

Abstract
Purpose
AIPL1 mutations cause the severe inherited blindness Leber congenital amaurosis (LCA). The similarity of AIPL1 to tetratricopeptide repeat (TPR) cochaperones that interact with the chaperone Hsp90 and the ability of AIPL1 to suppress the aggregation of NUB1 fragments in a chaperone-like manner suggest that AIPL1 might function as part of a chaperone heterocomplex facilitating retinal protein maturation. In this study the interaction of AIPL1 with molecular chaperones is revealed and functionally characterized.
Methods
AIPL1-interacting proteins were identified using a yeast two-hybrid system, and the effect of AIPL1 pathogenic mutations and sequence requirements mediating the identified interactions were investigated. The interactions were validated by a comprehensive set of biochemical assays, and the ability of the AIPL1-binding partners to cooperate with AIPL1 in the suppression of NUB1 fragment aggregation was assessed.
Results
AIPL1 interacts with the molecular chaperones Hsp90 and Hsp70. Mutations within the TPR domain of AIPL1 or removal of the chaperone TPR acceptor site abolished the interactions. Importantly, LCA-causing mutations in AIPL1 also compromised these interactions, suggesting that the essential function of AIPL1 in photoreceptors may involve the interaction with Hsp90 and Hsp70. Examination of the role of these chaperones in AIPL1 chaperone activity demonstrated that AIPL1 cooperated with Hsp70, but not with Hsp90, to suppress the formation of NUB1 inclusions.
Conclusions
These findings suggest that AIPL1 may cooperate with both Hsp70 and Hsp90 within a retina-specific chaperone heterocomplex and that the specialized role of AIPL1 in photoreceptors may therefore be facilitated by these molecular chaperones.
Mutations in the gene coding for AIPL1 cause Leber congenital amaurosis (LCA), the most severe form of inherited retinal dystrophy with the earliest age of onset.1 A combination of in vitro and in vivo studies has begun to reveal the potential roles of AIPL1 in retina. Yeast two-hybrid analysis first identified the interaction of AIPL1 with the proteasome-associated protein NUB1.2 NUB1 and a longer splice variant, NUB1L, target the unconjugated and protein-conjugated small ubiquitin-like proteins Nedd8 and FAT10 for proteasomal degradation.3-6 AIPL1 was shown to modulate the subcellular distribution of NUB1 from the nucleus toward the cytoplasm.7 N-terminal and C-terminal fragments of NUB1 form small perinuclear and large intranuclear inclusions, respectively, the formation of which is suppressed by AIPL1 in a chaperone-like manner.7 Subsequently, AIPL1 was shown to interact with proteins with an isoprenylation motif and to enhance protein farnesylation.8 Murine models of LCA with reduced or abolished AIPL1 expression revealed a posttranscriptional decrease in the levels of cGMP phosphodiesterase (PDE).9,10 However, the molecular mechanisms of disease pathogenesis caused by AIPL1 mutations have yet to be fully elucidated.
AIPL1 is a tetratricopeptide repeat (TPR) protein that shares 49% amino acid identity with human XAP2 (ARA9 or murine AIP).1,11-13 XAP2 is involved in the Hsp90-mediated targeted nuclear translocation and transactivation of the aryl hydrocarbon receptor (AhR).12-14 A large number of signal transduction proteins in addition to AhR, including the nuclear receptors of steroid hormones and proto-oncogenic protein kinases, require the coordinated activities of the Hsp70 and Hsp90 molecular chaperones and their associated cochaperones for folding and conformational activation. The Hsp90-associated multicomponent complex includes cochaperones (XAP2, PP5, and the large immunophilins CyP40, FKBP51, and FKBP52), which competitively recognize the highly conserved Hsp90 C-terminal MEEVD TPR acceptor structural motif through their TPR domains.15-21
TPR is a degenerate 34-amino acid motif.22 Crystal structures of a number of cochaperone TPR domains demonstrate that the overall architecture of the TPR domain is conserved.23-27 High-resolution crystal structures of the TPR domains of the cochaperone Hop, in complex with the C-terminal heptapeptide of Hsp70 (TIEEVD) and the C-terminal pentapeptide of Hsp90 (MEEVD), predicted that amino acids in the TPR domains are involved in tight electrostatic interactions with the main chain carboxylate of the ultimate aspartic acid residue in the conserved EEVD sequence of the bound peptide, forming a two-carboxylate clamp.24 A similar network of electrostatic interactions was shown to mediate the interaction of the FKBP52 TPR domain with the Hsp90 C-terminal pentapeptide.27 The conservation of the TPR domain in AIPL1, together with the similarity of AIPL1 to XAP2, has led to the suggestion that AIPL1 may function as a TPR cochaperone involved in Hsp90-mediated signal transduction.1,28,29 However, an association of AIPL1 with Hsp70/Hsp90-dependent protein folding machinery or a direct interaction of AIPL1 with either protein has yet to be demonstrated.
Here, we report the use of a cytoplasmic-based yeast two-hybrid system (CytoTrapXR; Stratagene, La Jolla, CA), followed by protein binding assays to identify and confirm novel interacting partners for AIPL1. This study is the first to describe the specific association of AIPL1 with Hsp90 and Hsp70. We also attempt to show the effects these proteins might have in cells by assessing their behavior in the suppression of NUB1 fragment inclusions. Overall, our data suggest that AIPL1 may use components of the Hsp70 and Hsp90 chaperone machineries to fulfill its important photoreceptor-specific functions.
MATERIALS AND METHODS
Plasmids
pGEX-2T-AIPL1, pCMV-Tag3C-AIPL1, pEGFP-C1-NUB1-N, and pEGFP-C2-NUB1-C have been described previously.7,30 pCMV5-70 was a kind gift from Harm H. Kampinga (University of Gröningen, Netherlands). Full-length human AIPL1 cDNA was cloned into pSos (Stratagene) to produce pSos-AIPL1. Mutations were engineered in pSos-AIPL1, pMyr-Hsp90α(204-733), and pMyr-Hsp70(262-650) using a mutagenesis kit (QuikChange Site-Directed Mutagenesis kit; Stratagene) according to the manufacturer’s instructions. All plasmid constructs were confirmed by sequencing (Big Dye Terminator; PerkinElmer Life Sciences, Wellesley, MA).
Antibodies
Rabbit polyclonal antisera anti-AIPL1 (Ab-hAIPL1) and anti-NUB1 (Ab-hNUB1) have been described previously.7,30 Monoclonal anti-c-myc clone 9E10 (mouse ascites fluid) and monoclonal anti-Hsc70/Hsp70 clone BRM-22 (mouse ascites fluid) were purchased from Sigma (St. Louis, MO). Mouse anti-Hsp70 monoclonal antibody (SPA810), rabbit anti-Hsc70 polyclonal antibody (SPA816), and rabbit anti-Hsp90 polyclonal antibody (SPA846) were purchased from Stressgen Bioreagents (Victoria, BC, Canada). Mouse anti-Sos was purchased from BD Biosciences (Palo Alto, CA). Horseradish peroxidase-coupled goat anti-rabbit or goat anti-mouse secondary antibodies were purchased from Pierce Biotechnology (Rockford, IL).
Yeast Two-Hybrid Screen
The two-hybrid system (CytoTrapXR; Stratagene) was used with modifications.31 A bovine retinal cDNA library fused to a myristoylation sequence in the pMyr vector (Stratagene) was cotransformed with the bait plasmid pSos-AIPL1 into the yeast Saccharomyces cerevisiae temperature-sensitive mutant strain cdc25Hα. Yeast were transformed using the lithium acetate method,32 left to recover at 24°C for 39 hours, and then transferred to 37°C. Yeast colonies that grew at the restrictive temperature of 37°C on synthetic media containing galactose (gal) but lacking leucine (−L) and uracil (−U) were considered putative positives and tested further. Positive colonies were cured of the pSos-AIPL1 plasmid and excluded from the screen if they were still able to grow at 37°C on gal (−U) plates. After this selection, pMyr-cDNA plasmids were isolated, sequenced, and retransformed with pSos-AIPL1 into cdc25Hα to confirm the interactions. Various pSos and pMyr constructs provided with the two-hybrid system (CytoTrap; Stratagene) were used as controls for positive and negative interactions. Cdc25Hα transformed with pSos-AIPL1, pMyr-Hsp90α(204-733), pMyr-Hsp70(262-650), or mutants thereof were grown in synthetic media, and protein expression was verified according to the manufacturer’s instructions by resolving equal amounts of yeast cell extracts by SDS-PAGE, followed by Western blotting with the anti-Sos, anti-Hsp90, and anti-Hsp70 antibodies, respectively. Protein concentrations were determined using the BCA Protein Assay Kit (Pierce).
Cell Culture
SK-N-SH neuroblastoma cells and COS7 kidney fibroblast-like cells were cultured in Dulbecco modified Eagle medium/F12 (1:1) (GlutaMAX1; Invitrogen, Paisley, UK) supplemented with 10% fetal bovine serum and penicillin/streptomycin (10,000 U/mL; 10,000 μg/mL). Y79 human retinoblastoma cells were maintained in suspension culture in RPMI 1640 and medium (GlutaMAX1; Invitrogen) supplemented with 20% fetal bovine serum and 50 μg/mL gentamicin (Sigma).
Size-Exclusion Chromatography
Y79 retinoblastoma cells resuspended in ice-cold PBS and protease inhibitor cocktail (PIC) for use with mammalian cell and tissue extracts (Sigma) were disrupted mechanically with a cell breaker (HGM Precision Engineering, Heidelberg, Germany) on ice and centrifuged at 13,000g and 4°C for 30 minutes. Cell lysates were filtered through a 0.2-μm filter (Nalgene, Rochester, NY) and applied to a gel filtration column (Superdex 200; GE Healthcare Life Sciences) equilibrated in PBS. The protein concentration of the Y79 lysates was determined using reagent (Protein Assay Dye Reagent Concentrate; Bio-Rad Laboratories, Hercules, CA) according to the manufacturer’s instructions. Adult human retina was homogenized in PBS and prepared for size-exclusion chromatography (SEC) as described for the Y79 lysates. Informed consent was provided for all samples, institutional review board approval was obtained, and the tenets of the Declaration of Helsinki were followed. SEC fractions were collected by isocratic elution (1 mL/min) in PBS, and aliquots of each fraction were resolved by SDS-PAGE. Endogenous NUB1, AIPL1, Hsp70, Hsc70, and Hsp90 were detected by Western blot analysis using enhanced chemiluminescence (GE Healthcare UK Ltd., Chalfont, St. Giles, UK). A calibration curve was obtained with standard proteins of known molecular mass (Amersham Pharmacia Biotech, Piscataway, NJ). The standards were thyroglobulin (Mr, 669 kDa), apoferritin (Mr, 443 kDa), β-amylase (Mr, 200 kDa), albumin (Mr, 150 kDa), and carbonic anhydrase (Mr, 66 kDa). Blue dextran (Mr, 2000 kDa) was used to determine the column exclusion volume. Human retinal homogenates and Y79 cell lysates were resolved on the precalibrated gel filtration column (Superdex 200; GE Healthcare Life Sciences).
Affinity Pull-Down Assays
Recombinant expression of GST and GST-AIPL1 was induced in a 250-mL culture volume of exponential growth phase Escherichia coli JM109 cells by the addition of isopropyl β-D-thiogalactoside (IPTG; Sigma) to a final concentration of 1 mM. Induction was continued overnight at 24°C before the E. coli cells were harvested and lysed by mild sonication on ice in PBS and PIC for use with bacterial cell extracts (Sigma). Cellular debris was removed by centrifugation (14,000g at 4°C for 30 minutes), and the supernatant was filtered through a 0.45-μm filter (Nalgene) and added to 1 mL of a 50% slurry of glutathione Sepharose 4B (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) in PBS. Affinity purification of GST and GST-AIPL1 was performed as previously described.33 The subsequent affinity pull-down assay was performed as previously described33 with the following modifications: purified GST and GST-AIPL1 were coupled to glutathione Sepharose 4B at a final concentration of 0.2 μM overnight at 4°C. The following morning, COS7 cells were lysed in n-dodecyl β-D-maltoside (DM; Sigma) lysis buffer (1% DM; 5 mM EDTA, pH 8; PIC for use with mammalian cell and tissue extracts; in 1× PBS), and the concentration of soluble protein was determined with reagent (Protein Assay Dye Reagent Concentrate; Bio-Rad Laboratories). Three hundred micrograms of COS7 cell extracts were precleared on glutathione Sepharose 4B for 1 hour at 4°C before addition to glutathione Sepharose 4B-coupled GST and GST-AIPL1. Endogenous Hsp70 and Hsp90 associated with GST-AIPL1 were detected with anti-Hsp70 and anti-Hsp90, respectively.
Immunoprecipitation
Neuroblastoma cells (5 × 105 SK-N-SH) were seeded per well in a six-well plate (BD Biosciences). Twenty-four hours later, pCMV5-70 (0.5 μg) was transiently transfected in the absence and in the presence of increasing amounts of pCMV-Tag3C-AIPL1 (0-1 μg), and with appropriate amounts of pCMV-Tag3C plasmid to maintain an equal amount (1.5 μg) of total DNA in each transfection, with the use of reagent (Lipofectamine Plus; Invitrogen). Twenty-four hours after transient transfection, the SK-N-SH cells were washed twice with ice-cold PBS and lysed in DM lysis buffer (300 μL/well) for 15 minutes on ice. The lysates were centrifuged (13,000g at 4°C for 30 minutes), and the supernatant was removed. Myc-AIPL1 was immunoprecipitated from the SK-N-SH lysates (250 μL) with anti-c-myc (2 μL mouse ascites fluid) overnight at 4°C. Protein G Sepharose 4 Fast Flow (GE Healthcare Bio-Sciences AB; 50 μL of 50% slurry) equilibrated in DM lysis buffer was added to the lysates for 3 hours at 4°C. The immune complex was washed five times in ice-cold DM lysis buffer, recovered by centrifugation, and eluted in sample buffer (0.0625 M Tris-HCl, pH 6.8; 2.5% SDS; 5% 2-mercaptoethanol; 10% glycerol; 0.25% bromophenol blue). Myc-AIPL1 and Hsp70 were detected in the input and immune complex with anti-AIPL1 and anti-Hsp70/Hsc70 or anti-Hsp70 by Western blot analysis. Where indicated, coimmunoprecipitation of Hsp70 with myc-AIPL1 from SK-N-SH cells was examined in the presence of increasing ionic strength or 5 mM adenosine triphosphate (ATP)/Mg2+ (Sigma).
NUB1 Inclusion Formation
Neuroblastoma cells (2 × 104 SK-N-SH) were seeded per well in eight-well chamber slides (Perminox; BD Biosciences). Twenty-four hours later, the cells were transiently transfected using reagent (Lipofectamine Plus; Invitrogen) with 50 ng pEGFP-C1-NUB1-N or pEGFP-C2-NUB1-C, in combination with pCMV-Tag3C-AIPL1, pCMV5-70, or both. pEGFP-C1-NUB1-N or pEGFP-C2-NUB1-C was cotransfected with the other plasmids at a ratio of 1:2 (50:100 ng). The total amount of DNA transfected (250 ng) was maintained constant by the addition of empty pCMV-Tag3C. Twenty-four hours after transient transfection, cells were fixed or treated with vehicle, 5 μM geldanamycin (GA; Sigma), or 50 μM MG132 (Biomol International, Exeter, UK) for 4 hours. After treatment with GA or MG132, the cells were fixed, and GFP-NUB1-N and GFP-NUB1-C inclusion formation was counted, as described previously.7 Unpaired Student’s t-test was used to determine levels of statistical significance. Cells were visualized with a laser scanning confocal microscope (LSM510; Carl Zeiss, Oberkochen, Germany). The percentage enhancement of the Hsp70 effect by AIPL1 in the presence of MG132 was calculated as the difference between the percentage inclusion suppression by both AIPL1 and Hsp70 and the percentage inclusion suppression by Hsp70 alone.
RESULTS
Identification and Characterization of AIPL1-Binding Proteins by Yeast Two-Hybrid Analysis
To identify potential binding partners of AIPL1, we used full-length human AIPL1 (amino acids 1-384) as bait to screen a bovine retinal cDNA library using the yeast two-hybrid system (CytoTrapXR; Stratagene). This system enables the identification of protein interactions in the cytoplasm based on the recruitment of the hSos-fusion to the membrane and subsequent complementation in a Ras temperature-sensitive mutant yeast strain, allowing cell growth at the restrictive temperature. Conventional yeast two-hybrid systems are based on the modular composition of transcription factors and require the nuclear localization of the interacting partners to obtain reporter gene expression. Each of these systems may identify novel interacting partners because of the conformational differences and stabilities of the fusion proteins expressed in each system, their subcellular targeting, and amenability to the different subcellular localizations. Because AIPL1 is localized predominantly in the cytoplasm of cells and photoreceptors,30 the yeast two-hybrid system (CytoTrapXR; Stratagene) was used in this study to identify novel proteins that interact with AIPL1 in the cytoplasm.
This screen confirmed NUB1 as an AIPL1-interacting partner (data not shown). However, proteins with a C-terminal CAAX prenylation motif8 were not detected as putative interactors in the yeast two-hybrid system (CytoTrapXR; Stratagene) screen for reasons that were unclear but that might have reflected the myristoylation membrane targeting of the bait in contrast to normal cellular sites for farnesylation. Importantly, a novel interaction between AIPL1 and molecular chaperones was identified. Ten cDNAs expressing Hsp90 and one expressing Hsp70 were identified after the selection of yeast growth at the restrictive temperature of 37°C in a screen of approximately 1 × 106 colonies. Nine of the Hsp90 cDNAs were identified as two different clones of Hsp90α (corresponding to human HSP90AA1) encoding amino acid residues 204 to 733 or 426 to 733 and designated Hsp90α(204-733) and Hsp90α(426-733), respectively. One clone was identified as Hsp90β (corresponding to human HSP90AB1), including residues 198 to 724 (Hsp90β[198-724]), and one clone was identified as Hsp70 (corresponding to human HSPA8) including residues 262 to 650 (Hsp70[262-650]).
The Hsp90α(204-733) and Hsp70(262-650) clones were used in directed two-hybrid interactions. Both Hsp90 and Hsp70 were able to grow at the restrictive temperature of 37°C when cotransformed with pSos-AIPL1 but were unable to grow when cotransformed with pSos alone (Fig. 1). The growth of Hsp90 was comparable with the growth of the pMyr-SB + pSos positive control; however, Hsp70 growth with pSos-AIPL1 was slower than Hsp90 or pMyr-SB (Fig. 1).
Figure 1.

Interaction of AIPL1 with Hsp90 and Hsp70 in vivo. Yeast (cdc25Hα) was cotransformed with pMyr-SB and pSos (positive control), and either empty pSos or pSos-AIPL1 with pMyr-Hsp70(262-650) or pMyr-Hsp90α(204-733) and were grown in selective media at 24°C or at 37°C.
The ability of various pSos-AIPL1 mutants to interact with Hsp90 and Hsp70 was assessed (Fig. 2). Sequence changes that have been associated with LCA pathogenesis were tested (H82Y, A197P, C239R, G262S, W278X, R302L).1,34,35 G262S and R302L were able to interact with both molecular chaperones, similar to the wild-type protein, whereas H82Y appeared to have very slightly reduced growth with Hsp70. In contrast, A197P, C239R, and W278X did not enable growth at 37°C (Figs. 2A, 2B). Each of the AIPL1 pathogenic mutant hSos fusion proteins was expressed at similar levels in equal amounts of cdc25Hα yeast cell extracts, with the exception of R302L, which was more abundantly expressed than wild-type AIPL1, and W278X, which could not be detected (Fig. 2C).
Figure 2.

Interaction of AIPL1 pathogenic, TPR, and domain-mapping mutants with Hsp90 or Hsp70 in vivo. (A) Yeast (cdc25Hα) was cotransformed with pMyr-SB and pSos (positive control), empty prey vector pMyr and pSos-AIPL1 (negative control), and pSos-AIPL1 or the pSos-AIPL1 mutants with pMyr-Hsp70(262-650) or (B) pMyr-Hsp90α(204-733). Serial dilutions (dilution factor 1-203) were tested for their ability to grow at 37°C or 24°C. (C) Yeast was transformed with pSos-AIPL1, the pSos-AIPL1 mutants, or pSos alone, and the protein expression was assessed by Western blot analysis. *Sequence changes associated with LCA pathogenesis.
Three additional sequence variants were engineered in specific functional domains. Lysine 265 was mutated to alanine (K265A). This residue is topologically equivalent to PP5 (K97A), murine AIP (K266A), CyP40 (K308A), FKBP51 (K352A), and FKBP52 (K354A). The lysine residue conserved at this position in the TPR domain of each protein is required for interaction with the C-terminal MEEVD sequence in Hsp90.36-39 A further change (E317X) was engineered based on the observed loss of Hsp90 binding in truncation mutants of FKBP51 (N404) and FKBP52 (N406) that removed sequences C-terminal to the TPR domain core structure.25,39 The topologically equivalent residue in AIPL1 was mutated (E317X) to test the requirement for sequences downstream of the AIPL1 TPR core domain. Finally, the C terminus of human AIPL1 (residues 329-384) is imperfectly conserved in primates but absent from nonprimates.40 The AIPL1 mutant Q329X was engineered to assess the importance of this region for the chaperone interactions.
The AIPL1 TPR mutant K265A was expressed at levels equivalent to wild-type AIPL1 (Fig. 2C). However, the interaction with Hsp90 and Hsp70 was reduced compared with the interaction with wild-type AIPL1 (Figs. 2A, 2B). The growth of the AIPL1 TPR mutant E317X with Hsp90 and Hsp70 was unaffected (Fig. 2A). Removal of the AIPL1 primate-specific region (Q329X) severely reduced the expression of AIPL1 in cdc25Hα cells and consequently failed to support growth at the restrictive temperature, suggesting that the primate-specific region of human AIPL1 may be required for the stable expression of human AIPL1 as part of an hSos-fusion protein (Fig. 2).
To probe further the TPR dependence of the AIPL1 interaction with Hsp90 and Hsp70, the C-terminal Hsp90 MEEVD (pMyr-Hsp90α(ΔMEEVD)) and Hsp70 TIEEVD (pMyr-Hsp70(ΔTIEEVD)) TPR acceptor motifs were truncated (Fig. 3). In directed two-hybrid interactions, the growth of yeast cotransformed with pSos-AIPL1 and pMyr-Hsp90α(ΔMEEVD) or Hsp70(ΔTIEEVD) was severely reduced compared with that of Hsp90α(204-733) or Hsp70(262-650), respectively, even though Hsp90α(204-733) and Hsp70(262-650) were expressed at levels similar to those of the wild-type proteins in cdc25Hα yeast cell extracts (Figs. 3A, 3B). These data support a TPR-mediated binding of AIPL1 to Hsp90 and Hsp70 and show that disease-causing mutations in AIPL1 can disrupt this interaction.
Figure 3.
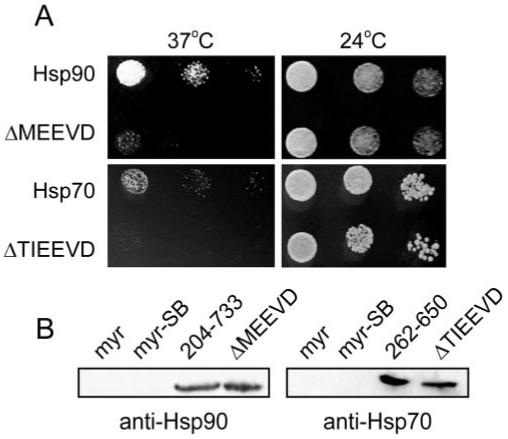
Interaction of AIPL1 with Hsp90 and Hsp70 on truncation of the chaperone C-terminal TPR acceptor site. (A) Yeast was cotransformed with pSos-AIPL and pMyr-Hsp90α(204-733), pMyr-Hsp90α(ΔMEEVD), pMyr-Hsp70(262-650), or pMyr-Hsp70(ΔTIEEVD). The ability of the yeast to grow at the restrictive temperature of 37°C and the permissive temperature of 24°C was assessed. (B) The expression of the chaperone wild-type and C-terminal truncation mutants was assessed by Western blot analysis.
Composition of the AIPL1 Chaperone Heterocomplex in Y79 Retinoblastoma Cell Lysates
To assess a potential heterocomplex of endogenous AIPL1 and the molecular chaperones, Y79 retinoblastoma cells were mechanically disrupted, and the crude cell fraction was separated by size exclusion chromatography. Y79 cells express endogenous AIPL1 and have been described to share several photoreceptor-like properties.41 The fractionation of AIPL1, NUB1, and the molecular chaperones Hsp90, Hsc70, and Hsp70 was examined.
AIPL1 in Y79 cell extracts was recovered in a major complex of 150 to 200 kDa (Fig. 4A). The elution of endogenous AIPL1 in a major complex of this size was confirmed by fractionation of human retinal homogenate (Fig. 4A). Two chaperone-associated fractionation peaks were detected in Y79 cell extracts (Fig. 4B). In the first major peak, endogenous AIPL1 cofractionated with the molecular chaperone Hsp70 (150-200 kDa; fraction number 25-26; Fig. 4B). In the second peak, the molecular chaperone Hsp90α/β fractionated to a molecular mass of approximately 400 to 650 kDa (fraction numbers 18-21; Fig. 4B). The AIPL1-binding protein NUB1 cofractionated with Hsp90α/β (fraction numbers 19-20). Longer exposures showed that AIPL1 and Hsp70 were detected in the Hsp90α/β high molecular mass fractions (Fig. 4B). Therefore, all the components examined were detected at varying levels in the major Hsp90α/β fractionation peak, including NUB1, AIPL1, and Hsc70/Hsp70. These data suggest the transient and dynamic association of the components of the AIPL1 chaperone heterocomplex in cells.
Figure 4.

Composition of the AIPL1 chaperone heterocomplex in Y79 retinoblastoma cells. (A) The soluble fraction from mechanically disrupted Y79 cells or from homogenized human retina, as indicated, was applied to a precalibrated gel filtration column. (B) Y79 soluble cell extracts were fractionated by SEC on a precalibrated gel filtration column, resolved by SDS-PAGE, and endogenous AIPL1, Hsp70, Hsc70, Hsp90, and NUB1 were detected by Western blot analysis. Fraction numbers and molecular weight markers (kDa) are shown above each panel.
Interaction of AIPL1 with the Molecular Chaperones In Vitro
In vitro biochemical assays were used to validate the interaction of AIPL1 with the molecular chaperones. The ability of GST-AIPL1 (Fig. 5A; Coomassie) to affinity trap Hsp90 and Hsp70 from COS7 cell lysates was examined. Hsp90 and Hsp70 were not trapped by the glutathione Sepharose or GST (Fig. 5A; Beads, GST). Endogenous Hsp90 and Hsp70 were affinity trapped by purified GST-AIPL1 (Fig. 5A; GST-AIPL1), suggesting that AIPL1 can associate with endogenous Hsp90 and Hsp70. Furthermore, Hsp70 was coimmunoprecipitated with AIPL1 in a concentration-dependent manner (Fig. 5B). myc-AIPL1 complexes were immunoprecipitated from transfected SK-N-SH cell lysates with the anti-c-myc 9E10 antibody. In the absence of AIPL1, Hsp70 was not detected in the immune complex (Fig. 5B). Hsp70 was specifically immunoprecipitated with increasing amounts of myc-AIPL1 (Fig. 5B). The interaction of Hsp70 with AIPL1 was disrupted by increasing ionic strength but was not affected by the presence of ATP/Mg2+, suggesting that wild-type AIPL1 was not recognized as a misfolded substrate for the Hsp70 chaperone function (Fig. 5C), consistent with the binding of AIPL1 to Hsp70 through its TPR motif.
Figure 5.
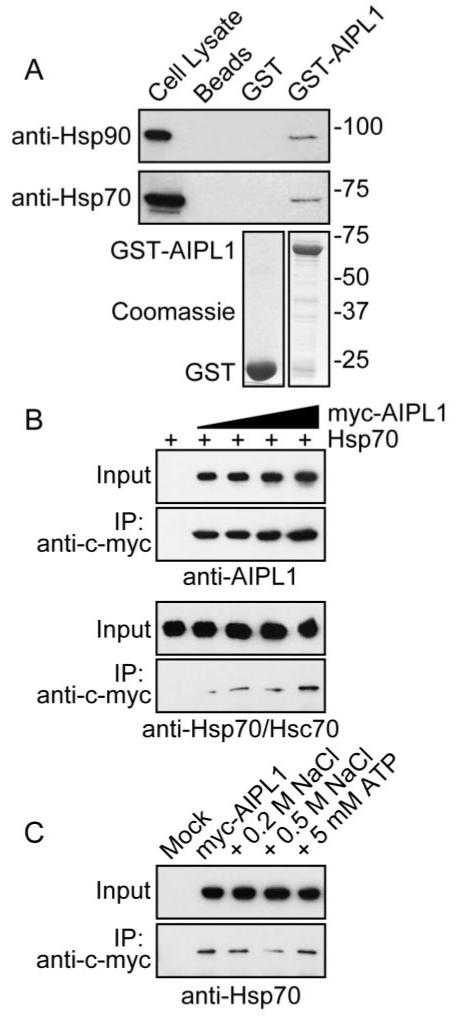
Characterization of AIPL1-chaperone interactions using in vitro protein-binding assays. (A) Affinity pull-down of endogenous Hsp90 and Hsp70 with GST-AIPL1. GST and GST-AIPL1 were affinity purified on glutathione Sepharose 4B (lower; Coomassie stain). COS7 cell lysates were applied to GST (GST) or GST-AIPL1 (GST-AIPL1) prebound to glutathione Sepharose 4B. Endogenous Hsp90 and Hsp70 pulled down with GST-AIPL1 were detected with anti-Hsp90 or anti-Hsp70 (upper; Western blot). Molecular weight markers are in kDa. (B) Coimmunoprecipitation of Hsp70 with myc-AIPL1. Myc-AIPL1 was immunoprecipitated from transfected SK-N-SH lysates with anti-c-myc 9E10. Myc-AIPL1 and Hsp70 were detected in the input and immune complex with anti-AIPL1 and anti-Hsp70/Hsc70, respectively. (C) Coimmunoprecipitation of Hsp70 with myc-AIPL1 from SK-N-SH cells was examined in the presence of increasing ionic strength or ATP/Mg2+.
Effect of GA on AIPL1 Chaperone Activity
To investigate the effect of these chaperones on AIPL1 function, the effect of chaperone inhibition and overexpression was tested on an in vitro AIPL1 assay previously established in our laboratory.7 AIPL1 is able to suppress the formation of inclusions arising from the aggregation of GFP-NUB1-N and GFP-NUB1-C fragments in a concentration-dependent manner.7 To probe the potential involvement of Hsp90 in this process, cells expressing these aggregation-prone fragments were treated with vehicle or the Hsp90 ATPase inhibitor GA in the absence and in the presence of AIPL1. GA treatment was limited to 4 hours to minimize the confounding influence of other molecular chaperones, such as Hsp70, that are induced after prolonged GA treatment.42 In the absence of AIPL1, there appeared to be an increase in the percentage of transfected cells with GFP-NUB1-N and GFP-NUB1-C inclusions after 4-hour treatment with vehicle or GA, suggesting that more inclusions were formed over time; however, this did not reach statistical significance (P > 0.1). In the presence of AIPL1, the formation of GFP-NUB1-N, and GFP-NUB1-C inclusions was suppressed, as shown previously (Fig. 6; AIPL1).7 The percentage of GFP-NUB1-N and GFP-NUB1-C inclusions in the presence of AIPL1 after treatment with vehicle or GA was not significantly different (P > 0.1). Therefore, the AIPL1-mediated suppression of the GFP-NUB1-N and GFP-NUB1-C inclusions did not appear to be affected by GA, suggesting that Hsp90 may not be involved in this process.
Figure 6.
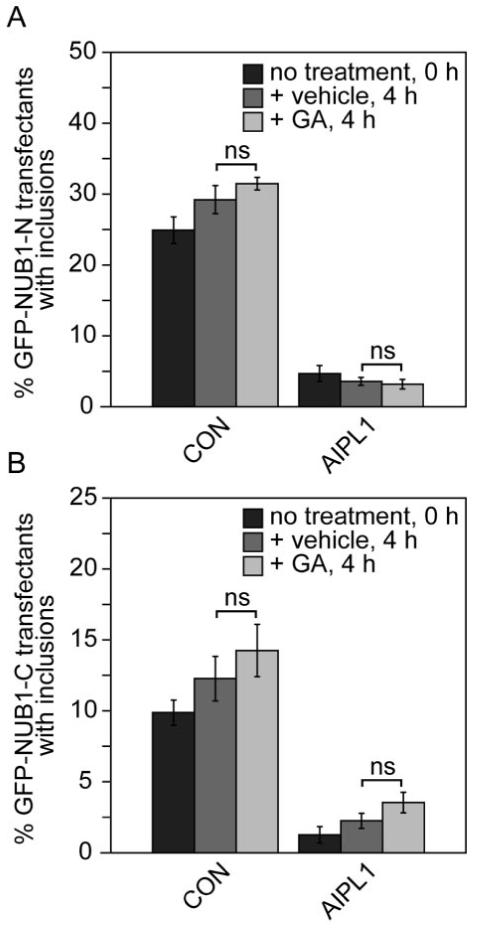
Effect of Hsp90 ATPase inhibitor GA on AIPL1 chaperone activity. Graphs of the inclusion incidence for the effect of GA on the AIPL1-mediated suppression of (A) GFP-NUB1-N inclusions and (B) GFP-NUB1-C inclusions. The percentage of GFP-NUB1-N or GFP-NUB1-C inclusions alone (CON) or in the presence of AIPL1 (AIPL1) was assessed in the absence of treatment (no treatment, 0 hours) and after 4-hour treatment with vehicle or 5 μM GA. Error bars are ± SD. Unpaired Student’s t-test was used to determine levels of statistical significance.
Cooperation of Hsp70 with AIPL1 in the Suppression of NUB1 Aggregation
Because Hsp70 is involved in the process of protein folding and degradation and because NUB1 fragments are likely targets for proteasomal degradation, we tested the hypothesis that AIPL1 and Hsp70 could cooperate to suppress the aggregation of the NUB1 fragments. We examined the AIPL1-mediated suppression of NUB1 fragment inclusion formation in the presence of Hsp70, with or without the proteasome inhibitor MG132 (Fig. 7).
Figure 7.

Participation of Hsp70 in AIPL1 chaperone activity. (A) Immunofluorescence confocal microscopy depicting the formation of GFP-NUB1-N (CON) (upper) or GFP-NUB1-C (CON) (lower) inclusions in the absence (−MG132) and in the presence (+MG132) of MG132 and cotransfected with AIPL1, Hsp70, or AIPL1 + Hsp70, as indicated. Scale bars, 10 μM. Cell counts depicting the percentage of (B) GFP-NUB1-N and (C) GFP-NUB1-C inclusions alone (CON) or in the presence of AIPL1, Hsp70, or AIPL1 + Hsp70 in the absence and presence of MG132. Error bars are ± SD. (D) Enhancement of the Hsp70 effect by AIPL1 and the AIPL1 mutants. Error bars are ± SD.
In the absence of MG132, GFP-NUB1-N formed multiple small inclusions that decorated the perinuclear region of transfected cells, whereas GFP-NUB1-C formed large intranuclear inclusions (Fig. 7A; −MG132; CON), as described previously.7 AIPL1 and Hsp70 were individually able to reduce the incidence of GFP-NUB1-N and GFP-NUB1-C inclusions; however, Hsp70 was less efficient than AIPL1 (Figs. 7B, 7C; −MG132; Hsp70; AIPL1). With the combination of AIPL1 and Hsp70, suppression of the GFP-NUB1-N and GFP-NUB1-C inclusions was already maximal and similar to the ability of AIPL1 alone to suppress inclusion formation (Figs. 7B, 7C; −MG132; AIPL1 + Hsp70).
In the presence of the proteasome inhibitor MG132, inclusions were observed in almost all the cells transfected with GFP-NUB1-N and GFP-NUB1-C (Figs. 7A, 7B; +MG132; CON). The dramatic increase in the GFP-NUB1-N and GFP-NUB1-C inclusions in the presence of MG132 supports previous observations that the GFP-NUB1-N and GFP-NUB1-C inclusions colocalize with ubiquitin and Hsp70 in cells, thus demonstrating that these fragments are targets for proteasomal degradation and form inclusions in response to aggregation and failed degradation.7 AIPL1 or Hsp70 alone was able to suppress the formation of GFP-NUB1-N and GFP-NUB1-C inclusions in the presence of MG132 (Figs. 7B, 7C; +MG132) to a lesser extent than in the absence of MG132. The suppression of GFP-NUB1-N and GFP-NUB1-C inclusions in the presence of MG132 was greater when both AIPL1 and Hsp70 were present. GFP-NUB1-N inclusions were observed in only approximately 30% of cells in the presence of AIPL1 and Hsp70 (Figs. 7A, 7B; +MG132; AIPL1 + Hsp70), and GFP-NUB1-C inclusions were observed in 23% of cells (Figs. 7A, 7C; +MG132; AIPL1 + Hsp70). Therefore, AIPL1 and Hsp70 together suppressed the aggregation of GFP-NUB1-N and GFP-NUB1-C more efficiently than either AIPL1 or Hsp70 alone, suggesting that AIPL1 and Hsp70 cooperate to suppress the formation of NUB1 fragment inclusions.
We next wanted to assess the ability of Hsp70 to cooperate with the AIPL1 mutants in the suppression of GFP-NUB1-N and GFP-NUB1-C inclusions. To determine the AIPL1 enhancement of the Hsp70 effect, the percentage suppression of inclusions mediated by Hsp70 alone was subtracted from the percentage inclusion suppression in the presence of both AIPL1 and Hsp70 (Fig. 7D). The data show that with the exception of W278X and E317X, each of the AIPL1 mutants was able to enhance the Hsp70 effect and to cooperate with Hsp70 in the suppression of GFP-NUB1-N inclusions (Fig. 7D). W278X and E317X were unable to enhance the Hsp70-mediated suppression of GFP-NUB1-N inclusions (enhancement of Hsp70 effect, 0%) and did not differ from the ability of Hsp70 alone to suppress inclusions. Interestingly, Q329X was significantly less efficient (P < 0.05) than wild-type AIPL1 in enhancing the Hsp70 effect, but it did not exhibit a complete absence of this activity as seen for the W278X and E317X C-terminal truncation mutants. In contrast, H82Y was significantly more efficient (P < 0.05) than wild-type AIPL1. These data correlate with previous studies on the effect of AIPL1 mutations on GFP-NUB1 aggregation that showed H82Y to be hyperactive and C-terminal truncations to have reduced activity (Q329X) or no activity (W278X, E317X).7,43
DISCUSSION
Full-length human AIPL1 was shown to interact with Hsp90 and Hsp70 by yeast two-hybrid analysis, and these interactions were validated by in vitro biochemical assays. Yeast two-hybrid analysis suggested that the AIPL1 interaction with Hsp70 appeared weaker than with Hsp90, and analysis of AIPL1 mutants showed that similar structural features were involved in the interaction of AIPL1 with both molecular chaperones.
Although the interaction of AIPL1 with Hsp90 and Hsp70 was expected based on its homology with certain well-characterized Hsp90-interacting immunophilins and the presence of a TPR domain, previous yeast two-hybrid studies did not identify an interaction of AIPL1 with either of these two chaperones.2,8 However, unlike conventional yeast two-hybrid systems, these (CytoTrap; Stratagene) interactions occurred in the cytoplasm rather than in the nucleus, eliminating the requirement for the bait fusion proteins to be targeted to the nucleus. The screen (CytoTrap; Stratagene) identified the well-characterized AIPL1 interactor NUB1 and the novel interactors Hsp70 and Hsp90, confirming the usefulness of system (CytoTrap; Stratagene) for the confirmation of previously detected interactions and the identification of novel interactors not amenable for detection by conventional nuclear two-hybrid systems.
Analysis of LCA disease-causing mutations in AIPL1 showed that A197P, C239R, and W278X disrupted the interaction with Hsp90 and Hsp70. AIPL1(W278X) undergoes misfolding and aggregation and has been shown to form SDS-insoluble inclusions.7 The AIPL1 pathogenic mutants A197P and C239R were expressed at levels comparable to wild-type AIPL1 in yeast but failed to interact with Hsp90 and Hsp70. In mammalian cells, A197P and C239R were similar to wild-type AIPL1 with respect to their subcellular distribution and their expression levels, but limited trypsin proteolysis suggested that A197P and C239R differ structurally compared with native AIPL1 protein.7,8 A structural model of the AIPL1 TPR domain predicts that A197 occurs in position 20 of helix B in TPR1 and that C239 occurs in position 10 of helix A in TPR2 (Fig. 8).8 These residues are highly conserved in the TPR motifs of the cochaperones, suggesting that these residues may be important for the TPR-mediated interaction of AIPL1 with the molecular chaperones (Fig. 8).
Figure 8.

Alignment of TPR domains of proteins that bind Hsp90 or Hsp70. Residues in bold are part of the TPR consensus and are required for the packing of adjacent TPR α-helices (helix A and helix B). TPR residues predicted to be involved in the formation of tight electrostatic interactions with the EEVD motif from the crystal structure of Hop TPR2A and TPR1 with the Hsp90 MEEVD and Hsp70 PTIEEVD, respectively, are outlined.24 Residues underlined are predicted to form a peptide-binding pocket through tight electrostatic interactions based on the crystal structure of FKBP52 with the Hsp90 MEEVD.27 The lysine residue, demarcated by an asterisk, is the TPR residue targeted in AIPL1(K265A). Circled residues are AIPL1 mutations in the TPR domain previously associated with pathogenesis and tested for Hsp90 binding in our yeast two-hybrid screen, including A197P, C239R, G262S, and W278X. The other mutations associated with pathogenesis tested in our screen (H82Y and R302L) are outside the region depicted.
The AIPL1 disease-associated amino acid substitutions H82Y, G262S, and R302L were able to interact with Hsp90 and Hsp70 with an apparent affinity similar to that of the wild-type fusion protein, suggesting that these substitutions do not severely disrupt the interaction of AIPL1 with the molecular chaperones. The H82Y and R302L amino acid changes are controversial in their disease-causing status.35,43 The AIPL1 H82Y mutation was first described as a heterozygous change in an LCA patient,34 and R302L may not be a disease-causing variant of AIPL1 but rather a rare benign variant given that nonconservative residues occur at this position in other species.35 Therefore, further patient/population DNA and protein analyses are needed to determine the status of these sequence changes.
Further analysis revealed the importance of the AIPL1 TPR domain in the interaction with molecular chaperones. The interaction of AIPL1(K265A) with Hsp90 and Hsp70 was diminished. Removal of the C-terminal Hsp90 MEEVD or Hsp70 TIEEVD TPR acceptor site also severely reduced the interaction with AIPL1. The identity of residues predicted to be involved in tight electrostatic interactions with the C-terminal TPR acceptor sites of Hsp90 and Hsp70 is almost perfectly conserved in the Hop, PP5, CyP40, FKBP51, FKBP52, and XAP2 TPR domains.36-39 Our data and the conservation of these residues in AIPL1 (Fig. 8) suggest that AIPL1 is a bona fide Hsp90 and Hsp70 TPR cochaperone.
The interaction of the AIPL1(E317X) with Hsp90 and Hsp70 was unaffected. In contrast, truncation of FKBP51 and FKBP52 at the topologically equivalent residue largely reduced Hsp90 binding and association with the progesterone receptor.39 These data suggest that sequences C-terminal to the AIPL1 TPR core domain are dispensable for the recognition of the molecular chaperones by AIPL1.
Removal of the AIPL1 primate-specific region severely reduced the expression of Q329X in yeast, suggesting that the primate-specific region of human AIPL1 may be required for the stable expression in our yeast two-hybrid screen. Interestingly, human AIPL1 lacking the last 56 amino acids was previously used in the GAL4 yeast two-hybrid screen and was able to interact with the putative interactors identified in that screen.8 This variation might have resulted from experimental differences between the GAL4 and CytoTrap (Stratagene) yeast two-hybrid systems, including differences in the fusion proteins expressed in each system and their subcellular targeting and stability. Evidence supporting the importance of the 56-amino acid polyproline-rich region in primate vision includes the finding that the mutations A336Δ2 and P351Δ12 cause LCA and autosomal dominant cone-rod dystrophy or juvenile retinitis pigmentosa, respectively.1,35
A number of other TPR cochaperones have been shown to interact with Hsp90 and Hsp70. The Hsp90-interacting TPR cochaperones XAP2 and CyP40 have been shown to interact with Hsc70.44,45 Conservation of the same network of electrostatic interactions directing the interaction of CyP40 with Hsp90 were required for the interaction with the C-terminal carboxylate of Hsc70, though CyP40, similar to AIPL1, showed preferential binding with Hsp90 over Hsc70.44
The interaction of AIPL1 with the molecular chaperones led us to assess whether these interactions might play a role in the AIPL1-mediated functional modulation of NUB1. Hsp70 functioned with AIPL1 in the chaperone-like suppression of NUB1 fragment aggregation on proteasomal inhibition. A chaperone-like activity has also been reported for FKBP52 and XAP2, each of which has been shown to suppress the thermal aggregation of rhodanese and citrate synthase.45-47 Hsc70 appeared to cooperate with XAP2 in this chaperone-like activity because the combination of XAP2 and Hsc70 together suppressed the aggregation of citrate synthase more efficiently than Hsc70 alone.45 Our data show that NUB1 fragments are substrates for proteasomal degradation and that wild-type AIPL1 was able to cooperate with Hsp70 to enhance the Hsp70-mediated proteasomal targeting of these misfolded fragments. We previously demonstrated that the AIPL1-mediated modulation of NUB1 nuclear translocation and the suppression of NUB1 inclusions require a C-terminal region of AIPL1.7 Here, we extend these findings and show that functional NUB1 modulation mediated by a C-terminal region of AIPL1 is similarly required for the effects seen in combination with Hsp70. Although the TPR-mediated interaction of AIPL1 with Hsp70 was reduced by the AIPL1 TPR mutations A197P, C239R, and K265A and the deletion of the Hsp70 TIEEVD motif, the cooperation of AIPL1 with Hsp70 in the suppression of NUB1 inclusions was affected by the AIPL1 C-terminal truncations. These data suggest that the distinct chaperone activities of AIPL1 and Hsp70 cooperate in the suppression of NUB1 inclusions dependent on a C-terminal region of AIPL1 and that this occurs independently of a TPR-mediated interaction of AIPL1 with Hsp70. The inhibition of the Hsp90 ATPase activity by GA did not affect the suppression of NUB1 fragment aggregation by AIPL1. Based on the model that GA treatment shifts the equilibrium of Hsp90 client proteins from Hsp90-mediated protein folding to Hsp70-mediated degradation,48 our data suggest that the NUB1 fragments are substrates for Hsp70-mediated protein degradation but are not true client proteins for the AIPL1-Hsp90 protein folding chaperone machinery. Although Hsp70-mediated proteasomal degradation targets misfolded proteins in general, the Hsp90 foldosome targets a specific subset of client proteins, including signal transduction nuclear receptors and specific signaling kinases, the identities of which are dependent on the Hsp90-associated cochaperone. Therefore, we suggest that though this model does not exclude NUB1 as a client for the AIPL1-Hsp90 folding machinery, a photoreceptor-specific AIPL1-Hsp90 client protein has yet to be identified.
In conclusion, although the physiological relevance of the interactions identified in this study requires further characterization in vivo, our data suggest that the specialized role of AIPL1 in photoreceptors may be modulated by the molecular chaperones Hsp90 and Hsp70, and this association may be important in the pathogenesis of LCA. Further experiments are needed to address whether AIPL1 functions directly as part of a photoreceptor-specific Hsp70-Hsp90 heterocomplex in vivo that could chaperone several proteins directly regulating their folding, stability, assembly into multiprotein complexes, or degradation. For example, murine models of LCA with partial or complete loss of AIPL1 have demonstrated a posttranscriptional decrease in the levels of the PDE holoenzyme, an essential component of visual phototransduction. AIPL1 may chaperone the biosynthesis or assembly of the PDE subunits or protect them from proteasomal degradation.9,10,28 It is possible that the regulation of PDE by AIPL1 may be mediated by the interaction of AIPL1 with the molecular chaperones Hsp70 and Hsp90. It will be important now to determine whether this is indeed the case and to characterize the association of AIPL1 with these molecular chaperones in photoreceptors and to dissect the potential involvement of these proteins in LCA pathogenesis.
Acknowledgments
The authors thank Tatyana Novoselova and Alison Hardcastle for practical assistance and advice with the CytoTrapXR yeast two-hybrid screening and H. H. Kampinga for providing the plasmid pCMV5 to 70. They also thank Action Medical Research and the Wellcome Trust for financial support.
Supported by Wellcome Trust Project Grant GR077929MA and Action Medical Research Grant SP4014.
Footnotes
Disclosure: J. Hidalgo-de-Quintana, None; R.J. Evans, None; M.E. Cheetham, None; J. van der Spuy, None
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
References
- 1.Sohocki MM, Bowne SJ, Sullivan LS, et al. Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat Genet. 2000;24:79–83. doi: 10.1038/71732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akey DT, Zhu X, Dyer M, et al. The inherited blindness associated protein AIPL1 interacts with the cell cycle regulator protein NUB1. Hum Mol Genet. 2002;11:2723–2733. doi: 10.1093/hmg/11.22.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kito K, Yeh ET, Kamitani T. NUB1, a NEDD8-interacting protein, is induced by interferon and down-regulates the NEDD8 expression. J Biol Chem. 2001;276:20603–20609. doi: 10.1074/jbc.M100920200. [DOI] [PubMed] [Google Scholar]
- 4.Kamitani T, Kito K, Fukuda-Kamitani T, Yeh ET. Targeting of NEDD8 and its conjugates for proteasomal degradation by NUB1. J Biol Chem. 2001;276:46655–46660. doi: 10.1074/jbc.M108636200. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka T, Kawashima H, Yeh ET, Kamitani T. Regulation of the NEDD8 conjugation system by a splicing variant, NUB1L. J Biol Chem. 2003;278:32905–32913. doi: 10.1074/jbc.M212057200. [DOI] [PubMed] [Google Scholar]
- 6.Hipp MS, Raasi S, Groettrup M, Schmidtke G. NEDD8 ultimate buster-1L interacts with the ubiquitin-like protein FAT10 and accelerates its degradation. J Biol Chem. 2004;279:16503–16510. doi: 10.1074/jbc.M310114200. [DOI] [PubMed] [Google Scholar]
- 7.van der Spuy J, Cheetham ME. The Leber congenital amaurosis protein AIPL1 modulates the nuclear translocation of NUB1 and suppresses inclusion formation by NUB1 fragments. J Biol Chem. 2004;279:48038–48047. doi: 10.1074/jbc.M407871200. [DOI] [PubMed] [Google Scholar]
- 8.Ramamurthy V, Roberts M, van den Akker F, Niemi G, Reh TA, Hurley JB. AIPL1, a protein implicated in Leber’s congenital amaurosis, interacts with and aids in processing of farnesylated proteins. Proc Natl Acad Sci U S A. 2003;100:12630–12635. doi: 10.1073/pnas.2134194100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramamurthy V, Niemi GA, Reh TA, Hurley JB. Leber congenital amaurosis linked to AIPL1: a mouse model reveals destabilization of cGMP phosphodiesterase. Proc Natl Acad Sci U S A. 2004;101:13897–13902. doi: 10.1073/pnas.0404197101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu X, Bulgakov OV, Wen XH, et al. AIPL1, the protein that is defective in Leber congenital amaurosis, is essential for the biosynthesis of retinal rod cGMP phosphodiesterase. Proc Natl Acad Sci U S A. 2004;101:13903–13908. doi: 10.1073/pnas.0405160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuzhandaivelu N, Cong YS, Inouye C, Yang WM, Seto E. XAP2, a novel hepatitis B virus X-associated protein that inhibits X transactivation. Nucleic Acids Res. 1996;24:4741–4750. doi: 10.1093/nar/24.23.4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carver LA, Bradfield CA. Ligand-dependent interaction of the aryl hydrocarbon receptor with a novel immunophilin homolog in vivo. J Biol Chem. 1997;272:11452–11456. doi: 10.1074/jbc.272.17.11452. [DOI] [PubMed] [Google Scholar]
- 13.Ma Q, Whitlock JP., Jr A novel cytoplasmic protein that interacts with the Ah receptor, contains tetratricopeptide repeat motifs, and augments the transcriptional response to 2,3,7,8-tetrachloro-dibenzo-p-dioxin. J Biol Chem. 1997;272:8878–8884. [PubMed] [Google Scholar]
- 14.Meyer BK, Pray-Grant MG, Vanden Heuvel JP, Perdew GH. Hepatitis B virus X-associated protein 2 is a subunit of the unliganded aryl hydrocarbon receptor core complex and exhibits transcriptional enhancer activity. Mol Cell Biol. 1998;18:978–988. doi: 10.1128/mcb.18.2.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Owens-Grillo JK, Czar MJ, Hutchison KA, Hoffmann K, Perdew GH, Pratt WB. A model of protein targeting mediated by immunophilins and other proteins that bind to hsp90 via tetratricopeptide repeat domains. J Biol Chem. 1996;271:13468–13475. doi: 10.1074/jbc.271.23.13468. [DOI] [PubMed] [Google Scholar]
- 16.Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 17.Dolinski KJ, Cardenas ME, Heitman J. CNS1 encodes an essential p60/Sti1 homolog in Saccharomyces cerevisiae that suppresses cyclophilin 40 mutations and interacts with Hsp90. Mol Cell Biol. 1998;18:7344–7352. doi: 10.1128/mcb.18.12.7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marsh JA, Kalton HM, Gaber RF. Cns1 is an essential protein associated with the hsp90 chaperone complex in Saccharomyces cerevisiae that can restore cyclophilin 40-dependent functions in cpr7Delta cells. Mol Cell Biol. 1998;18:7353–7359. doi: 10.1128/mcb.18.12.7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young JC, Obermann WM, Hartl FU. Specific binding of tetratricopeptide repeat proteins to the C-terminal 12-kDa domain of hsp90. J Biol Chem. 1998;273:18007–18010. doi: 10.1074/jbc.273.29.18007. [DOI] [PubMed] [Google Scholar]
- 20.Buchner J. Hsp90 & Co.—a holding for folding. Trends Biochem Sci. 1999;24:136–141. doi: 10.1016/s0968-0004(99)01373-0. [DOI] [PubMed] [Google Scholar]
- 21.Meyer BK, Perdew GH. Characterization of the AhR-hsp90-XAP2 core complex and the role of the immunophilin-related protein XAP2 in AhR stabilization. Biochemistry. 1999;38:8907–8917. doi: 10.1021/bi982223w. [DOI] [PubMed] [Google Scholar]
- 22.Lamb JR, Tugendreich S, Hieter P. Tetratrico peptide repeat interactions: to TPR or not to TPR? Trends Biochem Sci. 1995;20:257–259. doi: 10.1016/s0968-0004(00)89037-4. [DOI] [PubMed] [Google Scholar]
- 23.Das AK, Cohen PW, Barford D. The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein-protein interactions. EMBO J. 1998;17:1192–1199. doi: 10.1093/emboj/17.5.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheufler C, Brinker A, Bourenkov G, et al. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 25.Taylor P, Dornan J, Carrello A, Minchin RF, Ratajczak T, Walkinshaw MD. Two structures of cyclophilin 40: folding and fidelity in the TPR domains. Structure. 2001;9:431–438. doi: 10.1016/s0969-2126(01)00603-7. [DOI] [PubMed] [Google Scholar]
- 26.Sinars CR, Cheung-Flynn J, Rimerman RA, Scammell JG, Smith DF, Clardy J. Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes. Proc Natl Acad Sci U S A. 2003;100:868–873. doi: 10.1073/pnas.0231020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu B, Li P, Liu Y, et al. 3D structure of human FK506-binding protein 52: implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex. Proc Natl Acad Sci U S A. 2004;101:8348–8353. doi: 10.1073/pnas.0305969101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Spuy J, Cheetham ME. The chaperone function of the LCA protein AIPL1. In: Hollyfield JG, Anderson RE, LaVail MM, editors. Retinal Degenerative Diseases. New York: Plenum Press; 2005. p. 471. [Google Scholar]
- 29.van der Spuy J. Focus on molecules: the aryl hydrocarbon receptor interacting protein-like 1 (AIPL1) Exp Eye Res. 2006;83:1307–1308. doi: 10.1016/j.exer.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 30.van der Spuy J, Chapple JP, Clark BJ, Luthert PJ, Sethi CS, Cheetham ME. The Leber congenital amaurosis gene product AIPL1 is localized exclusively in rod photoreceptors of the adult human retina. Hum Mol Genet. 2002;11:823–831. doi: 10.1093/hmg/11.7.823. [DOI] [PubMed] [Google Scholar]
- 31.Evans RJ, Chapple JP, Grayson C, Hardcastle AJ, Cheetham ME. Assay and functional analysis of the ARL3 effector RP2 involved in X-linked retinitis pigmentosa. Methods Enzymol. 2005;404:468–480. doi: 10.1016/S0076-6879(05)04041-3. [DOI] [PubMed] [Google Scholar]
- 32.Gietz D, St Jean A, Woods RA, Schiestl RH. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van der Spuy J, Kana BD, Dirr HW, Blatch GL. Heat shock cognate protein 70 chaperone-binding site in the co-chaperone murine stress-inducible protein 1 maps to within three consecutive tetratricopeptide repeat motifs. Biochem J. 2000;345(pt 3):645–651. doi: 10.1042/0264-6021:3450645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heegaard S, Rosenberg T, Preising M, Prause JU, Bek T. An unusual retinal vascular morphology in connection with a novel AIPL1 mutation in Leber’s congenital amaurosis. Br J Ophthalmol. 2003;87:980–983. doi: 10.1136/bjo.87.8.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sohocki MM, Perrault I, Leroy BP, et al. Prevalence of AIPL1 mutations in inherited retinal degenerative disease. Mol Genet Metab. 2000;70:142–150. doi: 10.1006/mgme.2000.3001. [DOI] [PubMed] [Google Scholar]
- 36.Russell LC, Whitt SR, Chen MS, Chinkers M. Identification of conserved residues required for the binding of a tetratricopeptide repeat domain to heat shock protein 90. J Biol Chem. 1999;274:20060–20063. doi: 10.1074/jbc.274.29.20060. [DOI] [PubMed] [Google Scholar]
- 37.Bell DR, Poland A. Binding of aryl hydrocarbon receptor (AhR) to AhR-interacting protein: the role of hsp90. J Biol Chem. 2000;275:36407–36414. doi: 10.1074/jbc.M004236200. [DOI] [PubMed] [Google Scholar]
- 38.Ward BK, Allan RK, Mok D, et al. A structure-based mutational analysis of cyclophilin 40 identifies key residues in the core tetratricopeptide repeat domain that mediate binding to Hsp90. J Biol Chem. 2002;277:40799–40809. doi: 10.1074/jbc.M207097200. [DOI] [PubMed] [Google Scholar]
- 39.Cheung-Flynn J, Roberts PJ, Riggs DL, Smith DF. C-terminal sequences outside the tetratricopeptide repeat domain of FKBP51 and FKBP52 cause differential binding to Hsp90. J Biol Chem. 2003;278:17388–17394. doi: 10.1074/jbc.M300955200. [DOI] [PubMed] [Google Scholar]
- 40.Sohocki MM, Sullivan LS, Tirpak DL, Daiger SP. Comparative analysis of aryl-hydrocarbon receptor interacting protein-like 1 (Aipl1), a gene associated with inherited retinal disease in humans. Mamm Genome. 2001;12:566–568. doi: 10.1007/s003350020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reid TW, Albert DM, Rabson AS, et al. Characteristics of an established cell line of retinoblastoma. J Natl Cancer Inst. 1974;53:347–360. doi: 10.1093/jnci/53.2.347. [DOI] [PubMed] [Google Scholar]
- 42.Bagatell R, Paine-Murrieta GD, Taylor CW, et al. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of Hsp90-binding agents. Clin Cancer Res. 2000;6:3312–3318. [PubMed] [Google Scholar]
- 43.van der Spuy J, Munro PM, Luthert PJ, et al. Predominant rod photoreceptor degeneration in Leber congenital amaurosis. Mol Vis. 2005;11:542–553. [PubMed] [Google Scholar]
- 44.Carrello A, Allan RK, Morgan SL, et al. Interaction of the Hsp90 cochaperone cyclophilin 40 with Hsc70. Cell Stress Chaperones. 2004;9:167–181. doi: 10.1379/CSC-26R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yano M, Terada K, Mori M. AIP is a mitochondrial import mediator that binds to both import receptor Tom20 and preproteins. J Cell Biol. 2003;163:45–56. doi: 10.1083/jcb.200305051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bose S, Weikl T, Bugl H, Buchner J. Chaperone function of Hsp90-associated proteins. Science. 1996;274:1715–1717. doi: 10.1126/science.274.5293.1715. [DOI] [PubMed] [Google Scholar]
- 47.Pirkl F, Buchner J. Functional analysis of the Hsp90-associated human peptidyl prolyl cis/trans isomerases FKBP51, FKBP52 and Cyp40. J Mol Biol. 2001;308:795–806. doi: 10.1006/jmbi.2001.4595. [DOI] [PubMed] [Google Scholar]
- 48.Hohfeld J, Cyr DM, Patterson C. From the cradle to the grave: molecular chaperones that may choose between folding and degradation. EMBO Rep. 2001;2:885–890. doi: 10.1093/embo-reports/kve206. [DOI] [PMC free article] [PubMed] [Google Scholar]