

Abstract
Ept1, Ept2, Ept6, and Ept9 are quantitative trait loci mapped in crosses between the ACI and Copenhagen (COP) rat strains as genetic determinants of responsiveness of the pituitary gland to estrogens. We have developed four congenic rat strains, each of which carries, on the genetic background of the ACI rat strain, alleles from the COP rat strain that span one of these quantitative trait loci. Relative to the female ACI rats, female ACI.COP-Ept1 rats exhibited reduced responsiveness to 17β-estradiol (E2) in the pituitary gland, as evidenced by quantification of pituitary mass and circulating prolactin, and in the mammary gland, as evidenced by reduced susceptibility to E2-induced mammary cancer. The ACI.COP-Ept2 rat strain exhibited reduced responsiveness to E2 in the pituitary gland but did not differ from the ACI strain in regard to susceptibility to E2-induced mammary cancer. Interestingly, female Ept2 congenic rats exhibited increased responsiveness to E2 in the thymus, as evidenced by enhanced thymic atrophy. The ACI.COP-Ept6 rat strain exhibited increased responsiveness to E2 in the pituitary gland, which was associated with a qualitative phenotype suggestive of enhanced pituitary vascularization. The ACI.COP-Ept9 rat strain exhibited reduced responsiveness to E2 in the anterior pituitary gland, relative to the ACI rat strain. Neither Ept6 nor Ept9 impacted responsiveness to E2 in the mammary gland or thymus. These data indicate that each of these Ept genetic determinants of estrogen action is unique in regard to the tissues in which it exerts its effects and/or the direction of its effect on estrogen responsiveness.
THE ROLES OF estrogens in regulating the development and function of numerous tissues and cell types are well defined. However, the mechanisms through which estrogens regulate these processes are only poorly understood. In the anterior pituitary gland, estrogens regulate proliferation, survival, and/or function of several cell types, including the prolactin (PRL)-producing lactotroph (1,2,3,4). It is becoming increasingly clear that estrogen action in the pituitary gland and other tissues is subject to strong genetic control. Different rat strains exhibit marked quantitative and qualitative differences in the responsiveness of the pituitary gland to estrogens. When induction of pituitary growth is evaluated as the phenotype, the Fischer 344 (F344) and ACI rat strains are highly responsive to estrogens, the Copenhagen (COP) rat strain is moderately responsive, and the Brown Norway (BN) and Sprague Dawley rat strains are weakly responsive (5,6,7,8,9). These strain differences are being exploited in genetic and genomic studies directed at identifying the genes and regulatory pathways that determine the underlying phenotypes, and it is anticipated that this information will reveal novel insight into the mechanisms through which estrogens regulate proliferation and survival of specific cell populations. Multiple quantitative trait loci (QTL) that exert significant effects on the ability of administered estrogens to increase pituitary mass, a surrogate indicator of absolute lactotroph number, have been mapped in crosses between the F344 and BN strains, the ACI and COP strains, and the ACI and BN strains (10,11,12,13). Several of these QTL have been further evaluated in the context of congenic rat strains to begin to define the mechanisms through which they exert their effects on estrogen responsiveness in the pituitary gland (14,15,16).
Estrogen action in the mammary gland, uterus, thymus, and testis is also subject to genetic control. In the mammary gland of the ACI rat, estrogens induce a robust epithelial hyperplasia and a high incidence of mammary carcinoma (17,18). In contrast, the COP and BN rat strains are less responsive to estrogens with respect to induction of mammary hyperplasia and are resistant to induction of mammary cancer (19,20,21,22,23,24). A total of nine QTL have been mapped in intercrosses between the ACI and COP or BN rat strains that determine susceptibility to estrogen-induced mammary cancer (22,23,24). In the uterus of the BN rat, estrogens induce marked inflammation and pyometritis, responses that are rarely observed in ACI or F344 rats (25,26). Recent studies have localized QTL that determine susceptibility to these uterine phenotypes (25,26). In the thymus, estrogens inhibit thymocyte proliferation and induce thymic atrophy (27,28). This phenotype is more severe in BN rats than ACI rats, and three QTL have been mapped that impact the induction of thymic atrophy by estrogens (29). Finally, in the testis, estrogens inhibit spermatogenesis and induce marked testicular atrophy. The Lewis strain is more sensitive than the F344 strain with respect to estrogen-induced testicular atrophy, and two QTL impacting this phenotype have been mapped using a panel of recombinant inbred strains (30). The picture emerging from these genetic studies is that each of these estrogen-regulated phenotypes behaves as a complex genetic trait. Because the QTL impacting these different phenotypes only occasionally colocalize, it appears that the majority of these QTL function in a tissue- or cell-type specific context when exerting their effects on estrogen responsiveness.
In this study, we generated and characterized a set of congenic rat strains to evaluate the impact of four QTL, each of which was identified as a determinant of sensitivity to estrogen-induced pituitary growth, on estrogen action in the pituitary gland, mammary gland, and thymus. The resulting data indicate that these QTL exert their actions on the pituitary gland in a manner that is independent of gender and the chemical form of the estrogen used to induce pituitary growth. Moreover, the actions of these QTL are, for the most part, tissue specific. Finally, the impact of the individual QTL on estrogen-induced pituitary growth was observed to vary markedly as a function of the duration of estrogen treatment.
Materials and Methods
Procurement and care of animals
The Institutional Animal Care and Use Committee of the University of Nebraska Medical Center approved all procedures involving live animals. ACI/SegHsd rats were obtained from Harlan Sprague Dawley (Indianapolis, IN). COP rats were obtained from Charles River Laboratories (Wilmington, MA). Animals were housed in a barrier facility under controlled temperature, humidity, and 12-h light, 12-h dark conditions. This facility was accredited by the American Association for Accreditation of Laboratory Animal Care and operated in accordance with the standards outlined in the Guide for the Care and Use of Laboratory Animals (Department of Health and Human Services Publication 85-23). The animals were caged and fed as described previously (9,12).
Generation of congenic rat strains
To investigate the impact of individual Ept loci on estrogen-induced pituitary growth, mammary carcinogenesis, and other estrogen-regulated phenotypes, we generated four congenic rat strains, each of which encompasses the 95% confidence interval (CI) of the Ept1, Ept2, Ept6, or Ept9 QTL, by introgressing the corresponding region of rat chromosome RNO3, RNO6, or RNO10 from the donor COP strain onto the recipient ACI background (Fig. 1). This was done using the marker-assisted selective breeding strategy of iterative backcrossing with selection for donor alleles across the QTL of interest and selection for recipient alleles across all other chromosomes or chromosomal segments (25,31,32). Briefly, female COP rats and male ACI rats were crossed to generate F1 progeny carrying the ACI Y chromosome. Male F1 progeny were then backcrossed to ACI females. The resulting male N2 progeny were genotyped, and those that were heterozygous at each of seven to 12 markers spanning the desired Ept locus were then genotyped at markers on each of the other autosomes (Table 1). Chromosomal regions harboring Ept loci that were not being selected for were rigorously monitored by genotyping at markers spaced less than 20 million base pair (Mb) apart across the 95% CI of each nonselected QTL. Backcrossing with selection was repeated for six generations, at which time each marker undergoing selection across the desired Ept locus remained heterozygous, whereas those markers outside the QTL of interest were homozygous for ACI alleles. Selected N6 progeny were mated to generate founders that were homozygous for COP alleles at markers spanning the Ept locus of interest, and these founders were mated to propagate the congenic strain. Nomenclature of the resultant congenic strains is in accordance with the guidelines of the International Rat Genome Nomenclature Committee (http://rgd.mcw.edu/nomen/rules-for-nomen.shtml), which stipulate that the recipient strain be indicated first, followed by the donor strain and the markers that flank the region known to be homozygous for alleles from the donor strain. Thus, the four congenic strains are designated as follows: ACI.COP-(D6Rat80-D6Rat146)/Shul, referred to herein as ACI.COP-Ept1 or Ept1; ACI.COP-(D3Rat130-D3Rat114)/Shul, referred to as ACI.COP-Ept2 or Ept2; ACI.COP-(D3Mgh16-D3Rat119)/Shul, referred to as ACI.COP-Ept6 or Ept6; and ACI.COP-(D10Mgh8-D10Rat4)/Shul, referred to as ACI.COP-Ept9 or Ept9.
Figure 1.

Ept congenic intervals. The vertical line in each panel represents the physical distance along the indicated rat chromosome. The location of each polymorphic microsatellite marker at which genotype was defined is indicated to the right of the line, and the genome coordinate of each marker (Mb) is indicated to the left of the line. The solid black rectangles represent the 95% CI for each Ept locus, as defined previously by composite interval mapping (12). The stippled gray rectangles represent the regions of each chromosome known to be homozygous for COP alleles. The white extensions to these rectangles represent regions of undefined genotype. All remaining segments of each chromosome are homozygous for ACI alleles. A, RNO3 and locations of Ept2 and Ept6. B, RNO6 and location of Ept1. C, RNO10 and location of Ept9.
Table 1.
Markers used during generation of congenic rat strains
| Ept1 | Ept2 | Ept6 | Ept9 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Markers used for selection of donor alleles in desired Ept locus | |||||||||||
| D6Rat80 | D6Rat108 | D6Rat39 | D3Rat130 | D3Rat119 | D3Rat108 | D3Mgh9 | D3Mgh16 | D3Rat58 | D10Rat45 | D10Mgh8 | D10Rat27 |
| D6Rat146 | D6Rat155 | D6Rat26 | D3Rat37 | D3Arb18 | D3Rat26 | D3Rat124 | D3Rat52 | D3Rat277 | D10Rat21 | D10Mit7 | D10Rat11 |
| D6Mit3 | D6Rat153 | D6Rat103 | D3Rat70 | D3Rat133 | D3Rat21 | D3Rat49 | D10Rat4 | ||||
| D3Rat150 | D3Rat6 | D3Rat114 | |||||||||
| Markers used for selection for recipient alleles in nondesired Ept loci | |||||||||||
| D3Mgh16 | D3Rat277 | D3Rat130 | D3Mgh9 | D3Mgh16 | D3Rat58 | D3Rat130 | D3Rat119 | D3Rat108 | D3Mgh16 | D3Rat277 | D3Rat130 |
| D3Rat108 | D3Rat37 | D3Arb18 | D3Rat277 | D10Rat45 | D10Mgh8 | D3Rat37 | D3Arb18 | D3Rat26 | D3Rat108 | D3Rat37 | D3Arb18 |
| D3Rat26 | D3Rat24 | D3Rat70 | D10Rat27 | D10Mit7 | D10Rat11 | D3Rat70 | D3Rat133 | D3Rat21 | D3Rat26 | D3Rat21 | D3Rat150 |
| D3Rat133 | D3Mgh11 | D3Rat21 | D1Rat121 | D1Rat251 | D1Rat234 | D3Rat150 | D3Rat6 | D3Rat114 | D3Rat6 | D1Rat121 | D1Rat251 |
| D3Rat150 | D3Rat6 | D10Rat45 | D1Rat261 | D1Rat133 | D1Rat323 | D10Rat45 | D10Mgh8 | D10Rat27 | D1Rat234 | D1Rat261 | D1Rat133 |
| D10Mgh8 | D10Rat27 | D10Mit7 | D1Rat123 | D1Rat75 | D1Rat119 | D10Mit7 | D10Rat11 | D1Rat121 | D1Rat323 | D1Rat123 | D1Rat75 |
| D10Rat11 | D1Rat121 | D1Rat251 | D1Rat87 | D6Rat80 | D6Rat108 | D1Rat251 | D1Rat234 | D1Rat261 | D1Rat119 | D1Rat87 | D6Rat80 |
| D1Rat234 | D1Rat261 | D1Rat133 | D6Rat39 | D6Rat146 | D6Rat103 | D1Rat133 | D1Rat323 | D1Rat123 | D6Rat108 | D6Rat39 | D6Rat146 |
| D1Rat323 | D1Rat123 | D1Rat75 | D1Rat75 | D1Rat119 | D1Rat87 | D6Rat103 | |||||
| D1Rat119 | D1Rat87 | D6Rat80 | D6Rat108 | D6Rat39 | |||||||
| D6Rat146 | D6Rat103 | ||||||||||
| Markers used for selection of recipient alleles at background loci | |||||||||||
| D2Rat6 | D2Rat134 | D2Rat185 | D2Mit6 | D2Rat58 | D4Rat126 | D4Mgh7 | D4Rat71 | D4Rat43 | D4Rat103 | D5Rat7 | D5Rat84 |
| D5Rat95 | D5Rat37 | D5Rat46 | D7Rat44 | D7Rat36 | D7Rat101 | D8Rat26 | D8Rat105 | D8Rat5 | D9Rat131 | D9Mgh3 | D9Rat2 |
| D11Rat40 | D11Rat29 | D11Rat95 | D12Mgh2 | D12Rat5 | D12Mgh10 | D13Mgh2 | D13Rat33 | D13Rat24 | D14Rat23 | D14Rat22 | D14Rat51 |
| D14Rat88 | D15Rat78 | D15Rat102 | D15Rat8 | D16Rat21 | D16Rat16 | D16Rat93 | D17Rat6 | D17Rat51 | D17Rat79 | D18Rat30 | D18Mit9 |
| D18Rat57 | D19Rat34 | D19Mgh2 | D19Rat7 | D20Rat22 | D20Mgh1 | ||||||
Genotyping
Genomic DNA was isolated from tail clips using DNeasy columns according to the manufacturer’s protocol (QIAGEN, Valencia, CA). Genetic markers that are polymorphic between the ACI and COP rat strains were selected using the Rat Genome Database (http://rgd.mcw.edu). A total of 86–93 markers spanning the genome was used, depending upon the congenic rat strain, with marker densities of approximately 20 Mb across Ept intervals. Genotyping was performed by PCR amplification and polyacrylamide or agarose gel electrophoresis, as previously described (12,22).
Evaluation of estrogen-regulated phenotypes
Female rats were treated with 17β-estradiol (E2), released from sc SILASTIC brand silicon tubing implants (Dow Corning, Midland, MI), beginning at 9 wk of age as described by us previously (17,19,20,21,22,23,25). These implants continuously release E2 into the circulation, resulting in physiological levels of E2 normally observed in rats during pregnancy (17,19). Untreated control rats received empty SILASTIC brand silicon tubing implants. Each rat was examined by palpation once or twice per week to detect the presence of mammary tumors. The rats were euthanized by decapitation after 12 or 28 wk of treatment. At necropsy, the anterior pituitary gland was evaluated visually to determine whether the gland exhibited loss of normal symmetry and coloration, and photographed under a dissecting microscope. The gland was then removed, weighed, frozen in liquid nitrogen, and stored at −80 C or fixed in 10% neutral buffered formalin and processed for histological examination. All grossly discernable mammary tumors were harvested, a portion was fixed in Lillie’s solution, processed and embedded in paraffin for histological evaluation, and the remainder was frozen in liquid nitrogen and stored at −80 C. The thymus was dissected, weighed, frozen in liquid nitrogen, and stored at −80 C. The spleen was collected as a source of DNA, frozen in liquid nitrogen, and stored at −80 C. Trunk blood was collected, allowed to clot at 4 C, centrifuged, and the serum was stored at −80 C. The concentration of PRL in the serum was determined by RIA using the Rat Prolactin [125I] Biotrak Assay System (Amersham Biosciences, Piscataway, NJ).
Statistical analysis
Differences in pituitary mass, serum PRL concentration, thymus mass, and mammary tumor number between experimental groups were assessed using Kruskal-Wallis ANOVA and Mann-Whitney post hoc tests with Bonferroni adjustment for multiple comparisons. Mammary cancer latency, defined as the number of days of E2 treatment preceding the appearance of palpable mammary cancer, was evaluated using Kaplan-Meier plots and the log-rank test. Statistical analyses were conducted using SPSS software version 12 (SPSS, Inc., Chicago, IL). Statistical significance was defined at the P < 0.05 level.
Results
Impact of Ept loci on induction of pituitary growth
Six Ept loci, each of which exerted a significant effect on induction of pituitary growth by the synthetic estrogen diethylstilbestrol (DES), were previously mapped in male F2 rats generated in reciprocal intercrosses between the ACI and COP rat strains (12). Congenic rat strains have been generated to define the impact of each of the four Ept loci that exerted the greatest effect on trait variance in the (ACIxCOP)F2 and (COPxACI)F2 populations on induction of pituitary growth, as well as other estrogen-regulated phenotypes. To determine whether the actions of these Ept loci were specific to the gender of the experimental animals or the chemical form of the estrogen used to induce the different responses to hormone, we evaluated the ability of the naturally occurring estrogen E2 to induce pituitary growth in female rats of each of the parental and congenic rat strains. Female rats of the ACI and COP strains differed dramatically with respect to induction of pituitary growth by E2, an observation in agreement with data from our previous studies on estrogen-induced pituitary growth (9,12,20,33). After 12-wk E2 treatment, pituitary mass was increased 5.8-fold in female ACI rats, but only 3.3-fold in female COP rats (Fig. 2). This corresponds to a net increase of 44.0 mg pituitary mass in ACI rats compared with 21.2 mg in COP rats. Pituitary mass in E2-treated ACI rats was significantly greater than in E2-treated COP rats. Based on data from our previous mapping studies, we predicted that COP alleles at Ept1, Ept2, or Ept9, when introgressed onto the ACI genetic background, would inhibit induction of pituitary growth by E2, whereas COP alleles at Ept6 would enhance E2-induced pituitary growth. Although the inhibitory actions of COP alleles at Ept1, Ept2, and Ept9 on E2-induced pituitary growth were clearly apparent in female congenic rats, no impact of Ept6 was observed at this 12-wk time point (Fig. 2). Pituitary mass in E2-treated Ept1, Ept2, and Ept9 rats was significantly lower than that observed in E2-treated ACI rats, and did not differ from that observed in E2-treated COP rats. In the Ept1 congenic strain, 12-wk E2 treatment resulted in a 4.1-fold induction of pituitary mass, which corresponds to a net increase in pituitary mass of 24.7 mg, relative to untreated controls. E2 induced a 3.9-fold increase in pituitary mass in the Ept2 congenic strain, a net increase in pituitary mass of 26.1 mg, relative to untreated controls. In the Ept9 congenic strain, E2 increased pituitary mass 5.0-fold, a net increase of 31.2 mg, relative to untreated controls. The 6.0-fold induction of pituitary mass in the Ept6 congenic strain was indistinguishable from that observed in the parental ACI strain. Pituitary mass in untreated rats did not differ significantly between the different rat strains. The inhibitory actions of Ept1, Ept2, and Ept9 on induction of pituitary mass remained apparent when pituitary weight was normalized to body weight (data not shown).
Figure 2.
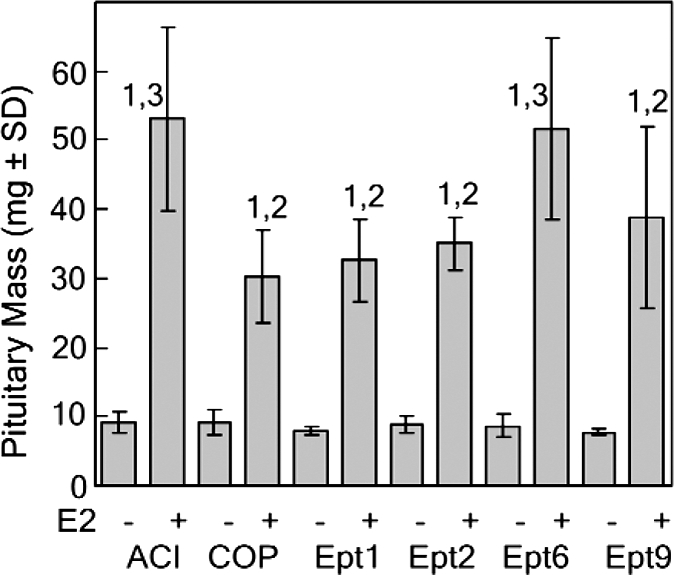
Impact of Ept loci on pituitary mass after 12-wk E2 treatment. Female rats of each strain were treated with E2, released from sc SILASTIC brand implants, beginning at 9 wk of age. Control rats received empty implants. The rats were euthanized 12 wk later, and the pituitary glands were removed and weighed. Each bar represents the mean pituitary mass ± sd of the mean. The untreated control groups included eight to 12 rats. The E2-treated groups included 10–25 rats. Numeral 1 indicates a statistically significant (P < 0.05) difference relative to the corresponding untreated control group. Numeral 2 indicates a statistically significant difference relative to ACI rats receiving the same treatment. Numeral 3 indicates a statistically significant difference relative to COP rats receiving the same treatment.
The impact of each of the Ept loci on induction of pituitary growth was also examined in a 28-wk experiment. Once again, the ACI and COP strains differed dramatically with respect to sensitivity to E2-induced pituitary growth. After 28-wk E2 treatment, pituitary mass in ACI rats was increased 15.7-fold, a net increase of 145 mg, relative to untreated controls (Fig. 3). In contrast, pituitary mass was increased only 3.5-fold in E2-treated COP rats, a net increase of 28.3 mg, relative to that observed in untreated COP rats. Although the inhibitory effect of Ept2 on E2-induced pituitary growth remained apparent after 28-wk treatment, the inhibitory effects of Ept1 and Ept9 were no longer observed at this time point. In the Ept2 congenic strain, 28-wk E2 treatment increased pituitary mass by 7.6-fold, a net increase of 65.9 mg, relative to controls. Pituitary mass in E2-treated Ept1 and Ept9 rats did not differ significantly from that in E2-treated ACI rats. These data strongly suggest that Ept2 restrains E2-stimulated pituitary growth by acting through a different mechanism than either Ept1 or Ept9. Interestingly, after 28-wk E2 treatment, pituitary mass in Ept6 congenic rats was significantly increased relative to ACI rats, an observation that is consistent with the prediction based on our original mapping data. In this congenic strain, E2 increased pituitary mass 25.2-fold, a net increase of 206.4 mg, over controls, strongly suggesting that one or more genes residing within the Ept6 congenic interval enhance pituitary growth over the extended period of E2 treatment.
Figure 3.

Impact of Ept loci on pituitary mass after 28-wk E2 treatment. Female rats of each strain were treated with E2, released from sc SILASTIC brand implants, beginning at 9 wk of age. Control rats received empty implants. The rats were euthanized 28 wk later, and the pituitary glands were removed and weighed. Each bar represents the mean pituitary mass ± sd of the mean. The untreated control groups included three to five rats. The E2-treated groups included seven to 21 rats. Numeral 1 indicates a statistically significant (P < 0.05) difference relative to the corresponding untreated control group. Numeral 2 indicates a statistically significant difference relative to ACI rats receiving the same treatment. Numeral 3 indicates a statistically significant difference relative to COP rats receiving the same treatment.
Data presented previously indicate that continuous treatment with E2 induces significant growth of the pituitary gland in each of the rat strains evaluated. However, clear quantitative differences in the extent to which pituitary mass increased in response to E2 were observed between strains. The different rat strains evaluated in this study also differed with respect to pituitary gland morphology and coloration. When evaluated visually, the pituitary glands of female ACI rats treated with E2 for 28 wk frequently exhibited a dark red appearance, indicative of vascular changes and/or hemorrhage, as well as a loss of gland symmetry (Fig. 4). In contrast, neither pituitary discoloration nor loss of symmetry was observed in the E2-treated COP rats. Pituitary gland discoloration was observed at varying incidence in E2-treated Ept1, Ept6, and Ept9 rats, but not Ept2 congenic rats, suggesting that COP alleles at Ept2 restrain the biological processes that contribute to the hemorrhagic changes seen in the ACI, Ept1, Ept6, and Ept9 strains. Interestingly, pituitary gland discoloration was evident in a subset of Ept6 congenic rats within 12-wk initiation of E2 treatment. Because discoloration was not observed at this time point in the ACI strain, it would appear that COP alleles at this locus enhance this qualitative phenotype.
Figure 4.

Impact of Ept loci on pituitary gross morphology. Each pituitary gland was evaluated visually at necropsy. Photographs of pituitary glands from representative untreated and E2-treated rats illustrate the impact of E2 on gland enlargement, morphology, and coloration. The bar graph to the right of each photograph indicates the percentage of animals in each group exhibiting the dark-red discoloration indicative of increased vascularization and/or hemorrhage. The absence of a red bar is indicative of a 0% incidence of discolored pituitary glands in that experimental group. Grossly evident discoloration was observed in a fraction of Ept6 rats treated with E2 for 12 wk, as well as in a fraction of the ACI, Ept1, Ept6, and Ept9 rats treated with E2 for 28 wk. Discoloration of the pituitary gland was not observed in E2-treated COP or Ept2 rats at either time point.
Histologically, the pituitary glands of untreated female rats were unremarkable, and no discernable differences were noted between strains (data not shown). The pituitary glands of E2-treated rats exhibited multiple histological indicators of hyperplasia, including enlarged nuclei and nucleoli, prominent juxtanuclear Golgi, scattered mitotic figures, and rare apoptotic cells. Glands from all strains also exhibited dilated thin-walled blood vessels, as well as varying degrees of acute multifocal hemorrhage and hemosiderin deposits.
Impact of Ept loci on circulating PRL
We previously demonstrated a significant positive correlation between pituitary mass and circulating PRL in DES-treated male ACI, COP, (ACIxCOP)F1, (ACIxCOP)F2, and [(ACIxCOP)F1xACI] backcross rats (9). A positive correlation between pituitary mass and circulating PRL was also observed in a panel of DES-treated recombinant inbred rat strains generated in reciprocal intercrosses between the Lewis and F344 strains (30). Pituitary mass has also been demonstrated to correlate strongly with pituitary DNA content (5,11). Together, these data suggest that pituitary mass in estrogen-treated rats is a surrogate indicator of the absolute number of PRL-producing lactotrophs present within the pituitary gland. We quantified circulating PRL levels to determine whether the four Ept loci exerted parallel effects on pituitary mass and circulating PRL. Circulating PRL levels were significantly lower in Ept1 and Ept2 congenic rats treated with E2 for 12 wk, relative to ACI rats, suggesting that the reduction in pituitary mass observed upon E2 treatment in the Ept1 and Ept2 congenic strains was associated with a parallel reduction in absolute lactotroph number (Fig. 5). In contrast, circulating PRL levels in E2-treated Ept6 and Ept9 rats did not differ significantly from that observed in treated ACI rats. When pituitary mass for each of the two inbred and four congenic rat strains was compared with circulating PRL, a clear positive correlation was observed (r = 0.89).
Figure 5.

Impact of Ept loci on circulating PRL after 12-wk E2 treatment. Female rats of each strain were treated as described in Fig. 2. Trunk blood was collected after decapitation, serum was isolated, and PRL was assayed by radioimmunochemistry. Each bar represents the mean PRL level ± sd of the mean. The untreated control groups included eight to 12 rats. The E2-treated groups included 10–25 rats. Numeral 1 indicates a statistically significant (P < 0.05) difference relative to the corresponding untreated control group. Numeral 2 indicates a statistically significant difference relative to ACI rats receiving the same treatment. Numeral 3 indicates a statistically significant difference relative to COP rats receiving the same treatment.
Impact of Ept loci on induction of thymic atrophy
Estrogens inhibit cell proliferation within the cortex of the thymus and thereby induce thymic atrophy (28). The sensitivity of the thymus to administered estrogens is genetically determined (28,29). For example, male BN rats are significantly more sensitive to DES-induced thymic atrophy than are male ACI rats, and three QTL that determine sensitivity to DES-induced thymic atrophy in a population of male (BNxACI)F2 rats have been mapped to RNO2 and RNO10 (29). Because this phenotype had not been evaluated in the COP rat strain, we compared the impact of administered E2 on thymic atrophy in the ACI, COP, and Ept congenic rat strains. Although E2 induced thymic atrophy in both the ACI and COP rat strains, the effect was much more pronounced in female COP rats than in ACI rats (Fig. 6). In ACI rats, 12-wk E2 treatment resulted in a 38% reduction in thymus mass, which corresponded to a net decrease in average mass of 83.3 mg (Fig. 6B). In contrast, E2 induced a 65% reduction in thymus mass in COP rats, which corresponded to a net decrease in average mass of 169.6 mg. The Ept1, Ept6, and Ept9 congenic strains resembled the ACI strain with respect to both the amount and percentage of thymus mass lost in response to E2 treatment. Thymus mass in each of these congenic strains decreased by 36–39% in response to E2, which corresponded to net decreases in mass of 63.2–77.1 mg. Interestingly, the Ept2 congenic strain more closely resembled the COP strain with respect to basal thymus mass and was more sensitive to E2-induced thymic atrophy than either of the ACI, Ept1, Ept6, or Ept9 congenic strains. E2 induced a 49% reduction in thymus mass in Ept2 congenic rats, which corresponded to a net decrease in mass of 123.8 mg (Fig. 6B). The differences in sensitivity to E2-induced thymic atrophy exhibited by these rat strains remained apparent when the weight of the thymus was normalized to body mass (data not shown). These data strongly suggest that the Ept2 congenic interval harbors one or more genes that influence basal thymus mass and sensitivity to E2-induced thymic atrophy. This quantitative trait locus has been designated Esta4 (estrogen-induced thymic atrophy 4), after the nomenclature convention used previously to designate QTL impacting this phenotype (29).
Figure 6.

Impact of Ept loci on thymic atrophy after 12-wk E2 treatment. Female rats of each strain were treated with E2, released from sc SILASTIC brand implants, beginning at 9 wk of age. Control rats received empty implants. The rats were euthanized 12 wk later, and the thymus was removed and weighed. A, Each bar represents the mean thymus mass ± sd of the mean. The untreated control groups included four to 12 rats. The E2-treated groups included four to 25 rats. Numeral 1 indicates a statistically significant (P < 0.05) difference relative to the corresponding untreated control group. Numeral 2 indicates a statistically significant difference relative to ACI rats receiving the same treatment. Numeral 3 indicates a statistically significant difference relative to COP rats receiving the same treatment. B, Each bar indicates the decrease in average thymus mass in E2-treated rats relative to untreated control rats of the same strain.
Impact of Ept loci on induction of mammary cancer
As discussed previously, estrogens induce pituitary lactotroph hyperplasia and increase circulating PRL. Because PRL stimulates cell proliferation within the mammary epithelium, it has been suggested that estrogens may indirectly contribute to mammary cancer development in rats through their stimulatory actions on the pituitary gland. To define further the genetic relationship between E2-induced pituitary growth and mammary cancer, we evaluated susceptibility of the Ept1, Ept2, Ept6, and Ept9 congenic rat strains to E2-induced mammary cancer. Like the ACI strain, each of these congenic rat strains exhibited a high incidence of mammary cancer when treated with E2 (Fig. 7). Whereas 95% of E2-treated ACI rats exhibited mammary cancer within 196 d (28 wk) of initiation of E2 treatment, the incidence of mammary cancer in the four congenic strains ranged from 75–88%. Median latency to the appearance of the first palpable mammary cancer in the Ept2, Ept6, and Ept9 congenic strains ranged from 139–148 d, similar to that of the highly susceptible ACI strain, which exhibited a median latency of 145 d. Interestingly, latency in the Ept1 strain was significantly (P = 0.048) prolonged relative to the other strains. In this congenic strain, median latency was 165 d. Median latency in the resistant COP strain could not be estimated because fewer than 50% of the E2-treated COP rats developed mammary cancer. When tumor number at necropsy was evaluated after 196-d E2 treatment, the ACI, Ept2, Ept6, and Ept9 strains exhibited similar mean numbers of mammary cancers (Table 2). Average tumor number in E2-treated Ept1 congenic rats was significantly reduced relative to the ACI strain. E2-treated COP rats exhibited an average of 0.14 mammary cancers per rat. Each of the tumors induced by E2 in the different rat strains was evaluated microscopically and classified as mammary adenocarcinoma. Age-matched untreated control rats from each strain did not develop mammary cancer over the 196-d time course. Together, these data indicate that the Ept1 congenic interval harbors a genetic variant (or variants) that influences the development of mammary cancer. This quantitative trait locus has been designated Emca10 (estrogen-induced mammary cancer 10).
Figure 7.

Impact of Ept loci on E2-induced mammary cancer. Female rats of each strain were treated with E2, released from sc SILASTIC brand implants, beginning at 9 wk of age. Control rats received empty implants. The rats were euthanized 28 wk later or as necessitated due to treatment-related morbidity. Each data point represents the time at which an animal in the population at risk exhibited the first palpable mammary cancer. Mammary cancer did not develop in untreated, ovary intact, female rats during the course of this experiment. A, Data from the Ept1 and Ept2 congenic strains are illustrated relative to data from the ACI and COP strains. B, Data from the Ept6 and Ept9 congenic strains are illustrated relative to data from the ACI and COP strains.
Table 2.
Mammary tumor number after 196 d of treatment
| Strain | Treatment | Mean | sd | No. |
|---|---|---|---|---|
| ACI | Untreated | 0.0 | 4 | |
| E2 | 7.0 | 5.3 | 21 | |
| COP | Untreated | 0.0 | 5 | |
| E2 | 0.1 | 0.4 | 7 | |
| Ept1 | Untreated | 0.0 | 3 | |
| E2 | 2.4 | 1.7 | 17 | |
| Ept2 | Untreated | 0.0 | 3 | |
| E2 | 4.5 | 3.0 | 17 | |
| Ept6 | Untreated | 0.0 | 3 | |
| E2 | 4.3 | 3.2 | 8 | |
| Ept9 | Untreated | 0.0 | 3 | |
| E2 | 4.8 | 3.6 | 12 |
Discussion
Data presented herein indicate that COP alleles for Ept1, Ept2, Ept6, and Ept9, when carried on the ACI genetic background, influence the responsiveness of the anterior pituitary gland to E2. When pituitary mass was evaluated as the phenotype, the Ept1, Ept2, Ept6, and Ept9 congenic rat strains each differed from the ACI strain at the 12 and/or 28-wk time point examined in this study. Overall, the direction of the effect of each congenic interval on pituitary mass was in agreement with predictions based on our published QTL mapping experiments in which pituitary mass in DES-treated male F2 rats from reciprocal crosses between the ACI and COP rat strains served as the phenotype (12). Thus, the current study confirms our previously published findings. Together, the data from the published QTL mapping studies and the studies described herein on the different Ept congenic rat strains indicate that the impacts of Ept1, Ept2, Ept9, and Ept6 on responsiveness of the anterior pituitary gland to estrogens are largely, if not wholly, independent of gender as well as the chemical form of the estrogen used to induce pituitary growth.
In this study the impacts of Ept1, Ept6, and Ept9 on pituitary growth varied as a function of the duration of E2 treatment. Although inhibitory effects of COP alleles at Ept1 and Ept9 were clearly observed after 12-wk E2 treatment, these effects were lost by 28 wk. By contrast, the ability of COP alleles at Ept6 to enhance E2-induced pituitary growth did not become apparent until the 28-wk time point. Interestingly, the ability of COP alleles at Ept2 to restrain E2-induced pituitary growth was observed at both the 12 and 28-wk time points. These data strongly suggest that the genes residing within the different Ept loci act on distinct processes to influence the growth response of the anterior pituitary gland to administered E2.
Ept2 and Ept6 both map to RNO3 (12). The congenic intervals for these two QTL overlap by approximately 20 Mb, with the Ept6 congenic interval extending into the Ept2 CI and including a region of RNO3 that resides under a statistically significant local LOD peak (Fig. 1). The Ept6 congenic strain did not exhibit the anticipated increase in pituitary mass in response to 12-wk E2 treatment. One possible reason for this observation is that COP alleles at the linked Ept2-associated determinant of pituitary responsiveness to estrogen may have counteracted the ability of COP alleles at Ept6 to enhance induction of pituitary growth. If true, then the actions of the Ept6 must outweigh the actions of the Ept2-associated determinant by 28-wk E2 treatment, when Ept6 congenic rats exhibited increased pituitary mass relative to ACI rats.
Relative to female ACI rats, female Ept1 congenic rats exhibited reduced pituitary mass when treated with E2 for 12 wk, but not when treated for 28 wk. The female Ept1 congenic rats also exhibited reduced susceptibility to E2-induced mammary cancer when compared with ACI rats. Although these data indicate that the Ept1 congenic interval harbors one or more genes that influence responsiveness to estrogens in both the anterior pituitary gland and the mammary gland, it is not currently known whether the same gene or genes within Ept1 impact both the pituitary growth and mammary cancer phenotypes. Because the actions of Ept1 on induction of pituitary mass were lost by 28-wk treatment, it is unlikely that the diminished pituitary mass and circulating PRL observed at the 12-wk time point directly contributed to the reduced susceptibility of the Ept1 congenic strain to mammary cancer. Supporting this assertion is the observation that female Ept2 congenic rats exhibited a dramatic and sustained diminution in pituitary mass and circulating PRL but did not exhibit reduced susceptibility to E2-induced mammary cancer. The mammary cancer quantitative trait locus residing within the Ept1 congenic interval has been designated Emca10, to distinguish it from the Ept1 determinant of pituitary responsiveness. A total of 14 Ept loci and 10 Emca loci have now been mapped in various crosses between the ACI and COP or BN rat strains (12,13,22,23,24). With the exceptions of Ept1 and Emca10, which may colocalize on RNO6, and Ept5 and Emca4, which map to the same region of RNO7, the remaining Ept and Emca loci segregate independently within the individual crosses in which they were identified. These data suggest that the genetic factors that impact estrogen responsiveness in the anterior pituitary are largely distinct from those that impact responsiveness to estrogens in the mammary gland.
When treated with E2, female Ept2 congenic rats exhibited reduced pituitary growth, relative to ACI rats, at both the 12 and 28-wk time points. The female Ept2 rats also exhibited enhanced E2-induced thymic atrophy when compared with female ACI rats. It is not currently known whether the same Ept2 associated gene(s) impacts both the pituitary growth and thymic atrophy phenotypes. E2-induced thymic atrophy was more severe in COP rats than ACI rats. Similarly, in a previous study, thymic atrophy was more severe in DES-treated male BN rats, which exhibit very little pituitary growth in response to estrogens, than in DES-treated male ACI rats (29). Because PRL is known to enhance thymocyte proliferation, survival, and maturation (34,35,36,37), these data might suggest that the high levels of circulating PRL in estrogen-treated ACI rats attenuate induction of thymic atrophy by estrogens in this rat strain. However, the data from the Ept1 and Ept2 congenic rat strains do not support this assertion. Whereas induction of pituitary growth and circulating PRL was significantly diminished in E2-treated Ept1 and Ept2 rats, the extent to which E2 induced thymic atrophy was exacerbated only in Ept2 rats. Therefore, it is concluded that the impact of Ept2 on E2-induced thymic atrophy is most probably independent of its effect of circulating PRL.
Since our initial description of the ACI rat model of E2-induced mammary cancer in 1997, the physiological relevance of this model to breast cancer in humans has become increasingly clear, and this model has gained wide use in the breast cancer research community. Because estrogens induce pituitary growth in ACI rats, morbidity relating to enlargement of the pituitary gland may diminish the usefulness of this mammary cancer model in long-term studies, such as those performed to evaluate agents for preventing breast cancer. In an attempt to reduce morbidity relating to pituitary enlargement, investigators have modified the method for delivering E2 so as to reduce circulating E2. Although this has been partially effective, the reduced level of E2 also prolongs the period of time required for mammary cancer to appear (38). Part of our rationale for performing the studies described herein was to determine whether the estrogen-induced pituitary growth and mammary cancer phenotypes could be genetically separated. Of the four congenic rat strains evaluated in this study, the Ept2 strain exhibited a sustained reduction in pituitary growth over a 196-d time course while fully retaining the unique susceptibility of parental ACI rat strain to mammary cancer. Therefore, the Ept2 congenic rat strain offers an advantage over the ACI strain for use in long-term mammary cancer studies.
Based on our previous mapping studies, we predicted that the Ept6 congenic rat strain would exhibit an increased pituitary growth response to E2, relative to the ACI rat strain. Although no such effect of Ept6 was observed at the 12-wk time point, pituitary mass was significantly increased, relative to that observed in ACI rats, in female Ept6 rats treated with E2 for 28 wk. Interestingly, the E2-treated female Ept6 congenic rats frequently exhibited a grossly apparent alteration in pituitary coloration, consistent with increased vascularization, at the 12 and 28-wk time points. Because this phenotype was observed more frequently in E2-treated Ept6 rats than in ACI rats, it appears that COP alleles at Ept6 enhance this phenotype. Although the effects of estrogen on pituitary angiogenesis have not been investigated in the ACI rat strain, a role for neovascularization in estrogen-induced pituitary growth in the F344 rat has long been established (39,40,41,42,43,44). Therefore, it would appear that Ept6 may impact E2-induced pituitary growth by acting upon pathways that regulate vascularization within the anterior pituitary gland.
Wendell and colleagues (8,45) have performed studies to identify genetic determinants of estrogen-induced pituitary growth in the F344 rat strain. QTL impacting pituitary mass in DES-treated F2 and backcross progeny generated in crosses between the F344 and BN rat stains have been mapped by these investigators to RNO2, RNO3, RNO5, and RNO9 (10,11,46). One of these QTL, Edpm3, colocalizes with Ept2 on RNO3. A congenic strain carrying BN alleles across Edpm3 was demonstrated to exhibit reduced DES-induced pituitary growth and cell proliferation relative to the F344 strain (14). Interestingly, Edpm3 rats unexpectedly exhibited a greater angiogenic response to DES than did F344 rats (15). The Edpm3 congenic interval (14) overlaps with both the Ept2 and Ept6 congenic intervals (Fig. 1). If one assumes that the angiogenic phenotypes exhibited by the Edpm3 and Ept6 congenic strains are manifestations of the same gene, then this gene must reside within the region of overlap between the Edpm3 and Ept6 congenic intervals. The absence of this phenotype in the Ept2 congenic strain would further suggest that the gene responsible for this phenotype resides proximal to the Ept2 congenic interval. Based on these data, we hypothesize that the gene on RNO3 that contributes to estrogen-induced angiogenesis in the pituitary gland resides between D3Mgh7 (36.556 Mb) and D3Rat130 (44.551 Mb).
In addition to acting directly on the pituitary gland to stimulate lactotroph proliferation, estrogens act through multiple indirect mechanisms to induce pituitary growth and pituitary tumor development (reviewed in Ref. 3). For example, estrogens act on the tuberoinfundibular dopaminergic neurons of the hypothalamus to inhibit the release of dopamine into the hypophysial portal circulation for transport to the anterior pituitary gland, where it acts via D2 dopamine receptors on the lactotroph to inhibit lactotroph proliferation and PRL gene expression (47,48,49). In addition, estrogens have induced neovascularization within the pituitary gland, resulting in the development of a direct arterial blood supply, which supplants the hypophysial portal blood supply and thereby circumvents regulation of the pituitary lactotroph by dopamine (39,40). Finally, gross enlargement of the anterior pituitary in response to estrogen treatment has physically damaged the mediobasal hypothalamus and disrupted the function of the tuberoinfundibular dopaminergic neurons residing there (3). Additional studies are required to determine whether the genetic determinants of pituitary responsiveness residing within the Ept loci identified in this study impact these or other processes.
In summary, this study conclusively demonstrates the existence of one or more genetic determinants of estrogen action on the pituitary gland within the Ept1 locus on RNO3, the Ept2 and Ept6 loci on RNO6, and the Ept9 locus on RNO10. The actions of Ept6 and Ept9 were observed only on the pituitary gland. However, actions of Ept1 were observed on the pituitary and mammary glands, and actions of Ept2 were observed on the pituitary gland and the thymus. The dynamic range of responsiveness to estrogens in the pituitary gland, mammary gland, and thymus observed when comparing the ACI, COP, and other inbred rat strains is remarkable. It is likely that orthologous genetic variants segregating in the human population exert similar actions on estrogen responsiveness in these and other target tissues. Identification of the genetic determinants of estrogen responsiveness will greatly enhance our understanding of estrogen action at the molecular level, and improve our ability to use in a safe and effective manner drugs that target estrogen receptor signaling.
Footnotes
This work was supported by National Institutes of Health Grants R01-CA68529 and R01-CA77876 (to J.D.S.). National Institutes of Health Cancer Center Support Grant P30-CA36727 supported shared resources within the University of Nebraska Medical Center, Eppley Cancer Center. B.S.S. was supported in part by training Grant DAMD17-00-1-0361 and individual postdoctoral fellowship DAMD17-03-1-0477 from the United States Army Breast Cancer Training Program.
Contributions: S.G.K. phenotypically characterized the four congenic rat strains. K.K.H., M.T.M., and S.G.K. developed the congenic rat strains. V.S. performed the prolactin assays. B.S.S. and K.A.G. provided both technical assistance and scientific input into performance of the research. R.D.M. and J.D.S. performed the histopathological evaluations of tissues. J.L.M. provided expertise in statistical analysis of data. J.D.S. conceived and directed the study and wrote the manuscript.
Disclosure Statement: The authors have nothing to disclose.
First Published Online April 17, 2008
Abbreviations: BN, Brown Norway; CI, confidence interval; COP, Copenhagen; DES, diethylstilbestrol; E2, estradiol; F344, Fischer 344; Mb, million base pair; PRL, prolactin; QTL, quantitative trait loci.
References
- Sarkar DK, Hentges ST, De A, Reddy RH 1998 Hormonal control of pituitary prolactin-secreting tumors. Front Biosci 3:d934–d943 [DOI] [PubMed] [Google Scholar]
- Gorski J, Wendell D, Gregg D, Chun TY 1997 Estrogens and the genetic control of tumor growth. Prog Clin Biol Res 396:233–243 [PubMed] [Google Scholar]
- Spady TJ, McComb RD, Shull JD 1999 Estrogen action in the regulation of cell proliferation, cell survival, and tumorigenesis in the rat anterior pituitary gland. Endocrine 11:217–233 [DOI] [PubMed] [Google Scholar]
- Sarkar DK 2006 Genesis of prolactinomas: studies using estrogen-treated animals. Front Horm Res 35:32–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiklund J, Wertz N, Gorski J 1981 A comparison of estrogen effects on uterine and pituitary growth and prolactin synthesis in F344 and Holtzman rats. Endocrinology 109:1700–1707 [DOI] [PubMed] [Google Scholar]
- Wiklund J, Rutledge J, Gorski J 1981 A genetic model for the inheritance of pituitary tumor susceptibility in F344 rats. Endocrinology 109:1708–1714 [DOI] [PubMed] [Google Scholar]
- Wiklund JA, Gorski J 1982 Genetic differences in estrogen-induced deoxyribonucleic acid synthesis in the rat pituitary: correlations with pituitary tumor susceptibility. Endocrinology 111:1140–1149 [DOI] [PubMed] [Google Scholar]
- Wendell DL, Herman A, Gorski J 1996 Genetic separation of tumor growth and hemorrhagic phenotypes in an estrogen-induced tumor. Proc Natl Acad Sci USA 93:8112–8116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spady TJ, Pennington KL, McComb RD, Shull JD 1999 Genetic bases of estrogen-induced pituitary growth in an intercross between the ACI and Copenhagen rat strains: dominant Mendelian inheritance of the ACI phenotype. Endocrinology 140:2828–2835 [DOI] [PubMed] [Google Scholar]
- Wendell DL, Gorski J 1997 Quantitative trait loci for estrogen-dependent pituitary tumor growth in the rat. Mamm Genome 8:823–829 [DOI] [PubMed] [Google Scholar]
- Wendell DL, Daun SB, Stratton MB, Gorski J 2000 Different functions of QTL for estrogen-dependent tumor growth of the rat pituitary. Mamm Genome 11:855–861 [DOI] [PubMed] [Google Scholar]
- Strecker TE, Spady TJ, Schaffer BS, Gould KA, Kaufman AE, Shen F, McLaughlin MT, Pennington KL, Meza JL, Shull JD 2005 Genetic bases of estrogen-induced pituitary tumorigenesis: identification of genetic loci determining estrogen-induced pituitary growth in reciprocal crosses between the ACI and Copenhagen rat strains. Genetics 169:2189–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull JD, Lachel CM, Murrin CR, Pennington KL, Schaffer BS, Strecker TE, Gould KA 2007 Genetic control of estrogen action in the rat: mapping of QTLs that impact pituitary lactotroph hyperplasia in a BN × ACI intercross. Mamm Genome 18:657–669 [DOI] [PubMed] [Google Scholar]
- Wendell DL, Pandey J, Kelley P 2002 A congenic strain of rat for investigation of control of estrogen-induced growth. Mamm Genome 13:664–666 [DOI] [PubMed] [Google Scholar]
- Pandey J, Wendell DL 2006 Angiogenesis and capillary maturation phenotypes associated with the Edpm3 locus on rat chromosome 3. Mamm Genome 17:49–57 [DOI] [PubMed] [Google Scholar]
- Pandey J, Bannout A, Wendell DL 2004 The Edpm5 locus prevents the ‘angiogenic switch’ in an estrogen-induced rat pituitary tumor. Carcinogenesis 25:1829–1838 [DOI] [PubMed] [Google Scholar]
- Shull JD, Spady TJ, Snyder MC, Johansson SL, Pennington KL 1997 Ovary intact, but not ovariectomized female ACI rats treated with 17β-estradiol rapidly develop mammary carcinoma. Carcinogenesis 18:1595–1601 [DOI] [PubMed] [Google Scholar]
- Li SA, Weroha SJ, Tawfik O, Li JJ 2002 Prevention of solely estrogen-induced mammary tumors in female ACI rats by tamoxifen: evidence for estrogen receptor mediation. J Endocrinol 175:297–305 [DOI] [PubMed] [Google Scholar]
- Spady TJ, Harvell DME, Snyder MC, Pennington KL, McComb RD, Shull JD 1998 Estrogen-induced tumorigenesis in the Copenhagen rat: disparate susceptibilities to development of prolactin-producing pituitary tumors and mammary carcinomas. Cancer Lett 124:95–103 [DOI] [PubMed] [Google Scholar]
- Harvell DM, Strecker TE, Tochacek M, Xie B, Pennington KL, McComb RD, Roy SK, Shull JD 2000 Rat strain specific actions of 17β-estradiol in the mammary gland: correlation between estrogen-induced lobuloalveolar hyperplasia and susceptibility to estrogen-induced mammary cancers. Proc Natl Acad Sci USA 97:2779–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull JD, Pennington KL, Reindl TM, Snyder MC, Strecker TE, Spady TJ, Tochacek M, McComb RD 2001 Susceptibility to estrogen-induced mammary cancer segregates as an incompletely dominant phenotype in reciprocal crosses between the ACI and Copenhagen rat strains. Endocrinology 142:5124–5130 [DOI] [PubMed] [Google Scholar]
- Gould KA, Tochacek M, Schaffer BS, Reindl TM, Murrin CR, Lachel CM, VanderWoude EA, Pennington KL, Flood LA, Bynote KK, Meza JL, Newton MA, Shull JD 2004 Genetic determination of susceptibility to estrogen-induced mammary cancer in the ACI rat: mapping of Emca1 and Emca2 to chromosomes 5 and 18. Genetics 168:2113–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer BS, Lachel CM, Pennington KL, Murrin CR, Strecker TE, Tochacek M, Gould KA, Meza JL, McComb RD, Shull JD 2006 Genetic bases of estrogen-induced tumorigenesis in the rat: mapping of loci controlling susceptibility to mammary cancer in a Brown Norway × ACI intercross. Cancer Res 66:7793–7800 [DOI] [PubMed] [Google Scholar]
- Shull JD 2007 The rat oncogenome: comparative genetics and genomics of rat models of mammary carcinogenesis. Breast Dis 28:69–86 [DOI] [PubMed] [Google Scholar]
- Gould KA, Pandey J, Lachel CM, Murrin CR, Flood LA, Pennington KL, Schaffer BS, Tochacek M, McComb RD, Meza JL, Wendell DL, Shull JD 2005 Genetic mapping of Eutr1, a locus controlling E2-induced pyometritis in the Brown Norway rat, to RNO5. Mamm Genome 16:854–864 [DOI] [PubMed] [Google Scholar]
- Pandey J, Gould KA, McComb RD, Shull JD, Wendell DL 2005 Localization of Eutr2, a locus controlling susceptibility to DES-induced uterine inflammation and pyometritis, to RNO5 using a congenic rat strain. Mamm Genome 16:865–872 [DOI] [PubMed] [Google Scholar]
- Luz NP, Marques M, Ayub AC, Correa PR 1969 Effects of estradiol upon the thymus and lymphoid organs of immature female rats. Am J Obstet Gynecol 105:525–528 [PubMed] [Google Scholar]
- Gould KA, Shull JD, Gorski J 2000 DES action in the thymus: inhibition of cell proliferation and genetic variation. Mol Cell Endocrinol 170:31–39 [DOI] [PubMed] [Google Scholar]
- Gould KA, Strecker TE, Hansen KK, Bynote KK, Peterson KA, Shull JD 2006 Genetic mapping of loci controlling sensitivity to diethylstilbestrol-induced thymic atrophy in the Brown Norway rat. Mamm Genome 17:451–464 [DOI] [PubMed] [Google Scholar]
- Tachibana M, Lu L, Hiai H, Tamura A, Matsushima Y, Shisa H 2006 Quantitative trait loci determining weight reduction of testes and pituitary by diethylstilbesterol in LEXF and FXLE recombinant inbred strain rats. Exp Anim 55:91–95 [DOI] [PubMed] [Google Scholar]
- Gould KA, Dietrich WF, Borenstein N, Lander ES, Dove WF 1996 Mom1 is a semi-dominant modifier of intestinal adenoma size and multiplicity in Min/+ mice. Genetics 144:1769–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeland E, Morel L, Achey K, Yui M, Longmate J 1997 Speed congenics: a classic technique in the fast lane (relatively speaking). Immunol Today 18:472–477 [DOI] [PubMed] [Google Scholar]
- Harvell DM, Buckles LK, Gould KA, Pennington KL, McComb RD, Shull JD 2003 Rat strain specific attenuation of estrogen action in the anterior pituitary gland by dietary energy restriction. Endocrine 21:175–183 [DOI] [PubMed] [Google Scholar]
- Gaufo GO, Diamond MC 1996 Prolactin increases CD4/CD8 cell ratio in thymus-grafted congenitally athymic nude mice. Proc Natl Acad Sci USA 93:4165–4169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mello-Coelho V, Savino W, Postel-Vinay MC, Dardenne M 1998 Role of prolactin and growth hormone on thymus physiology. Dev Immunol 6:317–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan N, Thellin O, Buckley DJ, Horseman ND, Buckley AR 2003 Prolactin suppresses glucocorticoid-induced thymocyte apoptosis in vivo. Endocrinology 144:2102–2110 [DOI] [PubMed] [Google Scholar]
- Carreno PC, Sacedon R, Jimenez E, Vicente A, Zapata AG 2005 Prolactin affects both survival and differentiation of T-cell progenitors. J Neuroimmunol 160:135–145 [DOI] [PubMed] [Google Scholar]
- Ravoori S, Vadhanam MV, Sahoo S, Srinivasan C, Gupta RC 2007 Mammary tumor induction in ACI rats exposed to low levels of 17β-estradiol. Int J Oncol 31:113–120 [PubMed] [Google Scholar]
- Elias KA, Weiner RI 1984 Direct arterial vasculature of estrogen-induced prolactin-secreting anterior pituitary tumors. Proc Natl Acad Sci USA 81:4549–4553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnet F, Elias KA, Fagin K, Neill A, Goldsmith P, Weiner RI 1984 Formation of a direct arterial blood supply to the anterior pituitary gland following complete or partial interruption of the hypophyseal portal vessels. Neuroendocrinology 39:251–255 [DOI] [PubMed] [Google Scholar]
- Schechter J, Ahmad N, Elias K, Weiner R 1987 Estrogen-induced tumors: changes in the vasculature in two strains of rat. Am J Anat 179:315–323 [DOI] [PubMed] [Google Scholar]
- Elias KA, Weiner RI 1987 Inhibition of estrogen-induced anterior pituitary enlargement and arteriogenesis by bromocriptine in Fischer 344 rats. Endocrinology 120:617–621 [DOI] [PubMed] [Google Scholar]
- Stepien H, Grochal M, Zielinski KW, Mucha S, Kunert-Radek J, Kulig A, Stawowy A, Pisarek H 1996 Inhibitory effects of fumagillin and its analogue TNP-470 on the function, morphology and angiogenesis of an oestrogen- induced prolactinoma in Fischer 344 rats. J Endocrinol 150:99–106 [DOI] [PubMed] [Google Scholar]
- Banerjee SK, Sarkar DK, Weston AP, De A, Campbell DR 1997 Over expression of vascular endothelial growth factor and its receptor during the development of estrogen-induced rat pituitary tumors may mediate estrogen-initiated tumor angiogenesis. Carcinogenesis 18:1155–1161 [DOI] [PubMed] [Google Scholar]
- Cracchiolo D, Swick JW, McKiernan L, Sloan E, Raina S, Sloan C, Wendell DL 2002 Estrogen-dependent growth of a rat pituitary tumor involves, but does not require, a high level of vascular endothelial growth factor. Exp Biol Med (Maywood) 227:492–499 [DOI] [PubMed] [Google Scholar]
- Sclafani RV, Wendell DL 2001 Suppression of estrogen-dependent MMP-9 expression by Edpm5, a genetic locus for pituitary tumor growth in rat. Mol Cell Endocrinol 176:145–153 [DOI] [PubMed] [Google Scholar]
- Shaw-Bruha CM, Happe HK, Murrin LC, Rodriguez-Sierra JF, Shull JD 1996 17β-Estradiol inhibits the production of dopamine by the tuberoinfundibular dopaminergic neurons of the male rat. Brain Res Bull 40:33–36 [DOI] [PubMed] [Google Scholar]
- DeMaria JE, Livingstone JD, Freeman ME 2000 Ovarian steroids influence the activity of neuroendocrine dopaminergic neurons. Brain Res 879:139–147 [DOI] [PubMed] [Google Scholar]
- Steyn FJ, Anderson GM, Grattan DR 2007 Expression of ovarian steroid hormone receptors in tuberoinfundibular dopaminergic neurons during pregnancy and lactation. J Neuroendocrinol 19:788–793 [DOI] [PubMed] [Google Scholar]