

Abstract
In this study, we evaluated the efficacy of vaccinia virus (VACV) containing mutations in the E3L virulence gene to protect mice against a lethal poxvirus challenge after vaccination by scarification. VACV strains with mutations in the E3L gene had significantly decreased pathogenicity, even in immune deficient mice, yet retained the ability to produce a potent Th1-dominated immune response in mice after vaccination by scarification, while protecting against challenge with wild type, pathogenic VACV. Initial experiments were done using the mouse-adapted, neurovirulent Western Reserve (WR) strain of vaccinia virus. Testing of the full E3L deletion mutation in the Copenhagen and NYCBH strains of VACV, which are more appropriate for use in humans, produced similar results. These results suggest that highly attenuated strains of VACV containing mutations in E3L have the potential for use as scarification administered vaccines.
Keywords: Vaccinia, Smallpox vaccine, E3L
1. Introduction
Poxviruses are a large and diverse group of viruses that can infect a wide range of organisms, including birds, insects, and many mammals, including humans. They are double-stranded DNA viruses with linear genomes generally ranging from 130–300 kbp. Poxviruses replicate in the cytoplasm of infected cells using both proteins that they encode and resources from the infected cell. Variola virus is the causative agent of smallpox, a devastating human disease [1].Vaccination against smallpox has thus far relied upon cross-protective antigens from other orthopoxviruses such as vaccinia virus (VACV), which itself causes relatively mild symptoms in humans during natural infections [1]. The use of VACV as a vaccine has resulted in successful eradication of smallpox, but recent concerns about the potential use of variola virus as a bioweapon has renewed interest in developing an improved vaccine. Indeed, the current smallpox vaccine has resulted in a significant number of severe side effects, including encephalitis, eczema vaccinatum, generalized and progressive vaccinia infections, as well as many other less severe adverse reactions. There have also been recent reports of cardiac complications following vaccination [2]. In future vaccination campaigns, these complications are likely to be compounded by the growing numbers of individuals with compromised immune functions due to HIV infection, organ transplants, and cancer treatments [3]. In addition to its use as a smallpox vaccine, VACV is being developed as a vector for vaccination against heterologous antigens.
The risks associated with currently available strains of VACV have prompted generation of a variety of attenuated strains of VACV. These include modified Ankara strain (MVA) [4], a mutant of the Lister strain of VACV deleted for the essential viral UDG gene [5], NYVAC [6] and LC16m8 [7]. MVA and LC16M8 were generated by passaging through alternative hosts, while NYVAC and the Lister mutant were engineered to contain attenuating deletions. All of these attenuated strains, except LC16m8 have very limited capacity for replication in human cells [6, 8–10]. Although non-replicating vectors have the advantage of increased safety, high doses of these vectors (107 to 108 pfu) are often required to induce a strong immune response [9, 11, 12]. Most of these vaccinations also require multiple immunizations [13] often defeating the purpose of using live virus vaccines which usually induce potent immune response with a single dose.
The vaccinia virus E3L gene products are potent inhibitors of the innate immune response [14–16]. The proteins encoded by the VACV E3L gene contain a C-terminal consensus dsRNA-binding domain that can bind to and sequester viral dsRNA [17] produced in infected cells, probably as a result of convergent transcription of the viral genome [18]. Free dsRNA can act as a pathogen associated molecular pattern (PAMP), leading to pro-inflammatory signal transduction, expression of interferon and other pro-inflammatory genes, and activation of antiviral proteins already induced by interferon [19, 20]. The E3L encoded proteins also contain an N-terminal consensus Z-nucleic acid binding domain [21]. This domain is necessary for virulence in the mouse model.
In an effort to develop replication-competent but attenuated strains of VACV we have constructed and tested a variety of VACV mutants with deletions in the E3L interferon resistance gene. These mutant virus strains are highly attenuated in both immune deficient and immune competent mice, after either intra-nasal or intra-cranial infection. Despite attenuation these viruses can induce a protective immune response after intra-nasal vaccination [22]. In the present study we demonstrate that these viruses can also induce protective takes after administration by the more standard route of immunization by scarification. We also demonstrate that vaccination by scarification induces potent cell mediated immunity to VACV and primes for production of neutralizing and comet inhibition antibodies.
2. Materials and Methods
2.1. Recombinant virus construction
Viruses used in these studies were constructed in the Western Reserve strain of vaccinia virus (a mouse-adapted neurovirulent strain developed by serial passage of vaccinia virus strain in the brains of mice [23]), in the Copenhagen strain (VC2, kindly provided by Virogenetics) or in the New York City Board of Health strain (ACAM2000, kindly provided by Acambis) [24].VACVΔE3L was constructed by in vivo recombination resulting in the replacement of the E3L gene with the E. coli lacZ gene [25]. Vaccinia virus mutants with N or C terminal sections of the E3L gene deleted or a foreign gene inserted were generated by in vivo recombination of E3L mutant genes into VACVΔE3L. VACVE3LΔ7C, which is deleted of the last 7 C terminal amino acids, was constructed as previously described [26]. VACVE3LΔ54N, which is deleted of the first 54 N terminal amino acids, was constructed as previously described [27]. The wtVACV used in this study is a revertant of VACVΔE3L, wtVACV03. Unless otherwise noted, the WR strain of VACV was used in all experiments.
2.2. Cell culture
BHK-21 (Baby Hamster Kidney) and RK-13 (Rabbit Kidney) cells were maintained in Eagle’s Minimum Essential Medium (MEM-Gibco, BRL) supplemented with 10% Fetal Bovine Serum (FBS-Hyclone), 50 µg/ml of gentamycin, and 0.1mM non-essential amino acid solution (Gibco-BRL). Both BHK and RK-13 cells were incubated at 37°C in 5% CO2.
2.3. Preparation of virus stocks
All virus stocks were prepared in BHK cells, as previously described [27]. Viruses were partially purified by centrifugation through a 36% sucrose pad.
2.4. Mice
SCID (Severe Combined Immune Deficient) mice were obtained from either the Jackson Laboratory (CBySmn.CB17-Prkdcscid/J) or Charles River Laboratories (Fox Chase SCID mouse, CB17/lcr-PrKdcSCID/CrL). BALB/c and C57BL/6 mice were obtained from either The Jackson Laboratory or Charles River Laboratories. muMT B cell deficient mice were obtained from the Jackson laboratory. All mice were housed in the Arizona State University Animal Resource Center according to the university’s Institutional Animal Care and Use regulations. Mice were 4–5 weeks of age when used in experiments unless otherwise indicated. Mice were either of both sexes or were females only as indicated in individual experiments.
2.5. Scarification infection of mice
BALB/c, C57BL/6 or SCID mice were anesthetized using a intraperitoneally (IP) injected cocktail containing 7.5 mg/ml xylazine, 2.5 mg/ml acepromazine maleate and 37.5 mg/ml ketamine at approximately 1.5 µl/gm of body weight. Hair on a 1-inch strip of the dorsal surface of the base of the tail was then removed with Nair Hair Remover and the skin rinsed thoroughly with sterile water. 15 scratches were then made horizontally across the tail skin with a sterile 26-gauge syringe needle. The skin was then infected by placing 10µl of virus suspended in 1mM Tris pH 8.8 on the skin using a gel loading tip and then using the side of the tip to rub the virus into the scratches. Mock-infected animals received 1mM Tris pH 8.8 containing no virus. Animals were monitored for weight loss, signs of illness and severity of skin pocks. Animals that received vaccination boosts were scarified a second time one month after the initial infection using the same procedure.
2.6. Infection of mice
BALB/c mice were manually restrained and injected IP in the lower right abdominal quadrant with 100 µl of virus suspended in 1mM Tris pH 8.8. Mock-infected animals received 100µl 1mM Tris pH 8.8 containing no virus.
2.8. Recovery of virus from tissues
Tissues were harvested from chemically euthanized animals at 5 days post-infection, as previously described [27]. Briefly, tissues were removed, placed in cryogenic vials, and quick-frozen in liquid nitrogen. Each animal’s ovaries were combined upon removal. Harvested tail tissue consisted of the entire scarified skin area down to the level of the connective tissues. Tissues were homogenized either by using a liquid nitrogen cooled mortar and pestle or by using a mechanical tissue homogenizer. The samples were freeze-thawed 3 times. Samples were then centrifuged at 4°C for 10 minutes at 1000xg to remove cell debris. Virus content in the supernatants was assessed by titration in RK-13 cells and the amount of virus per gram of tissue was calculated.
2.9. Challenge experiments
Either C57BL/6 or BALB/c mice were vaccinated by scarification and then challenged one month later intranasally (IN) with 106 pfu of Western Reserve strain wild type vaccinia virus (wtVACV). Mice were monitored for weight loss every other day and signs of illness every day for 2 weeks after challenge. Data points shown on graphs are the average percent weight loss for the mice remaining in each experimental group.
2.10. Harvest of blood samples
Blood samples for anti-vaccinia antibody titering were harvested by heart puncture in anesthetized mice. Blood was then placed in 1.5 ml microfuge tubes that had been lightly coated with petroleum jelly. The blood was then allowed to clot on ice for 1–2 hours and was subsequently centrifuged at 1000xg for 10 minutes at 10°C. Serum was then removed and stored at −20°C until antibody assays were performed.
2.11. Antibody assays
Neutralizing antibody assays were performed on serum from mice vaccinated by scarification with 1 × 106 pfu E3L gene mutants of vaccinia virus. Blood was harvested 14 days after vaccination. Serial dilutions of serum samples were made. Each dilution was mixed with a known quantity of wtVACV(WR) vaccinia virus and incubated at 37 C for 1 hour. The mixture was then used to infect RK-13 cell titer plates. The neutralizing antibody titer is the inverse of the serum dilution at which a 50% plaque reduction occurred as calculated by linear regression.
For comet inhibition assays, serum from vaccinated animals was diluted as indicated and used to treat monolayers of BSC-40 cells that had been infected with approximately 30 pfu of IHD-J strain of VACV. Plaques were stained after 44 hours of infection.
2.12. Harvest and processing of splenocytes
Spleens were removed from chemically euthanized animals and splenocytes were dissociated into media using a 70µm cell strainer. Red blood cells were removed from the splenocytes by lysis with an ammonium chloride lysing reagent from BD Pharmingen. Media used for splenocyte processing was RPMI 1640 media (Cellgro #10-040-CV) supplemented with 10%low IgG fetal bovine serum (Hyclone #SH30151) and penicillin/streptomycin antibiotic. Splenocytes were then resuspended in RPMI-1640 modified media (ATCC, Vitacell #30-2001) supplemented with 10% low IgG fetal bovine serum (Hyclone #SH30151), 0.05mM 2-mercaptoethanol and penicillin/streptomycin antibiotic. Splenocytes for all immunological assays were used in this media for consistency. Splenocytes were adjusted to a concentration of 8 ×105 cells/100µl prior to use.
2.13. Secreted Cytokine Assays
Mice were infected by scarification with 1 × 106 pfu VACV E3L mutants or wtVACV or mock-infected with 1 mM Tris pH 8.8. 8 days post-infection spleens were harvested from the mice, processed to remove red blood cells and adjusted to the appropriate concentration as indicated above. A20 cells (ATCC #TIB-208) were used as antigen presenting cells. These cells were maintained and used in RPMI-1640 modified media (ATCC, Vitacell #30-2001) supplemented with 10% low IgG fetal bovine serum (Hyclone #SH30151), 0.05mM 2-mercaptoethanol and penicillin/streptomycin antibiotic. These cells were infected with VACV WT (WR strain) at an MOI of 1 for 14 hours. The cells were then washed 3 times in media to remove free virus and used as antigen presenting cells. 100µl of splenocytes from each animal (8 × 105 cells) were placed in wells of a 96 well plate. 3 × 105 vaccinia virus antigen presenting cells (infected A20 cells) in 100µl of media were then placed in the wells with the splenocytes and incubated at 37°C, 5% CO2 for 36 hours. The cells and media were then removed from the wells and centrifuged to pellet the cells. The supernatants were used in the secreted cytokines assay.
The secreted cytokines assay was done using a Cytometric Bead Array from BD Pharmingen (#551287). This assay detects mouse Th1/ Th2 cytokines and included analysis for IL-2, IL-4, IL-5, IFN-γ, and TNF-α. The assay was performed as outlined in the instruction manual included with the kit. Samples were analyzed using a flow cytometer and BD CBA software.
2.14. Intracellular Cytokine Staining Assays
Mice were infected by scarification with 1 × 106 pfu VACV E3L mutants or wtVACV or mock-infected with 1 mM Tris pH 8.8. 8 days post-infection spleens were harvested from the mice, processed to remove red blood cells and adjusted to the appropriate concentration as indicated above. A20 cells (ATCC #TIB-208) were used as antigen presenting cells. These cells were maintained and used in RPMI-1640 modified media (ATCC, Vitacell #30-2001) supplemented with 10% low IgG fetal bovine serum (Hyclone #SH30151), 0.05mM 2-mercaptoethanol and penicillin/streptomycin antibiotic. The A20 cells were infected with wtVACV(WR) at an MOI of 1 for 8 hours. The cells were then washed 3 times in media to remove free virus and used as antigen presenting cells. 100µl of splenocytes from each animal (8 × 105 cells) were placed in wells of a 96 well plate. 3 × 105 vaccinia virus antigen presenting cells (infected A20 cells) in 100µl of media were then placed in the wells with the splenocytes and incubated at 37°C, 5% CO2 for 20 hours. 20µg/ml Brefeldin A (Sigma #B7651) was then added to the wells. Cells were incubated for 4 hours. After this incubation, plates were centrifuged at 500 × g for 4 minutes and supernatants were removed by aspiration. The cells were resuspended in 50µl staining buffer (1 X PBS, 1% low IgG serum, and 0.1% sodium azide). The cells were stained for cell surface markers using anti-CD4 (BD Pharmingen #553047, FITC-conjugated) and anti-CD8 (BD Pharmingen #553034, CyChrome-conjugated) antibodies added for 20 minutes at 4°C. The cells were then washed three times with staining buffer and resuspended in 200µl fixing solution (4% paraformaldehyde in 1 X PBS, adjusted to pH 7.2). Fixing was performed for 15 minutes at room temperature followed by two washes with staining buffer. The cells were resuspended in 200µl staining buffer and stored overnight at 4°C. The next day the plates were centrifuged, the staining buffer was removed, and the cells were resuspended in 50µl permeabilization buffer (1XPBS, 1% low IgG serum, 0.1% sodium azide, and 0.1% saponin (Sigma #S-4521). PE conjugated anti-IFN-γ antibody (eBiosciences #12-7311) was then added as the intracellular cytokine stain. The antibody was added to each well at a concentration of 0.335 µg/50ul permeabilization buffer. The plate was incubated for 30 minutes at 4°C and then washed three times in permeabilization buffer. Cells were resuspended in 400µl of staining buffer and analyzed by flow cytometry.
2.15. Statistical analysis
All statistical analyses were performed using Sigmastat 3.5© (Systat Software, Inc). Details of each analysis are indicated in individual figure legends and in the body of the text.
3. Results
3.1. Virulence of E3L mutant VACVs in SCID mice after scarification
Scarification with vaccinia virus is the commonly used method of smallpox vaccination. However, administration of vaccinia virus to immune compromised individuals by scarification can lead to severe complications. To evaluate attenuation of viruses containing mutations in E3L, we assayed for virulence after delivery by scarification in SCID mice. Most of the studies performed in this manuscript were done using the mouse adapted WR strain of VACV. Infection of mice with this mouse-adapted virus may be more relevant to human disease than infection of mice with a human VACV strain (i.e., Copenhagen or NYCBH). Nonetheless, all of the work with the WR strain was confirmed by infection of mice with the Copenhagen and NYCBH strains of VACV. The virus strains tested (Figure 1A) included a mutant virus with a full deletion of the E3L gene (VACVΔE3L) and a mutant with the Ambystoma tigrinum virus eIF2α homologue gene inserted in place of E3L (VACVΔE3L::vIF2αH; for simplicity sake we will call this virus VACV-vIF2αH) (Talasela et al., in preparation). This ATV gene is a homologue of the cellular eIF2α gene [28] and of the vaccinia virus interferon resistance gene K3L [29]. Both mutants were sensitive to the effects of interferon in cell culture, but while VACVΔE3L has a narrow host range [27], VACV-vIF2αH has a wider host range, at least in cell cultures (Talasa et al., in preparation). Also tested were VACV E3L mutants with deletions of the N terminus. This region contains a consensus Z-nucleic acid (Z-NA) binding domain [21]. Although deletions of N terminal residues of the E3L have little effect on the cell culture phenotype of VACV, they do affect pathogenicity in mice [27, 30]. Both VACVE3LΔ83N and VACVE3LΔ54N contain deletion of key residues required for binding Z-form nucleic acid, but VACVE3LΔ54N also produces an unstable protein that turns over rapidly within the cell (data not shown). The C terminus of the E3L protein contains a dsRNA binding domain that is associated with interferon resistance, host range, and pathogenesis [31, 32]. A VACV E3L mutant with the 7 C terminal amino acids removed was also tested as part of these studies. This E3L protein binds dsRNA with lower affinity than the wtE3L protein [26].
Fig. 1. Infection of SCID mice by scarification.
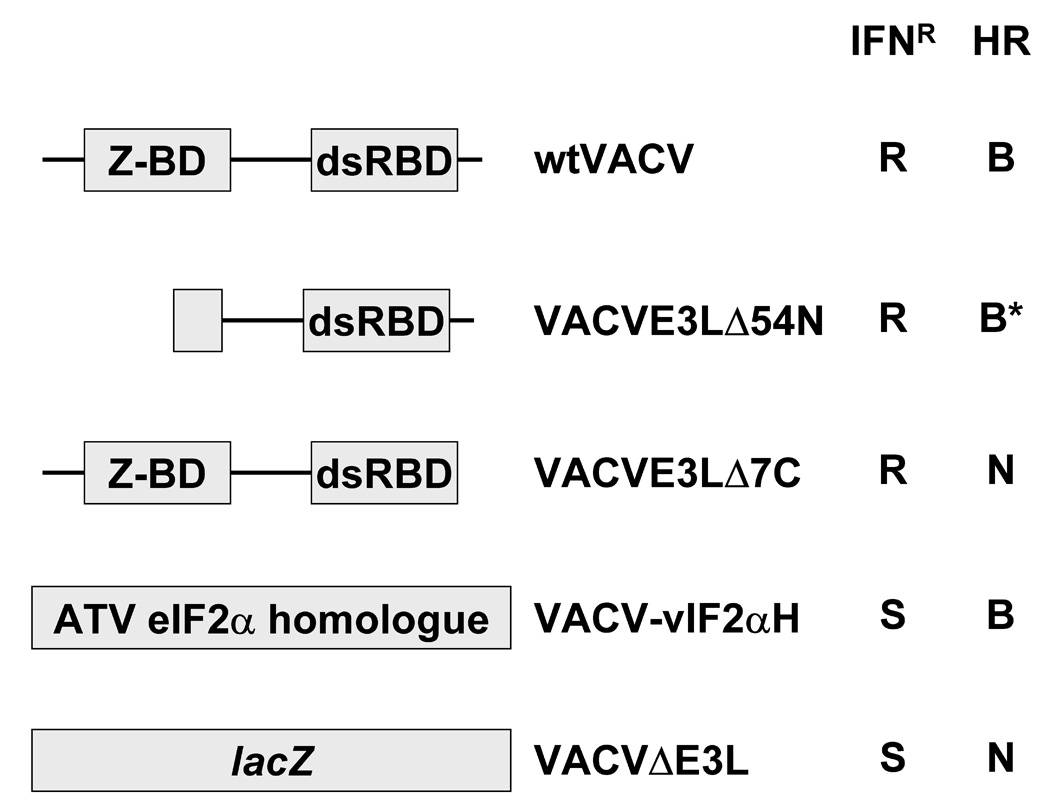
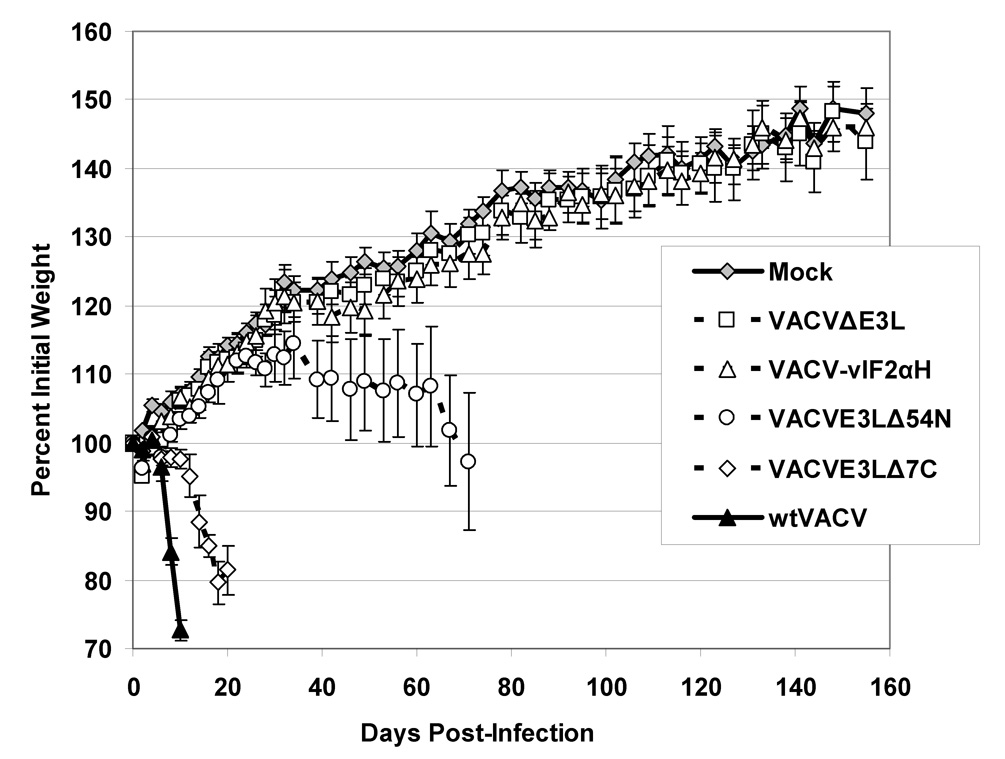
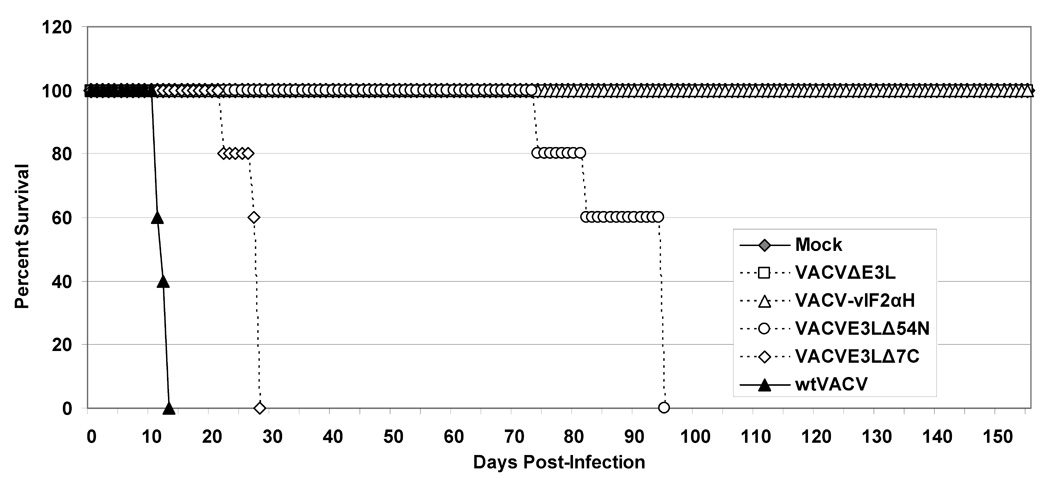
(A) Z-DNA binding domains (Z-DBD) and double stranded RNA binding domains (dsRBD) of E3L proteinare shown. Viruses deleted of the entire E3L gene had insertions of either the ATVeIF2α homologue gene (2aH) or the marker gene lacZ. Interferon resistance (IFNR) and host range (HR) of the viruses in cell culture systems are shown. R = interferon resistant, S = interferon sensitive, B = broad host range, and N = narrow host range. *While VAVCE3LΔ54N forms plaques in HeLa cells, it does so at a lower efficiency than wtVACV. (B and C) Infection of SCID mice. Groups of 5 female SCID mice were infected by tail scarification with 106pfu of either wtVACV or the indicated VACV E3L mutants and monitored for weight loss (B) and mortality (C) for 5 months after infection. Lines ending prematurely in panel B indicate mortality. Error bars indicate the standard error of the mean. Two-Way Repeated Measure ANOVA tests were performed on the weight loss data to determine differences between the groups. Survival analysis was performed using the log rank test. Outcomes were defined as either “censor” (mice which were still alive after 160 days) or “failure” (mice which either died or were euthanized, from causes related from the scarification).
Figure 1B and C shows the results of infecting SCID mice by scarification with either wtVACV or the E3L mutants described above. Figure 1B shows the percent weight change data after tail scarification infection of SCID mice over a 5-month period and Figure 1C shows the percent survival of the scarified mice. wtVACV infected SCID mice lost weight and died rapidly, with an average time to death of 12.0 days. VACVE3LΔ7C infected mice also lost weight, although somewhat more slowly than wtVACV infected mice, and died, with an average time to death of 26.6 days. A Two-Way Repeated Measure ANOVA was performed on the weight data through day 10 when all mice were still alive. Pairwise comparisons of groups at each time point (Student-Newman-Keuls method) showed that at day 4 post scarification, the group infected with wtVACV was different from all the others (P <0.05). By day 10, both wtVACV and VACVE3LΔ7C groups were different from all other groups (P <0.05). Although in short-term experiments (data not shown) VACVE3LΔ54N did not cause any weight loss or mortality in SCID mice, the data in Figure 1B and C shows that VACVE3LΔ54N caused illness and death over longer time periods with an average time to death of 88.2 days. A second Two-Way Repeated Measure ANOVA was performed, excluding the wt-VACV and VACVE3LΔ7C groups through day 78. Pairwise comparisons of groups at each time point showed that by day 46, VVE3LΔ54N was different from the mock, VVΔE3L and VACV-vIF2αH groups (P <0.05). Mice infected with VACVE3LΔ7C and VACVE3LΔ54N developed widespread pocks and scabbing on their paws and tails prior to their deaths. At day 28 for VACVE3LΔ7C infected mice and day 95 for VACVE3LΔ54N infected mice, the remaining 3 mice in each cage were euthanized for humane reasons. VACVΔE3L and VACV-vIF2αH caused no significant weight loss (Figure 1B, as determined by a Two-Way Repeated measure ANOVA performed on only the mock, VACVΔE3L, and VACV-vIF2αH infected groups (P=0.69)), or mortality out to 5 months after infection (Figure 1C). However, several mice in these two cages had mild recurrent pocks in the area of scarification with pocks in the VACV-vIF2αH infected mice clearing completely at about 4 months after infection and 2 out of the 5 VACVΔE3L infected mice still having single pocks on their tails at 5 months after infection. These results demonstrate that E3L deletion mutants caused varying degrees of pathogenicity in an immune deficient animal model with several mutants causing almost no disease beyond a local reaction to the virus.
3.2. Virus titers in ovary tissue after intraperitoneal infection with E3L mutant VACVs
As an additional measure of pathogenesis, the ability of the VACV E3L mutants to replicate in the ovaries of intraperitoneally infected mice was examined. Figure 2 shows the virus concentrations recovered from the ovaries of BALB/c mice. Each mouse was infected with 106pfu of the indicated virus. At 5 days PI, both ovaries were harvested from each mouse, homogenized together, and virus was titered in RK-13 cells. wtVACV infection resulted in the recovery of approximately 109 pfu/gram of tissue, or 4 logs higher titers than any of the VACV E3L mutants. VACVE3LΔ7C and VACVE3LΔ54N both yielded average concentrations of between 104 and 105 pfu/gram of ovary tissue. Interestingly, no virus was detected in 1 out of 4 VACVE3LΔ7C infected mice and 2 out of 4 VACVE3LΔ54N infected mice. Only one VACV-vIF2ΔH infected mouse had a measurable level of virus in its ovaries (103 pfu/gram of tissue) and no virus was recovered from any of the VACVΔE3L infected mice. Pairwise comparisons of a One-Way ANOVA of ranks (Student-Newman-Keuls Method) a One-Way ANOVA of ranks (Student-Newman-Keuls Method) showed that all the groups were different from the wtVACV group (P<0.05).
Fig. 2. Virus titers recovered from the ovaries of mice infected by intraperitoneal injection.
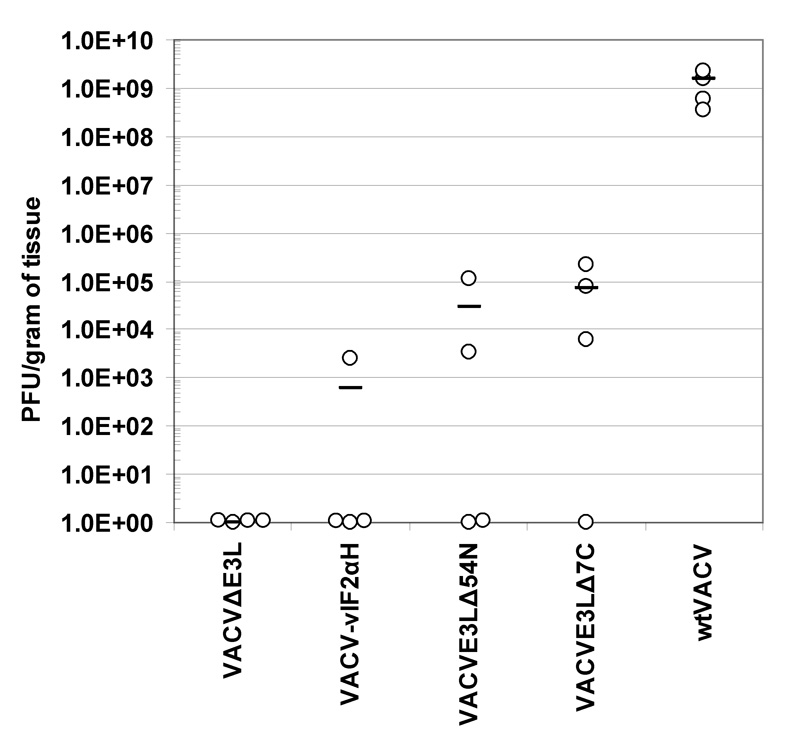
Four female BALB/c mice per group were infected IP with 106 pfu of wtVACV or VACV E3L mutants. Ovaries were removed 5 days post-infection. Both ovaries were harvested and combined for titering from each mouse. A one-way ANOVA on ranks followed by pairwise comparisons, using the Student-Newman-Keuls Method was used to analyze the data.
3.3. Pock severity and viral titers from tail tissue scarified using E3L mutant VACVs
Infection with wtVACV infection caused severe pocks on the scarified tails of BALB/c mice, with significant swelling and tissue destruction as the pocks developed and healed (summarized in Table 1). Pocks induced by scarification with VACVE3LΔ7C were also destructive to tissue although the damage was not as severe as that caused by wtVACV (Table 1). Pocks induced by VACVE3LΔ54N were much milder than the pocks induced by either wtVACV or VACVE3LΔ7C and while there was significant scabbing, the pocks did not generally result in any tail swelling or tissue destruction and healed rapidly. VACVΔE3L and VACV-vIF2αH both caused pocks that were barely visible and mostly consisted of a rough, flaky appearance of the infected skin. These pocks did not cause any tissue destruction and healed quickly. Additionally, as seen in Table 1, vaccination takes (ie. the formation of a pock) were extremely reliable with all of the vaccinia virus E3L mutants inducing visible lesions at a dose of 106 pfu and most at a dose as low as 105 pfu, similar to wtVACV.
Table 1. Summary of the results of scarification vaccinations of BALB/c mice.
The number of “takes”, ie pocks or skin disturbances, are shown by virus construct and virus dose (104, 105, and 106 pfu). Increased plus marks indicate increased severity of tissue damage associated with takes.
| Pock Formation | ||||
|---|---|---|---|---|
| Virus | 104 | 105 | 106 | Local Symptoms |
| wtVACV | 0/5 | 5/5 | 5/5 | ++++ |
| VACVE3LΔ7C | 0/5 | 5/5 | 5/5 | +++ |
| VACVE3LΔ54N | 0/5 | 5/5 | 5/5 | ++ |
| VACV-vIF2αH | 0/5 | 5/5 | 5/5 | + |
| VACVΔE3L | 0/5 | 3/5 | 5/5 | + |
Tissue replication in the skin of vaccinated animals was also monitored. Skin from the scarified tails of BALB/c mice was removed 5 days post-infection, homogenized and titered on RK-13 cells. The results are shown in Figure 4. Virus concentrations from the skin of mice scarified with wtVACV and VACVE3LΔ7C were in excess of 109 pfu/gram of tissue. VACVE3LΔ54N tissue concentrations were approximately one log lower at 108 pfu/gram of tissue. VACVΔE3L and VACV-vIF2αH yielded very similar tissue virus concentrations, averaging between 106 and 107 pfu/gram of tissue, which is approximately 3 logs less than that seen in wtVACV infections. Pairwise comparisons between groups showed that all the groups except the VACVE3LΔ7C group were different from the wtVACV group. In addition, the both the VACVΔE3L and the VACV-vIF2αH were different from the VACVE3LΔ4N group (P<0.05). Thus, there is a good correlation between growth of virus on the skin and severity of skin lesions.
Fig. 4. Weight change after challenge of BALB/c mice vaccinated by scarification.
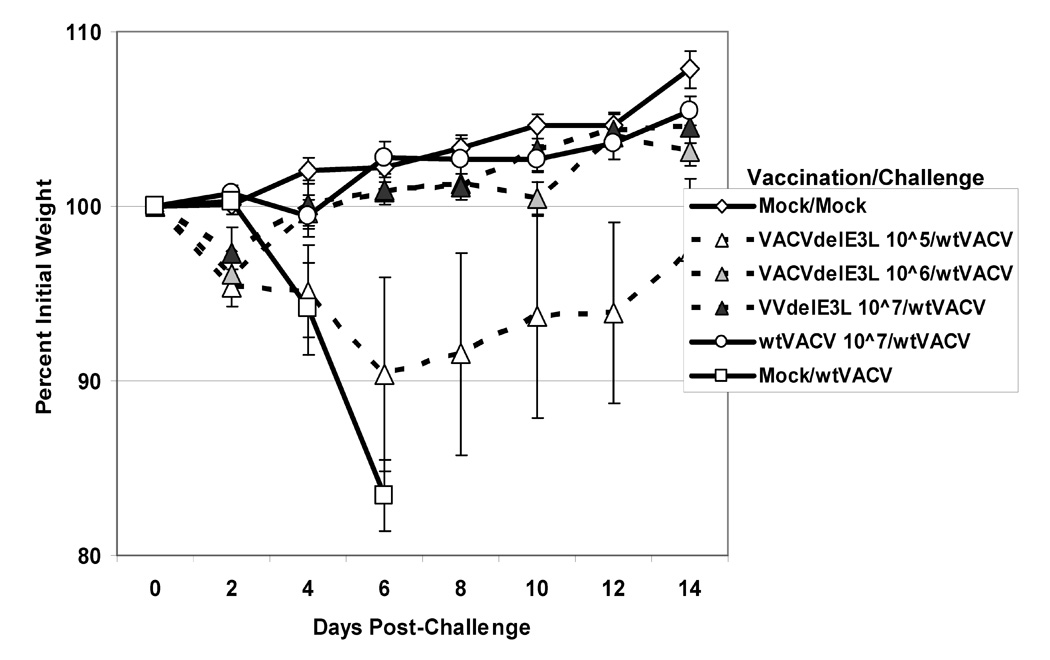
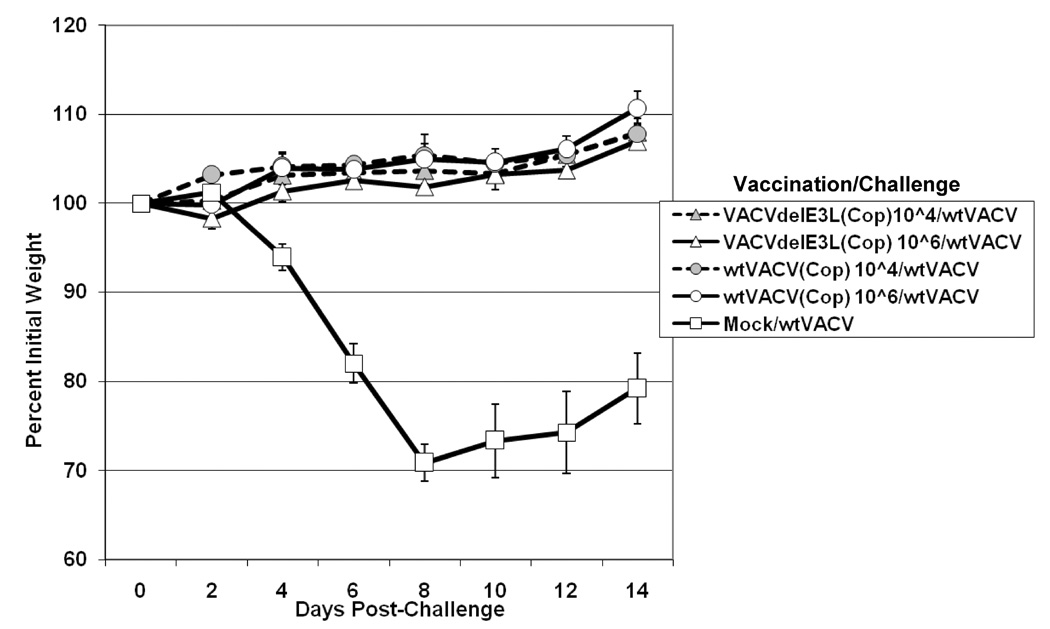
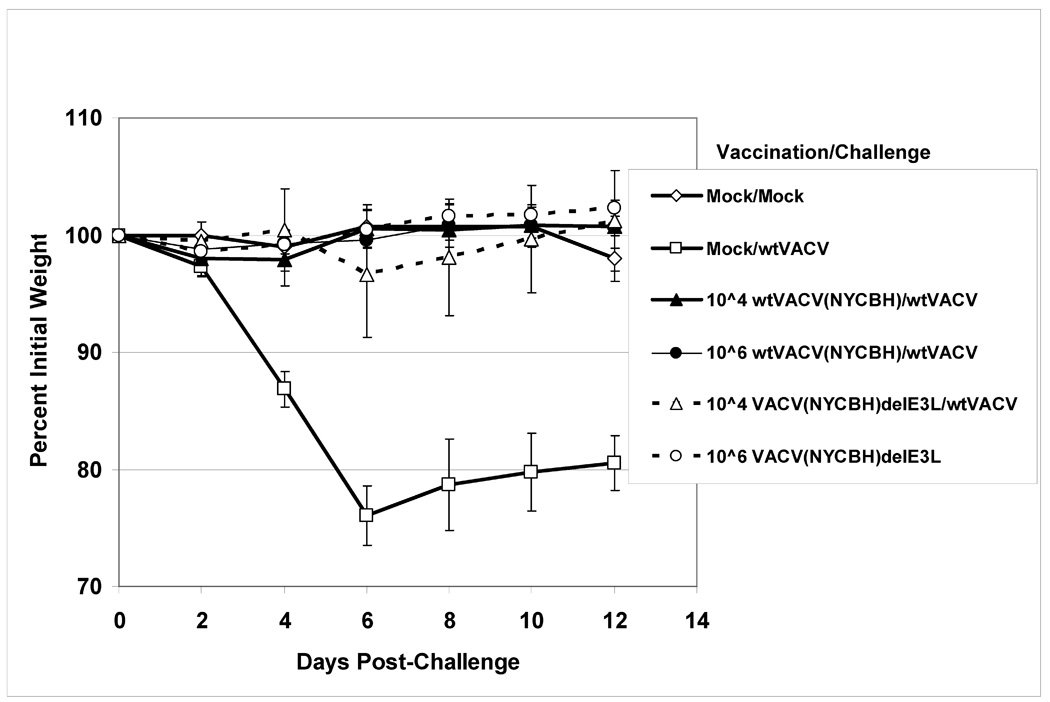
Groups of 5 Balb/C female mice were scarified with varying doses of either VACVΔE3L or wtVACV in either a WR strain (panel A), Copenhagen strain (panel B), or NYCBH strain (panel C). One month after vaccination the mice were challenged IN with 106 pfu of wtVACV(WR). After challenge, mice were monitored for weight loss every other day and monitored every day for signs of illness. Mock/Mock mice were mock vaccinated and mock challenged. Mock/WT mice were mock vaccinated and challenged with 106 pfu of wtVACV. Lines ending prematurely indicate mortality. A Two-Way Repeated Measure ANOVA was performed over the entire length of the study to determine which groups were different from one another, and pairwise comparisons were performed (Student-Newman-Keuls Method) to compare each group inidividually with every other group. For WR strain vaccinated mice comparisons were repeated through day 6, to allow for pairwise comparisons of groups prior to any animal deaths.
3.4. Scarification vaccination in BALB/c mice
To determine if these highly attenuated viruses could induce a protective immune response against wtVACV, animals were vaccinated by scarification with 105, 106, and 107 pfu of mutant VACVs or wtVACV as a positive control. With the exception of one mouse vaccinated with 105 pfu of VACVΔE3L, all mice in the experiment developed pocks after vaccination. Animals were challenged 30 days post-vaccination with 106 pfu of wtVACV. Figure 4A shows weight loss of animals vaccinated with VACVΔE3L or wtVACV and then challenged with wtVACV. Except for the VACVΔE3L 105 cage, which contained one mouse that didn’t develop a pock during vaccination, all other cages of mice were substantially protected from weight loss after challenge (Figure 6A and data not shown). A Two-Way Repeated Measure ANOVA was performed over the entire length of the study, with pairwise comparisons to compare each group with every other group. Only the mock vaccinated/VACV challenged group, and the group vaccinated with 105 pfu of VVΔE3L were different from the mock vaccinated/mock challenged animals. Cages of mice vaccinated with 106 pfu of VACVΔE3L showed minor weight differences from mock vaccinated/mock challenged mice on days 2, 10, and 14 after challenge. In the cage of mice vaccinated with 105 pfu of VACVΔE3L one mouse lost significant weight, while the other four mice were fully protected from disease. For all of the other mutant viruses, all of the animals developed pocks at the three doses tested and all of the animals were protected from weight loss (data not shown). Thus, development of a pock, no matter how mild, correlated with protection from disease upon challenge with wtVACV. In contrast, all of the mock vaccinated animals lost 12–23% of their initial body weights by day 6 and one animal died. To determine if protection was durable, these experiments were repeated with a 3-month interval between vaccination and challenge, with the results again demonstrating effective vaccination as measured by a failure of challenge with wtVACV to induce weight loss (data not shown).
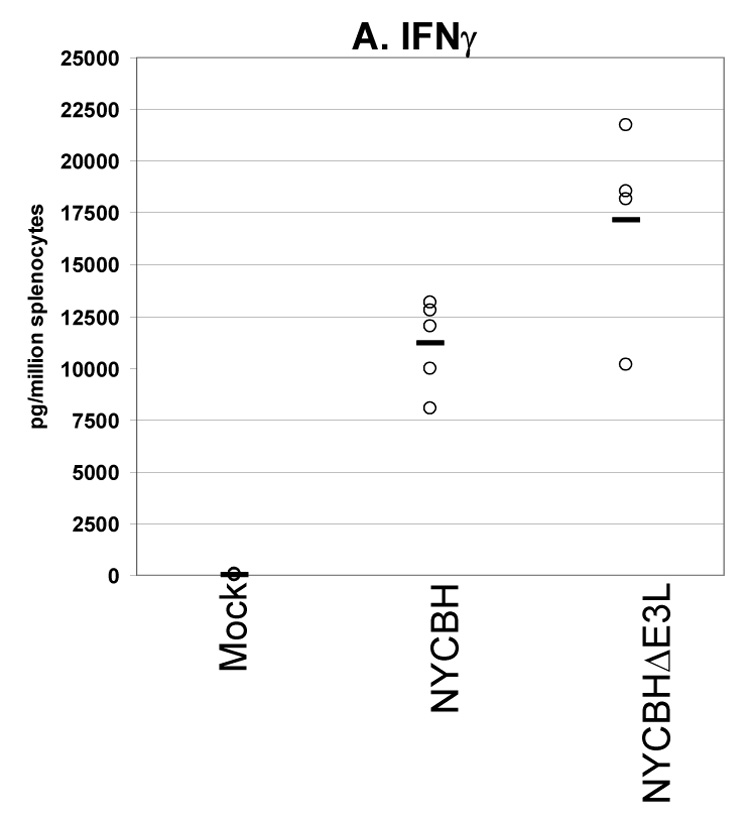
Fig. 6. Cytokines secreted from splenocytes from VACV(NYCBH) vaccinated mice.
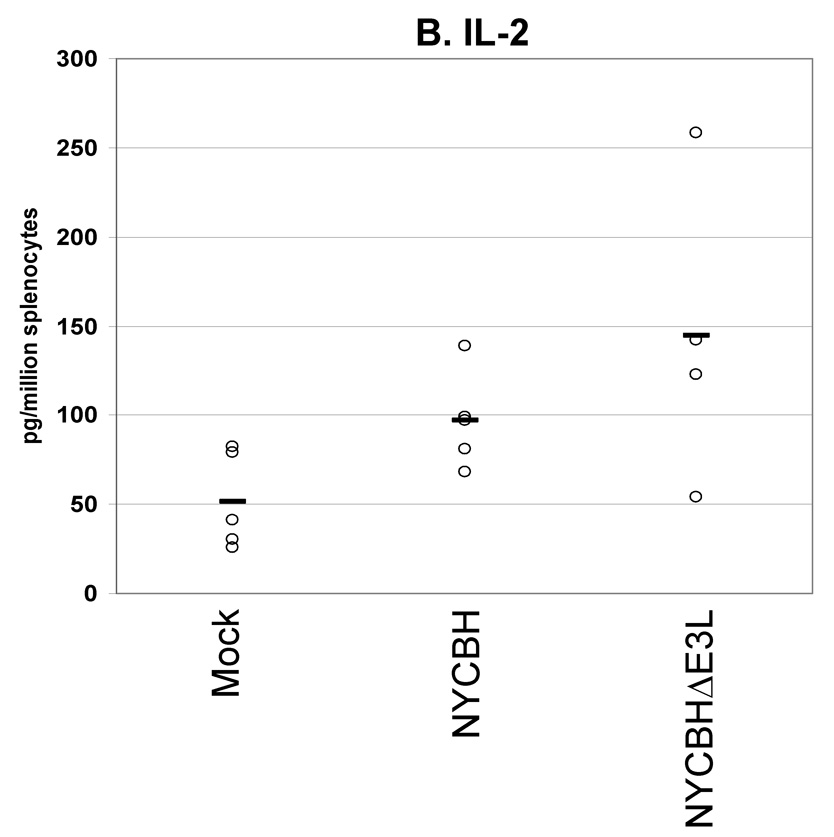
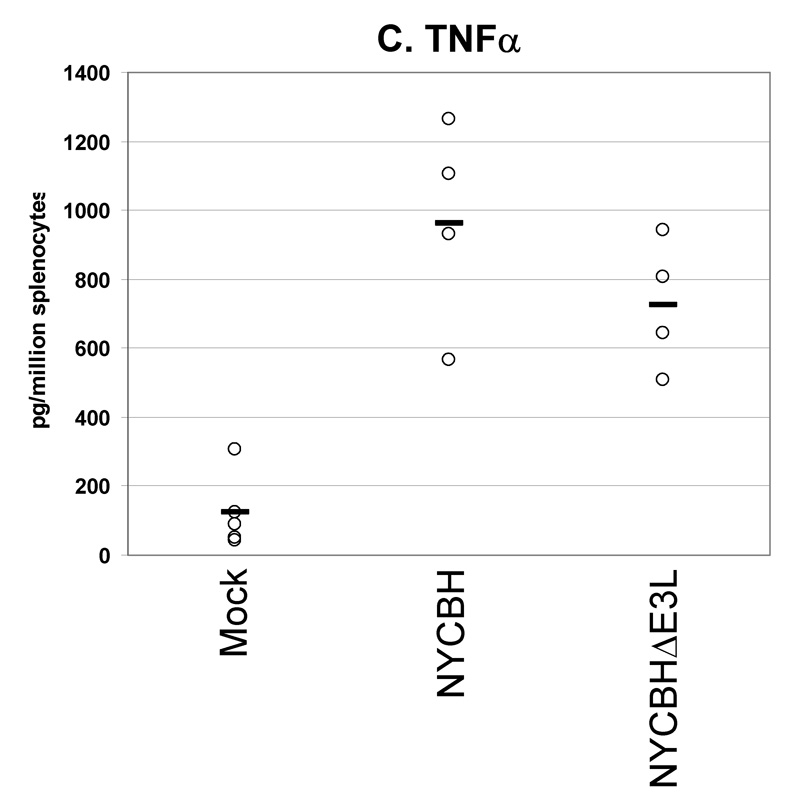
Groups of 4 female BALB/c mice were mock infected or were vaccinated by scarification with 1×106 pfu VACVΔE3L(NYCBH) or wtVACV(NYCBH) as indicated. Splenocytes were harvested 8 days post-infection and exposed to wtVACV infected A20 antigen presenting cells for 36 hours. The cytokines (A) IFN-γ, (B) IL-2, and (C) TNF-α were measured in the cell culture supernatants. Symbols represent individual mice used in each experiment. Bars indicate the average value for each group of mice. A one-way ANOVA on ranks followed by pairwise comparisons (Student-Newman-Keuls Method) was performed on all of the data.
The WR strain of vaccinia virus is a mouse-adapted, neurovirulent strain and is not appropriate for use in humans. To determine if E3L mutations in strains appropriate for use in humans had the potential for a safe attenuated vaccine, we constructed similar E3L mutations in a Copenhagen and NYCBH background [1]. Both wtVACV(COP) and wtVACV(NYCBH) are highly attenuated in mouse models, compared to wtVACV(WR). wtVACV(COP) is attenuated by at least 3 logs compared to wtVACV(WR) and wtVACV(NYCBH) is attenuated a further 4 logs [22]. Deletion of E3L further attenuated VACV(COP) by over 6 logs, and further attenuated VACV(NYCBH) by approximately 3 logs [22].
To test the vaccinating efficacy of E3L mutants in these background strains more appropriate for human use, mice were vaccinated by scarification with 104 or 106 pfu of wtVACV(COP), wtVACV(NYCBH), or with VACVΔE3L in each background. One month after vaccination the mice were challenged intranasally with 106 pfu of wtVACV(WR). Figure 4B shows percent weight loss data from mice vaccinated with VACVΔE3L(COP) and wtVACV(COP) and challenged one month later, and Figure 4C shows similar experiments for animals vaccinated with NYCBH viruses. Vaccination with any of the four viruses, at 104 or at 106 pfu, fully protected against disease. Thus, in a virus background appropriate for use in humans, VACVΔE3L protects mice from weight loss after challenge with wtVACV.
3.5. Immune responses to vaccination with E3L mutant VACVs
A secreted cytokines assay was used to quantify the levels and types of cytokines being produced by splenocytes from mice vaccinated with VACV E3L mutants or wtVACV. VACV-infected antigen-presenting cells were used to restimulate splenocytes harvested from mice 8 days after vaccination. This assay measured the cytokines released into cell culture media after 36 hours of exposure to antigen-presenting cells (Figures 5, A–E). Splenocytes from mice vaccinated with VACV E3L mutants and wtVACV produced primarily Th1 type immune responses dominated by the production of IFN-γ, TNF-α, and IL-2. The production of Th2 type cytokines, IL-4 and IL-5, was very low.
Fig. 5. Cytokines secreted from splenocytes from VACV(WR) vaccinated mice.
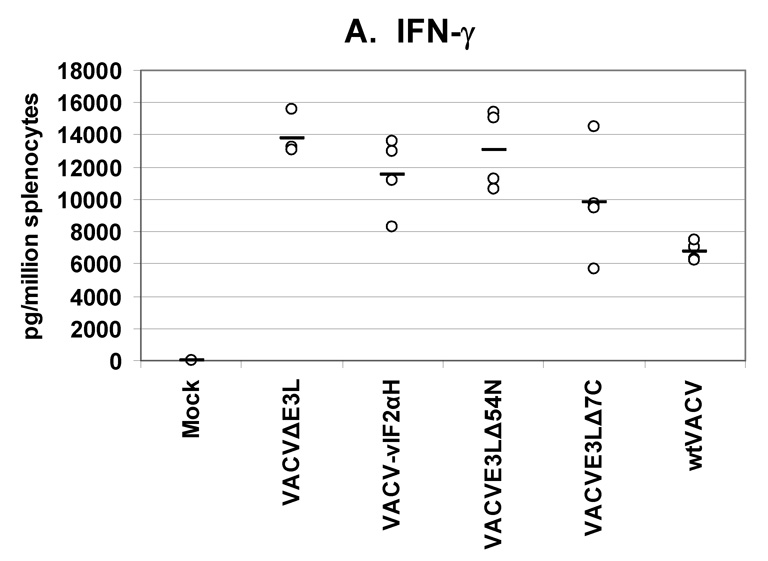
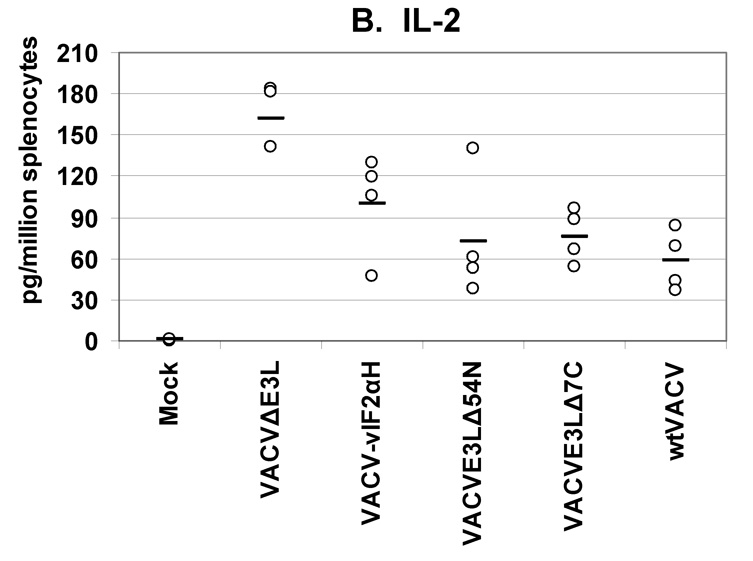
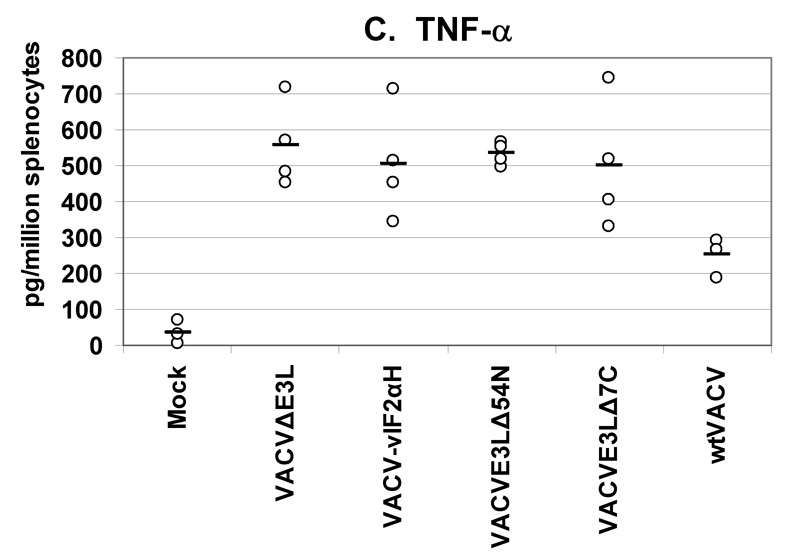
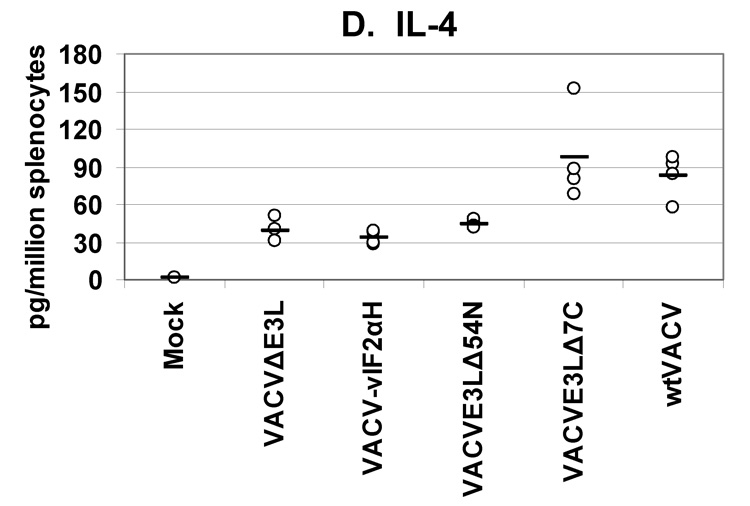
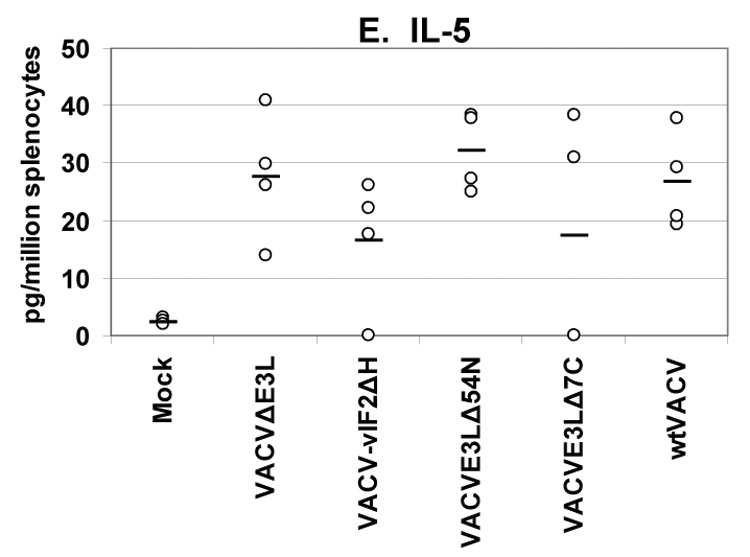
Groups of 4 female BALB/c mice were either mock infected or were infected by scarification with 1×106 pfu VACV E3L mutants or with wtVACV as indicated. Splenocytes were harvested 8 days post-infection and exposed to wtVACV infected A20 antigen presenting cells for 36 hours. The cytokines (A) IFN-γ, (B) IL-2, (C) TNF-α, (D) IL-4, and (E) IL-5 were measured in the cell culture supernatants. Symbols represent individual mice used in each experiment. Bars indicate the average value for each group of mice. A one-way ANOVA on ranks followed by pairwise comparisons (Student-Newman-Keuls Method) was performed on all of the data.
Splenocytes from mice vaccinated with the VACV E3L mutants produced higher IFN-γ than splenocytes from mice vaccinated with wtVACV or mock vaccinated animals (P<0.05). VACVΔE3L produced the highest average level, approximately 14,000 pg/million splenocytes, compared to an average of just over 6000 pg/million cells for wtVACV (Figure 5A). Splenocytes from animals vaccinated with VACVΔE3L produced higher average levels of IL-2 (150 pg/million splenocytes) than splenocytes from wtVACV (60 pg/million cells) and the other VACV E3L mutants (P<0.05, Figure 5B). IL-2 levels for VACV-vIF2αH vaccinated mice were elevated but were not statistically different from the levels seen in wtVACV vaccinated mice. TNF-α levels were similar for all of the VACV E3L mutants, averaging 500 pg/million cells, while splenocytes from mice vaccinated with wtVACV produced 250 pg/million cells on average (Figure 5C). IL-4 and IL-5 levels were very low overall. IL-4 levels produced by the splenocytes of mice vaccinated with VACVΔE3L, VACVE3LΔ54N and VACV-vIF2αH were lower than levels seen after wtVACV vaccination (Figure 5D) (P<0.05). IL-5 levels from the vaccinated mice (Figure 5E) showed no particular pattern.
We also performed a secreted cytokines assay to examine the production of cytokines by splenocytes from BALB/c mice vaccinated with wtVACV(NYCBH) and VACVΔE3L(NYCBH) (Figure 6, A–C). 8 days after vaccination splenocytes were harvested and restimulated with VACV-infected antigen presenting cells. Consistent with previous experiments using the wtVACV(WR) strain, vaccination with VACVΔE3L(NYCBH) produced higher levels of IFN-γ than the wt strain. Comparable levels of IL-2 and TNF-α were secreted from splenocytes of animals vaccinated with either virus.
To determine the percent of CD4+ and CD8+ splenocytes secreting IFNγ in response to vaccination, intracellular cytokine staining was performed. Mice were vaccinated by scarification with VACV E3L mutants or wtVACV and splenocytes were harvested 8 days later. Vaccinia virus infected antigen-presenting cells were used to restimulate splenocytes. After 20 hours of restimulation brefeldin A was then added for an additional 4 hours to block cytokine secretion. Cells were then surface stained for CD4 and CD8, permeabilized, and stained intracellularly for IFNγ. There were no marked differences between levels of IFNγ positive CD4+ splenocytes from mice vaccinated with VACV E3L mutants or wtVACV (P>0.05, data not shown). For IFNγ positive CD8+ splenocytes, only splenocytes from animals vaccinated with VVE3LΔ54N were markedly lower than splenocytes from wtVACV vaccinated animals (P<0.05, data not shown). CD4+ cells averaged about 5 percent IFNγ positive and CD8+ cells averaged around 10 percent IFNγ positive, after subtracting background from mock vaccinated mice.
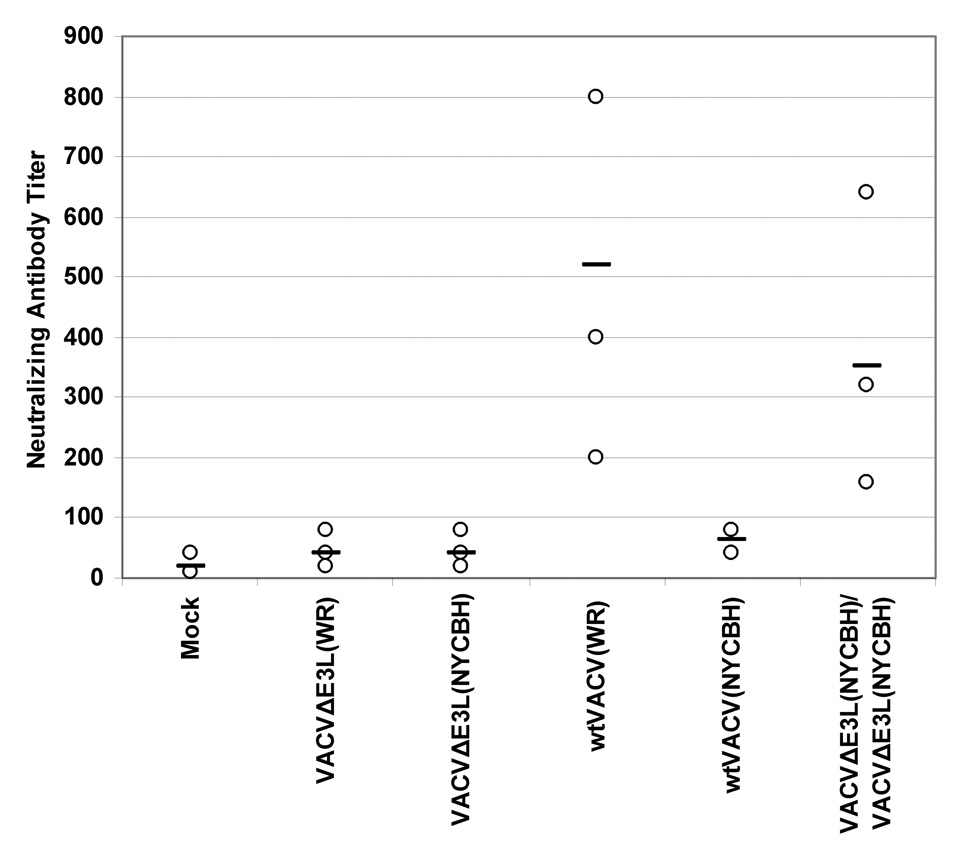
Since high anti-vaccinia antibody titers have in the past been considered a sign of successful vaccination [1], we compared the neutralizing antibody titers elicited by each of the VACV E3L mutants to antibody titers after vaccination with wtVACV. Blood was drawn 14 days after the mice were scarified and the serum was assayed for the presence of neutralizing antibodies (Figure 7A). Surprisingly, blood from mice vaccinated with VACV E3L mutants showed low concentrations of neutralizing antibodies against wtVACV. While vaccination with wtVACV produced an average neutralizing antibody titer in excess of 500, VACVE3LΔ7C vaccinated mice produced less than half that amount. Mice vaccinated with VACVΔE3L,VACV-vIF2αH, and VACVE3LΔ54N all had neutralizing antibody titers below 200, significantly less than that generated by vaccination with wtVACV (P<0.05). As many vaccines require boosting with additional doses to be effective, we tested the effect of a single boost 1 month after the initial vaccination on subsequent antibody production when using VACVΔE3L as the vaccinating agent. Mice vaccinated with VACVΔE3L and then boosted with VACVΔE3L developed very high antibody titers, in excess of 1000, a level which is twice as high as that induced by a single dose of wtVACV (Fig. 7A).
Fig. 7. Antibody titers in mice vaccinated by scarification.
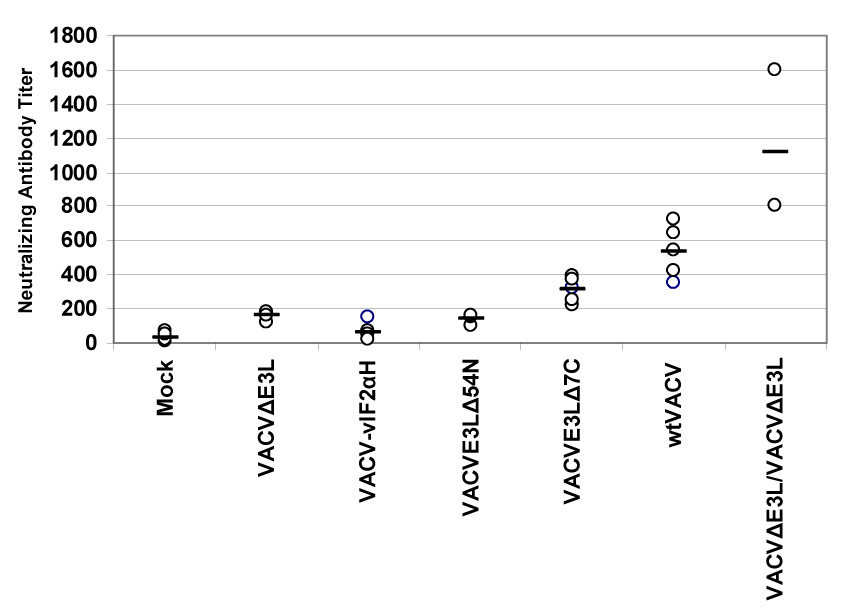
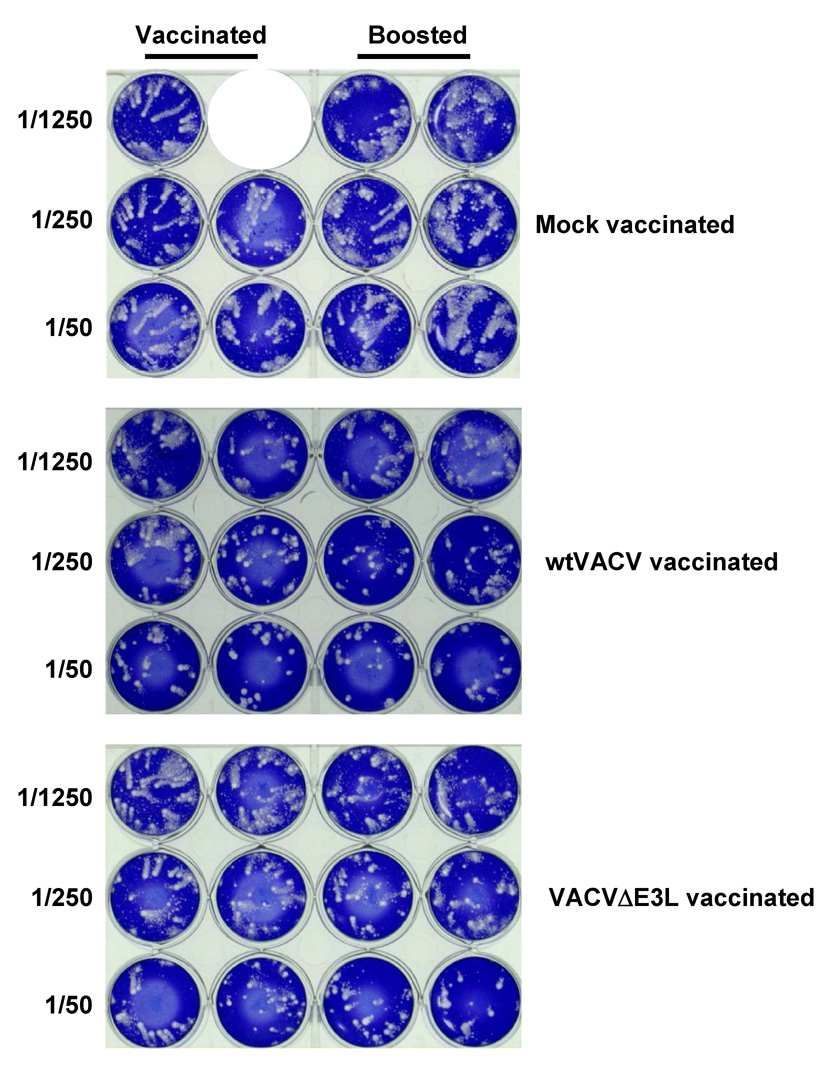
Groups of female BALB/c mice were vaccinated by scarification with 1×106 pfu of VACV E3L mutants or wtVACV in a WR strain background (panels A and B), or with VACVΔE3L or wtVACV in a NYCBH background (panel C), as indicated and blood was harvested 14 days later. Also shown is data from mice vaccinated with VACVΔE3L then boosted with VACVΔE3L 1 month later. Neutralizing antibody titers in serum (panels A and C) were determined by plaque reduction assays on RK-13 cells. The neutralizing antibody titer is the inverse of the serum dilution at which a 50% plaque reduction occurred. Comet inhibition assays were also performed (panel B), as described in Materials and Methods.
Antiserum that can inhibit extra-cellular virus (EV) release from cells has been suggested to be important for protection against pathogenic poxviruses [33]. We therefore also assayed for the ability of serum from vaccinated mice to inhibit comet formation, as a measure of inhibition of release of EV [33]. As shown in Figure 7B, serum from one mouse vaccinated with a single dose of wtVACV substantially inhibited comet formation at a dilution of 1/250, while serum from the other mouse only inhibited comet formation at a dilution of 1/50. Serum from one mouse vaccinated with a single does of VAVCΔE3L failed to inhibit comet formation, while serum from the second mouse inhibited at a dilution of 1/50. Boosting increased comet formation inhibitory activity in both wtVACV and VACVΔE3L vaccinated mice. Thus, as with plaque neutralizing activity, a boost was necessary to induce substantial comet formation inhibitory activity in mice vaccinated with VACVΔE3L.
We also evaluated neutralizing antibody production in mice vaccinated with VACVΔE3L(NYCBH) (Fig. 7C). For comparison, mice were also vaccinated with wtVACV(WR). As in previous experiments mice vaccinated with virus strains deleted of the E3L gene failed to produce significant levels of antibodies upon primary vaccination. However, we again found that by performing a single boost of the vaccinated mice 1 month after vaccination with a second dose, antibody titers increased significantly. Interestingly, while vaccination with wtVACV(WR) produced high levels of antibodies, wtVACV(NYCBH) infection did not.
Previously published data suggests that neutralizing antibodies play a central role in protecting against poxvirus infection [34, 35], while our data suggests that mice could be protected from challenge with wtVACV in the absence of neutralizing antibodies or comet formation inhibiting antibodies. To ask if antibodies were necessary for protection in this model system, B cell −/− mice were vaccinated and challenged. As shown in Figure 8, vaccination of B cell deficient mice with VACVDE3L fully protected against challenge with wtVACV. These results demonstrate that in this model and antibody response is not necessary for protection against challenge.
Fig. 8. Challenge of B cell −/− vaccinated mice.
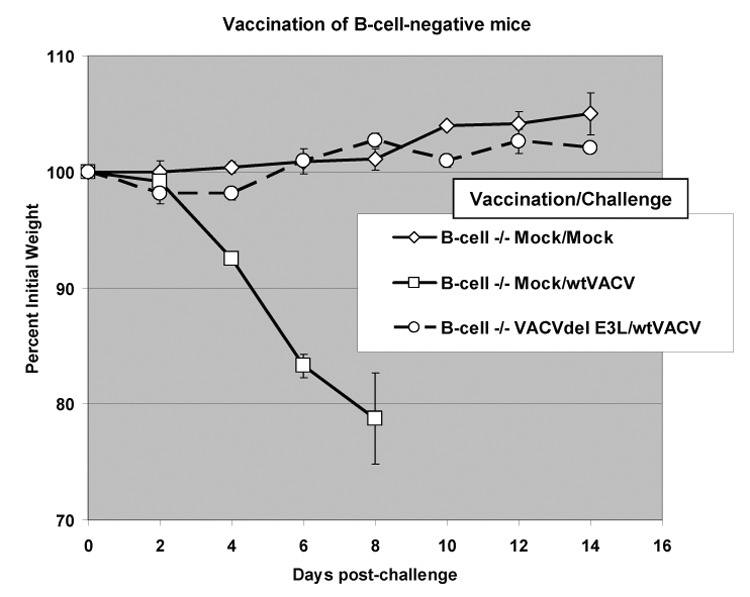
B cell −/− mice were vaccinated and challenged as described in Fig. 4.
4. Discussion
The E3L gene of vaccinia virus contributes to its pathogenesis [27, 30]. Both the N and C terminal domains of the E3L protein have been found to be necessary for full virulence in mouse models [27]. In this report we demonstrate that when administered by scarification, viruses containing mutations in E3L can induce potent Th1 dominated immune responses that are protective against intra-nasal challenge with wtVACV. These viruses are highly attenuated when administered by scarification even in SCID mice, with VACVΔE3L and VACV-vIF2αH, causing little detectable pathology or disease even after 6 months of infection. Thus, these viruses have the potential for use as replication-competent, attenuated vaccines for protection against smallpox and as replication-competent, attenuated vaccine vectors for protection against heterologous diseases.
We have evaluated four viruses for pathogenicity and efficacy: VACVE3LΔ7C, VACVE3LΔ54N, VACVΔE3L and VACV-vIF2αH. VACVE3LΔ7C, VACVE3LΔ54N are replication competent, partially attenuated viruses. Both replicate to near wt titers in the nasal mucosa [22] and in the dermis, but are attenuated by 4–5 logs after IN infection of wt mice [22]. Both have reduced virulence in SCID mice, when infected either IN [22]or by scarification, and both replicate to intermediate titers in ovaries after IP infection. For VACVE3LΔ7C this is likely due to the decrease in affinity that E3LΔ7C protein has for dsRNA [17, 31]. The properties of VACVE3LΔ54N are likely due to a combination of properties of E3LΔ54N: lack of a functional Z-DNA-binding domain [27, 30], and rapid turn-over of E3LΔ54N protein (Fig. 2). Induction of pathogenesis after a long latent period by VACVE3LΔ54N suggests that this virus is replicating at low level for a long period of time in the SCID mice. Such prolonged replication could lead to selection of a more pathogenic variant. Since E3L proteins with larger deletions in the N-terminus than Δ54N are stable, a mutation that yields a stable protein could lead to a more pathogenic virus that could have caused disease after a long latent period. Both VACVΔE3L and VACV-vIF2αH, are much more highly attenuated than VACVE3LΔ7C and VACVE3LΔ54N. Neither virus caused detectable weight loss in SCID mice after IN [22] or scarification infection, even up to six months after infection. VACVΔE3L was not detectable in ovaries after IP infection. For VACV-vIF2αH, virus was detected in the ovaries of only one of three mice, although at 6 logs lower titer than wtVACV. Both viruses did replicate either after IN infection [22] or scarification infection, although titers were 3–4 logs lower than in wtVACV infected mice. The detection of VACVΔE3L replication in the nasal mucosa and skin but not in ovaries is surprising, since ovaries are thought to be the most sensitive organ to detect VACV replication. It is possible that the unique sensitivity of VACVΔE3L and VACV-vIF2αH to IFN is compromising replication in the ovaries to a greater extent than in the nasal mucosa and in the skin. While the ATV eIF2α homologue partially complements deletion of E3L in cells in culture (Talasela et al., manuscript in preparation), there was very little decrease in attenuation detected in virus containing the eIF2α homologue in place of E3L. Again, it is possible that the IFN-sensitivity of VACV-vIF2αH (Talasela et al., manuscript in preparation), may be limiting virus replication and pathogenicity in the whole animal.
Somewhat surprisingly, all of the attenuated viruses tested in this study produced detectable skin reactions (takes) that no matter how mild, were protective against challenge with wtVACV. All of the viruses produced takes in all of the animals tested, when administered by scarification at 106 pfu. At 105 pfu all of the viruses except VACVΔE3L produced takes in all of the animals tested: VACVΔE3L produced takes in 7 of 10 animals vaccinated at this dose. The severity of the take directly correlated with pathogenicity of the virus tested and the extent of virus replication in the dermis, with the order being: wtVACV>VACVE3LΔ7C>VACVE3LΔ54N>VACV-vIF2αH=VACVΔE3L. For VACVΔE3L, which gave takes on about 70% of the animals at 105 pfu, those animals that had a take were fully protected, while the animals that didn’t have a take were largely unprotected. Thus, the very mild skin lesions produced by the highly attenuated viruses, VACVΔE3L and VACV- vIF2αH, were fully protective against a challenge with wtVACV, even when the animals were challenged three months after vaccination (data not shown).
In order to correlate protection with immune response, we characterized induction of neutralizing antibodies, comet formation inhibiting antibodies and induction of cell mediated immunity. At 2 weeks post vaccination, high titers of neutralizing antibodies were detected in serum from animals immunized with wtVACV and VACVE3LΔ7C. The less pathogenic viruses, VACVΔE3L, VACV-vIF2αH and VACVE3LΔ54N, as well as wtVACV (NYCBH), produced lower levels of neutralizing antibodies than VACVE3LΔ7C(WR) and wtVACV(WR). The ability to induce neutralizing antibodies correlated best with the replication capacity of these viruses, as measured either on the skin after scarification, or in ovaries after IP infection. Thus, it may be that the dose of viral antigen is the key determinant of induction of neutralizing antibodies. However, infection with the more highly attenuated viruses apparently primed for induction of both neutralizing and comet formation inhibiting antibodies, because high titers of these antibodies were detected after a single boost with VACVΔE3L. Despite inducing a poor primary antibody response these viruses all protected against challenge with wtVACV. Thus, either the low levels of neutralizing antibodies were sufficient for protection, or the small number of antibody producing cells generated during primary vaccination were sufficient to expand and protect the animals upon challenge, or other components of the immune system, likely cell mediated immunity, were compensating for this low antibody response. Interestingly, B cell deficient mice vaccinated with E3L deletion mutant viruses are also protected against challenge with pathogenic vaccinia virus despite an inability to produce antibodies. These results are in contrast to other previously reported results [34, 35], demonstrating a critical role for antibodies in protection against challenge of mice with vaccinia virus.
To begin to evaluate induction of cell mediated immunity, we measured cytokines released from ex vivo restimulated splenocytes from vaccinated animals. These assays provided the first information that would begin to explain why VACVΔE3L and VACV-vIF2αH are protective in vaccination experiments despite the absence of substantial levels of neutralizing antibodies. Restimulated splenocytes from mice vaccinated with viruses deleted of all or part of the E3L gene produced roughly twice as much IFN-γ and TNF-α as splenocytes from wtVACV vaccinated mice. Splenocytes from VACVΔE3L vaccinated mice also secreted more IL-2. Splenocytes from all of the vaccinated animals secreted very low levels of IL-4 and IL-5. Thus, these highly attenuated viruses induce potent Th1 dominated immune responses.
Intracellular cytokine staining was also used to identify which cell types were activated during vaccination. Animals vaccinated with either wtVACV or any of the attenuated viruses described in this manuscript had similar percentages of CD4+/IFNγ+ T cells (about 5% of CD4+ T cells), and similar percentages of CD8+/IFNγ+ T cells (about 10% of the CD8+ T cells). The fact that these experiments showed similar percentages of IFN-γ positive CD4+ and CD8+ cells from the spleens of both VACVΔE3L vaccinated mice and wtVACV vaccinated mice indicates that it is possible that T cells are not the only cells from the spleens of vaccinated mice that are producing IFN-γ, or that T cells from VACVΔE3L vaccinated mice are secreting more IFNγ on a per cell basis than splenocytes from wtVACV vaccinated animals.
VACVΔE3L induces a potent Th1 dominated cell mediated immune response despite replicating poorly on the skin of mice. We believe this is due to induction of pro-inflammatory signal transduction and pro-inflammatory gene expression in cells infected with VACVΔE3L [36]. The E3L gene products can bind to double helical nucleic acids and sequester viral induced PAMPs. Thus, infection with wtVACV does not lead to activation of the p38 MAP kinase pathway, and does not lead to activation of the IKK complex [36]. Consistent with this, we detected no activation of the pro-inflammatory transcription factors ATF-2, NF-κB or IRF-3 and very little increased pro-inflammatory gene expression in cells infected with wtVACV [36]. Virus deleted for E3L leads to signaling through the p38 MAP kinase pathway leading to phosphorylation of ATF-2, leads to phosphorylation of IKKα/βand IKKε, and leads to nuclear accumulation and activation of both NF-κB and IRF-3 [36]. Microarray experiments have shown that infection with VACVΔE3L induces upregulation of a large number of proinflammatory gene products [36]. We believe that this massive increase in virus-induced signaling leading to activation of proinflammatory transcription factors and proinflammatory gene expression is responsible for the efficacy of VACVΔE3L as a vaccine despite its attenuation and limited replication capacity in infected mice.
Most of the work described in this manuscript was done with the WR strain of VACV. VACV(WR) is a highly neurovirulent, mouse adapted strain of VACV, and while it is relevant for studies in the mouse model, it is not appropriate for use in humans [23]. To test attenuation and efficacy of attenuating mutations in E3L in strains of vaccinia virus more appropriate for use in humans, experiments were performed to test vaccination efficacy and immunological characteristics in two additional strains. The strains chosen were the Copenhagen and New York City Board of Health strains, both of which have been previously used in human vaccination campaigns [1]. These strains were found to more attenuated than the VACV(WR) in the mouse models tested [22]. Introducing the E3L deletion mutation into these viruses led to further attenuation in mice. VACVΔE3L in both a Copenhagen and a NYCBH background were fully able to induce very mild protective pocks in vaccinated mice. As was seen with VACVΔE3L(WR), VACVΔE3L(NYCBH) did not induce the synthesis of neutralizing antibodies, but did prime for synthesis of neutralizing antibodies. Splenocytes from VACVΔE3L(NYCBH) vaccinated mice also secreted somewhat more IFNγ, similar to splenocytes from VACVΔE3L(WR) vaccinated mice.
Fig. 3. Virus concentrations in tail tissue after scarification.
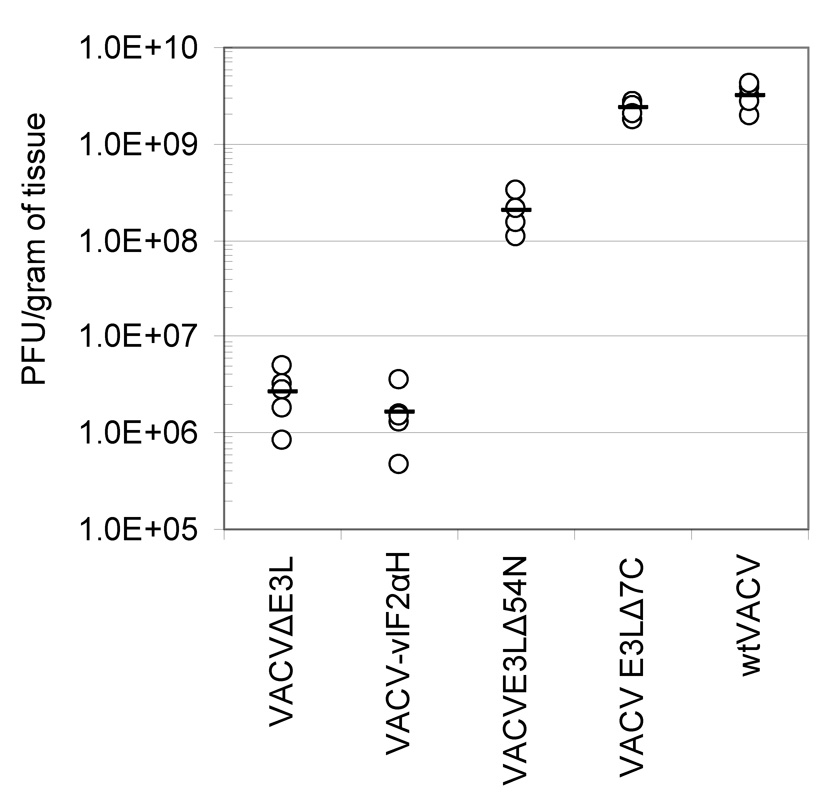
Five female BALB/c mice per group were infected by scarification with 106 pfu wtVACV and VACV E3L mutants. Tail tissue was harvested 5 days after scarification for determination of virus concentration. A one-way ANOVA on ranks followed by pairwise comparisons, using the Student-Newman-Keuls Method was used to analyze the data.
Table 2.
Splenocyte cytokine secretion (pg/106 spenocytes)
| IFNγ | TNFα | IL-2 | IL-4 | IL-5 | |
|---|---|---|---|---|---|
| Mock | 0.0+/−0.0 | 34.4+/−27.0 | 0.7+/−0.8 | 1.4+/−0.3 | 2.3+/−0.5 |
| VACVΔE3L(WR) | 13776.1+/−1208.3 | 556.7+/−118.3 | 161.9+/−23.8 | 38.1+/−9.8 | 27.7+/−11.2 |
| VACV-vIF2αH(WR) | 11508.8+/−2383.7 | 505.8+/−155.4 | 100.4+/−36.9 | 33.4+/−5.1 | 16.5+/−11.5 |
| VACVE3LΔ54N(WR) | 13069.7+/−2474.6 | 532.8+/−31.9 | 72.7+/− 45.6 | 44.4+/−3.4 | 32.1+/−6.9 |
| VACVE3LΔ7C(WR) | 9841.6+/−3594.4 | 499.6+/−180.4 | 76.2+/−19.6 | 96.9+/−37.2 | 17.3+/−20.2 |
| wtVACV(WR) | 6746.8+/−634.3 | 252.9+/−44.8 | 58.1+/−22.2 | 83.0+/−18.0 | 26.8+/−8.5 |
| VACVÄE3L(NYCBH) | 17159.0+/−4926.8 | 725.3+/−188.8 | 144.3+/−84.7 | NT* | NT |
| wtVACV(NYCBH) | 11204.4+/−2143.7 | 960.2+/−259.7 | 96.8+/−26.8 | NT | NT |
Not tested
Acknowledgements
The work was supported by the following grants from the NIH: I52347, AI66326 and AI 66501.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fenner F, Henderson DA, Arita I, Jezek Z, Ladnyi ID. Geneva, Switzerland: World Health Organization; Smallpox and its eradication. 1988
- 2.Cassimatis DC, Atwood JE, Engler RM, Linz PE, Grabenstein JD, Vernalis MN. Smallpox vaccination and myopericarditis: a clinical review. J Am Coll Cardiol. 2004;43:1503–1510. doi: 10.1016/j.jacc.2003.11.053. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15120802. [DOI] [PubMed] [Google Scholar]
- 3.Fulginiti VA, Papier A, Lane JM, Neff JM, Henderson DA. Smallpox vaccination: a review, part II. Adverse events. Clin Infect Dis. 2003;37:251–271. doi: 10.1086/375825. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12856218. [DOI] [PubMed] [Google Scholar]
- 4.Meyer H, Sutter G, Mayr A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J Gen Virol. 1991;72(Pt 5):1031–1038. doi: 10.1099/0022-1317-72-5-1031. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=2033387. [DOI] [PubMed] [Google Scholar]
- 5.Coulibaly S, Bruhl P, Mayrhofer J, Schmid K, Gerencer M, Falkner FG. The nonreplicating smallpox candidate vaccines defective vaccinia Lister (dVV-L) and modified vaccinia Ankara (MVA) elicit robust long-term protection. Virology. 2005;341:91–101. doi: 10.1016/j.virol.2005.06.043. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16061267. [DOI] [PubMed] [Google Scholar]
- 6.Tartaglia J, Perkus ME, Taylor J, Norton EK, Audonnet JC, Cox WI, Davis SW, van der Hoeven J, Meignier B, Riviere M, et al. NYVAC: a highly attenuated strain of vaccinia virus. Virology. 1992;188:217–232. doi: 10.1016/0042-6822(92)90752-b. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=1566575. [DOI] [PubMed] [Google Scholar]
- 7.Kenner J, Cameron F, Empig C, Jobes DV, Gurwith M. LC16m8: an attenuated smallpox vaccine. Vaccine. 2006;24:7009–7022. doi: 10.1016/j.vaccine.2006.03.087. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17052815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tartaglia J, Cox WI, Pincus S, Paoletti E. Safety and immunogenicity of recombinants based on the genetically-engineered vaccinia strain, NYVAC. Dev Biol Stand. 1994;82:125–129. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7958466. [PubMed] [Google Scholar]
- 9.Ramirez JC, Gherardi MM, Esteban M. Biology of attenuated modified vaccinia virus Ankara recombinant vector in mice: virus fate and activation of B- and T-cell immune responses in comparison with the Western Reserve strain and advantages as a vaccine. J Virol. 2000;74:923–933. doi: 10.1128/jvi.74.2.923-933.2000. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10623755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenwirth B, Kuhn EM, Heeney JL, Hurpin C, Tartaglia J, Bonnet MC, Moingeon P, Erdile L. Safety and immunogenicity of ALVAC wild-type human p53 (vCP207) by the intravenous route in rhesus macaques. Vaccine. 2001;19:1661–1670. doi: 10.1016/s0264-410x(00)00416-3. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11166889. [DOI] [PubMed] [Google Scholar]
- 11.Meseda CA, Garcia AD, Kumar A, Mayer AE, Manischewitz J, King LR, Golding H, Merchlinsky M, Weir JP. Enhanced immunogenicity and protective effect conferred by vaccination with combinations of modified vaccinia virus Ankara and licensed smallpox vaccine Dryvax in a mouse model. Virology. 2005;339:164–175. doi: 10.1016/j.virol.2005.06.002. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15993917. [DOI] [PubMed] [Google Scholar]
- 12.Vollmar J, Arndtz N, Eckl KM, Thomsen T, Petzold B, Mateo L, Schlereth B, Handley A, King L, Hulsemann V, et al. Safety and immunogenicity of IMVAMUNE, a promising candidate as a third generation smallpox vaccine. Vaccine. 2006;24:2065–2070. doi: 10.1016/j.vaccine.2005.11.022. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16337719. [DOI] [PubMed] [Google Scholar]
- 13.Amara RR, Villinger F, Altman JD, Lydy SL, O'Neil SP, Staprans SI, Montefiori DC, Xu Y, Herndon JG, Wyatt LS, et al. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science. 2001;292:69–74. doi: 10.1126/science.1058915. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11393868. [DOI] [PubMed] [Google Scholar]
- 14.Shors ST, Beattie E, Paoletti E, Tartaglia J, Jacobs BL. Role of the vaccinia virus E3L and K3L gene products in rescue of VSV and EMCV from the effects of IFN-alpha. J Interferon Cytokine Res. 1998;18:721–729. doi: 10.1089/jir.1998.18.721. [DOI] [PubMed] [Google Scholar]
- 15.Xiang Y, Condit RC, Vijaysri S, Jacobs B, Williams BR, Silverman RH. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J Virol. 2002;76:5251–5259. doi: 10.1128/JVI.76.10.5251-5259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang HW, Watson JC, Jacobs BL. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci U S A. 1992;89:4825–4829. doi: 10.1073/pnas.89.11.4825. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=1350676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang HW, Jacobs BL. Identification of a conserved motif that is necessary for binding of the vaccinia virus E3L gene products to double-stranded RNA. Virology. 1993;194:537–547. doi: 10.1006/viro.1993.1292. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=8099244. [DOI] [PubMed] [Google Scholar]
- 18.Colby C, Jurale C, Kates JR. Mechanism of synthesis of vaccinia virus double-stranded ribonucleic acid in vivo and in vitro. J Virol. 1971;7:71–76. doi: 10.1128/jvi.7.1.71-76.1971. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=5543434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pestka S, Langer JA, Zoon KC, Samuel CE. Interferons and their actions. Annu Rev Biochem. 1987;56:727–777. doi: 10.1146/annurev.bi.56.070187.003455. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=2441659. [DOI] [PubMed] [Google Scholar]
- 20.Sen GC, Lengyel P. The interferon system. A bird's eye view of its biochemistry. J Biol Chem. 1992;267:5017–5020. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=1371992. [PubMed] [Google Scholar]
- 21.Kim YG, Muralinath M, Brandt T, Pearcy M, Hauns K, Lowenhaupt K, Jacobs BL, Rich A. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc Natl Acad Sci U S A. 2003;100:6974–6979. doi: 10.1073/pnas.0431131100. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12777633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vijaysri S, Jentarra G, Heck MC, Mercer AA, McInnes CJ, Jacobs BL. Vaccinia viruses with mutations in the E3L gene as potential replication-competent, attenuated vaccines: Intra-nasal vaccination. Vaccine. 2008;26:664–676. doi: 10.1016/j.vaccine.2007.11.045. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18096276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ludvikova V, Kutinova L, Simonova V, Otavova M. Evaluation of various virulence tests with low virulence vaccinia virus in mice. Biologicals. 1994;22:187–190. doi: 10.1006/biol.1994.1025. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7917234. [DOI] [PubMed] [Google Scholar]
- 24.Monath TP, Caldwell JR, Mundt W, Fusco J, Johnson CS, Buller M, Liu J, Gardner B, Downing G, Blum PS, et al. ACAM2000 clonal Vero cell culture vaccinia virus (New York City Board of Health strain)--a second-generation smallpox vaccine for biological defense. Int J Infect Dis. 2004;8 Suppl 2:S31–S44. doi: 10.1016/j.ijid.2004.09.002. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15491873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kibler KV, Shors T, Perkins KB, Zeman CC, Banaszak MP, Biesterfeldt J, Langland JO, Jacobs BL. Double-stranded RNA is a trigger for apoptosis in vaccinia virus-infected cells. J Virol. 1997;71:1992–2003. doi: 10.1128/jvi.71.3.1992-2003.1997. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9032331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He Y, Tan SL, Tareen SU, Vijaysri S, Langland JO, Jacobs BL, Katze MG. Regulation of mRNA translation and cellular signaling by hepatitis C virus nonstructural protein NS5A. J Virol. 2001;75:5090–5098. doi: 10.1128/JVI.75.11.5090-5098.2001. http://jvi.asm.org/cgi/content/full/75/11/5090 http://jvi.asm.org/cgi/content/abstract/75/11/5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brandt TA, Jacobs BL. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J Virol. 2001;75:850–856. doi: 10.1128/JVI.75.2.850-856.2001. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11134298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Essbauer S, Bremont M, Ahne W. Comparison of the eIF-2alpha homologous proteins of seven ranaviruses (Iridoviridae) Virus Genes. 2001;23:347–359. doi: 10.1023/a:1012533625571. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11778703. [DOI] [PubMed] [Google Scholar]
- 29.Davies MV, Elroy-Stein O, Jagus R, Moss B, Kaufman RJ. The vaccinia virus K3L gene product potentiates translation by inhibiting double-stranded-RNA-activated protein kinase and phosphorylation of the alpha subunit of eukaryotic initiation factor 2. J Virol. 1992;66:1943–1950. doi: 10.1128/jvi.66.4.1943-1950.1992. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=1347793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brandt T, Heck MC, Vijaysri S, Jentarra GM, Cameron JM, Jacobs BL. The N-terminal domain of the vaccinia virus E3L-protein is required for neurovirulence, but not induction of a protective immune response. Virology. 2005;333:263–270. doi: 10.1016/j.virol.2005.01.006. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15721360. [DOI] [PubMed] [Google Scholar]
- 31.Chang HW, Uribe LH, Jacobs BL. Rescue of vaccinia virus lacking the E3L gene by mutants of E3L. J Virol. 1995;69:6605–6608. doi: 10.1128/jvi.69.10.6605-6608.1995. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7666567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shors T, Kibler KV, Perkins KB, Seidler-Wulff R, Banaszak MP, Jacobs BL. Complementation of vaccinia virus deleted of the E3L gene by mutants of E3L. Virology. 1997;239:269–276. doi: 10.1006/viro.1997.8881. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9434718. [DOI] [PubMed] [Google Scholar]
- 33.Vanderplasschen A, Hollinshead M, Smith GL. Antibodies against vaccinia virus do not neutralize extracellular enveloped virus but prevent virus release from infected cells and comet formation. J Gen Virol. 1997;78(Pt 8):2041–2048. doi: 10.1099/0022-1317-78-8-2041. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9267005. [DOI] [PubMed] [Google Scholar]
- 34.Belyakov IM, Earl P, Dzutsev A, Kuznetsov VA, Lemon M, Wyatt LS, Snyder JT, Ahlers JD, Franchini G, Moss B, Berzofsky JA. Shared modes of protection against poxvirus infection by attenuated and conventional smallpox vaccine viruses. Proc Natl Acad Sci U S A. 2003;100:9458–9463. doi: 10.1073/pnas.1233578100. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12869693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Panchanathan V, Chaudhri G, Karupiah G. Correlates of protective immunity in poxvirus infection: where does antibody stand? Immunol Cell Biol. 2008;86:80–86. doi: 10.1038/sj.icb.7100118. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17923850. [DOI] [PubMed] [Google Scholar]
- 36.Langland JO, Kash JC, Carter V, Thomas MJ, Katze MG, Jacobs BL. Suppression of proinflammatory signal transduction and gene expression by the dual nucleic acid binding domains of the vaccinia virus E3L proteins. J Virol. 2006;80:10083–10095. doi: 10.1128/JVI.00607-06. [DOI] [PMC free article] [PubMed] [Google Scholar]