast±SE | SCNT blast±SE | IVF blast±SE |
|---|---|---|---|---|---|---|---|---|
| BLUE E3 | NS | 0.206±0.371 | 1.323±0.456 b | 1.904±0.388 c | 0.343±0.292 | 0.237±0.285 | 1.060±0.696 a | |
| CC C1 | NS | 0.375±0.565 | 1.450±0.607 | 2.055±0.545 b | 0.345±0.455 | 0.324±0.293 | n.d. | |
| CCC B6 | Multiple | 0.632±0.898 | 1.386±0.459 | 0.782±0.374 | 1.345±0.585 | 0.808±0.297 | 0.723±0.000 | |
| EE A11 | Membrane bound O-acyltransferase domain containing 5 | MBOA T5 | 0.173±0.354 | 0.876±0.201 b | 1.262±0.208 c | 0.560±0.243 b | 0.476±0.241 a | 1.232±0.199 c |
| EE A12 | NS | 0.146±0.361 | 1.072±0.207 b | 1.469±0.277 b | 0.315±0.245 | 0.264±0.248 | 0.935±0.386 a | |
| EE H2 | Multiple | 0.173±0.409 | 1.131±0.284 c | 1.608±0.580 b | 0.338±0.355 | 0.334±0.235 a | 0.966±0.301 c | |
| EE H8 | Homo sapiens arylhydrocarbon receptor nuclear translocator | ARNT | 0.241±0.405 | 1.250±0.228 b | 1.331±0.219 c | 0.548±0.270 a | 0.471±0.247 | 0.963±0.443 b |
| FF E4 | Multiple immune components | 0.441±0.353 | 0.743±0.184 b | 0.888±0.218 b | 0.872±0.210 b | 0.992±0.244 b | 0.864±0.181 a | |
| G B8 | Canis familiaris simitCoatomer zeta-1 subunit | COPZ 1 | 0.232±0.412 | 0.818±0.215 b | 0.299±0.830 | 0.379±0.308 | 0.262±0.241 | n.d |
| N E12 | Homo sapiens serine/threonine protein kinase Kp78 (ribosomal) | MARK 3 | 1.335±0.465 | 0.974±0.252 | 1.001±0.272 | 1.512±0.285 | 1.144±0.377 | 0.816±0.334 |
| HH A7 | NS | 0.558±0.673 | 2.033±0.773 a | 0.619±0.427 | 2.539±2.283 a | 0.354±0.677 | 4.885±1.886 b | |
| L E8 | CpG Island plus others | 0.464±0.450 | 0.804±0.253 | 1.033±0.229 a | 0.485±0.242 | 0.552±0.242 | 0.721±0.446 | |
| LL D3 | NS | 0.555±0.718 | 0.893±0.687 | 1.344±0.777 | 0.180±0.338 | 0.842±0.000 | n.d. | |
| NN F4 | Mus musculus RIKEN cDNA 2810429O05 gene | 0.349±0.581 | 0.000±0.000 | 0.000±0.000 | 0.190±0.535 | 0.401±0.303 | n.d. | |
| PINK E2 | NS | 0.027±0.399 | 1.578±0.329 c | 1.642±0.185 d | 0.379±0.285 a | 0.216±0.259 a | 0.788±0.340 c | |
| PINK E9 | NS | 0.041±0.349 | 0.807±0.207 c | 0.955±0.174 d | 0.546±0.238 c | 0.366±0.251 b | 0.804±0.190 d | |
| PINK E10 | Multiple | 0.050±0.399 | 0.829±0.183 d | 0.932±0.170 d | 0.587±0.229 c | 0.500±0.243 c | 0.752±0.246 c | |
| PP C2 | NS | 0.091±0.405 | 0.972±0.239 b | 0.964±0.261 c | 0.186±0.320 | 0.193±0.238 | 0.961±0.000 a | |
| PP D6 | NS | 0.208±0.406 | 1.110±0.179 b | 1.188±0.170 c | 0.884±0.213 b | 0.695±0.237 a | 1.478±0.209 b | |
| PP E2 | Multiple | 0.135±0.401 | 1.205±0.428 b | 1.268±0.187 d | 0.567±0.327 | 0.315±0.243 a | 1.187±0.319 c | |
| PP E4 | PREDICTED: Bos taurus similarto malignant T cell amplified sequence 1 | MCTS 1 | 0.503±0.430 | 1.493±0.182 b | 1.044±0.173 a | 0.846±0.235 | 0.667±0.260 | 0.908±0.223 |
| PP E5 | PREDICTED: Canis familiaris Similar to Methyltransferase-like | 0.183±0.353 | 1.422±0.187 c | 1.091±0.172 c | 0.770±0.287 a | 0.658±0.250 a | 1.566±0.382 b | |
| PP E6 | PREDICTED: Bos taurus similar to Paired box protein Pax-3 | PAX3 | 0.134±0.403 | 1.774±0.184 c | 1.225±0.169 c | 0.686±0.223 a | 0.583±0.245 a | 1.271±0.213 b |
| PP G1 | NS | 0.118±0.409 | 0.409±0.254 c | 0.798±0.188 d | 0.402±0.308 a | 0.245±0.238 a | 1.095±0.000 b | |
| PP H6 | Homo sapiens FRG1 (FRG1) gene, complete cds (multiple) | FRG1 | 0.104±0.351 | 1.379±0.255 c | 1.131±0.169 c | 0.911±0.202 c | 0.876±0.237 b | 1.314±0.190 c |
| Q A2 | Homo sapiens serine/threonine protein kinase Kp78 (ribosomal) | MARK 3 | 0.268±0.417 | 0.768±0.242 a | 0.607±0.177 a | 0.556±0.210 | 0.498±0.239 | 0.632±0.248 |
| Q H5 | PREDICTED: Bos taurus similar to Forkhead box protein J2 | FOXJ2 | 0.067±0.399 | 0.961±0.220 c | 1.092±0.225 c | 0.425±0.231 c | 0.298±0.235 b | 1.299±0.699 c |
| QQ D3 | NS | 0.642±0.366 | 0.740±0.258 | 0.632±0.176 | 0.738±0.234 | 0.663±0.234 | 2.111±0.912 a | |
| T A6 | NS | 0.217±0.890 | 1.347±0.000 | 1.485±0.873 | 0.772±0.000 | 1.206±0.781 | n.d. | |
| TT G8 | 790G17 on chromosome 1q21.1-21.3 | 0.440±0.661 | 0.737±0.613 | 3.954±0.000 a | 0.289±0.523 | 0.235±0.511 | n.d. | |
| W E3 | NS | 0.147±0.405 | 1.470±0.262 c | 1.134±0.229 c | 0.444±0.285 a | 0.322±0.241 a | 2.809±0.000 b | |
| W F1 | NS | 0.038±0.349 | 1.162±0.219 c | 1.147±0.178 c | 0.760±0.250 c | 0.340±0.236 b | 1.990±0.191 d |
Figure 7.
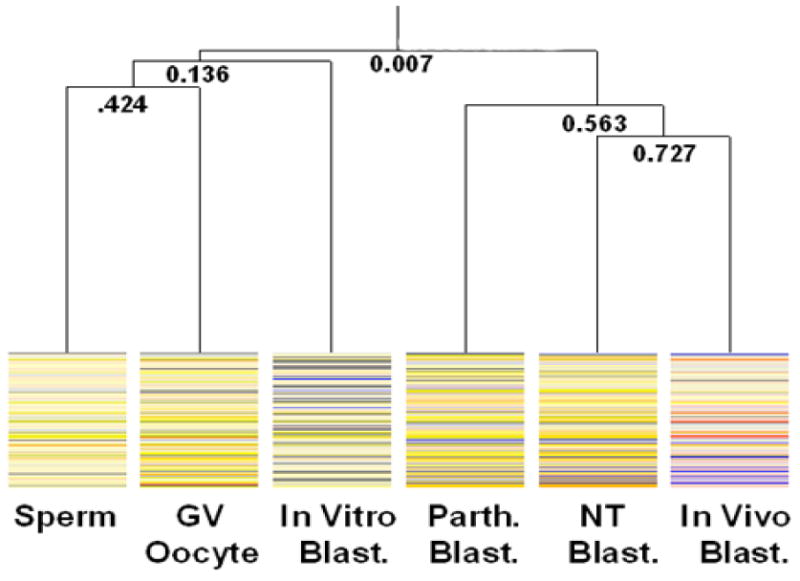
Condition Tree of gametes and blastocyst stage embryos. Sperm, GV oocytes, and in vitro-produced blastocysts (In Vitro Blast.) had the highest similarity to each other and grouped together. The second group included SCNT- (NT Blast.), and in vivo-produced blastocysts. Parthenogenetic-produced blastocysts (Parth. Blast.) also clustered with this group. This clustering pattern suggests that the methylation remodeling events of the SCNT-produced blastocysts more accurately mimics the remodeling of the in vivo-produced blastocysts than the remodeling that occurs in the in vitro-produced blastocysts. This condition tree only includes the data where there was at least one difference between the samples, thus the top branch shows a very low correlation. Hypermethylated spots, as compared to the reference, are shown in red and those that are hypomethylated are shown in blue.
Additional validation of the hierarchical clustering was performed by using bootstrap analysis (Not shown here, but on our web site http://animalsciences.missouri.edu/faculty/prather/ ). Unfortunately, the TIGR Multiple Array Viewer software used to do the bootstrap analysis does not include the same correlation analysis that is used by the GeneSpring software. Specifically, the Standard Correlation used in the GeneSpring software is commonly referred to as Pearson correlation around zero. The TIGR Multiple Array Viewer does not contain this correlation procedure so the Pearson Correlation analysis was substituted. Therefore, caution should be used in attempting to extrapolate the bootstrapping results to the clustering generated by using GeneSpring. The strongest support is shown for the clustering of the SCNT-, Parthenogenetic-, and in vivo-produced blastocysts.
Identification of Putatively Imprinted Genes
Imprinted genes are thought to be resistant to the passive and active demethylation events that occur immediately after fertilization. The methylation status of imprinted genes is subsequently maintained from the gamete to the somatic cells. ANOVA of the sperm and GV stage oocyte samples identified 28 genomic regions where the methylation status was significantly different (p<0.05) as measured by PDMH microarray analysis (Table 7).
Table 7.
Differential methylation in sperm and oocytes samples as measured by using DMH microarray analysis. The normalized oocyte and sperm ratios (reference/sample) were analyzed by using ANOVA (p<0.05). A significant difference in methylation at the gamete stage identifies putatively imprinted genomic regions.
| Clone I.D. | P-value | OOCYTE ± SE | SPERM ± SE |
|---|---|---|---|
| B C3 | 0.040 | 1.937 ± 0.188 | 1.0127 ± 0.183 |
| B G12 | 0.045 | 2.543 ± 0.337 | 0.8317 ± 0.197 |
| B G7 | 0.037 | 0.555 ± 0.265 | 1.156 ± 0.257 |
| BLUE D9 | 0.002 | 3.127 ± 0.217 | 1.185 ± 0.170 |
| C B9 | 0.039 | 2.002 ± 0.183 | 1.136 ± 0.167 |
| D E1 | 0.045 | 2.147 ± 0.201 | 1.153 ± 0.169 |
| EE G10 | 0.039 | 1.457 ± 0.229 | 0.560 ± 0.180 |
| EEE D4 | 0.006 | 2.825 ± 0.223 | 1.074 ± 0.191 |
| F F3 | 0.045 | 0.935 ± 0.236 | 2.500 ± 0.244 |
| III B12 | 0.012 | 1.097 ± 0.236 | 0.403 ± 0.232 |
| JJ G10 | 0.037 | 0.805 ± 0.186 | 1.060 ± 0.173 |
| NN E1 | 0.028 | 1.451 ± 0.182 | 1.007 ± 0.168 |
| NN E2 | 0.039 | 0.664 ± 0.185 | 0.870 ± 0.167 |
| NN G12 | 0.016 | 0.726 ± 0.186 | 0.971 ± 0.167 |
| NN G6 | 0.005 | 1.474 ± 0.187 | 0.759 ± 0.173 |
| NN G9 | 0.044 | 0.647 ± 0.187 | 1.710 ± 0.204 |
| OO G8 | 0.028 | 0.664 ± 0.210 | 0.927 ± 0.167 |
| PINK H4 | 0.001 | 2.644 ± 0.191 | 1.164 ± 0.170 |
| PP D10 | 0.002 | 2.044 ± 0.193 | 0.909 ± 0.170 |
| PP D9 | 0.039 | 1.155 ± 0.187 | 0.738 ± 0.194 |
| PP G3 | 0.000 | 1.871 ± 0.227 | 0.116 ± 0.166 |
| PP H3 | 0.044 | 0.592 ± 0.184 | 0.790 ± 0.170 |
| RR G7 | 0.044 | 2.12 ± 0.190 | 0.788 ± 0.195 |
| S G2 | 0.011 | 1.465 ± 0.185 | 0.843 ± 0.202 |
| T G6 | 0.012 | 2.291 ± 0.188 | 1.039 ± 0.170 |
| TT G10 | 0.005 | 2.012 ± 0.192 | 1.053 ± 0.173 |
| X G12 | 0.012 | 0.634 ± 0.210 | 0.893 ± 0.16) |
| X G9 | 0.039 | 0.604 ± 0.186 | 1.562 ± 0.172 |
Discussion
We show that the epigenetic remodeling of blastocysts produced by in vitro techniques is incomplete relative to the in vivo-produced blastocyst. An unexpected result of this study identified less similarity in methylation of in vitro-produced blastocysts compared to the in vivo-produced blastocysts than the methylation of parthenogenetic- and in vitro-produced blastocysts. Although unexpected, these results are consistent with a recent study that showed that gene expression in the bovine SCNT-produced blastocysts was more similar to in vivo-produced blastocysts than the patterns in in vivo-produced and in vitro-produced blastocysts (Smith et al. 2005). The development rate of blastocysts produce in vitro is comparable to parthenogenetic- and higher than SCNT-produced blastocysts. Producing offspring by embryo transfer is more efficient using in vitro-produced blastocysts compared to using SCNT. This suggests that factors other than DNA methylation are responsible for the decreased developmental potential. Indeed, the pattern of gene expression between in vitro- and in vivo-produced blastocysts is quite similar (Whitworth et al. 2004) even though it appears here that the pattern of DNA methylation is quite different.
Clones were organized according to similar methylation profiles in the gametes and blastocysts by Self Organizing Map analysis. BLAST analysis identified sequence homology with 10 clones that were hypermethylated in the in vivo-produced blastocysts relative to the other samples (GAD2, DDX10, WNT8B, SIX6, ATF2, PPOX, ZCSL2, HIST2H2BE, TBC1D2, SF3A3: Table 5). The regions sequenced corresponded primarily to areas immediately around the start site of the gene. Accordingly, down-regulated expression of these genes is expected based on the premise that hypermethylation interferes with the assembly and binding of transcription factors. BLAST analysis identified sequence homology with clones that were hypomethylated in the in vivo-produced blastocysts relative to the other samples (ARNT, Zeta-1 COP, MLF1, MARK3, MCTS1, methyltransferase-like (LOC533379), FRG1, FOXJ2: Table 6). Hypomethylation of regulatory regions associated with these genes would generally be expected to result in up-regulated expression.
Correlation analysis of the methylation profiles showed that the sperm and GV oocyte were most similar. Imprinted regions are thought to be resistant to the active and passive demethylation processes in early embryonic development and to tissue specific epigenetic remodeling. The methylation profile of the in vitro-produced blastocysts clustered with the sperm and GV oocyte instead of with the other blastocysts. This clustering pattern suggests there was incomplete methylation remodeling in the in vitro-produced blastocysts relative to the other blastocysts.
Twelve regions were analyzed by using bisulfite analysis to validate the microarray results. The reference/sample ratios were consistent with the microarray data in 7 of 8 of the regions that were highly methylated. When low levels of methylation were observed, the bisulfite analysis results were consistent with the microarray data in 10 of the 16 clones. Overall, the bisulfite analysis results validated the microarray results for 17 of the 24 genomic regions. While the correlation of bisulfite and PDMH analysis is not 100%, it should be noted that these results are not the ends, and that this tool permits the identification of genomic regions that justify further attention to confirm their importance in regulating development. Furthermore, of the 24 regions that were analyzed, there were no regions where the microarray results and the bisulfite sequencing results were contradictory. Specifically, none of the regions were identified as hypomethylated by using the microarray analysis and also identified as hypermethylated by using bisulfite sequencing, and visa versa. These results suggest there was cross-reactivity in the PDMH whereby differential methylation was identified but bisulfite sequencing showed the sample and the reference to be essentially unmethylated. Amplification of repeated sequences would explain the observed cross-reactivity but sequencing did not generally identify repeated sequences.
There are two possible explanations for the differences between the two techniques. The detection procedure used to validate the PDMH in this study (bisulfite sequencing) may not have been sufficient to detect rare, highly methylated strands. The PCR based approach to target production of the PDMH analysis could have amplified a low abundance strand to produce a detectable signal after hybridization with the arrays. Also, the low quantity of template DNA available from the blastocysts necessitated the high number of cycles in the amplification step. Additional optimization of the target production or hybridization could minimize this potentially confounding effect.
The hypomethylation detected in the in vivo-produced blastocysts distinguishes the methylation profiles from the other samples. Hypomethylation of the TE relative to the ICM may indicate a regulatory mechanism for the expression of genes that are critical in implantation(Morgan et al. 2005; Santos et al. 2002). The pig is similar to most other mammals in that the genomes are demethylated during preimplantation development (Dean et al. 2001; Kang 2001). Defects in DNA remodeling and subsequent transcriptional abnormalities in SCNT- and in vitro-produced blastocysts may account for the high pregnancy loss that occurs around day 25-45 of gestation in the pig (Lai and Prather 2003). In this study, the in vivo-produced blastocysts were different from the other blastocysts in that there was no time in culture. For example, previous studies have shown that epigenetic changes caused by in vitro preimplantation culture results in aberrant methylation and expression of a number of imprinted genes (Doherty et al. 2000; Mann et al. 2004; Sasaki et al. 1995). Further, the authors suggest that the epigenetic remodeling and reprogramming of the trophoblast may be more sensitive to the disruptive effect of the culture media than the ICM.
Although the maintenance of imprinted porcine genes was not analyzed in this study, we identified 16 putatively imprinted genes by using PDMH. Ten regions in the GV oocyte and 6 regions in the sperm were found to maintain the hypermethylation or hypermethylation status from the gamete to the in vivo-produced blastocyst. Bisulfite analysis or pyrosequencing and transcriptional profiling are needed to confirm that these regions are imprinted. Monitoring the methylation status of imprinted genes in cells or embryos cultured in vitro will be important in the identification of in vitro culture conditions which support the development of IVF and SCNT embryos.
Our results show that there appear to be fewer hypomethylated regions in parthenogenetic-, SCNT-, and in vitro-produced blastocysts as compared to in vivo-produced blastocysts. Additionally, blastocysts produced by in vitro fertilization also appear to lack the methylation events that occur in parthenogenetic-, SCNT- and in vivo-produced blastocysts. Although the methylation profile of the in vitro-produced blastocysts is less similar to the in vivo-produced blastocyst than those of the parthenogenetic- or SCNT-produced blastocysts, blastocysts produced by IVF have greater developmental potential to produce offspring after embryo transfer.
Recently, the transcriptional expression patterns of bovine SCNT- and in vivo-produced blastocysts were shown to have greater similarity to each other than the similarity of the expression patterns shared by in vitro- and in vivo-produced blastocysts (Smith 2005). Expression microarray analysis was used to analyze the gene expression profiles of SCNT donor cells, and of individual bovine SCNT-produced blastocysts and blastocysts produced by using AI procedures. Hierarchical clustering showed that the AI- and SCNT-produced blastocysts were more similar to each other than to the IVF-produced blastocysts. Similar observations have been made in the activation of rRNA synthesis for embryos produced in vitro or by SCNT (Bjerregaard et al. 2006). Asynchronous rRNA transcription was observed in the in vitro-produced blastocysts from the 4-cell stage through the blastocyst stage. Conversely, the cells in SCNT-produced embryos that developed to the 16-cell stage and the blastocyst stage were shown to be transcriptionally active. Activation of rRNA transcription observed in SCNT-produced blastocysts is consistent with the activation of rRNA transcription for in vivo-produced blastocysts. The similarity of transcriptional activity in blastocysts produced by SCNT and in vivo procedures is surprising given the low developmental competence of the SCNT-produced blastocysts after embryo transfer. These results suggest that developmental potential may be controlled by a relatively small number of genes since analogous transcriptional activity in the blastocysts produced by SCNT and in vivo procedures do not result in analogous developmental potential.
In conclusion, the use of PDMH permitted global analysis of differential methylation of the pig genome. While the resources weren't available to sequence each clone on the array we could demonstrate the utility of the tool and to justify additional sequencing. The genomic regions that were identified will be useful as markers for understanding the changes in DNA methylation during pig embryogenesis. The use of PDMH permits us to conclude that CpG methylation remodeling that occurs in the development of the in vivo-derived blastocyst does not occur in blastocysts produced by using in vitro techniques. Specifically, the methylation events that occur in the development of parthenogenetic and SCNT-produced blastocysts are more similar to the in vivo-produced blastocysts than the methylation remodeling events in the in vitro-produced blastocysts. Recently it was shown that in parthenogenetic-, NT-and in vitro-produced blastocysts (all produced by using in vitro-matured oocytes), only the in vitro-produced blastocysts failed to replicate the hypermethylation of specific regions found in the in vivo-produced blastocysts (Gioia et al. 2005). These results suggest that DNA methylation in sperm may be resistant to epigenetic remodeling directed by in vitro-matured oocytes. Conversely, in vitro-produced oocytes appear to be able to direct remethylation in the parthenogenetic- and SCNT-produced blastocyst. This type of analysis will be instrumental in identifying factors (oocyte maturation media, embryo culture media, selection of donor cells used in SCNT) that are critical in the efficient production of embryos by using in vitro techniques. Identifying the specific effects of aberrant methylation, as it relates to transcriptional reprogramming is necessary to fully understand how incorrect genomic remodeling can interfere with the development of the early embryo. Additional research is needed to identify the specific aberrant methylation events that have a direct influence on the developmental deficits observed when producing offspring by using in vitro techniques.
Supplementary Material
Acknowledgments
The authors would like to acknowledge surgical skills of Tom Cantley and David Wax; August Rieke and Lonnie Dowell for gathering samples and supplying animals; and Nathan Bivens of the University of Missouri-Columbia DNA core for his expert assistance with the sequencing.
Funding for this project is from the National Center for Research Resources at the National Institutes of Health R01 RR13438 and Food for the 21st Century at the University of Missouri.
References
- Abeydeera LR. In vitro production of embryos in swine. Theriogenology. 2002;57(1 Special Issue SI):257–273. doi: 10.1016/s0093-691x(01)00670-7. [DOI] [PubMed] [Google Scholar]
- Abeydeera LR, Day BN. Fertilization and Subsequent Development in Vitro of Pig Oocytes Inseminated in a Modified Tris-Buffered Medium with Frozen-Thawed Ejaculated Spermatozoa. Biology of Reproduction. 1997;57(4):729–734. doi: 10.1095/biolreprod57.4.729. [DOI] [PubMed] [Google Scholar]
- Archer GS, Dindot S, Friend TH, Walker S, Zaunbrecher G, Lawhorn B, Piedrahita JA. Hierarchical phenotypic and epigenetic variation in cloned swine. Biology of Reproduction. 2003;69(2):430–436. doi: 10.1095/biolreprod.103.016147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaujean N, Hartshorne G, Cavilla J, Taylor J, Gardner J, Wilmut I, Meehan R, Young L. Non-conservation of mammalian preimplantation methylation dynamics. Current Biology. 2004;14(7):R266–R267. doi: 10.1016/j.cub.2004.03.019. [DOI] [PubMed] [Google Scholar]
- Bjerregaard B, Pedersen HG, Jakobsen AS, Rickords LF, Lai L, Cheong HT, Samuel M, Prather RS, Strejcek F, Rasmussen ZR, Laurincik J, Schellander K, Niemann H, Maddox-Hyttel P, Thomsen PD. Activation of ribosomal RNA genes in porcine embryos produced in vitro or by somatic cell nuclear transfer. Molecular Reproduction & Development. 2006 doi: 10.1002/mrd.20594. in press. [DOI] [PubMed] [Google Scholar]
- Carter DB, Lai L, Park KW, Samuel M, Lattimer JC, Jordan KR, Estes DM, Besch-Williford C, Prather RS. Phenotyping of transgenic cloned pigs. Cloning and Stem Cells. 2002;4:131–145. doi: 10.1089/153623002320253319. [DOI] [PubMed] [Google Scholar]
- Conway KL. Birth weight of bovine calves produced by nuclear transfer (cloning) and their offspring (embryo transfer) Dissertation Abstr Int. 1996;57:3462. [Google Scholar]
- Dean W, Santos F, Stojkovic M, Zakhartchenko V, Walter J, Wolf E, Reik W. Conservation of methylation reprogramming in mammalian development: Aberrant reprogramming in cloned embryos. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(24):13734–13738. doi: 10.1073/pnas.241522698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty AS, Mann MRW, Tremblay KD, Bartolomei MS, Schultz RM. Differential effects of culture on imprinted H19 expression in the preimplantation mouse embryo. Biology of Reproduction. 2000;62(6):1526–1535. doi: 10.1095/biolreprod62.6.1526. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Brown PO. DNA arrays for analysis of gene expression. In: Weissman SM, editor. CDNA Preparation and Characterization Methods Enzymol. Vol. 303. Academic Press Inc; San Diego, CA 92101: 1999. pp. 179–205. [DOI] [PubMed] [Google Scholar]
- Gioia L, Barboni B, Turriani M, Capacchietti G, Pistilli MG, Berardinelli P, Mattioli M. The capability of reprogramming the male chromatin after fertilization is dependent on the quality of oocyte maturation. Reproduction. 2005;130(1):29–39. doi: 10.1530/rep.1.00550. [DOI] [PubMed] [Google Scholar]
- Huang THM, Perry MR, Laux DE. Methylation profiling of CpG islands in human breast cancer cells. Human Molecular Genetics. 1999;8(3):459–470. doi: 10.1093/hmg/8.3.459. [DOI] [PubMed] [Google Scholar]
- Jiang L, Jobst P, Ayares D, Lai L, Samuel M, Prather RS, Tian XC. Expression of growth-regulating imprinted genes in decreased and surviving cloned pigs. Cloning and Stem Cells. 2007 doi: 10.1089/clo.2006.0041. accepted 11-14-06. [DOI] [PubMed] [Google Scholar]
- Kang YK. Typical demethylation events in cloned pig embryos. Clues on species-specific differences in epigenetic reprogramming of a cloned donor genome. Journal of Biological Chemistry. 2001;276(43):39980–39984. doi: 10.1074/jbc.M106516200. [DOI] [PubMed] [Google Scholar]
- Lai L, Prather RS. Production of cloned pigs by using somatic cells as donors. Cloning & Stem Cells. 2003;5:233–242. doi: 10.1089/153623003772032754. [DOI] [PubMed] [Google Scholar]
- Machaty Z, Day BN, Prather RS. Development of Early Porcine Embryos in Vitro and in Vivo. Biology of Reproduction. 1998;59(2):451–455. doi: 10.1095/biolreprod59.2.451. [DOI] [PubMed] [Google Scholar]
- Mann MRW, Chung YG, Nolen LD, Verona RI, Latham KE, Bartolomei MS. Disruption of imprinted gene methylation and expression in cloned preimplantation stage mouse embryos. Biology of Reproduction. 2003;69(3):902–914. doi: 10.1095/biolreprod.103.017293. [DOI] [PubMed] [Google Scholar]
- Mann MRW, Lee SS, Doherty AS, Verona RI, Nolen LD, Schultz RM, Bartolomei MS. Selective loss of imprinting in the placenta following preimplantation development in culture. Development. 2004;131(15):3727–3735. doi: 10.1242/dev.01241. [DOI] [PubMed] [Google Scholar]
- Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Embryogenesis - Demethylation of the zygotic paternal genome. Nature. 2000a;403(6769):501–502. doi: 10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- Mayer W, Smith A, Fundele R, Haaf T. Spatial separation of parental genomes in preimplantation mouse embryos. Journal of Cell Biology. 2000b;148(4):629–634. doi: 10.1083/jcb.148.4.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan HD, Santos F, Green K, Dean W, Reik W. Epigenetic reprogramming in mammals [Review] Human Molecular Genetics. 2005;14(Special Issue 1):R47–R58. doi: 10.1093/hmg/ddi114. [DOI] [PubMed] [Google Scholar]
- Rickman DS, Herbert CJ, Aggerbeck LP. Optimizing spotting solutions for increased reproducibility of cDNA microarrays - art. no. e109. Nucleic Acids Research. 2003;31(18):E109. doi: 10.1093/nar/gng109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougier N, Bourchis D, Gomes DM, Niveleau A, Plachot M, Paldi A, Viegaspequignot E. Chromosome Methylation Patterns During Mammalian Preimplantation Development. Genes & Development. 1998;12(14):2108–2113. doi: 10.1101/gad.12.14.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos F, Hendrich B, Reik W, Dean W. Dynamic reprogramming of DNA methylation in the early mouse embryo. Developmental Biology. 2002;241(1):172–182. doi: 10.1006/dbio.2001.0501. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Fergusonsmith AC, Shum ASW, Barton SC, Surani MA. Temporal and Spatial Regulation of H19 Imprinting in Normal and Uniparental Mouse Embryos. Development. 1995;121(12):4195–4202. doi: 10.1242/dev.121.12.4195. [DOI] [PubMed] [Google Scholar]
- Shi W, Dirim F, Wolf E, Zakhartchenko V, Haaf T. Methylation reprogramming and chromosomal aneuploidy in in vivo fertilized and cloned rabbit preimplantation embryos. Biology of Reproduction. 2004;71(1):340–347. doi: 10.1095/biolreprod.103.024554. [DOI] [PubMed] [Google Scholar]
- Smith SL, Everts RE, Tian XC, Du FL, Sung LY, Rodriguez-Zas SL, Jeong BS, Renard JP, Lewin HA, Yang XZ. Global gene expression profiles reveal significant nuclear reprogramming by the blastocyst stage after cloning. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(49):17582–17587. doi: 10.1073/pnas.0508952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamashiro KLK, Wakayama T, Akutsu H, Yamazaki Y, Lachey JL, Wortman MD, Seeley RJ, D'Alessio DA, Woods SC, Yanagimachi R, Sakai RR. Cloned mice have an obese phenotype not transmitted to their offspring. Nature Medicine. 2002;8(3):262–267. doi: 10.1038/nm0302-262. [DOI] [PubMed] [Google Scholar]
- Whitworth K, Springer GK, Forrester LJ, Spollen WG, Ries J, Lamberson WL, Bivens N, Murphy CN, Mathialigan N, Green JA, Prather RS. Developmental expression of 2,489 genes during pig embryogenesis: An EST project. Biology of Reproduction. 2004;71:1230–1243. doi: 10.1095/biolreprod.104.030239. [DOI] [PubMed] [Google Scholar]
- Wilmut I, Beaujean N, de Sousa PA, Dinnyes A, King TJ, Paterson LA, Wells DN, Young LE. Somatic cell nuclear transfer. Nature. 2002;419(6907):583–586. doi: 10.1038/nature01079. [DOI] [PubMed] [Google Scholar]
- Yoshioka K, Suzuki C, Itoh S, Kikuchi K, Iwamura S, Rodriguez-Martinez H. Production of piglets derived from in vitro-produced Blastocysts fertilized and cultured in chemically defined media: Effects of theophylline, adenosine, and cysteine during in vitro fertilization. Biology of Reproduction. 2003;69(6):2092–2099. doi: 10.1095/biolreprod.103.020081. [DOI] [PubMed] [Google Scholar]
- Zhang SQ, Kubota C, Yang L, Zhang YQ, Page R, O'Neill M, Yang XZ, Tian XC. Genomic imprinting of H19 in naturally reproduced and cloned cattle. Biology of Reproduction. 2004;71(5):1540–1544. doi: 10.1095/biolreprod.104.031807. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.