

Abstract
Human pluripotent stem cells would be invaluable for in vitro studies of aspects of human embryogenesis. With the goal of establishing pluripotent stem cell lines, gonadal ridges and mesenteries containing primordial germ cells (PGCs, 5–9 weeks postfertilization) were cultured on mouse STO fibroblast feeder layers in the presence of human recombinant leukemia inhibitory factor, human recombinant basic fibroblast growth factor, and forskolin. Initially, single PGCs in culture were visualized by alkaline phosphatase activity staining. Over a period of 7–21 days, PGCs gave rise to large multicellular colonies resembling those of mouse pluripotent stem cells termed embryonic stem and embryonic germ (EG) cells. Throughout the culture period most cells within the colonies continued to be alkaline phosphatase-positive and tested positive against a panel of five immunological markers (SSEA-1, SSEA-3, SSEA-4, TRA-1–60, and TRA-1–81) that have been used routinely to characterize embryonic stem and EG cells. The cultured cells have been continuously passaged and found to be karyotypically normal and stable. Both XX and XY cell cultures have been obtained. Immunohistochemical analysis of embryoid bodies collected from these cultures revealed a wide variety of differentiated cell types, including derivatives of all three embryonic germ layers. Based on their origin and demonstrated properties, these human PGC-derived cultures meet the criteria for pluripotent stem cells and most closely resemble EG cells.
Keywords: alkaline phosphatase/embryoid body/embryonic stem cell/embryonic germ cell
Pluripotent stem cells have been derived from two embryonic sources. Embryonic stem (ES) cells are derived from the inner cell mass of preimplantation embryos (1, 2), and embryonic germ (EG) cells are derived from primordial germ cells (PGCs) (3, 4). Both ES and EG cells are pluripotent and demonstrate germ-line transmission in experimentally produced chimeras (5, 6). Mouse ES and EG cells share several morphological characteristics such as high levels of intracellular alkaline phosphatase (AP), and presentation of specific cell surface glycolipids (7, 8) and glycoproteins (9). These properties are characteristic of, but not specific for, pluripotent stem cells. Other important characteristics include growth as multicellular colonies, normal and stable karyotypes, the ability to be continuously passaged, and the capability to differentiate into cells derived from all three embryonic germ layers. Pluripotent stem cell lines that share most of these characteristics also have been reported for chicken (10), mink (11), hamster (12), pig (13, 14), rhesus monkey (15), and common marmoset (16).
The pluripotency of ES and EG cells can be demonstrated in vitro and in vivo. Embryoid bodies (EBs) are differentiated cell aggregates first described as arising in human (17) and mouse (18–20) teratomas and teratocarcinomas. These aggregates range from a cluster of pluripotent stem cells enclosed by a layer of endoderm to complex structures closely resembling an embryo during early development. EBs from mouse pluripotent stem cells grown on feeder layers or in suspension may contain a variety of cell types. This property has been used as evidence of cell pluripotency (1, 21) and as a source of differentiating cells. With the proper combinations of growth and differentiation factors, mouse ES (22, 23) and EG (S.W., unpublished results) cultures can generate cells of the hematopoietic lineage and cardiomyocytes (24, 25). In addition, mouse ES cells have been used to generate in vitro cultures of neurons (26), skeletal muscle (27), and vascular endothelial cells (28). ES and EG cells from some species can form teratocarcinomas when injected into histocompatible or immunologically compromised mice. This property alone may not be a definitive test of stem cell pluripotency, as it has been demonstrated that rat and mouse visceral (yolk sac) endoderm are capable of forming highly differentiated teratomas containing cells of all three embryonic germ layers (29, 30). Perhaps the most definitive in vivo test of developmental potential would be a demonstrated contribution to all cell lineages in a chimeric animal, but this test is not practical or possible for all species and cannot be done with human cells.
We report here the establishment of cultures from human PGCs. These cultures have morphological, immunohistochemical, and karyotypic features consistent with those of previously described pluripotent stem cells and have a demonstrated ability to differentiate in vitro into derivatives of the three embryonic germ layers.
MATERIALS AND METHODS
Collection of Tissue, Establishment, and Maintenance of Cultures.
Gonadal ridges and mesenteries of 5- to 9-week postfertilization human embryos (obtained as a result of therapeutic termination of pregnancy by using a protocol approved by the Joint Committee on Clinical Investigation of the Johns Hopkins University School of Medicine) were mechanically disaggregated then incubated in 0.05% trypsin-0.5 mM EDTA (GIBCO/BRL) or 0.25% trypsin at 37°C for 5–10 min, or incubated in a combination of 0.01% hyaluronidase type V (Sigma), 0.1% collagenase type IV (Sigma), and 0.002% DNase type I (Sigma) at 37°C for 2 hr (31). Cells initially were cultured and subsequently passaged on a mouse STO fibroblast feeder layer mitotically inactivated with 5,000 rads (1 rad = 0.01 Gy) γ-radiation. Cells were grown in DMEM (GIBCO/BRL) supplemented with 15% fetal bovine serum (HyClone), 0.1 mM nonessential amino acids (GIBCO/BRL), 0.1 mM 2-mercaptoethanol (Sigma), 2 mM glutamine (GIBCO/BRL), 1 mM sodium pyruvate (GIBCO/BRL), 100 units/ml of penicillin (GIBCO/BRL), 100 μg/ml of streptomycin (GIBCO/BRL), 1,000 units/ml of human recombinant leukemia inhibitory factor (hrLIF, Genzyme), 1 ng/ml of human recombinant basic fibroblast growth factor (hrbFGF, Genzyme), and 10 μM forskolin (Sigma). Cultures were grown in 5% or 8% CO2, 95% humidity and were routinely passaged every 7 days after disaggregation with 0.05% trypsin/0.53 mM EDTA (GIBCO/BRL) or 0.25% trypsin at 37°C for 5–10 min.
Initial Characterization.
Cells were fixed for detection of AP activity in 66% acetone/3% formaldehyde and then stained with naphthol/FRV-alkaline AP substrate (Sigma). For immunocytochemistry, cells were fixed in 3% paraformaldehyde in Dulbecco’s PBS (GIBCO/BRL). Cell surface glycolipid- and glycoprotein-specific mAbs were used at 1:15 to 1:50 dilution. MC480 (SSEA-1), MC631 (SSEA-3), and MC813–70 (SSEA-4) antibodies were supplied by the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City). TRA-1–60 and TRA-1–81 were a gift of Peter Andrews (University of Sheffield, U.K.). Antibodies were detected by using biotinylated anti-mouse secondary antibody, strepavidin-conjugated horseradish peroxidase, and 3-amino-9-ethylcarbazole chromagen (BioGenex). Cells prepared for cytogenetic analysis were incubated in growth media with 0.1 μg/ml of Colcemid for 3–4 hr, trypsinized, resuspended in 0.075 M KCl, and incubated for 20 min at 37°C, then fixed in 3:1 methanol/acetic acid.
Immunohistochemistry.
EBs were collected from cultures and either immediately embedded or replated into single wells of a 96-well tissue culture plate and cultured for 14 days in the absence of hrLIF, hrbFGF, and forskolin, before embedding. For embedding, EBs were placed into a small drop of molten 1% low melting point agarose (FMC) prepared in PBS, and cooled to 42°C. Solidified agarose containing EBs then were fixed in 3% paraformaldehyde in PBS and embedded in paraffin. Individual 6-μm sections were placed on slides (ProbeOn Plus, Fisher Scientific) and immunohistochemical analysis was carried out by using a BioTek-Tech Mate 1000 automated stainer (Ventana-BioTek Solutions, Tucson, AZ). Antibodies used on paraffin sections included: HHF35 (muscle-specific actin, Dako), M 760 (desmin, Dako), CD34 (Immunotech, Luminy, France), Z 311, (S-100, Dako), sm311 (pan-neurofilament, Sternberger Monoclonals, Baltimore, MD), A 008 (alpha-1-fetoprotein, Dako), CKERAE1/AE3 (pan-cytokeratin, Boehringer Mannheim), OV-TL 12/30 (cytokeratin 7, Dako), and Ks20.8 (cytokeratin 20, Dako). Primary antibodies were detected by using biotinylated anti-rabbit or anti-mouse secondary antibody, strepavidin-conjugated horseradish peroxidase, and diaminobenzidine chromagen (Ventana-BioTek Solutions). Slides were counterstained with hematoxylin.
RESULTS
Of 38 human PGC cultures initiated, 36 (≈95%) demonstrated morphological, biochemical, and/or immunocytochemical characteristics consistent with previously characterized pluripotent stem cell lines. High levels of AP activity are associated with ES and EG cultures, as well as with PGCs in vivo (32). As seen in Fig. 1, cells that closely resemble individual stationary and migratory mouse PGCs (33) are readily detected in the initial plating of human PGCs. In the presence of an irradiated STO cell feeder layer, hrbFGF, forskolin, and hrLIF, solitary PGCs gave rise to large tightly compacted multicellular colonies resembling early passage mouse ES and EG cell colonies (Fig. 2). This morphology is in contrast to the flattened and loosely associated colonies typical of human embryonal carcinoma (34) and rhesus ES cells (15). The conversion efficiency from PGCs, which have limited in vitro survival and proliferative capacity in the mouse (35, 36), to cells that can be passaged at least 20 times was variable and did not depend on the embryonic stage or sex of the embryo. Generation and passage of derived cultures were less successful when mouse embryo fibroblasts, human fetal fibroblasts, or gelatin-coated tissue culture dishes were substituted for STO cells, or when hrLIF or hrbFGF were withdrawn. Unlike mouse pluripotent stem cells (ES and EG), these human cells were more resistant to disaggregation by trypsin/EDTA-based reagents.
Figure 1.

AP activity of individual human PGCs in culture. (A) Stationary and (B) migratory PGCs in a primary culture, growing on a feeder layer of mitotically inactivated mouse STO fibroblasts. (Bars represent 10 μm.)
Figure 2.

Colony morphology. (A) Human PGC-derived cell colony growing on a feeder layer of mitotically inactivated mouse STO fibroblasts. (B) Mouse ES colony growing on a feeder layer of mitotically inactivated mouse fibroblasts. Hoffman modulation optics. (Bars represent 100 μm.)
Like ES and EG cells from other species, the human PGC-derived cells possess high levels of AP activity (Fig. 3A). The percentage of visible AP-positive cells within a colony varied from ≈20% to >90%. Human PGC-derived cells were further characterized with a bank of five mAbs: SSEA-1, SSEA-3, SSEA-4, TRA-1–60, and TRA-1–81 (7, 8). As seen in Fig. 3 B–F, colonies stained strongly for four of the five antibodies, whereas colonies stained with the secondary antibody alone gave no signal (data not shown). The antibody recognizing SSEA-3 antigen stained the cells inconsistently and weakly. As with the results of AP staining, the percentage of cells within a colony that stained positive was variable.
Figure 3.

Expression of cell surface markers by human PGC-derived cell colonies. (A) AP. (B) SSEA-1. (C) SSEA-3. (D) SSEA-4. (E) TRA-1–60. (F) TRA-1–81. (Bars represent 100 μm.)
Karyotypic analyses carried out at passage 8–10 (60–70 days in culture) indicated apparently normal human chromosomes at the 300 band level of resolution (37). Of five different cultures examined, three were XX and two were XY. Karyotypes of each sex are shown in Fig. 4.
Figure 4.

Karyotype of human PGC-derived cell cultures. (A) XX, eight passages. (B) XY, 10 passages.
In the human PGC-derived cultures, a small percentage (≈1–20%) of colonies spontaneously generate EBs in the presence of hrLIF. Table 1 indicates antibody reactivity of the EBs. Immunohistochemical analysis of serial sections represents a sampling of the cells within an EB so the absence of a particular marker is not significant. However, most of the markers were represented in all of the EBs.
Table 1.
Immunohistochemical analysis of six human embryoid bodies
| Epitope | Marker | Embryoid body
|
|||||
|---|---|---|---|---|---|---|---|
| BF1 | KF1 | RI3 | RI4 | RI5 | BF2 | ||
| Muscle actin | Mesoderm | + | + | + | + | + | + |
| Desmin | + | + | + | ND | + | + | |
| CD34 | + | + | − | + | − | − | |
| S-100 | Ectoderm | + | + | + | − | + | + |
| Neurofilament, ps | ND | + | − | + | + | + | |
| Alpha fetoprotein | Endoderm | + | + | + | + | + | + |
| Cytokeratin, ps | + | + | + | + | + | + | |
| Cytokeratin 7 | + | + | ND | ND | + | + | |
ND, not determined because of tissue loss. +, − indicate positive and negative antibody reactivity, respectively. ps, pan-specific antibody reactivity. Embryoid bodies were formed in the presence of hrLIF, hrbFGF, and forskolin. RI3, RI4, RI5, and BF2 were subsequently cultured 2 weeks in the absence of these factors.
Immunohistochemical analysis of the EBs demonstrated that PGC-derived cells can differentiate into a variety of cell types, including derivatives of the three embryonic germ layers. Three distinct mesodermal derivatives were seen. These were antimuscle specific actin-reactive myocytes with prominent eccentric nuclei and cytoplasmic filaments (Fig. 5A), antidesmin-reactive mesenchymal cells (Fig. 5B), and anti-CD34-reactive vascular endothelium (Fig. 5C). Ectodermal derivatives include cells suggestive of neuroepithelia with nuclear localized anti-S-100-reactivity (Fig. 5 D and E) and antineurofilament-reactive cells (Fig. 5F). Endodermal derivatives include anti-α-1-fetoprotein-reactive cells, which appear within the interior of some EBs as well as form the exterior layer (Fig. 5G). Several types of anticytokeratin-reactive epithelia were seen, including nests of relatively undifferentiated cells (Fig. 5H) and simple cuboidal epithelial layers (Fig. 5I).
Figure 5.
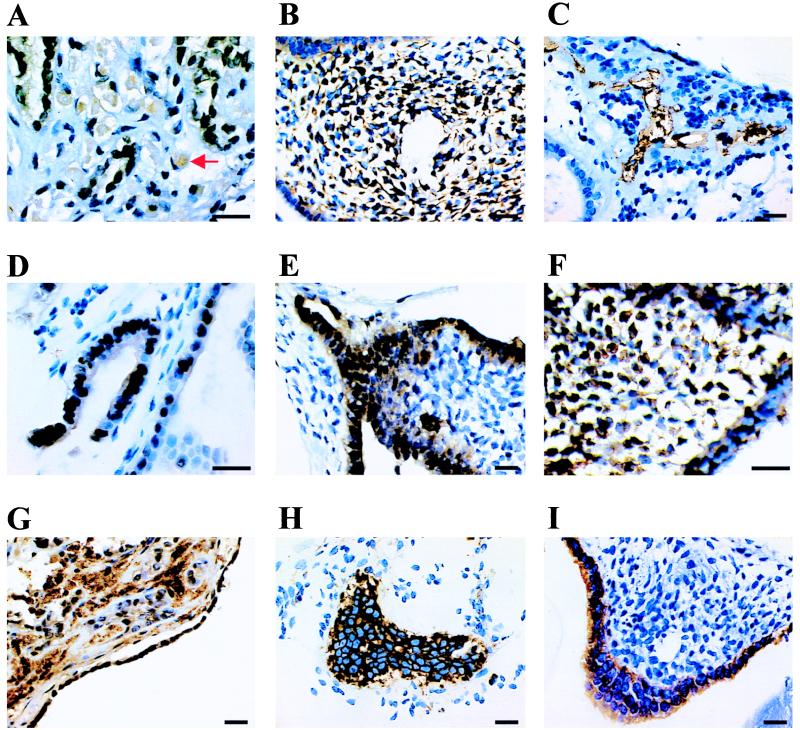
Immunohistological analysis of human EB sections. EB culture designation, antibody epitope, and objective power are as follows: (A) BF1, muscle-specific actin, ×100. Arrow indicates a cell with eccentric nuclei and cytoplasmic filaments. (B) RI5, desmin, ×60. (C) BF1, CD34, ×60. (D) BF1, S-100, ×100. (E) RI5, S-100, ×60. (F) RI, pan-neurofilament, ×100. (G) BF1, α-1-fetoprotein, ×60. (H) BF1, pan-cytokeratin, ×60. (I) RI5, cytokeratin 7, ×60. (Bars represent 20 μm.)
DISCUSSION
The human cultures described here satisfy the criteria used to define pluripotent stem cells. These include presentation of a series of markers commonly used to identify pluripotent stem cells, morphological similarity to mouse ES and EG cells, normal and stable karyotype maintained over at least 10 passages, and demonstrated ability to differentiate into a wide variety of cell types.
The histological profile of these human cells (AP+, SSEA-1+, SSEA-3+, SSEA-4+, TRA-1–60+, and TRA-1–81+) differs from undifferentiated human embryonal carcinoma (EC) and rhesus ES cells, which are SSEA-1 negative (15, 38). The fact that differentiation of the human EC line NTERA2 leads to increased expression of SSEA-1 may suggest that this marker is indicative of differentiation in the human PGC-derived cultures. However, NTERA2 differentiation is accompanied by the loss of the other markers (39, 40), which we do not observe. A second possibility is that SSEA-1 reactivity reflects an intrinsic difference between the relatively flat and loosely associated human EC and rhesus ES colonies and the multilayered and tightly compacted colonies formed by mouse ES and EG cells that are SSEA-1 positive.
Human cell cultures derived and grown as described have been continuously maintained for more than 20 passages. Maintaining high colony density and derivation of clonal cell lines is complicated by the difficulties associated with disaggregation of colonies to single cells. The highly compacted nature of these colonies suggests strong cell–cell adhesion. These interactions are notably more resistant to trypsin than mouse ES and EG colonies. Alternative disaggregation enzymes are currently under investigation.
The most useful and important property of these cells is their ability to differentiate in vitro into ectodermal, endodermal, and mesodermal derivatives. As with mouse ES and EG cells, human pluripotent stem cells can form EBs. These structures appear to recapitulate the normal developmental processes of early embryonic stages and promote the cell–cell interaction required for cell differentiation. Typically, mouse EBs are surrounded by a layer of visceral or parietal endoderm and contain a heterogeneous mixture of cell types. Morphologically, human EBs resemble those generated from mouse ES and EG cultures. Many have an outer layer that stains positive for α-1-fetoprotein (Fig. 5G) although the constituent cells of this layer do not always resemble those seen in mouse EBs. Identification of cell derivatives of the three embryonic germ layers in human EBs suggests that these cells are pluripotent and are capable of in vitro differentiation.
Although the properties of the cultures described in this paper are consistent with those of pluripotent stem cells, the cultures have a lower plating efficiency than most mouse EG and ES cell cultures, which may reflect difficulties associated with complete cell disaggregation. Although many of the PGC-derived cultures have been passaged 20–25 times, the immortality of these cells remains to be demonstrated.
Human pluripotent stem cells, with their potential to differentiate into a wide variety of cell types in culture, would be invaluable for studies of some aspects of human embryogenesis and for transplantation therapies. They may serve to define the culture conditions and differential gene expression necessary for cell-type-specific differentiation and for the isolation of lineage-restricted stem cells that could serve as a source of cells for transplantation. Genetic modification of these pluripotent stem cells may allow the generation of universal donor cells or cells that have been customized to meet individual requirements. Clearly, these goals warrant investigations on the isolation, study, and use of human pluripotent stem cells.
Acknowledgments
We thank Gail Stetten, Clara Moore, and Sarah South for assistance with the karyotype analyses, Mahmud Siddiqi for technical assistance and a critical reading of the manuscript, and Elizabeth Perlman for technical advice. We acknowledge the financial support of the Geron Corporation.
ABBREVIATIONS
- AP
alkaline phosphatase
- EG
embryonic germ
- ES
embryonic stem
- PGC
primordial germ cell
- EB
embryoid body
- hrLIF
human recombinant leukemia inhibitory factor
- hrbFGF
human recombinant basic fibroblast growth factor
References
- 1.Evans M J, Kaufman M H. Nature (London) 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 2.Martin G R. Proc Natl Acad Sci USA. 1981;78:7634–7638. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsui Y, Toksoz D, Nishikawa S, Nishikawa S, Williams D, Zsebo K, Hogan B L. Nature (London) 1991;353:750–752. doi: 10.1038/353750a0. [DOI] [PubMed] [Google Scholar]
- 4.Resnick J L, Bixler L S, Cheng L, Donovan P J. Nature (London) 1992;359:550–551. doi: 10.1038/359550a0. [DOI] [PubMed] [Google Scholar]
- 5.Stewart C, Gadi I, Bhatt H. Dev Biol. 1994;161:626–628. doi: 10.1006/dbio.1994.1058. [DOI] [PubMed] [Google Scholar]
- 6.Labosky P, Barlow D, Hogan B. Development (Cambridge, UK) 1994;120:3197–3204. doi: 10.1242/dev.120.11.3197. [DOI] [PubMed] [Google Scholar]
- 7.Solter D, Knowles B. Proc Natl Acad Sci USA. 1978;75:5565–5569. doi: 10.1073/pnas.75.11.5565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kannagi R, Cochran N, Ishigami F, Hakomori S, Andrews P, Knowles B, Solter D. EMBO J. 1983;2:2355–2361. doi: 10.1002/j.1460-2075.1983.tb01746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andrews P, Banting G, Damjanov I, Arnaud D, Avner P. Hybridoma. 1984;3:347–361. doi: 10.1089/hyb.1984.3.347. [DOI] [PubMed] [Google Scholar]
- 10.Pain B, Clark M E, Shen M, Nakazawa H, Sakurai M, Samarut J, Etches R J. Development (Cambridge, UK) 1996;122:2339–2348. doi: 10.1242/dev.122.8.2339. [DOI] [PubMed] [Google Scholar]
- 11.Sukoyan M A, Vatolin S Y, Golubitsa A N, Zhelezova A I, Semenova L A, Serov O L. Mol Reprod Dev. 1993;36:148–158. doi: 10.1002/mrd.1080360205. [DOI] [PubMed] [Google Scholar]
- 12.Doetschman T, Williams P, Maeda N. Dev Biol. 1988;127:224–227. doi: 10.1016/0012-1606(88)90204-7. [DOI] [PubMed] [Google Scholar]
- 13.Wheeler M B. Reprod Fertil Dev. 1994;6:563–568. doi: 10.1071/rd9940563. [DOI] [PubMed] [Google Scholar]
- 14.Shim H, Gutierrez-Adan A, Chen L, BonDurant R, Behboodi E, Anderson G. Biol Reprod. 1997;57:1089–1095. doi: 10.1095/biolreprod57.5.1089. [DOI] [PubMed] [Google Scholar]
- 15.Thomson J A, Kalishman J, Golos T G, Durning M, Harris C P, Becker R A, Hearn J P. Proc Natl Acad Sci USA. 1995;92:7844–7848. doi: 10.1073/pnas.92.17.7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomson J A, Kalishman J, Golos T G, Durning M, Harris C P, Hearn J P. Biol Reprod. 1996;55:254–259. doi: 10.1095/biolreprod55.2.254. [DOI] [PubMed] [Google Scholar]
- 17.Peyron A. Bull Assoc franç étude cancer. 1939;28:658–681. [Google Scholar]
- 18.Stevens L C. J Natl Cancer Inst. 1958;20:1257–1270. doi: 10.1093/jnci/20.6.1257. [DOI] [PubMed] [Google Scholar]
- 19.Stevens L C. J Natl Cancer Inst. 1959;23:1249–1295. [PubMed] [Google Scholar]
- 20.Stevens L C. Dev Biol. 1970;21:364–382. doi: 10.1016/0012-1606(70)90130-2. [DOI] [PubMed] [Google Scholar]
- 21.Doetschman T C, Eistetter H, Katz M, Schmidt W, Kemler R. J Embryol Exp Morphol. 1985;87:27–45. [PubMed] [Google Scholar]
- 22.Wiles M V, Keller G. Development (Cambridge, UK) 1991;111:259–267. doi: 10.1242/dev.111.2.259. [DOI] [PubMed] [Google Scholar]
- 23.Keller G, Kennedy M, Papayannopoulou T, Wiles M V. Mol Cell Biol. 1993;13:473–486. doi: 10.1128/mcb.13.1.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klug M, Soonpaa M, Field L. Am J Physiol. 1995;269:H1913–H1921. doi: 10.1152/ajpheart.1995.269.6.H1913. [DOI] [PubMed] [Google Scholar]
- 25.Rohwedel J, Sehlmeyer U, Shan J, Meister A, Wobus A. Cell Biol Int. 1996;20:579–587. doi: 10.1006/cbir.1996.0076. [DOI] [PubMed] [Google Scholar]
- 26.Bain G, Kitchens D, Yao M, Huettner J, Gottlieb D. Dev Biol. 1995;168:342–357. doi: 10.1006/dbio.1995.1085. [DOI] [PubMed] [Google Scholar]
- 27.Rohwedel J, Maltsev V, Bober E, Arnold H-H, Hescheler J, Wobus A. Dev Biol. 1994;164:87–101. doi: 10.1006/dbio.1994.1182. [DOI] [PubMed] [Google Scholar]
- 28.Wang R, Clark R, Bautch V. Development (Cambridge, UK) 1992;114:303–316. doi: 10.1242/dev.114.2.303. [DOI] [PubMed] [Google Scholar]
- 29.Sobis H, Verstuyf A, Vandeputte M. Development (Cambridge, UK) 1991;111:75–78. doi: 10.1242/dev.111.1.75. [DOI] [PubMed] [Google Scholar]
- 30.Sobis H, Verstuyf A, Vandeputte M. Int J Dev Biol. 1993;37:155–168. [PubMed] [Google Scholar]
- 31.Belldegrun A, Kasid A, Uppenkamp M, Topalian S, Rosenberg S. J Immunol. 1989;142:4520–4526. [PubMed] [Google Scholar]
- 32.Chiquoine A D. Anat Rec. 1954;118:135–146. doi: 10.1002/ar.1091180202. [DOI] [PubMed] [Google Scholar]
- 33.Donovan P J, Stott D, Cairns L A, Heasman J, Wylie C C. Cell. 1986;44:831–838. doi: 10.1016/0092-8674(86)90005-x. [DOI] [PubMed] [Google Scholar]
- 34.Andrews P W, Bronson D L, Benham F, Strickland S, Knowles B B. Int J Cancer. 1980;26:269–280. doi: 10.1002/ijc.2910260304. [DOI] [PubMed] [Google Scholar]
- 35.Dolci S, Williams D E, Ernst M K, Resnick J L, Brannan C I, Lock L F, Lyman S D, Boswell H S, Donovan P J. Nature (London) 1991;352:809–811. doi: 10.1038/352809a0. [DOI] [PubMed] [Google Scholar]
- 36.Matsui Y, Zsebo K, Hogan B L. Cell. 1992;70:841–847. doi: 10.1016/0092-8674(92)90317-6. [DOI] [PubMed] [Google Scholar]
- 37.Mitelman F. An International System for Human Cytogenetic Nomenclature. Basel: Karger; 1995. [Google Scholar]
- 38.Wenk J, Andrews P W, Casper J, Hata J, Pera M F, von Keitz A, Damjanov I, Fenderson B A. Int J Cancer. 1994;58:108–115. doi: 10.1002/ijc.2910580118. [DOI] [PubMed] [Google Scholar]
- 39.Andrews P W, Goodfellow P N, Shevinsky L H, Bronson D L, Knowles B B. Int J Cancer. 1982;29:523–531. doi: 10.1002/ijc.2910290507. [DOI] [PubMed] [Google Scholar]
- 40.Damjanov I, Fox N, Knowles B B, Solter D, Lange P H, Fraley E E. Am J Pathol. 1982;108:225–230. [PMC free article] [PubMed] [Google Scholar]