

Summary
Tandem mass spectrometry is currently used in newborn screening programmes to quantify the level of amino acids and acylcarnitines in dried blood spots for detection of metabolites associated with treatable diseases. We have developed assays for lysosomal enzymes in re-hydrated dried blood spots in which a set of substrates is added and the set of corresponding enzymatic products are quantified using tandem mass spectrometry with the aid of mass-differentiated internal standards. We have developed a multiplex assay of the set of enzymes that, when deficient, cause the lysosomal storage disorders Fabry, Gaucher, Hurler, Krabbe, Niemann–Pick A/B and Pompe diseases. These diseases were selected because treatments are now available or expected to emerge shortly. The discovery that acarbose is a selective inhibitor of maltase glucoamylase allows the Pompe disease enzyme, acid α-glucosidase, to be selectively assayed in white blood cells and dried blood spots. When tested with dried blood spots from 40 unaffected individuals and 10–12 individuals with the lysosomal storage disorder, the tandem mass spectrometry assay led to the correct identification of the affected individuals with 100% sensitivity. Many of the reagents needed for the new assays are commercially available, and those that are not are being prepared under Good Manufacturing Procedures for approval by the FDA. Our newborn screening assay for Krabbe disease is currently being put in place at the Wadsworth Center in New York State for the analysis of ~1000 dried blood spots per day.
Summary We have developed tandem mass spectrometry for the direct assay of lysosomal enzymes in rehydrated dried blood spots that can be implemented for newborn screening of lysosomal storage disorders. Several enzymes can be analysed by a single method (multiplex analysis) and in a high-throughput manner appropriate for newborn screening laboratories.
Introduction
Over the past 5 years or so we have been developing the use of tandem mass spectrometry (tandem MS) as a diagnostic platform for the early detection and confirmation of genetic disorders. Tandem MS as a diagnostic tool offers a number of key advantages, as will become apparent from studies and summary points described in this review. The confirmation of a genetic disease in humans continues to rest on the identification of an altered protein in the form of diminished or altered enzyme activity, or an alteration or absence of a structural protein. For some common genetic diseases for which there has been selection or genetic drift in certain ethnic groups, DNA technology can be utilized in an efficient manner for their early detection. The most common approach for individuals who unexpectedly present with clinical symptoms remains, however, the measurement of protein function from a biological sample. Various technologies have been used to measure protein function, including spectrophotometric, fluorometric, radiometric or immunological assays. With the exception of immunological assays, these assay methods are generally not multiplexable; that is, they are applied to assay the function of one protein at a time. Immunological assays may be multiplexable, but they provide protein abundance data, which is not useful in cases where nonfunctional protein is produced.
Assays for enzyme deficiencies: Lysosomal storage disorders
Our focus has been the development of the tandem mass spectrometer as a common platform for measuring enzyme function. This has been applied to blood samples obtained from individuals suspected or confirmed to have a particular genetic disease. We were originally successful in demonstrating, as a proof of principle, that the tandem mass spectrometer could be used to measure the four enzymes responsible for the Sanfilippo syndrome and that it could be performed in a multiplexed fashion in which all four enzymes in a fibroblast lysate are assayed in a single sample infusion into the tandem mass spectrometer (Gerber et al 2001a,b). We subsequently demonstrated that, for several of the lysosomal storage disorders, blood samples could be utilized for the quantitative measurement of the activities of enzymes responsible for Gaucher disease, Fabry disease, mucopolysaccharidosis-I (MPS I), Pompe disease, Krabbe disease and Niemann–Pick A or B diseases (Li et al 2004a,b). In each of these cases, the technology proved so sensitive that activity could actually be measured in dried blood spots that were submitted for newborn screening (after they were rehydrated in suitable buffers). Thus, the technology has been extended from the theoretical concept of a common platform used for diagnosis to a sensitive technology that can used for large-scale screening of newborns independently of selection by clinical symptoms. This is of clinical importance because the disorders of Gaucher, Fabry, MPS I and Pompe currently have available, for clinical use or trials, enzyme replacement therapy that either has been demonstrated to improve (Gaucher, Fabry; Scott et al 2003; Shah and Elliott 2005) or is expected to improve (MPS I, Pompe; Desnick 2004; Miebach 2005) the clinical status of affected individuals. In the case of Krabbe and Niemann–Pick disease, there is evidence that presymptomatic detection and intervention by the use of umbilical stem cell transplantation markedly improves the clinical course of the disease (Escolar et al 2005; Krivit 2004).
Tandem mass spectrometry for the multiplex analysis of lysosomal enzymes in dried blood spots
The laboratory of Nestor Chamoles has shown over the past several years that many lysosomal enzymes remain active in dried blood spots and can be assayed with fluorometric or radiometric substrates after rehydration in a suitable reaction buffer (see, for example, Chamoles et al (2002)). Such blood spots are prepared shortly after birth by spotting a drop of newborn blood onto a filter paper card and allowing the liquid to dry on the card. It is now commonplace in many countries to send dried blood spots to newborn screening laboratories for tandem MS analysis of amino acids and acylcarnitines (see, for example, Chace and Kalas (2005), Wilcken et al (2003)). Quantification of these compounds allows screening for up to about 30 treatable metabolic diseases. Based on all of this information, we decided to develop a multiplex tandem MS assay of several lysosomal enzymes using dried blood spots as the input sample.
The detection of lysosomal enzyme activities in dried blood spots faces the problem of low protein content and hence low concentration of enzymatic products to be analysed. We developed a new methodology that consists of the following steps: (1) A small punch (typically a few millimetres in diameter) is taken from the dried blood spot and rehydrated in a buffer to extract the enzymes of interest. (2) The substrate for the enzyme is added, and the mixture is allowed to incubate under defined conditions to allow enzymatic conversion of substrate to product. (3) The sample is submitted to a simple workup involving passage through a solid-phase extraction cartridge to remove the relatively large amounts of buffer salts and detergent (if present), as these would interfere with the electrospray ionization process in the mass spectrometer. (4) The sample is infused directly into the electrospray ionization mass spectrometer operating in tandem mode, and the amount of enzymatically-generated product is selectively detected along with an internal standard to determine the absolute amount of reaction product. The internal standard is chemically identical to or similar to the reaction product but is distinguished in the mass spectrometer from the reaction product by having a different mass. It is anticipated that this procedure would be easily transferable to newborn screening laboratories, many of which routinely use tandem MS to analyse metabolites in dried blood spots.
Our method for use of tandem MS to assay lysosomal enzymes in dried blood spots is illustrated in Fig. 1 for our assay of Krabbe disease (Li et al 2004a). Krabbe disease is caused by deficiency in the enzyme galactocerebroside-β-galactosidase. The Krabbe substrate is a synthetic galacto-sylceramide (from Avanti Polar Lipids, Inc. Alabaster, Alabama, USA) containing an 8-carbon fatty acyl chain in the ceramide moiety. This substrate is very similar in structure to that of the natural substrates, which contain longer fatty acyl chains. The dried blood spot punch is incubated with reaction buffer containing detergent and Krabbe substrate. Action of the Krabbe enzyme generates the Krabbe product, whose mass is different from that of the substrate (Fig. 1). At the end of the reaction, internal standard is added. This standard is similar in structure to the product, the only difference is that it contains a 10-carbon fatty acyl chain instead of an 8-carbon chain. The reaction mixture is extracted with ethyl acetate, and the extract is passed through a small plug of silica gel. Product and internal standard elute immediately from the silica, whereas buffer salts and detergent are retained. The eluant is concentrated to dryness with a stream of air (or in a vacuum desiccator), the residue is dissolved in methanol, and the solution is infused directly into the tandem mass spectrometer. All of these steps are simple liquid transfers and can be carried out using 96-well plates. The solid-phase extraction step is also carried out using a 96-well plate, a filter plate in this case, and a simple suction manifold attached to a water aspirator.
Fig. 1.
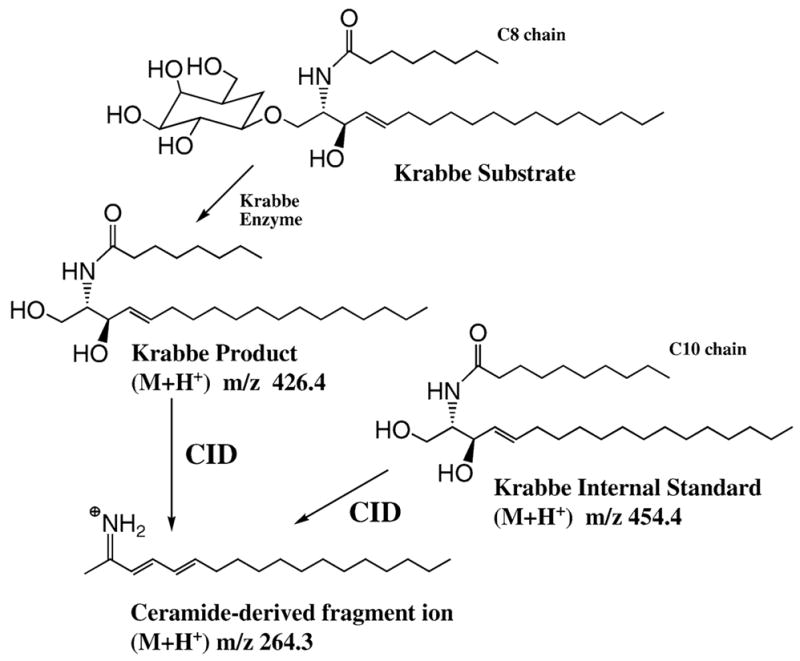
Tandem MS assay of galactocerebroside-β-galactosidase for the analysis of Krabbe disease. See text for discussion
The tandem MS analyses were carried out on a tandem quadrupole mass spectrometer (Sciex API III+), symbolized as Q1-q2-Q3, which is a type of instrument commonly used in clinical laboratories. Q1-q2-Q3 mass spectrometers are currently available from ABI-Sciex, ThermoElectron (Finnigan), or Waters (Micromass). Precursor analyte ions that are produced by electrospray ionization are selected by the first quadrupole mass filter (Q1); these are allowed to dissociate in a collision multipole (q2), and their dissociation products detected after passing through the second quadrupole mass filter (Q3). As shown in Fig. 1 the Krabbe product and internal standard ions undergo collision-induced dissociation (CID) to give fragment ions of m/z 264.3. However, the product and internal standard parent ions differ in mass, and thus are selectively quantified. Since ion intensities from electrospray are proportional to analyte concentrations (Kebarle and Ho 1997), the measured relative intensities of product and internal standard reporter fragment ions are proportional to product and internal standard concentrations. The proportionalities (response factors in the electrospray ionization source) are determined from calibration curves for product and internal standard. In this case, the Krabbe product and internal standard have near-identical ionization efficiencies, not surprisingly given the similarity in their structure.
We showed that the amount of Krabbe product increases linearly with the incubation time, that the product formed at fixed time increases linearly with the amount of blood (surface area of the dried blood spot punch), and that the reaction velocity displays hyperbolic kinetics with respect to substrate concentration (Li et al 2004a). We also optimized the reaction conditions with respect to pH and detergent concentration (Li et al 2004a). The coefficient of variance measured for 14 different punches from the same dried blood spot was 9.6%. Figure 2 shows typical tandem MS data obtained using our Krabbe enzyme assay.
Fig. 2.
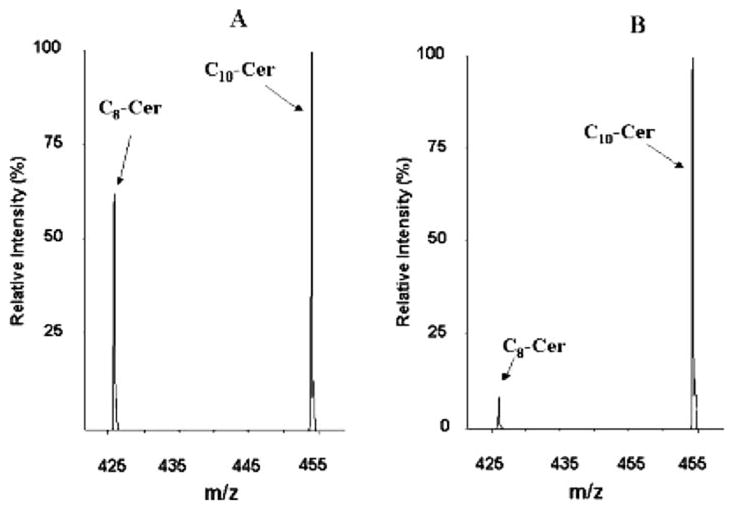
Tandem MS data obtained with the Krabbe assay. Panel (A) is from a dried blood spot from a healthy patient, and panel (B) is from a Krabbe patient. The product-to-internal standard ratio (C8-Cer to C10-Cer) is much higher for the Former
We also developed assays for Gaucher and Niemann–Pick Types A/B diseases using the same strategy as for Krabbe disease (Li et al 2004a). The enzyme deficient in Gaucher disease is β-glucocerebrosidase, and we used a substrate containing a ceramide with a 12-carbon fatty acyl chain with an α-glucosyl headgroup. The product is the 12-carbon fatty acyl ceramide, and the internal standard is the 14-carbon fatty acyl ceramide. The enzyme deficient in Niemann–Pick Types A/B disease is sphingomyelinase, and we used a substrate containing a ceramide with a 6-carbon fatty acyl chain and a phosphorylcholine head group. The product is the 6-carbon fatty acyl ceramide, and the internal standard is the ceramide with a 4-carbon fatty acyl chain. Since all products and internal standards have different masses, the Krabbe, Gaucher and Niemann–Pick-A/B enzymes can be analysed together by a single infusion into the tandem mass spectrometer (Li et al 2004b). All substrates and internal standards are available from Avanti Polar Lipids, Inc.
We also developed a tandem MS assay for analysis of Fabry disease, caused by the deficiency of the enzyme acid α-galactosidase A (Li et al 2004b). The assay is illustrated in Fig. 3. The Fabry product has three design features that facilitate the tandem MS analysis: (1) It is sufficiently hydrophobic that it elutes under the same conditions during the silica gel solid-phase extraction step as the ceramides formed in the three enzymatic reactions described above. (2) It contains a BOC (t-butoxycarbonyl) group which readily fragments in the collision cell of the mass spectrometer to give a major fragment ion. Directing the fragmentation along a single pathway increases the sensitivity of the assay. (3) It contains a benzoyl group, which provides a practical site of heavy isotope incorporation. The internal standard contains a[2H5]benzoyl group, which is prepared using inexpensive [2H5]benzoyl chloride.
Fig. 3.
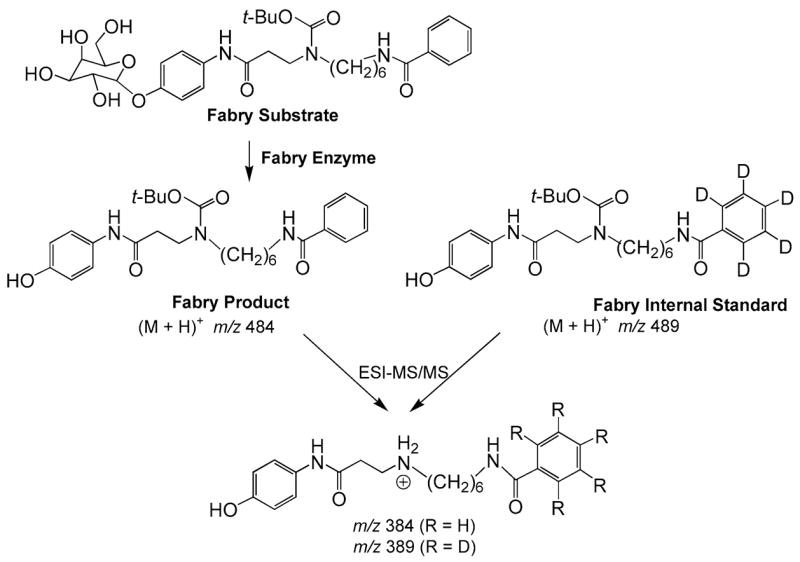
Tandem MS assay of α-galactosidase A for the analysis of Fabry disease. See text for discussion
Pompe disease is caused by deficiency of acid α-glucosidase. Our assay developed for this disease is similar to that used for Fabry disease. The Pompe substrate contains an α-linked glucosyl group and the hydrocarbon chain in the leaving group contains 8 carbons instead of 6 present in the Fabry substrate. Thus, the Pompe product and internal standard are distinguished in the mass spectrometer from the corresponding Fabry species.
The assay for Pompe disease in dried blood spots has been known to suffer from interference from another acid α-glucosidase, known as maltase glucoamylase, which is present in neutrophils (blood also contains two other α-glucosidases but these have a pH optimum near neutral pH and do not interfere with our Pompe assay carried out under acidic conditions). According to our measurements (Li et al 2004b), the Pompe enzyme constitutes only about 30% of total acid α-glucosidase activity in a detergent extract from human neutrophils. Thus, purified lymphocytes, fibroblasts, or muscle tissue have been recommended for the biochemical diagnosis of Pompe disease (Koster et al 1974; Shin et al 1985). Although an immunocapture procedure has been reported to allow a specific assay of the Pompe enzyme (Umapathysivam et al 2001), antibody availability and expense may be prohibitive for newborn screening. Furthermore, the development of a tandem MS assay for Pompe disease would allow this assay to be multiplexed with assays of the other lysosomal storage disorders using the same analytical method. We solved the maltase glucoamylase interference problem by finding that the commercially available tetrasaccharide acarbose is an efficient and specific inhibitor of maltase glucoamylase, as expressed by the measured Ki values, 142 and 0.14 mmol/L for the Pompe enzyme and maltase glucoamylase, respectively (Li et al 2004b). By including acarbose in the Pompe assay buffer, we have been able to spot Pompe patients with 100% sensitivity as shown by the data presented in Fig. 5 below.
Fig. 5.
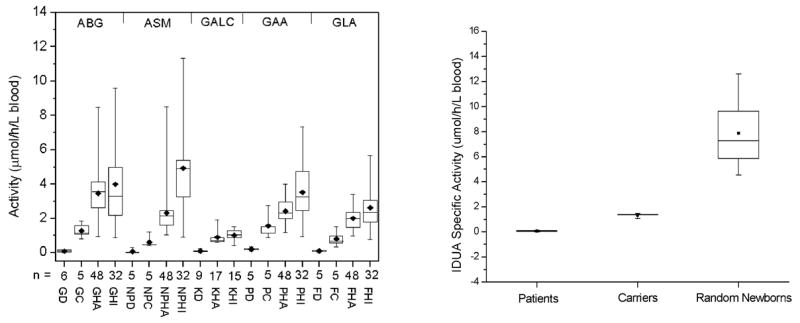
Tandem MS assays of Gaucher, Niemann–Pick-A/B, Krabbe, Pompe and Fabry diseases (left panel). GD, GC, GHA and GHI are Gaucher disease, carrier, healthy adult, and healthy infant patients, respectively. NPD, NPC, NPHA and NPHI are for Niemann–Pick-A/B samples. KD, KHA and KHI are for Krabbe samples. PD, PC, PHA and PHI are for Pompe samples. FD, FC, FHA and FHI are for Fabry samples. The right panel shows Hurler disease assay results
We also developed an assay for α-L-iduronidase for the analysis of mucopolysaccharidosis type I (MPS I, Hurler disease) (Wang et al 2005). The assay is similar to those for the other lysosomal storage disorders described above and is shown in Fig. 4. The Hurler substrate is our structurally most complex substrate prepared to date. We were able to prepare the compound starting from commercially availably heparin. Degradation of heparin with nitrous acid gives a synthetic intermediate that can readily be elaborated into the Hurler substrate. We have prepared this substrate on the gram scale, sufficient for 100 000 assays. Reagent costs, even for this structurally complex substrate, are minimal (estimated to be $0.01–0.02 per assay).
Fig. 4.

Tandem MS assay of α-L-iduronidase for the analysis of mucopolysaccharidosis Type I. See text for discussion
We tested our set of six lysosomal enzyme assays using dried blood spots from 40 normal patients and from 10–15 patients previously diagnosed with the lysosomal storage disorders. Results are shown in Fig. 5. In all cases, the enzyme activities measured using dried blood spots from patients previously diagnosed with the lysosomal storage disease are well separated from the activities measured with healthy patient dried blood spots. Our assay method using acarbose has been further examined by Dr Helmut Kallwass at Genzyme Corporation on 150 normal patient dried blood spots and blood spots from 24 infantile Pompe patients and 58 late-onset patients. All patients had low levels of α-glucosidase activity that were well resolved from the activities of normal patients (Kallwass et al 2005).
We also examined the stability of the six enzymes in dried blood spots that had been stored for a number of years at room temperature (Li et al 2004b; Wang et al 2005). All enzymes except the Krabbe enzyme were found to be stable over a 3-year storage period (less than 10% decrease in activity). In the case of the Krabbe enzyme, activity decreased by 9% over a 10-day storage period at room temperature. These results establish that collection of dried blood spots and shipment to newborn screening laboratories at room temperature will not be a problem.
Developments towards tandem MS newborn screening for lysosomal storage disorders
The tandem MS assays that we have developed for lysosomal storage disorders may soon move into newborn screening laboratories. In response to the success of early bone marrow transplantation for the treatment of Krabbe disease (Escolar et al 2005), the Hunter’s Hope Foundation met with State Representatives in Albany, New York (http://www.huntershope.org/news/pressreleases/hhalbanypr.asp). As a result, the New York State screening laboratory at the Wadsworth Center is establishing our Krabbe disease assay (Li et al 2004a) and intend to use it as part of their newborn screening assay panel (under the direction of Dr. Kenneth Pass; (http://www.wadsworth.org/educate/passkrabbe.htm). Dr Pass’ laboratory is currently equipping the laboratory for tandem MS assays on ~1000 dried blood spots per day. It must be stressed that the Krabbe disease enzyme assay carried out with dried blood spots in a newborn screening programme is only the first step. Newborns with low enzyme activity found in a screen will be further evaluated with a collection of follow-up tests to confirm the diagnosis of Krabbe disease before a decision is made to initiate treatment.
Over the past two years, we have been in communication with Dr Joan Keutzer at Genzyme Corporation to bring our tandem MS assays for Krabbe disease and other treatable lysosomal diseases into practice in newborn screening laboratories. Some of the reagents are commercially available from Avanti Polar Lipids, Inc. Pilot lots of the reagents for assaying Fabry disease and Pompe disease have been synthesized by Genzyme. We have been consulting with Genzyme to optimize the synthesis of our substrate for MPS I on the multi-gram scale, and we are close to accomplishing this goal. These reagents are being manufactured under Good Manufacturing Practice (GMP), which is now required by the FDA for newborn screening reagents (http://www.fda.gov/cdrh/oivd/guidance/1301.html). Genzyme is planning to provide these reagents to newborn screening laboratories worldwide and has also agreed to provide assay training. No Intellectual Property protection for these assays have been sought (the assays have been published and are thus in the public domain), which will ensure that reagent costs will remain close to the manufacturing costs.
Development of tandem MS assays for additional enzymes
Given the recent developments in the use of enzyme replacement therapy and cord blood transplantation for the treatment of other lysosomal storage disorders (Harmatz et al 2004; http://www.biomarinpharm.com, Muenzer et al 2002), we are currently developing tandem MS assays for Hunter syndrome, Maroteaux–Lamy syndrome and metachromic leukodystrophy. A tandem MS assay for Tay-Sachs disease has recently been developed (M. H. Gelb, unpublished).
We are also developing tandem MS enzyme assays for other genetic diseases that have been difficult to diagnose at the biochemical level. One example is the enzymes that comprise the haem biosynthetic pathway. Deficiency of these enzymes give rise to the various types of porphyrias. Direct enzyme assays are rarely carried out in clinical laboratories because of a lack of robust and easily executed assays. More often, diagnoses are evaluated by measuring the level of porphyrin precursors in urine samples. Such analyses require 24 h urine collection during periods in which severe symptoms occur. This compromises rapid diagnosis and is also difficult because of problems with patient compliance.
Summary of the advantages of tandem MS for clinical enzyme activity profiling
In closing, we emphasize the following points to summarize the advantages of using tandem MS for providing early diagnosis information.
Tandem MS provides a powerful means of direct assay of the enzyme of interest. While the alternative approach of spotting a deficient enzyme activity by looking for an accumulated metabolite is also useful (for example in the screening for aminoacidurias and fatty acid oxidation diseases), for many diseases, including many lysosomal storage disorders, a suitable biomarker has not been identified despite extensive efforts by several laboratories. For example, the tetrasaccharide composed of four glucose residues was once considered as a possible indicator of Pompe disease, but subsequent studies have led to the conclusion that this marker is not adequate (Rozaklis et al 2002; and personal conversations with D. Millington, Duke University).
Tandem MS can quantify metabolites in complex samples without the need for time-consuming chromatographic steps.
Highly accurate quantitative information is obtained with the use of a suitable internal standard.
Tandem MS is highly multiplexable. For example, if 20 amino acids and dozens of fatty acyl carnitines can be detected in a single injection into the tandem mass spectrometer, it should be possible to quantify the products of 50–100 enzymes in a single run.
Tandem MS is an extremely sensitive technique, requiring only a few microlitres of blood or a few thousand fibroblasts.
Tandem MS is a fast technique, requiring less than 1–2 min per patient sample.
Reagents for tandem MS assays are needed only in minute amounts, and thus costs are kept to a minimum.
Tandem MS allows the use of the natural substrate or a close analogue. There are known cases of mutant enzymes that display low activity on the artificial substrate but not on the natural substrate.
It is our opinion that most enzymes can be assayed using tandem MS. Even assays for isomerases, for which the substrate and product have the same mass, can be developed as we have shown (Li et al 2003). In those few cases where the product cannot be analysed directly by tandem MS, it should be possible to develop a simple derivatization procedure to provide an analyte that is readily quantified by tandem MS.
Acknowledgments
We are grateful to J. Keutzer, H. Kallwass, K. Zhang and W.-L. Chuang at Genzyme Corporation for continued support and helpful discussions. This work was also supported by a grant from the National Institutes of Health (DK067859).
All studies with human subjects were approved by the IRB at the University of Washington and the Laboratory of Neurochemistry.
Footnotes
Competing interests: None declared
Presented at the 42nd Annual Meeting of the SSIEM, Paris, 6–9 September 2005
The authors M.H.G, F.T. and C.R.C. are co-PIs that direct the research described at the University of Washington. The contribution of N.A.C. was to provided many of the dried blood spots used in this study. The research at the University of Washington was funded by grants from the National Institutes of Health (DK67859) and from Genzyme Corporation. All authors are independent of the sponsors.
References
- Chace DH, Kalas TA. A biochemical perspective on the use of tandem mass spectrometry for newborn screening and clinical testing. Clin Biochem. 2005;38:296–309. doi: 10.1016/j.clinbiochem.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Chamoles NA, Blanco M, Gaggioli D, Casentini C. Tay–Sachs and Sandhoff diseases: enzymatic diagnosis in dried blood spots on filter paper: retrospective diagnoses in newborn-screening cards. Clin Chim Acta. 2002;318:133–137. doi: 10.1016/s0009-8981(02)00002-5. [DOI] [PubMed] [Google Scholar]
- Desnick RJ. Enzyme replacement and enhancement therapies for lysosomal diseases. J Inherit Metab Dis. 2004;27:385–410. doi: 10.1023/B:BOLI.0000031101.12838.c6. [DOI] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale MJ, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N Engl J Med. 2005;352:2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- Gerber SA, Scott CR, Turecek F, Gelb MH. Direct profiling of multiple enzyme activities in human cell lysates by affinity chromatography/electrospray ionization mass spectrometry: application to clinical enzymology. Anal Chem. 2001a;73:1651–1657. doi: 10.1021/ac0100650. [DOI] [PubMed] [Google Scholar]
- Gerber SA, Turecek F, Gelb MH. Design and synthesis of substrate and internal standard conjugates for profiling enzyme activity in the Sanfilippo syndrome by affinity chromatography/electrospray ionization mass spectrometry. Bioconj Chem. 2001b;12:603–615. doi: 10.1021/bc010007l. [DOI] [PubMed] [Google Scholar]
- Harmatz P, Whitley CB, Waber L, et al. Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux–Lamy syndrome) J Pediatr. 2004;144:574–580. doi: 10.1016/j.jpeds.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Kallwass H, Carr C, Chamoles NA, Keutzer J. Diagnosis of Pompe disease by enzyme assay in dried blood spots. J Inherit Metab Dis. 2005;28(Supplement 1):193. [Google Scholar]
- Kebarle P, Ho Y. On the mechanism of eletrospray mass spectrometry. In: Cole RB, editor. Electrospray Ionization Mass Spectrometry, Fundamental, Instrumentation & Applications. New York: Wiley; 1997. pp. 4–63. [Google Scholar]
- Koster JF, Slee RG, Hulsmann WC. The use of leucocytes as an aid in the diagnosis of glycogen storage disease type II (Pompe’s disease) Clin Chim Acta. 1974;51:319–325. doi: 10.1016/0009-8981(74)90319-2. [DOI] [PubMed] [Google Scholar]
- Krivit W. Allogeneic stem cell transplantation for the treatment of lysosomal and peroxisomal metabolic diseases. Semin Immunol. 2004;26:119–132. doi: 10.1007/s00281-004-0166-2. [DOI] [PubMed] [Google Scholar]
- Li Y, Ogata Y, Freeze HH, Scott CR, Turecek F, Gelb MH. Affinity capture and elution/electrospray ionization mass spectrometry assay of phosphomannomutase and phosphomannose isomerase for the multiplex analysis of congenital disorders of glycosylation types Ia and Ib. Anal Chem. 2003;75:42–48. doi: 10.1021/ac0205053. [DOI] [PubMed] [Google Scholar]
- Li Y, Brockman K, Turecek F, Scott CR, Gelb MH. Tandem mass spectrometry for the direct assay of enzymes in dried blood spots: application to newborn screening for Krabbe disease. Clin Chem. 2004a;50:638–640. doi: 10.1373/clinchem.2003.028381. [DOI] [PubMed] [Google Scholar]
- Li Y, Scott CR, Chamoles NA, et al. Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin Chem. 2004b;50:1785–1796. doi: 10.1373/clinchem.2004.035907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miebach E. Enzyme replacement therapy in mucopolysaccharidosis type I. Acta Paediatr Suppl. 2005;94:58–60. doi: 10.1111/j.1651-2227.2005.tb02114.x. [DOI] [PubMed] [Google Scholar]
- Muenzer J, Lamsa JC, Garcia A, Dacosta J, Garcia J, Treco DA. Enzyme replacement therapy in mucopolysaccharidosis type II (Hunter syndrome): a preliminary report. Acta Paediatr Suppl. 2002;91:98–99. doi: 10.1111/j.1651-2227.2002.tb03115.x. [DOI] [PubMed] [Google Scholar]
- Rozaklis T, Ramsay SL, Whitfield PD, Ranieri E, Hopwood JJ, Miekle PJ. Determination of oligosaccharides in Pompe disease by electrospray ionization tandem mass spectrometry. Clin Chem. 2002;48:131–139. [PubMed] [Google Scholar]
- Scott CR, Gelb MH, Turecek F, Li Y, Ogata Y, Wang D. Development of tandem mass spectrometry for the detection of lysosomal storage diseases from newborn blood spots. Am J Hum Genet. 2003;73:1660. [Google Scholar]
- Shah JS, Elliott PM. Fabry disease and the heart: an overview of the natural history and the effect of enzyme replacement therapy. Acta Paediatr Suppl. 2005;94:11–14. doi: 10.1111/j.1651-2227.2005.tb02103.x. [DOI] [PubMed] [Google Scholar]
- Shin YS, Endres W, Unterreithmeier J, Rieth M, Schaub J. Diagnosis of Pompe’s disease using leukocyte preparations. Kinetic and immunological studies of 1,4-α-glucosidase in human fetal and adult tissues and cultured cells. Clin Chem Acta. 1985;148:9–19. doi: 10.1016/0009-8981(85)90295-5. [DOI] [PubMed] [Google Scholar]
- Umapathysivam K, Hopwood JJ, Meikle PJ. Determination of acid alpha-glucosidase activity in blood spots as a diagnostic test for Pompe disease. Clin Chem. 2001;47:1378–1383. [PubMed] [Google Scholar]
- Wang D, Eadala B, Sadílek M, et al. Tandem mass spectrometry for newborn screening of mucopolysaccharidosis type I using dried blood spots. Clin Chem. 2005;51:898–900. doi: 10.1373/clinchem.2004.047167. [DOI] [PubMed] [Google Scholar]
- Wilcken B, Wiley V, Hammond J, Carpenter K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med. 2003;348:2304–2312. doi: 10.1056/NEJMoa025225. [DOI] [PubMed] [Google Scholar]