

Abstract
Protein kinase C (PKC)-θ mediates the critical TCR signals required for T cell activation. Previously, we have shown that in response to TCR stimulation, PKC-θ−/− T cells undergo apoptosis due to greatly reduced levels of the anti-apoptotic molecule, Bcl-xL. In this study, we demonstrate that PKC-θ-regulated expression of Bcl-xL is essential for T cell-mediated cardiac allograft rejection. Rag1−/− mice reconstituted with wild-type T cells readily rejected fully mismatched cardiac allografts, whereas Rag1−/− mice reconstituted with PKC-θ −/− T cells failed to promote rejection. Transgenic expression of Bcl-xL in PKC-θ −/− T cells was sufficient to restore cardiac allograft rejection, suggesting that PKC-θ-regulated survival is required for T cell-mediated cardiac allograft rejection in this adoptive transfer model. In contrast to adoptive transfer experiments, intact PKC-θ −/− mice displayed delayed, but successful cardiac allograft rejection, suggesting the potential compensation for PKC-θ function. Finally, a subtherapeutic dose of anti-CD154 Ab or CTLA4-Ig, which was not sufficient to prevent cardiac allograft rejection in the wild-type mice, prevented heart rejection in the PKC-θ −/− mice. Thus, in combination with other treatments, inhibition of PKC-θ may facilitate achieving long-term survival of allografts.
T cell activation is a critical step in the initiation of adaptive immunity, because it is only via the T cell activation process that naive T cells differentiate into armed effector T cells that mediate the actual immune responses. Biochemical signaling events initiated by engagement of the TCR and costimulatory molecules instruct the T cell activation process. Protein kinase C (PKC)4 has long been known to mediate TCR signals, because phorbol ester (PKC activator) together with ionomycin (a Ca2+ mobilizer) mimics the signals for T cell activation (1). Among the 11 members of PKC family, PKC-θ is the only isoform translocating to the immunological synapse and mediating the signals essential for T cell activation and survival (1–5). The unique function of PKC-θ in T cells is also confirmed by in vivo studies using PKC-θ-deficient mice that illustrated the essential role of PKC-θ in the development of T cell-driven immune responses. For example, PKC-θ is reported to be required for the development of both Th1-dependent experimental autoimmune encephalomyelitis and Th2-dependent airway hyperresponsiveness (6–9). The unique function of PKC-θ in T cells is also reflected by the fact that T cells obtained from mice deficient in other isoforms of PKC do not display T cell defects similar to those observed in PKC-θ −/− mice (4, 10, 11). In contrast to PKC-θ-deficient mice, mice deficient in PKC-θ have defects in the activation of B cells, but not T cells (12, 13). These results clearly demonstrated that T and B cells, two important components of adaptive immunity, use different isoforms of PKC to mediate signals required for their activation. The highly specific role of PKC-θ in T cells is attributed to its ability to stimulate signaling pathways such as NF-κB, AP-1, and NF-AT critical for T cell activation. T cells specifically deficient in PKC-θ display defective activation of NF-κB, AP-1, and NF-AT, whereas the active form of PKC-θ, but not of other isoforms of PKC, selectively enhances the activation of these three transcription factors (2, 3, 14–16). PKC-θ regulates these three signaling pathways in T cells most likely via activating different downstream signaling molecules. Li et al. (17) reported that stress-activated protein kinase is required for PKC-θ-mediated activation of AP-1, but not for the activation of NF-κB. PKC-θ regulates Ca2+/calcineurin-dependent NF-AT pathway via stimulation of phospholipase Cγ1 (14, 15). In contrast, PKC-θ-mediated activation of NF-κB, but not of AP-1, in T cells is dependent on CARMA1/Bcl10/MALT1 complexes (18–21). Interestingly, PKC-θ depends on the same downstream adaptor molecules CARMA1 and Bcl10 for the activation of NF-κB in B cells (22–27). Thus, selective use of the specific isoform of PKC, but not of downstream molecules, may determine the specificity in the activation of NF-κB pathway in different cell types.
Inhibition of T cell activation is the key to control unwanted immunological attacks on transplanted tissues. Transplanted tissues induce strong alloreactive responses that are usually 100-fold greater than the immune responses elicited by conventional Ags. Potent immunosuppression is thus required to prevent allograft rejection (28). Because PKC-θ is a critical signaling molecule required for T cell activation and survival, it is a potential drug target for controlling T cell-mediated allograft rejection. However, the role of PKC-θ in allograft rejection has not been determined. Using an acute cardiac allograft rejection model, we demonstrated that PKC-θ-regulated T cell survival plays a critical role in mediating allograft rejection. Furthermore, we showed that in combination with a subtherapeutic dose of anti-CD154 Ab, inhibition of PKC-θ activity could achieve long-term survival of cardiac allografts.
Materials and Methods
Mice
Female BALB/c (H-2d), C57BL/6 (H-2b), and C57BL/6-Rag1−/− (H-2b) mice were purchased from The Jackson Laboratory. PKC-θ −/− mice were originally described by Sun et al. (2), and backcrossed to C57BL/6 for 15 generations. C57BL/6 Bcl-xL transgenic (Bcl-xLTg) (H-2b) mice have been previously described (29). PKC-θ −/− mice and Bcl-xLTg mice were crossed to generate PKC-θ −/−/Bcl-xLTg (H-2b) mice. Animals were housed under specific pathogen-free conditions in the animal facility of the Biological Resource Laboratory at the University of Illinois following the university guidelines.
Proliferation assays
Enriched T cells (0.3 × 105 cells/well) were cultured in 96-well plates in the absence or presence of 10 ng/ml mouse rIL-2 (R&D Systems), and stimulated for 48 h in the presence or absence of anti-CD3 and anti-CD28 Abs. After 72 h of stimulation, cultures were pulsed with 1 μCi/well [3H]thymidine for 8 h. For MLR, responder T cells were purified from C57BL/6 spleens of different genotypes of C57BL/6 mice, and incubated with mitomycin C-treated BALB/c splenocytes at different ratios in 96-well flat-bottom plates for 3 days. Proliferation of the T cells was then monitored by pulsing with 1.0 μCi of [3H]thymidine for 8 h. Cells were then harvested and counted for thymidine incorportation, as we have described previously (4, 11, 15).
Adoptive transfer
Spleens were removed from 8- to 12-wk-old wild-type (WT), PKC-θ −/−, and PKC-θ −/−/Bcl-xLTg mice. A single-cell suspension was obtained by passing the splenocytes through a 70-μm cell strainer (BD Discovery Labware), and the viable washed cells were counted using trypan blue exclusion. RBC were lysed with ACK lysis buffer (Sigma-Aldrich). Afterward, T cells were isolated using a T cell isolation kit (R&D Systems or Miltenyi Biotec), according to manufacturer’s protocol. T cell purity was always >95% determined by flow cytometry. A total of 10 × 106 cells was suspended in 200 μl of PBS, and was administered i.v. in the tail vein of Rag1−/− mice that received cardiac allografts from BALB/c mice.
Heterotopic vascularized cardiac transplantation
Heart transplantation was performed using microsurgical techniques, as described previously (30). The donor aorta was sutured to the recipient aorta in an end to side fashion using 10-0 sutures (USSC), and the inferior vena cava to the recipient inferior vena cava in the same manner. Tolerance was induced by using an anti-CD154 mAb purified in our laboratory (MR1, 1 mg/mouse × 1 or 3) following the previous protocol (30), or by using human CTLA4-Ig (Orencia) purchased from Bristol-Myers Squibb. Graft function was assessed daily by palpation, with rejection defined as the absence of beating. The day the graft stopped beating was defined as the time of rejection. Graft rejection was also verified by autopsy and pathological examination. Loss of cardiac graft function within 48 h of transplant was considered a technical failure, and those cases were excluded from further analysis.
Histological examination
Transplanted mice were sacrificed for histopathological examinations. The grafts were frozen rapidly in liquid nitrogen. Sections (4–6 μm) were stained with H&E for the assessment of cellular infiltration. For detecting expression of CD4+ and CD8+ on infiltrated cells, immunohistochemistry was performed with a standard avidinbiotin peroxidase complex method. Cryosections were stained with the following primary mAbs: anti-CD4 (GK1.5) and anti-CD8 (Ly-2; BD Pharmingen). Biotinylated goat anti-rat IgG (Jackson ImmunoResearch Laboratories) and HRP-streptavidin (Zymed Laboratories) were then added at room temperature. Immunostaining was developed by diaminobenzidine and counterstained with Mayer’s hematoxylin.
Results
PKC-θ −/− T cells displayed greatly reduced MLR
Alloantigen-induced activation of T cells initiates potent immune responses against allografts. Our previous studies have demonstrated a requirement for PKC-θ in the activation of T cells induced by cross-linking anti-CD2/28 (2, 4, 15, 31). We first determined whether PKC-θ also plays a role in the activation of alloreactive T cells in response to alloantigens in vitro. T cells were purified from WT and PKC-θ−/− mice on a C57BL/6 background. T cells were activated with anti-CD3 (1 μg/ml) and anti-CD28 (2 μg/ml) Abs, and the proliferation of the T cells was analyzed by [3H]thymidine incorporation (Fig. 1a). Both the proliferation and IL-2 production (Fig. 1b) of the PKC-θ−/− T cells were markedly reduced compared with that of the WT T cells. Furthermore, exogenous IL-2 only partially rescued defective proliferation of the PKC-θ−/− T cells, confirming the critical role of PKC-θ in T cell activation. For MLR analysis, T cells were stimulated by mitomycin C-treated BALB/c splenocytes at different ratios (Fig. 1c). Indeed, PKC-θ−/− T cells also showed greatly decreased proliferation in response to allo-Ag stimulation. This was further confirmed by the reduced IL-2 production from alloantigen-activated PKC-θ−/− T cells (Fig. 1d). IFN-γ is a critical cytokine often associated with T cell-mediated allograft rejection. We therefore also measured IFN-γ produced by alloreactive T cells (Fig. 1e). PKC-θ−/− T cells produced much less IFN-γ when compared with WT T cells. These results suggest that PKC-θ is required for the activation of alloreactive T cells.
FIGURE 1.
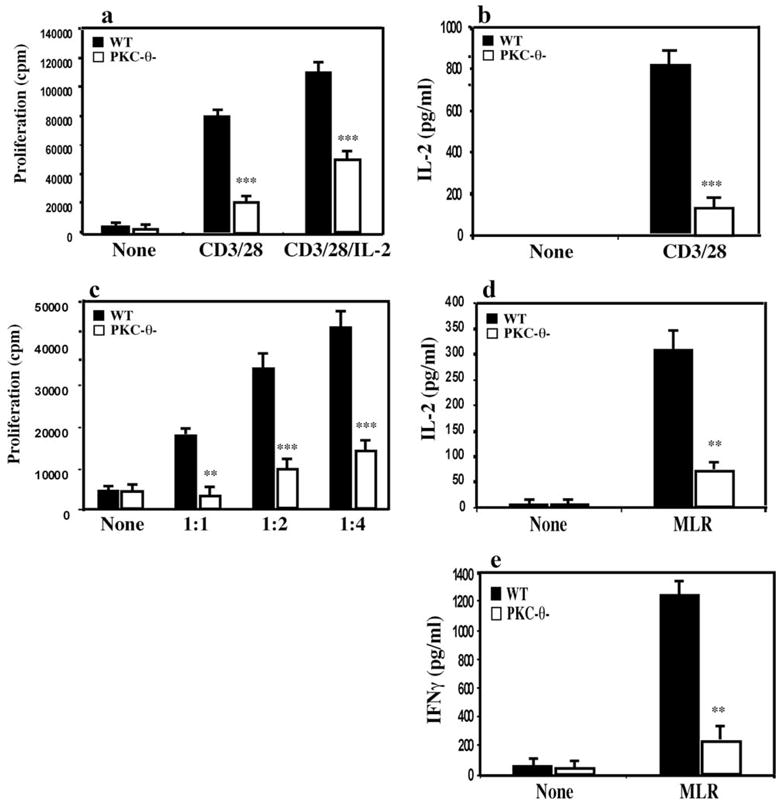
PKC-θ−/− T cells had greatly reduced MLR responses. a, T cells purified from WT (■) and PKC-θ−/− mice (□) were left in medium (None) or were stimulated with plate-bound anti-CD3 (1 μg/ml) and soluble anti-CD28 (2 μg/ml) Abs with or without exogenous IL-2. After 72 h, the proliferation of the T cells was monitored by [3H]thymidine incorporation. b, T cells purified from WT (■) and PKC-θ−/−mice (□) were left in medium (None) or subject to stimulation with plate-bound anti-CD3 (1 μg/ml) and soluble anti-CD28 (2 μg/ml) Abs for 72 h. IL-2 concentrations in the supernatant were then measured by ELISA. c, Purified WT (■) and PKC-θ−/− (□) T cells alone (None) or mixed with mitomycin C-treated BALB/c stimulators (splenocytes) at 1:1, 1:2, or 1:4 ratios. After 72 h, the proliferation of the T cells was monitored by pulse chasing with [3H]thymidine for 8 h. d, T cells from WT (■) and PKC-θ−/−mice (□) were left in medium (None) or subject to MLR stimulation for 72 h. IL-2 concentration in the supernatants was then measured by ELISA. e, Same treatment as described in d, IFN-γ concentrations in the supernatant were measured by ELISA. Data are representative of at least three independent experiments (*, p < 0.05; **, p < 0.001; ***, p < 0.0001).
Rag1−/− mice reconstituted with PKC-θ−/− T cells failed to reject cardiac allografts
To evaluate the function of PKC-θ specifically in T cell-mediated allograft rejection in vivo, we chose a model that requires adoptive transfer of T cells to Rag1−/− mice. The PKC-θ−/− mice have been backcrossed to C57BL/6 mice for 15 generations. A total of 10 × 106 purified T cells from spleens of WT or PKC-θ−/− mice was adoptively transferred to each of the C57BL/6 Rag1−/− mice that received transplanted hearts from BALB/c donors. Rag1−/− mice without the adoptively transferred T cells did not reject cardiac allografts (data not shown), because Rag1−/− mice lack both B and T cells that are the essential components of the adaptive immunity. One hundred percent of the Rag1−/− mice reconstituted with WT T cells rejected cardiac allografts 13 days after transplantation (Fig. 2). In contrast, 100% of the transplanted hearts survived for >100 days in Rag1−/− mice reconstituted with PKC-θ−/− T cells (mean survival time, >100 vs 13 days; p < 0.005). This result suggests that PKC-θ is required for T cell-mediated cardiac allograft rejection in the adoptive transfer model.
FIGURE 2.
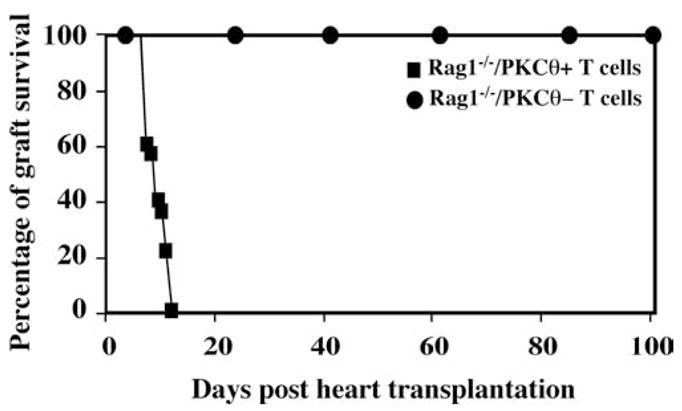
Rag1−/− mice reconstituted with PKC-θ−/−T cells failed to reject cardiac allografts. Cardiac allografts removed from BALB/c donors were transplanted in the abdominal cavity of the C57BL/6 Rag1−/−recipients. A total of 10 × 106 WT (square, n = 6) and PKC-θ−/− (circle, n = 6) purified T cells was then adoptively transferred to the syngeneic Rag1−/− recipients by i.v. injection. Cardiac allograft survival was assessed daily by monitoring the heartbeat of the grafts. The percentage of the surviving hearts was calculated at different days posttransplantation.
Transgenic expression of Bcl-xL in PKC-θ−/− T cells restored cardiac allograft rejection
We, as well as by others, have shown that PKC-θ mediates the TCR signals required for T cell activation as well as for enhancing T cell survival via up-regulation of Bcl-xL, an anti-apoptotic molecule (4, 5, 32). We therefore investigated whether PKC-θ-regulated survival plays a role in T cell-mediated allograft rejection. PKC-θ−/− mice were crossed to Bcl-xLTg mice. The Bcl-xL transgene is specifically targeted to T cell compartments using a Lckproximal promoter (29). To ensure that Bcl-xLTg prevents T cell apoptosis resulting from the lack of PKC-θ, apoptosis was compared between T cells obtained from spleens of WT, Bcl-xL Tg, PKC-θ−/−, and PKC-θ−/−/Bcl-xLTg mice (Fig. 3a). Purified CD4+ T cells from different genotypes of mice were cultured in medium or stimulated with anti-CD3/CD28 Abs, and the apoptotic cells were then detected by annexin V staining, as we described previously (4, 33, 34). Consistent with our published results (4), without stimulation, PKC-θ−/− CD4+ T cells displayed slightly more apoptosis than WT T cells. However, in response to anti-CD3 and anti-CD28 stimulation, PKC-θ−/− CD4+ T cells underwent accelerated apoptosis (~65% of apoptotic cells) compared with WT cells (~30% of apoptotic cells). Because PKC-θ−/− T cells displayed reduced Bcl-xL levels upon activation (4), we next tested whether overexpression of Bcl-xL can restore survival and proliferative defects. Bcl-xLTg restored the survival of the PKC-θ−/− T cells to almost WT levels (~30%), suggesting that Bcl-xLTg is sufficient to prevent apoptosis due to the lack of PKC-θ. Next, we determined the effects of Bcl-xLTg on the reduced MLR by PKC-θ−/− T cells (Fig. 3b). WT and PKC-θ−/− T cells were stimulated by splenocytes obtained from BALB/c mice. Consistent with previous observation, PKC-θ−/− T cells had greatly reduced MLR. Bcl-xLTg partially restored PKC-θ−/− T cell-mediated MLR, but not to the WT levels. Similarly, transgenic expression of Bcl- xLTg increased IL-2 (Fig. 3c) and IFN-γ (Fig. 3d) production by PKC-θ−/− T cells during the MRL, but again not to the levels produced by WT T cells. As a control, Bcl-xL Tg alone did not have obvious effects on above assays. These results suggest that Bcl-xLTg specifically restores PKC-θ-mediated T cell survival. However, PKC-θ also mediates other functions essential for T cell activation in addition to enhancing T cell survival.
FIGURE 3.
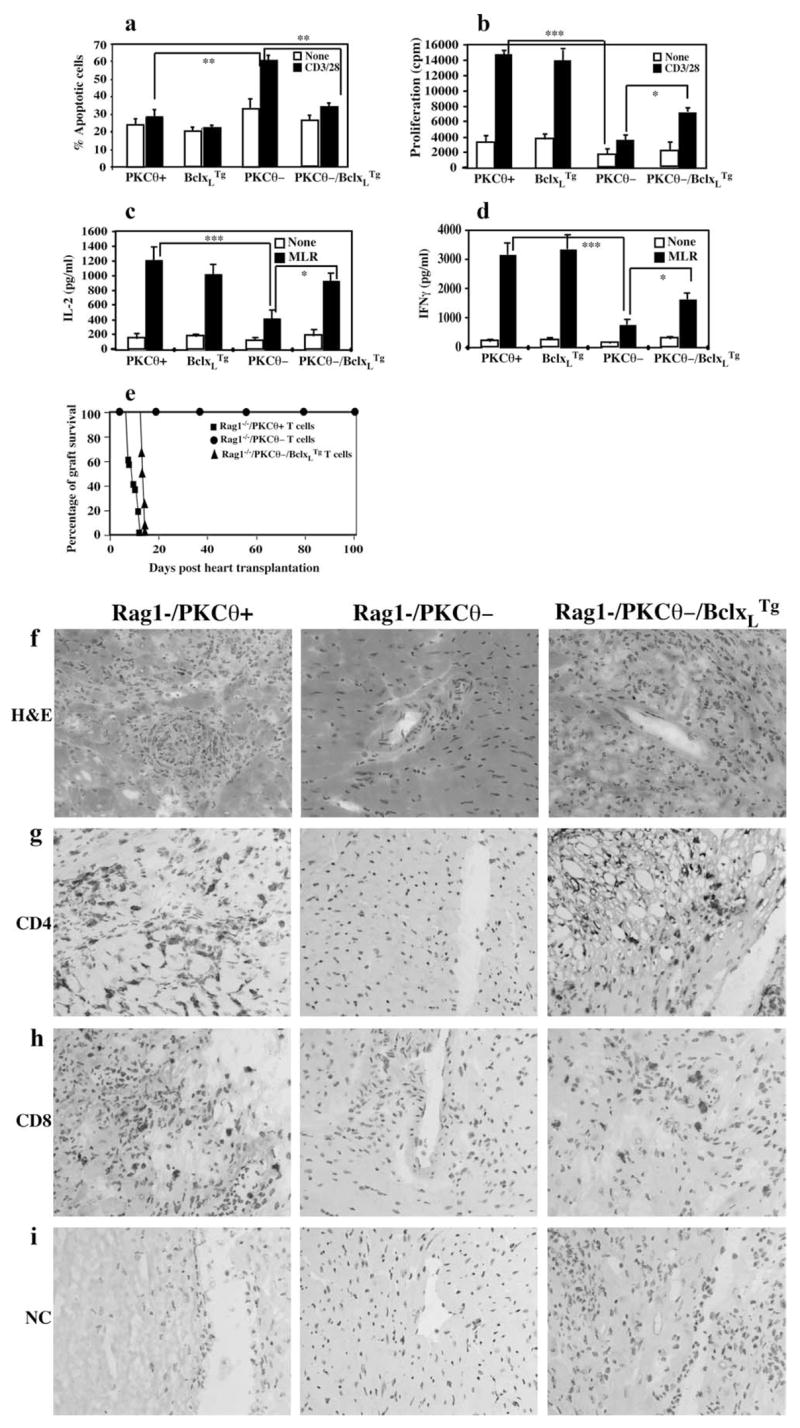
Transgenic expression of Bcl-xL in PKC-θ−/− T cells restored cardiac allograft rejection. a, Bcl-xLTg prevented the apoptosis of the PKC-θ−/− T cells. T cells purified from WT, Bcl-xLTg, PKC-θ−/−, and PKC-θ−/− /Bcl-xLTg mice were left in medium (□) or subjected to stimulation with plate-bound anti-CD3 (1 μg/ml) and soluble anti-CD28 (2 μg/ml) Abs (CD3/28) for 36 h. Apoptotic cells were then detected by flow cytometric analysis of annexin V-staining cells. Percentage of annexin V-positive cells was calculated. b, T cells obtained from WT, Bcl-xLTg, PKC-θ−/−, and PKC-θ−/− /Bcl-xLTg mice were either left alone (□) in medium or stimulated with BALB/c splenocytes (MLR, ■). Proliferation of the activated T cells was monitored by [3H]thymidine incorporation. c, Same treatment as described in b, and the IL-2 concentration in the supernatants was measured by ELISA. d, The same treatment as described in b, and the IFN-γ concentrations in the supernatant were measured by ELISA. e, Rag1−/− mice reconstituted with PKC-θ−/− /Bcl-xLTg T cells reject cardiac allografts. T cells purified from WT (n = 6), PKC-θ−/− (n = 6), and PKC-θ−/− /Bcl-xLTg (n = 5) mice were adoptively transferred into syngeneic Rag1−/− mice that received transplanted hearts from BALB/c donors. Survival of the cardiac allografts was assessed over time. f–i, Immunohistochemical analyses of the tissue sections of the cardiac allografts removed from Rag1−/− mice reconstituted with T cells obtained from WT (Rag1−/PKCθ+), PKC-θ−/− (Rag1-/PKCθ+), and PKC-θ−/− /Bcl-xLTg (Rag1-/PKCθ-/Bcl-xLTg) mice. Cryosections were prepared from cardiac allografts removed from recipients. Immunohistochemical analyses of the sections were performed with H&E staining (f), anti-CD4 Ab (g), anti-CD8 Ab (h), and isotype control Ab (i). The sections were examined using regular light microscope (×400) (*, p < 0.05; **, p < 0.001; ***, p < 0.0001).
To determine the effects of Bcl-xLTg on PKC-θ−/− T cell-mediated allograft rejection, cardiac allograft rejection was examined in Rag1−/−mice reconstituted with PKC-θ−/−/Bcl-xLTg T cells (Fig. 3e) (mean survival time, >100 vs 12 days; p < 0.005). Our previous experiments have demonstrated that Bcl-xLTg does not change the course of cardiac allograft rejection (35). Although cardiac allografts survived for >100 days in Rag1−/− mice reconstituted with PKC-θ−/− T cells, 100% of the Rag1−/− mice reconstituted with PKC-θ−/−/Bcl-xLTg T cells rejected cardiac allografts 15 days posttransplantation, similar to the Rag1−/− mice reconstituted with WT T cells. Sections of the transplanted hearts removed from recipients were examined by immunohistochemical analyses (Fig. 3, f–i). A large number of infiltrating cells and damaged muscle tissues were observed in the rejected hearts removed from Rag1−/− mice reconstituted with WT T cells (Fig. 3f, left panel), but not in the surviving hearts removed from PKC-θ−/− T cell-reconstituted Rag1−/− mice (Fig. 3f, middle panel). In agreement, infiltrating CD4+ (Fig. 3g, left panel) and CD8+ (Fig. 3h, left panel) cells were clearly detected in the rejected hearts, but not in the surviving hearts (middle panels of Fig. 3, g and h). As negative controls, no stained cells were observed in the sections treated with isotype control Ab (Fig. 3i), indicating the specificity of the anti-CD4 and the anti-CD8 Abs. Furthermore, hearts removed from the Rag1−/− mice reconstituted with PKC-θ−/− /Bcl- xLTg T cells also showed damaged muscle tissues (Fig. 3f, right panel) and infiltrating CD4+ (Fig. 3g, right panel) and CD8+ (Fig. 3h) cells, similar to the phenotypes observed in the rejected hearts. These results suggest that forced expression of Bcl-xLTg is sufficient to restore PKC-θ−/− T cell-mediated cardiac allograft rejection. PKC-θ-regulated survival of T cells thus plays a critical role in promoting acute rejection of cardiac allografts in the adoptive transfer model.
Intact PKC-θ−/− mice rejected cardiac allografts in a delayed manner
In addition to using adoptive transfer model, we investigated the fate of heart allografts in PKC-θ−/− mice (Fig. 4a). WT mice readily rejected heart allografts 7–10 days after transplantation. In contrast to the adoptive transfer experiments, PKC-θ−/− mice acutely rejected allogeneic hearts, although rejection occurred in a delayed fashion, 15–26 days after transplantation (mean survival time, 8.5 vs 19.5 days; p < 0.005). We hypothesized that impaired T cell function in the absence of PKC-θ can be partially compensated by other cell types present in spleen. To test this hypothesis, we adoptively transferred unsorted splenocytes from PKC-θ−/− mice into Rag1−/− mice, and monitored heart rejection (Fig. 4b). Mice reconstituted with either unsorted splenocytes or purified T cells from WT mice rejected cardiac allografts with a similar time course (mean survival time, 8.5 vs 9.5 days; p > 0.05). In clear contrast to Rag1−/− mice reconstituted with purified PKC-θ−/− T cells, Rag1−/− mice reconstituted with unsorted PKC-θ−/− splenocytes readily rejected allogeneic hearts within 17–27 days posttransplantation, a time course of rejection comparable to that in intact PKC-θ−/− mice (mean survival time, 20.5 vs 19.5 days; p > 0.05). Altogether, these results suggest that PKC-θ-regulated T cell function plays a critical role in cardiac allograft rejection. However, its absence can be partially compensated by other cells in the spleen.
FIGURE 4.
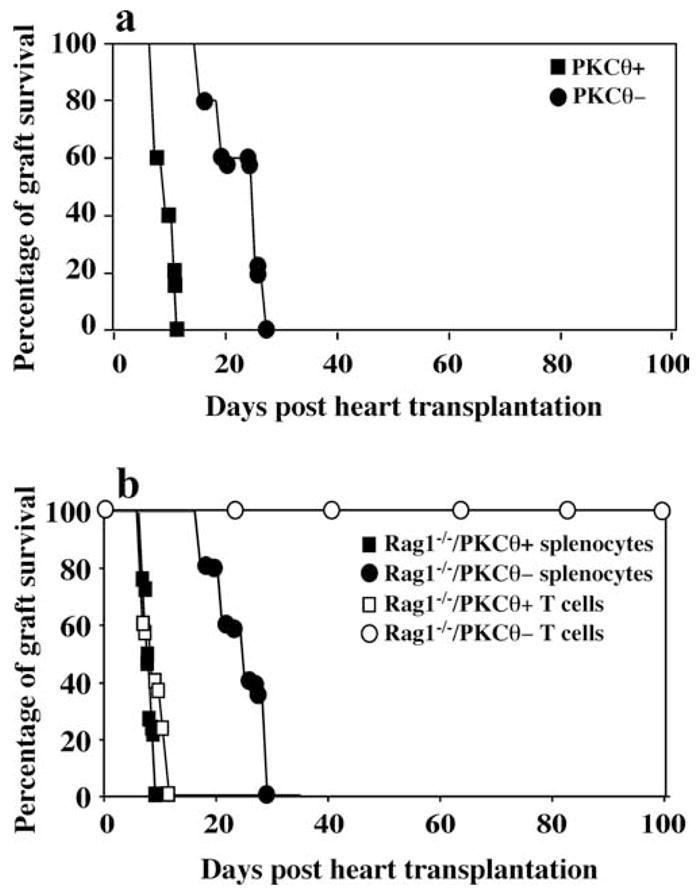
Delayed rejection of cardiac allografts by PKC-θ−/− mice. a, Hearts from BALB/c donors were transplanted to WT (PKCθ+, n = 6) or PKC-θ−/− (PKCθ−, n = 8) mice. Survival of the cardiac allografts was assessed over time. b, Unsorted splenocytes or purified T cells from both WT (n = 8) and PKC-θ−/− (n = 8) mice were adoptively transferred into Rag1−/− mice that received hearts from BALB/c donors. Survival of the cardiac allografts was assessed over time.
Treatment with a subtherapeutic dose of anti-CD154 mAb or CTLA4-Ig prevented cardiac allograft rejection in PKC-θ−/− mice, but not in WT mice
Because PKC-θ−/− mice eventually rejects the cardiac allografts, we seeked to determine whether additional therapies would enable long-term allograft survival in PKC-θ−/− mice. Anti-CD154 mAb treatment can promote long-term allograft survival (36). We determined the effects of different doses of anti-CD154 mAb on the cardiac allografts in our model (Fig. 5a). Without treatment, recipient mice readily rejected hearts 7–10 days after transplantation, whereas 3 doses (3 × 1 mg) of anti-CD154 mAb prevented rejection of hearts in WT mice (mean survival time, >100 vs 8.5 days; p < 0.002). A single dose of anti-CD154 mAb treatment only delayed rejection to 10–31 days posttransplantation (mean survival time, >19.75 vs 8.5 days; p < 0.05). We therefore investigated whether a single dose of anti-CD154 mAb could prevent heart rejection in PKC-θ−/− mice (Fig. 5b). Indeed, 100% of the transplanted hearts survived for >100 days in PKC-θ−/− mice treated with one dose of anti-CD154 mAb (mean survival time, >100 vs 20 days; p < 0.002). In addition, we also tested effects of CTLA4-Ig that inhibit the interactions between B7 and CD28 (Fig. 5c). One dose (0.2 mg/mouse) of CTLA4-Ig did not obviously change the course of rejection in WT mice, but allowed cardiac allografts survive for >100 days (mean survival time, >100 vs 12.5 days; p < 0.002). These results confirmed that PKC-θ plays a role in the regulation of alloreactive immune responses in vivo. These results suggest that drugs highly specific for PKC-θ may be likely to facilitate long-term allograft survival.
FIGURE 5.
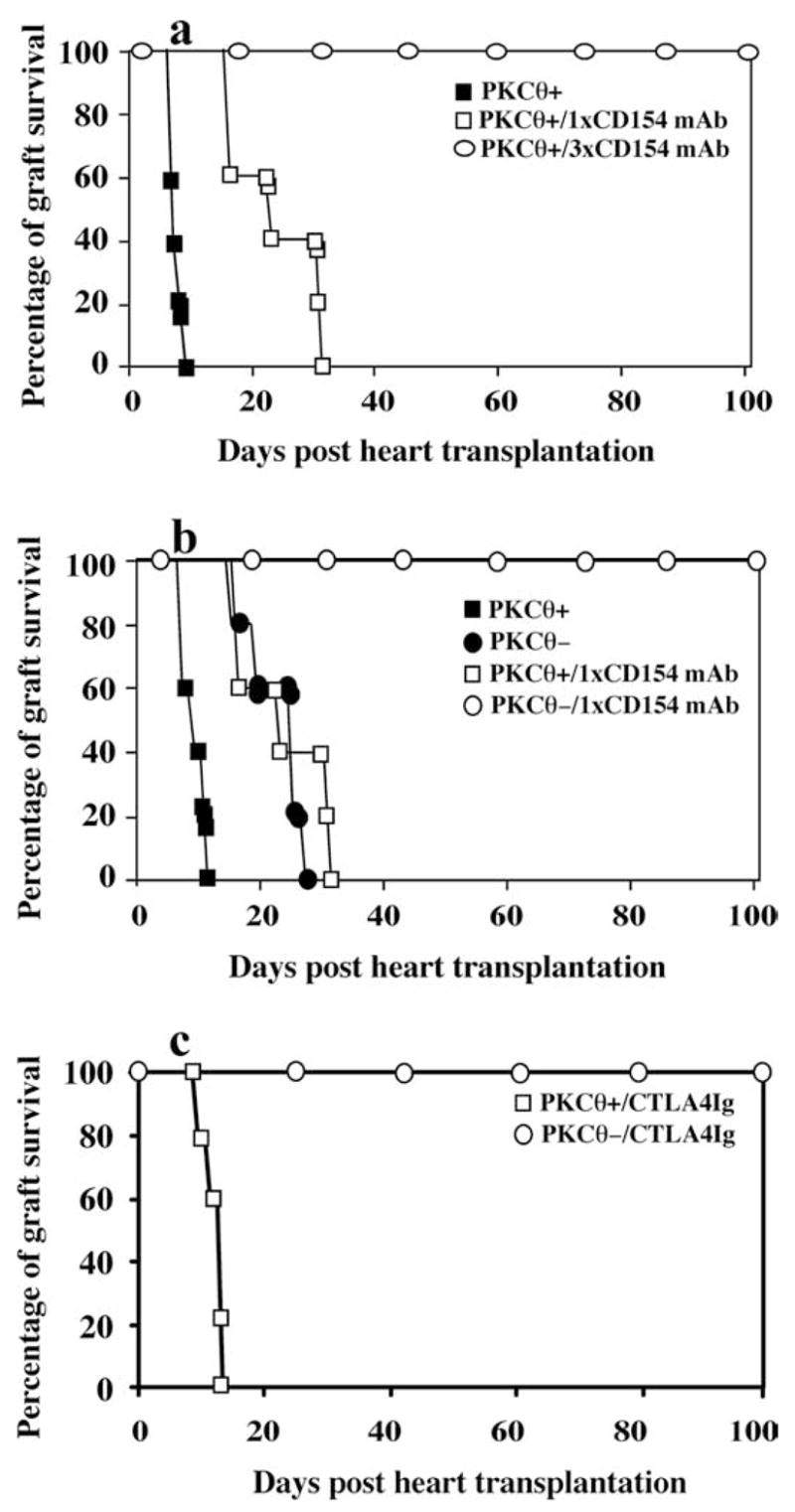
Treatment of PKC-θ−/− mice with a subtherapeutic dose of anti-CD154 mAb (mAb) or CTLA4-Ig prevented cardiac allograft rejection. a, Hearts from BALB/c donors were transplanted to WT mice treated with 0 mg (PKCθ+, n = 6), or once with 1 mg of anti-CD154 mAb (PKCθ+/1 × CD154 mAb, n = 6) or three times with 1 mg of anti-CD154 mAb (PKCθ+/3 × CD154 mAb, n = 8). Survival of the cardiac allografts was assessed over time. b, Hearts from BALB/c donors were transplanted to WT (n = 6) mice and PKC-θ−/− (n = 8) mice that were either not treated or treated once with 1 mg of anti- CD154 mAb (1× CD154 mAb). Survival of the cardiac allografts was assessed over time. c, Hearts from BALB/c donors were transplanted to WT (n = 5) mice and PKC-θ−/− (n = 6) mice that were treated with one dose of CTLA4-Ig (0.2 mg/mouse). Survival of the cardiac allografts was assessed over time.
Discussion
Activated T cells are susceptible to apoptosis (37, 38). TCRs deliver signals that are required not only for T cell activation, but also for enhancing cell survival (39, 40). Such survival signals ensure the completion of the T cell activation process essential for differentiating naive T cells into effectors that mediate actual immune responses (38). During T cell activation, the survival of the T cells is enhanced by IL-2, which acts as an extrinsic survival factor. In addition, activated T cells substantially up-regulate Bcl-xL that intrinsically increases their ability to resist apoptosis (38, 41, 42). Previous studies, including our own, have shown that PKC-θ is a critical signaling molecule mediating T cell survival by up-regulating Bcl-xL (4, 5, 32). In this study, we show that PKC-θ regulated T cell survival plays a critical role in acute cardiac allograft rejection.
Our adoptive transfer studies indicate that PKC-θ regulates the immune responses required for allograft rejection at least in part via affecting T cell survival. Our data show that in the absence of PKC-theta;, T cell responses to alloantigens are significantly reduced, suggesting a potential role for PKC-θ in allograft rejection. Indeed, PKC-θ−/− T cell-reconstituted Rag1−/− mice failed to reject cardiac allografts, confirming the notion that PKC-θ regulates T cell-mediated allograft rejection. This result also suggests that blocking PKC-θ function therapeutically may facilitate the survival of allografts. Because PKC-θ-mediated signals are required for both T cell activation and survival (2, 4, 31), it was important to distinguish whether PKC-θ-regulated survival was responsible for driving allograft rejection. PKC-θ enhances T cell survival by up-regulating Bcl-xL, which is supported by our data showing the restoration of the PKC-θ−/− T cell survival after transgenic expression of Bcl-xL. Furthermore, PKC-θ−/− T cell-mediated MLR and allograft rejection were also restored by Bcl-xLTg. The PKC-θ-regulated survival capacity of the T cells therefore influences the outcome of allografts. PKC-θ is believed to mediate CD28 costimulatory signals (1, 31, 43). Costimulatory blockade can promote allograft survival in many animal models (44–46). Such blockade has been shown to enhance apoptosis of the alloreactive T cells, resulting in a reduction in the number of reactive effector T cells in vivo. Furthermore, if the apoptosis is inhibited, costimulatory blockade fails in promoting tolerance to allografts (44–46). Because PKC-θ is a critical molecule regulating CD28-mediated survival signals, blockade of PKC-θ signals may thus facilitate the survival of allografts by reducing the alloreactive effector T cells.
Alloreactive responses responsible for the acute allograft rejection are usually 100-fold more powerful than the immune responses to nominal Ags most likely due to the higher precursor frequency of alloreactive T cells. A recent in vivo study showed that the precursor frequency of alloreactive T cells could be as high as 1 in 20 peripheral T cells (47), whereas the frequency of the Ag-specific T cells to a nominal Ag is roughly 1 in 106 peripheral T cells. Thus, the frequency of alloreactive T cells is five orders of magnitude greater than the frequency of the T cells specific for an Ag (48). It is proposed that the acquisition of transplantation tolerance is determined by the size of the alloreactive T cell pool (49). Our results can be explained by the fact that PKC-θ-regulated survival affects the size of the alloreactive T cell pool, which in turn influences allograft rejection. As PKC-θ−/− T cells undergo apoptosis in response to TCR stimulation (4, 5), it is predicted that the alloreactive PKC-θ−/− T cells are also susceptible to apoptosis in response to alloantigen stimulation, resulting in a reduced pool of the alloreactive T cells in the absence of PKC-θ. The failed allograft rejection observed in the PKC-θ−/− T cell-reconstituted Rag1−/− mice may thus result from the reduced number of allo reactive T cells. It is likely that Bcl-xLTg increases the number of effector alloreactive T cells by restoring the survival of PKC-θ−/− T cells, resulting in restoration of allograft rejection.
Our data do not favor the possibilities that anergy or T regulatory (Treg) cells are responsible for the failed allograft rejection. Previously, we have shown that the defective PKC-θ−/− T cell activation cannot be rescued by phorbol ester and ionomycin stimulation (2). Phorbol ester and ionomycin have been used to overcome anergy because they can bypass TCRs and directly stimulate T cells (50). Exogenous IL-2 can partially rescue the defective PKC-θ−/− T cell activation (Fig. 1a), because IL-2 can actually inhibit the apoptosis of the PKC-θ−/− T cells (4). Furthermore, anti-apoptotic Bcl-xL transgene restored cardiac allograft rejection, suggesting that it is the survival, but not likely the anergy, responsible for the failed cardiac allograft rejection. We have also examined Treg cells in PKC-θ−/− mice, and demonstrated that PKC-θ−/− mice have markedly reduced (5- to 10-fold) Treg cells (data not shown). Furthermore, Bcl-xL transgene that restored cardiac allograft rejection did not have effects on the reduced Treg cells, suggesting that the greatly reduced Treg cells are not likely to play a major role in the failed cardiac allograft rejection. What is then responsible for the difference in complete acceptance of the adoptive transfer model and the rejection of intact PKC-θ−/− mice? Stimulation of T cells in vivo depends on APCs to provide costimulatory signals. PKC-θ has been shown to mediate the CD28 costimulatory signals to stimulate T cell activation (43, 51). CD28 costimulatory signals are also responsible for up-regulation of Bcl-xL during T cell activation (38, 40, 41). We thus hypothesize that although PKC-θ mediates the important costimulatory signals required for T cell survival, its function can be overcomed or compensated by overwhelming costimulatory stimulation. This hypothesis explains why splenocytes capable of providing costimulatory stimulation restored cardiac allograft rejection even in the absence of PKC-θ. This hypothesis also predicts that further reduction of costimulatory signals in intact PKC-θ−/− mice can improve and may even achieve long-term survival of the cardiac allografts. Indeed, CTLA4-Ig treatment prevented heart rejection in PKC-θ−/− mice, suggesting that inhibition of CD28-mediated costimulatory signals is sufficient to prevent heart rejection.
Although allograft rejection remains a major problem in transplantation, immunosuppressive drugs that inhibit immune system make transplantation possible. Because PKC-θ is a critical molecule regulating T cell activation, drugs highly specific for PKC-θ are considered potentially useful for treating allograft rejection and autoimmunity (31, 52). Our results indicate that the inhibition of PKC-θ itself may not be sufficient for establishing long-term survival of allografts. In contrast to what we observed in Rag1−/− mice reconstituted with PKC-θ−/− T cells, cardiac allografts are rejected by PKC-θ−/− mice, although in a delayed manner. The function of PKC-θ can be partially compensated by unknown mechanisms. Our results also demonstrate that in combination with a subtherapeutic dose of anti-CD154 Ab, cardiac allograft rejection can be prevented in PKC-θ−/− mice. One of the functions of CD154 on T cells is to stimulate CD40 on dendritic cells, resulting in the up-regulation of B7 family members (53, 54). Up-regulation of B7 molecules leads to enhanced stimulation of CD28-mediated survival signals (55, 56). Blockade of CD40-CD154 interactions has been found to be an extremely effective approach to prevent allograft rejection (57). It is not clear whether the reduced survival capacity due to blocking CD154 contributes to the tolerance of cardiac allograft in PKC-θ−/− mice. Our results suggest that sub-therapeutic doses of the immunosuppressive drug, when combined with PKC-θ-specific inhibitor, may effectively prevent allograft rejection. Current immunosuppressive drugs have severe side effects not associated with their immunosuppressive action. For example, cyclosporine A and tacrolimus are nephrotoxic (58). In addition, the diabetogenic effect of tacrolimus (59) is also a major clinical problem associated with immunosuppressive drug use. It is possible that combining treatment of lower doses of immunosuppressive drugs with the PKC-θ inhibitors may prevent allograft rejection with significantly reduced side effects.
Acknowledgments
We thank Lianli Ma for the help with heart transplantation, and Dr. Mark Dizik for critically reading the manuscript.
Footnotes
This work was supported by grants from American Cancer Society of Illinois Division, Schweppe Foundation, UIC University of Illinois Cancer Center, and UIC Institutional Review Board, and National Institutes of Health Grant R01-AI053147.
Abbreviations used in this paper: PKC, protein kinase C; Bcl-xLTg, Bcl-xL transgenic; Treg, T regulatory; WT, wild type.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Altman A, Isakov N, Baier G. Protein kinase Cθ: a new essential superstar on the T-cell stage. Immunol Today. 2000;21:567–573. doi: 10.1016/s0167-5699(00)01749-7. [DOI] [PubMed] [Google Scholar]
- 2.Sun Z, Arendt CW, Ellmeier W, Schaeffer EM, Sunshine MJ, Gandhi L, Annes J, Petrzilka D, Kupfer A, Schwartzberg PL, Littman DR. PKC-θ is required for TCR-induced NF-κB activation in mature but not immature T lymphocytes. Nature. 2000;404:402–407. doi: 10.1038/35006090. [DOI] [PubMed] [Google Scholar]
- 3.Pfeifhofer C, Kofler K, Gruber T, Tabrizi NG, Lutz C, Maly K, Leitges M, Baier G. Protein kinase Cθ affects Ca2+ mobilization and NFAT cell activation in primary mouse T cells. J Exp Med. 2003;197:1525–1535. doi: 10.1084/jem.20020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manicassamy S, Gupta S, Huang Z, Sun Z. Protein kinase C-θ-mediated signals enhance CD4+ T cell survival by up-regulating Bcl-xL. J Immunol. 2006;176:6709–6716. doi: 10.4049/jimmunol.176.11.6709. [DOI] [PubMed] [Google Scholar]
- 5.Barouch-Bentov R, Lemmens EE, Hu J, Janssen EM, Droin NM, Song J, Schoenberger SP, Altman A. Protein kinase C-θ is an early survival factor required for differentiation of effector CD8+ T cells. J Immunol. 2005;175:5126–5134. doi: 10.4049/jimmunol.175.8.5126. [DOI] [PubMed] [Google Scholar]
- 6.Marsland BJ, Soos TJ, Spath G, Littman DR, Kopf M. Protein kinase Cθ is critical for the development of in vivo T helper (Th)2 cell but not Th1 cell responses. J Exp Med. 2004;200:181–189. doi: 10.1084/jem.20032229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salek-Ardakani S, So T, Halteman BS, Altman A, Croft M. Differential regulation of Th2 and Th1 lung inflammatory responses by protein kinase Cθ. J Immunol. 2004;173:6440–6447. doi: 10.4049/jimmunol.173.10.6440. [DOI] [PubMed] [Google Scholar]
- 8.Salek-Ardakani S, So T, Halteman BS, Altman A, Croft M. Protein kinase Cθ controls Th1 cells in experimental autoimmune encephalomyelitis. J Immunol. 2005;175:7635–7641. doi: 10.4049/jimmunol.175.11.7635. [DOI] [PubMed] [Google Scholar]
- 9.Tan SL, Zhao J, Bi C, Chen XC, Hepburn DL, Wang J, Sedgwick JD, Chintalacharuvu SR, Na S. Resistance to experimental autoimmune encephalomyelitis and impaired IL-17 production in protein kinase Cθ-deficient mice. J Immunol. 2006;176:2872–2879. doi: 10.4049/jimmunol.176.5.2872. [DOI] [PubMed] [Google Scholar]
- 10.Baier G. The PKC gene module: molecular biosystematics to resolve its T cell functions. Immunol Rev. 2003;192:64–79. doi: 10.1034/j.1600-065x.2003.00018.x. [DOI] [PubMed] [Google Scholar]
- 11.Manicassamy S, Sun Z. The critical role of protein kinase C-θ in Fas/FasL-mediated apoptosis. J Immunol. 2007;178:312–319. doi: 10.4049/jimmunol.178.1.312. [DOI] [PubMed] [Google Scholar]
- 12.Su TT, Guo B, Kawakami Y, Sommer K, Chae K, Humphries LA, Kato RM, Kang S, Patrone L, Wall R, et al. PKC-θ controls IκB kinase lipid raft recruitment and activation in response to BCR signaling. Nat Immunol. 2002;3:780–786. doi: 10.1038/ni823. [DOI] [PubMed] [Google Scholar]
- 13.Saijo K, Mecklenbrauker I, Santana A, Leitger M, Schmedt C, Tarakhovsky A. Protein kinase Cβ controls nuclear factor κB activation in B cells through selective regulation of the IκB kinase α. J Exp Med. 2002;195:1647–1652. doi: 10.1084/jem.20020408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altman A, Kaminski S, Busuttil V, Droin N, Hu J, Tadevosyan Y, Hipskind RA, Villalba M. Positive feedback regulation of PLCγ1/Ca2+ signaling by PKCθ in restimulated T cells via a Tec kinase-dependent pathway. Eur J Immunol. 2004;34:2001–2011. doi: 10.1002/eji.200324625. [DOI] [PubMed] [Google Scholar]
- 15.Manicassamy S, Sadim M, Ye RD, Sun Z. Differential roles of PKC-θ in the regulation of intracellular calcium concentration in primary T cells. J Mol Biol. 2006;355:347–359. doi: 10.1016/j.jmb.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 16.Baier-Bitterlich G, Uberall F, Bauer B, Fresser F, Wachter H, Grunicke H, Utermann G, Altman A, Baier G. Protein kinase C-θ isoenzyme selective stimulation of the transcription factor complex AP-1 in T lymphocytes. Mol Cell Biol. 1996;16:1842–1850. doi: 10.1128/mcb.16.4.1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Hu J, Vita R, Sun B, Tabata H, Altman A. SPAK kinase is a substrate and target of PKCθ in T-cell receptor-induced AP-1 activation pathway. EMBO J. 2004;23:1112–1122. doi: 10.1038/sj.emboj.7600125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno-Garcia ME, Ovechkina YL, Rawlings DJ. Phosphorylation of the CARMA1 linker controls NF-κB activation. Immunity. 2005;23:561–574. doi: 10.1016/j.immuni.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 19.Matsumoto R, Wang D, Blonska M, Li H, Kobayashi M, Pappu B, Chen Y, Lin X. Phosphorylation of CARMA1 plays a critical role in T cell receptor-mediated NF-κB activation. Immunity. 2005;23:575–585. doi: 10.1016/j.immuni.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 20.Wang D, You Y, Case SM, McAllister-Lucas LM, Wang L, DiStefano PS, Nunez G, Bertin J, Lin X. A requirement for CARMA1 in TCR-induced NF-κB activation. Nat Immunol. 2002;3:830–835. doi: 10.1038/ni824. [DOI] [PubMed] [Google Scholar]
- 21.Lee KY, D’Acquisto F, Hayden MS, Shim JH, Ghosh S. PDK1 nucleates T cell receptor-induced signaling complex for NF-κB activation. Science. 2005;308:114–118. doi: 10.1126/science.1107107. [DOI] [PubMed] [Google Scholar]
- 22.Hara H, Wada T, Bakal C, Kozieradzki I, Suzuki S, Suzuki N, Nghiem M, Griffiths EK, Krawczyk C, Bauer B, et al. The MAGUK family protein CARD11 is essential for lymphocyte activation. Immunity. 2003;18:763–775. doi: 10.1016/s1074-7613(03)00148-1. [DOI] [PubMed] [Google Scholar]
- 23.Jun JE, Wilson LE, Vinuesa CG, Lesage S, Blery M, Miosge LA, Cook MC, Kucharska EM, Hara H, Penninger JM, et al. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity. 2003;18:751–762. doi: 10.1016/s1074-7613(03)00141-9. [DOI] [PubMed] [Google Scholar]
- 24.Egawa T, Albrecht B, Favier B, Sunshine MJ, Mirchandani K, O’Brien W, Thome M, Littman DR. Requirement for CARMA1 in antigen receptor-induced NF-κB activation and lymphocyte proliferation. Curr Biol. 2003;13:1252–1258. doi: 10.1016/s0960-9822(03)00491-3. [DOI] [PubMed] [Google Scholar]
- 25.Ruland J, Duncan GS, Wakeham A, Mak TW. Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity. 2003;19:749–758. doi: 10.1016/s1074-7613(03)00293-0. [DOI] [PubMed] [Google Scholar]
- 26.Xue L, Morris SW, Orihuela C, Tuomanen E, Cui X, Wen R, Wang D. Defective development and function of Bcl10-deficient follicular, marginal zone and B1 B cells. Nat Immunol. 2003;4:857–865. doi: 10.1038/ni963. [DOI] [PubMed] [Google Scholar]
- 27.Ruefli-Brasse AA, French DM, Dixit VM. Regulation of NF-κB-dependent lymphocyte activation and development by paracaspase. Science. 2003;302:1581–1584. doi: 10.1126/science.1090769. [DOI] [PubMed] [Google Scholar]
- 28.Pattison JM, Krensky AM. New insights into mechanisms of allograft rejection. Am J Med Sci. 1997;313:257–263. doi: 10.1097/00000441-199705000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Chao DT, Linette GP, Boise LH, White LS, Thompson CB, Korsmeyer SJ. Bcl-xL and Bcl-2 repress a common pathway of cell death. J Exp Med. 1995;182:821–828. doi: 10.1084/jem.182.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yin D, Dujovny N, Ma L, Varghese A, Shen J, Bishop DK, Chong AS. IFN-γ production is specifically regulated by IL-10 in mice made tolerant with anti-CD40 ligand antibody and intact active bone. J Immunol. 2003;170:853–860. doi: 10.4049/jimmunol.170.2.853. [DOI] [PubMed] [Google Scholar]
- 31.Manicassamy S, Gupta S, Sun Z. Selective function of PKC-θ in T cells. Cell Mol Immunol. 2006;3:263–270. [PubMed] [Google Scholar]
- 32.Saibil SD, Jones RG, Deenick EK, Liadis N, Elford AR, Vainberg MG, Baerg H, Woodgett JR, Gerondakis S, Ohashi PS. CD4+ and CD8+ T cell survival is regulated differentially by protein kinase Cθ, c-Rel, and protein kinase B. J Immunol. 2007;178:2932–2939. doi: 10.4049/jimmunol.178.5.2932. [DOI] [PubMed] [Google Scholar]
- 33.Xie H, Huang Z, Sadim MS, Sun Z. Stabilized β-catenin extends thymocyte survival by up-regulating Bcl-xL. J Immunol. 2005;175:7981–7988. doi: 10.4049/jimmunol.175.12.7981. [DOI] [PubMed] [Google Scholar]
- 34.Xie H, Sadim MS, Sun Z. RORγt recruits steroid receptor coactivators to ensure thymocyte survival. J Immunol. 2005;175:3800–3809. doi: 10.4049/jimmunol.175.6.3800. [DOI] [PubMed] [Google Scholar]
- 35.Zhou P, Balin SJ, Mashayekhi M, Hwang KW, Palucki DA, Alegre ML. Transplantation tolerance in NF-κB-impaired mice is not due to regulation but is prevented by transgenic expression of Bcl-xL. J Immunol. 2005;174:3447–3453. doi: 10.4049/jimmunol.174.6.3447. [DOI] [PubMed] [Google Scholar]
- 36.Larsen CP, Elwood ET, Alexander DZ, Ritchie SC, Hendrix R, Tucker-Burden C, Cho HR, Aruffo A, Hollenbaugh D, Linsley PS, Winn KJ, Pearson TC. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996;381:434–438. doi: 10.1038/381434a0. [DOI] [PubMed] [Google Scholar]
- 37.Boehme SA, Lenardo MJ. Propriocidal apoptosis of mature T lymphocytes occurs at S phase of the cell cycle. Eur J Immunol. 1993;23:1552–1560. doi: 10.1002/eji.1830230724. [DOI] [PubMed] [Google Scholar]
- 38.Radvanyi LG, Shi Y, Vaziri H, Sharma A, Dhala R, Mills GB, Miller RG. CD28 costimulation inhibits TCR-induced apoptosis during a primary T cell response. J Immunol. 1996;156:1788–1798. [PubMed] [Google Scholar]
- 39.Weiss A, Littman DR. Signal transduction by lymphocyte antigen receptors. Cell. 1994;76:263–274. doi: 10.1016/0092-8674(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 40.Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-xL. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 41.Van Parijs L, Ibraghimov A, Abbas AK. The roles of costimulation and Fas in T cell apoptosis and peripheral tolerance. Immunity. 1996;4:321–328. doi: 10.1016/s1074-7613(00)80440-9. [DOI] [PubMed] [Google Scholar]
- 42.Noel PJ, Boise LH, Green JM, Thompson CB. CD28 costimulation prevents cell death during primary T cell activation. J Immunol. 1996;157:636–642. [PubMed] [Google Scholar]
- 43.Lin X, O’Mahony A, Mu Y, Geleziunas R, Greene WC. Protein kinase C-θ participates in NF-κB activation induced by CD3-CD28 costimulation through selective activation of IκB kinase β. Mol Cell Biol. 2000;20:2933–2940. doi: 10.1128/mcb.20.8.2933-2940.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Li XC, Zheng XX, Wells AD, Turka LA, Strom TB. Blocking both signal 1 and signal 2 of T-cell activation prevents apoptosis of alloreactive T cells and induction of peripheral allograft tolerance. Nat Med. 1999;5:1298–1302. doi: 10.1038/15256. [DOI] [PubMed] [Google Scholar]
- 45.Wells AD, Li XC, Li Y, Walsh MC, Zheng XX, Wu Z, Nunez G, Tang A, Sayegh M, Hancock WW, et al. Requirement for T-cell apoptosis in the induction of peripheral transplantation tolerance. Nat Med. 1999;5:1303–1307. doi: 10.1038/15260. [DOI] [PubMed] [Google Scholar]
- 46.Wekerle T, Kurtz J, Sayegh M, Ito H, Wells A, Bensinger S, Shaffer J, Turka L, Sykes M. Peripheral deletion after bone marrow transplantation with costimulatory blockade has features of both activation-induced cell death and passive cell death. J Immunol. 2001;166:2311–2316. doi: 10.4049/jimmunol.166.4.2311. [DOI] [PubMed] [Google Scholar]
- 47.Suchin EJ, Langmuir PB, Palmer E, Sayegh MH, Wells AD, Turka LA. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol. 2001;166:973–981. doi: 10.4049/jimmunol.166.2.973. [DOI] [PubMed] [Google Scholar]
- 48.Li XC, Strom TB, Turka LA, Wells AD. T cell death and transplantation tolerance. Immunity. 2001;14:407–416. doi: 10.1016/s1074-7613(01)00121-2. [DOI] [PubMed] [Google Scholar]
- 49.Chiffoleau E, Walsh PT, Turka L. Apoptosis and transplantation tolerance. Immunol Rev. 2003;193:124–145. doi: 10.1034/j.1600-065x.2003.00037.x. [DOI] [PubMed] [Google Scholar]
- 50.Blackman MA, Finkel TH, Kappler J, Cambier J, Marrack P. Altered antigen receptor signaling in anergic T cells from self-tolerant T-cell receptor β-chain transgenic mice. Proc Natl Acad Sci USA. 1991;88:6682–6686. doi: 10.1073/pnas.88.15.6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Coudronniere N, Villalba M, Englund N, Altman A. NF-κB activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-θ. Proc Natl Acad Sci USA. 2000;97:3394–3399. doi: 10.1073/pnas.060028097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Isakov N, Altman A. Protein kinase Cθ in T cell activation. Annu Rev Immunol. 2002;20:761–794. doi: 10.1146/annurev.immunol.20.100301.064807. [DOI] [PubMed] [Google Scholar]
- 53.Grewal IS, Flavell RA. A central role of CD40 ligand in the regulation of CD4+ T-cell responses. Immunol Today. 1996;17:410–414. doi: 10.1016/0167-5699(96)10030-x. [DOI] [PubMed] [Google Scholar]
- 54.Koch F, Stanzl U, Jennewein P, Janke K, Heufler C, Kampgen E, Romani N, Schuler G. High level IL-12 production by murine dendritic cells: up-regulation via MHC class II and CD40 molecules and down-regulation by IL-4 and IL-10. J Exp Med. 1996;184:741–746. doi: 10.1084/jem.184.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang Y, Wilson JM. CD40 ligand-dependent T cell activation: requirement of B7-CD28 signaling through CD40. Science. 1996;273:1862–1864. doi: 10.1126/science.273.5283.1862. [DOI] [PubMed] [Google Scholar]
- 56.Maxwell JR, Campbell JD, Kim CH, Vella AT. CD40 activation boosts T cell immunity in vivo by enhancing T cell clonal expansion and delaying peripheral T cell deletion. J Immunol. 1999;162:2024–2034. [PubMed] [Google Scholar]
- 57.Wekerle T, Kurtz J, Ito H, Ronquillo JV, Dong V, Zhao G, Shaffer J, Sayegh MH, Sykes M. Allogeneic bone marrow transplantation with costimulatory blockade induces macrochimerism and tolerance without cytoreductive host treatment. Nat Med. 2000;6:464–469. doi: 10.1038/74731. [DOI] [PubMed] [Google Scholar]
- 58.Myers BD, Newton L. Cyclosporine-induced chronic nephropathy: an obliterative microvascular renal injury. J Am Soc Nephrol. 1991;2:S45–S52. doi: 10.1681/ASN.V22s45. [DOI] [PubMed] [Google Scholar]
- 59.Wijnen RM, Ericzon BG. Update of tacrolimus in pancreas transplantation. Diabet Med. 1997;14:911–918. doi: 10.1002/(SICI)1096-9136(199711)14:11<911::AID-DIA513>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]