

Abstract
The time-of-flight neutron Laue technique has been used to determine the location of hydrogen atoms in the enzyme D-xylose isomerase (XI). The neutron structure of crystalline XI with bound product, D-xylulose, shows, unexpectedly, that O5 of D-xylulose is not protonated but is hydrogen-bonded to doubly protonated His54. Also, Lys289, which is neutral in native XI, is protonated (positively charged), while the catalytic water in native XI has become activated to a hydroxyl anion which is in close proximity to C1 and C2, the molecular site of isomerization of xylose. These findings impact our understanding of the reaction mechanism.
D-xylose isomerase (XI) from Streptomyces rubiginosus binds two divalent metal ions and catalyzes the conversion of the aldo-sugars D-xylose or D-glucose to the keto-sugars D-xylulose and D-fructose, respectively. During the reaction hydrogen (H) transfer occurs. The ring of the cyclic substrate (D-xylose or D-glucose) is opened catalytically to give a linear-chain molecule that the enzyme can further act on. Isomerization from aldose to ketose involves the removal of H from C2 of the substrate and its transfer to C1 (1-3). Three possible mechanisms for this movement, involving a cis-ene diol intermediate (4, 5), a hydride shift (1, 2, 6, 7), or a metal-mediated hydride shift (3, 8), have been suggested.
XI is a crucial enzyme in sugar metabolism, with important commercial applications of topical interest, notably in the production of biofuels and high fructose corn syrup. Understanding the transfer of H or protons during the catalytic reaction will aid in protein engineering efforts to improve the industrial performance of XI. Since we first determined the structure of XI (9), structures relevant to its reaction mechanism have been studied extensively by X-ray crystallographic techniques. However, H atom positions have been difficult to locate by X-ray diffraction in complexes of XI with various substrate and product analogues, even at the high resolution of 0.94 Å (10). This problem of locating H atoms in proteins has been successfully addressed using neutron crystallography, but rarely with proteins as large as XI (which is a 172 kDa homotetramer) because of difficulties in obtaining large enough diffraction-quality crystals (11).
We have exploited the recently developed time-of-flight neutron Laue diffraction method (12, 13) to make neutron diffraction studies of XI a possibility, since large crystals had been obtained. Initially, we determined the 1.8 Å neutron structure of the native enzyme XI (10). We now present results of the 2.2 Å neutron structure of XI in complex with the reaction product D-xylulose; we designate this complex as “XI-xylulose”. In this study the enzyme was deuterated so that accessible and exchangeable H atoms (bound to O or N) were replaced by D, and the D-xylulose that was bound to the enzyme was perdeuterated (all H replaced by D during synthesis).
Neutron diffraction studies of this combination of a deuterated enzyme with perdeuterated product, described here, were done in order to reveal exactly where H atoms are located in the complex so that their transfer during the catalyzed reaction could be inferred. We stress that the ligand in the complex that we report here (D-xylulose) corresponds to that of the product of the reaction.
The great advantage of neutron crystallography is that D atoms (neutron scattering length 6.67 × 10−15 m) appear as strong positive peaks in neutron scattering density maps, thereby revealing the locations of isotopically exchanged H atoms, while H atoms (neutron scattering length −3.74 × 10−15 m) appear as negative troughs (and therefore can be distinguished from D). Both C and O (6.65 and 5.80 × 10−15 m, respectively) scatter about the same as D (6.67 × 10−15 m), while N (9.36 × 10−15 m) scatters more (14). The neutron scattering powers of Mg2+ (5.37 × 10−15 m) and Mn2+ (−3.75 × 10−15 m) are of opposite sign, so that a mixture of these, commonly found in XI, can give no apparent scattering.
Crystals of XI were grown and then soaked with buffers containing D2O rather than H2O (15). In this way all labile and accessible Hs in the enzyme were replaced by Ds. The crystals were then soaked with D2O solutions of perdeuterated D-xylose. This D-xylose became bound in the active site where it was enzymatically converted to D-xylulose. These crystals (grown at pH 7.6, pD 8.0) were used for the room temperature neutron data collection for XI-xylulose on the PCS at LANSCE (11).
The structure, for which O, N, C atomic coordinates had already been reported (8, 9, 10, 15), was determined with the software suite CNS (16), modified for neutron refinement (nCNS) (17). The metal ions were fixed in the same octahedral holes at the positions M1 (called the structural metal) and M2 (called the catalytic metal) as were found in other XI X-ray crystal structures (coordinates from 4XIS (8) were used). Mg2+, while not a relatively strong scatterer of neutrons, is a relatively strong scatterer of X rays and is therefore more accurately located using X-ray crystallographic techniques. Omit and difference nuclear density Fourier maps confirmed the exact location of a metal ion at the M1 position. However there was little density in those maps at the M2 position, possibly indicating a mixture of Mn2+ and Mg2+ or greater mobility of the metal ion at this position. Experimental details are given in the supplementary information.
Several interesting differences are observed when the neutron structures of XI-xylulose and native XI are compared. First, the catalytic metal ion-bound water molecule is D2O in native XI but a hydroxyl anion, OD−, in XI-xylulose (Figures 1a and b). Secondly, Lys289 has only two D atoms in native XI, while it is positively charged, having three D atoms, in XI-xylulose (Figure 2). Thirdly, in both the neutron structures of native XI and XI-xylulose, His54 is doubly protonated on N with the D on Nδ1 bound to deprotonated Asp57. Fourthly, in XI-xylulose His220 is only singly protonated on N and therefore can bind more firmly to the metal ion than it does in native XI where His220 is doubly protonated on N.
FIGURE 1.
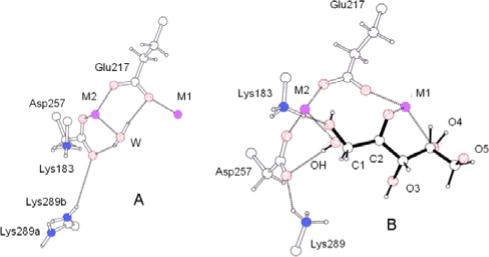
Environment of the catalytic, metal ion-bound, water molecule; D2O in native XI (a), OD−, in XI-xylulose (b).
FIGURE 2.
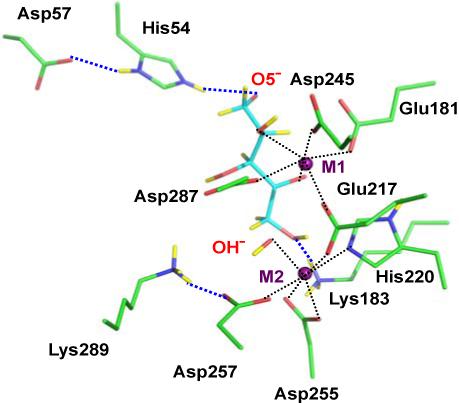
The active site of XI-xylulose. C, O, N, and D atoms are colored green, red, blue and yellow, respectively. H atoms have been left out for clarity.
The terminal hydroxyl group, O5 of the xylulose, is, to our surprise, deprotonated and binds to doubly protonated His54 in an Nε2-H+...O5− interaction (Figure 3). We carefully verified this negatively charged state of O5. Neither omit nor difference Fourier maps indicated the presence of a D atom on O5. Adjacent C4 has clear density for covalently bound D atoms in the Fourier maps, eliminating the possibility that O5 is disordered; the B-factor of O5 is found to be similar to that found for other atoms in D-xylulose. Asp57 is not protonated and is bound to His54 via protonated Nδ1, stabilizing the position of the latter. O5 accepts an H-bond from His54 and from a water molecule, but there are no acceptors nearby for it to donate an H to.
FIGURE 3.
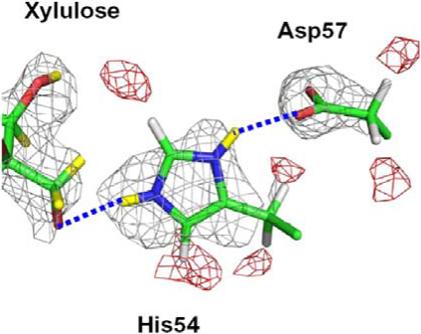
2FO-FC nuclear density, positive in grey (+1.2σ) and negative in red (−2.0σ), showing His54 doubly protonated. The displacement of the negative density troughs slightly away from the actual H atom positions is typical for nuclear density maps and is due to the superposition of positive and negative density features of different magnitudes.
The surroundings of the catalytic water molecule (in native XI) or hydroxyl group (in XI-xylulose) are of particular interest because both are near the C1-C2 bond of xylulose and therefore are presumed to play a significant role in the isomerization reaction. In native XI, the M2-bound water donates H bonds to Asp257 and Glu217 forming two metal ion-carboxylate-water motifs. The O atom of the D2O points one of its lone electron pairs toward M2 and the other in the direction where the C1-C2 bond of the linear substrate in a complex would be located. On the other hand, in the XI-xylulose structure, the M2-bound water is deprotonated to OD−. The D of the hydroxyl group points towards a carboxylate O of Asp257, as found for D2O in the native enzyme structure. O of the hydroxyl group appears to remain bound to M2 and now lies close to the C1 of the bound product xylulose. Lys289 is well ordered unlike the structure in 4XIS and in the native XI structure, and its Nζ has 3 D atoms. One of these is involved in an H bond to a carboxylate O of Asp257 (Figure 2). Since Lys289 only has two protons on Nζ in native XI but three in XI-xylulose, this might suggest that it is somehow involved in proton transfer during the reaction. Nζ of Lys183 has 3 D atoms in both native XI and XI-xylulose. One of these D atoms is H-bonded to O1 of xylulose in the complex.
O2 of xylulose is deprotonated in XI-xylulose, confirming that C2-O2 has become a carbonyl group. O2 also interacts with Cε1 of His220 by means of a C-H...O bond. The nuclear density and also the planar geometry of C2 indicate that C2 has lost, and C1 has gained, a D atom. O1, O3 and O4 of xylulose are protonated with their Ds pointing towards Nε2 of His220, solvent, and an O of Glu181, respectively. In this neutron structure of XI-xylulose, M1 appears to bind six O atoms in Glu181, Glu217, Asp245, Asp287 and O2 and O4 of xylulose (Figure 2), showing that xylulose has replaced two metal ion-bound water molecules that were found in the native unliganded structure. M2 also appears to bind six groups; His220, Glu217, Asp257, Asp255 (bidentate) and the hydroxyl group O atom.
Unlike the situation in native XI, His220 is only singly N-protonated in XI-xylulose, thereby making the nonprotonated N able to bind more firmly to M2. In native XI, Nε2 of the His54-Asp57 pair donates an H bond to a water molecule whereas in XI-xylulose this water has been replaced by O5 of xylulose (Figure 3). This strong salt-bridge-type Ne2-H+...O5− interaction, assists in holding the substrate in the active site, ready for the isomerization reaction.
These observations have important implications for our understanding of the reaction mechanism of XI. The main aims of this enzyme are to open the cyclic sugar that has bound to the metal ion at M1, and then to move H from C2 to C1 of substrate. It would appear that the deprotonation of the catalytic water is important in this. The finding that O5 is deprotonated suggests that, if a ring-opening stage exists, that it may differ in detail from the conventional Lewis acid-catalyzed reactions observed for other enzymes (18). Ionization of O5 is unlikely to have any significance with respect to the mechanism of isomerization because it is distant from the site of the chemical reaction, but it may be important, along with the metals cations, in stabilizing multiply charged intermediate ligands.
In XI-xylulose the hydroxyl anion is only 2.8 Å away from C1 and there is nothing close to C2. This is consistent with our hypothesis that this hydroxyl group is involved in an interaction with C1 and/or C2. It is possible that this interaction involves an H of the M2-bound water molecule protonating C1. This would be consistent with the formation of an ene-diol intermediate. However, there is biochemical evidence that the C1 and C2 H atoms do not exchange with solvent during the reaction and this questions the ene-diol mechanism for this enzyme (5). The hydroxyl group is also only 2.9 Å away from O1 (which is not bound to M2). Another possibility is that the M2-bound water hydroxyl group is involved in protonation of O1, and then that the resulting hydroxyl anion stabilizes the positive charge on C1 in the resulting carbocation intermediate.
In summary we suggest that the linear sugar substrate (xylose or glucose), after a ring-opening stage involving the His54-Asp57 pair, is extended and tethered at either end by an H bond from Lys183 and by an interaction with His54 Nε2 (to O5). At some stage the M2-bound water is deprotonated and Lys289 is protonated. Meanwhile, O2 of xylulose is deprotonated and the resulting unstable intermediate is stabilized by removing H from C2 and adding H to C1, either through a hydride shift or through an ene-diol intermediate. The precise role of the hydroxyl group in this proton transfer remains to be determined.
ACKNOWLEDGMENT
† AKK, HLC, MM, PL and JPG were supported by grants from the National Institutes of Health (CA06927, CA10925 and GM071939). The PCS is funded by the Office of Biological and Environmental Research of the Department of Energy.
Abbreviations
- XI
D-xylose isomerase
- PCS
protein crystallography station
- LANSCE
Los Alamos Neutron Science Center
- PDB
protein data bank accession code.
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2−3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
Coordinates and structure factors have been deposited in the Protein Data Bank under the accession code 3CWH.
Supporting Information Available: Details of crystallization, neutron crystallographic data collection, and structure refinement. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
REFERENCES
- 1.Collyer CA, Blow DM. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1362–1366. doi: 10.1073/pnas.87.4.1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collyer CA, Henrick K, Blow DM. J. Mol. Biol. 1990;212:211–235. doi: 10.1016/0022-2836(90)90316-E. [DOI] [PubMed] [Google Scholar]
- 3.Fenn TD, Ringe D, Petsko GA. Biochemistry. 2004;43:6464–6474. doi: 10.1021/bi049812o. [DOI] [PubMed] [Google Scholar]
- 4.Wohl A, Neuberg C. Berichte. 1900;33:3095–3110. [Google Scholar]
- 5.Rose IA, O'Connell EL, Mortlock RP. Biochem. Biophys. Acta. 1969;178:376–379. doi: 10.1016/0005-2744(69)90405-7. [DOI] [PubMed] [Google Scholar]
- 6.Ramachander S, Feather MS. Arch. Biochem. Biophys. 1977;178:576–580. doi: 10.1016/0003-9861(77)90228-4. [DOI] [PubMed] [Google Scholar]
- 7.Farber GK, Petsko GA, Ringe D. Protein Eng. 1989;1:459–466. doi: 10.1093/protein/1.6.459. [DOI] [PubMed] [Google Scholar]
- 8.Whitlow M, Howard JH, Finzel BC, Poulos TL, Winborne E, Gilliland GL. PROTEINS: Structure, Function, and Genetics. 1991;9:153–173. doi: 10.1002/prot.340090302. [DOI] [PubMed] [Google Scholar]
- 9.Carrell HL, Rubin BH, Hurley TJ, Glusker JP. J. Biol. Chem. 1984;259:3230–3236. [PubMed] [Google Scholar]
- 10.Katz AK, Li X, Carrell HL, Hanson BL, Langan P, Coates L, Schoenborn BP, Glusker JP, Bunick GJ. Proc. Natl. Acad. Sci. U.S.A. 2006;103:8342–8347. doi: 10.1073/pnas.0602598103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niimura N, Bau R. Acta Cryst. 2008;A64:12–22. doi: 10.1107/S0108767307043498. [DOI] [PubMed] [Google Scholar]
- 12.Langan P, Greene G, Schoenborn BP. J. Appl. Cryst. 2004;37:24–31. [Google Scholar]
- 13.Langan P, Greene G. J. Appl. Cryst. 2004;37:253. [Google Scholar]
- 14.Sears VF. Neutron News. 1992;3(3):29–37. [Google Scholar]
- 15.Hanson BL, Langan P, Katz AK, Li X, Harp JM, Glusker JP, Schoenborn BP, Bunick GJ. Acta Cryst. 2004;D60:241–249. doi: 10.1107/S0907444903025873. [DOI] [PubMed] [Google Scholar]
- 16.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 17.Mustyakimov M, Langan P. nCNS: An Open Source Distribution Patch for CNS for Macromolecular Structure Refinement. 2007 http://mnc.lanl.gov
- 18.Sugiura M, Hagio H, Kobayashi S. Helvet. Chim. Acta. 2002;85:3678–3691. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.