

Abstract
X-linked lymphoproliferative syndrome (XLP) is an inherited immunodeficiency characterized by increased susceptibility to Epstein–Barr virus (EBV). In affected males, primary EBV infection leads to the uncontrolled proliferation of virus-containing B cells and reactive cytotoxic T cells, often culminating in the development of high-grade lymphoma. The XLP gene has been mapped to chromosome band Xq25 through linkage analysis and the discovery of patients harboring large constitutional genomic deletions. We describe here the presence of small deletions and intragenic mutations that specifically disrupt a gene named DSHP in 6 of 10 unrelated patients with XLP. This gene encodes a predicted protein of 128 amino acids composing a single SH2 domain with extensive homology to the SH2 domain of SHIP, an inositol polyphosphate 5-phosphatase that functions as a negative regulator of lymphocyte activation. DSHP is expressed in transformed T cell lines and is induced following in vitro activation of peripheral blood T lymphocytes. Expression of DSHP is restricted in vivo to lymphoid tissues, and RNA in situ hybridization demonstrates DSHP expression in activated T and B cell regions of reactive lymph nodes and in both T and B cell neoplasms. These observations confirm the identity of DSHP as the gene responsible for XLP, and suggest a role in the regulation of lymphocyte activation and proliferation. Induction of DSHP may sustain the immune response by interfering with SHIP-mediated inhibition of lymphocyte activation, while its inactivation in XLP patients results in a selective immunodeficiency to EBV.
Keywords: Epstein–Barr virus/immunodeficiency/lymphoma
Epstein–Barr Virus (EBV) is a ubiquitous DNA virus associated in developed countries with infectious mononucleosis (IM) (1). After primary exposure, EBV infects mature B cells by binding to the C3b/CR2/CD21 cell-surface receptor, leading to their transformation and proliferation. In immunocompetent individuals, this is followed by the activation of reactive T cells, and then the classical features of IM (2–5). Neutralizing antibodies against EBV are detected in most individuals, indicating prior infection, and lifelong persistence of virus-infected B cells may be demonstrated by their outgrowth after in vitro treatment of peripheral blood lymphocytes (PBLs) with cyclosporine. The proliferation of latent virus-infected B cells is suppressed by EBV-specific T cells (6), and their uncontrolled proliferation is a common complication in individuals whose T cell immunity is compromised following bone marrow and solid-organ transplantation or development of AIDS (7).
XLP is an X-linked syndrome in which affected boys display unique vulnerability to infection by EBV (8). Remarkably, XLP patients do not display increased susceptibility to other members of the herpes family of DNA viruses, such as herpes simplex virus or cytomegalovirus. No specific immunological defect has been demonstrated in XLP-affected boys before EBV infection, although subtle abnormalities in IgM levels and IgG subclasses have been reported (9). Exposure to EBV results in fulminant IM characterized by massive proliferation of EBV-infected B cells and reactive cytotoxic T cells, which may result in fatal infiltration of the liver and other organs (10, 11). Patients who recover often develop severe hypogammaglobulinemia, and many go on to develop a monoclonal proliferation of EBV-infected B cells, typically an immunoblastic lymphoma (12). Less common manifestations of XLP syndrome include the development of hypogammaglobulinemia, aplastic anemia, or inflammatory conditions such as necrotizing vasculitis or lymphomatoid granulomatosis (11, 13).
The gene for XLP was initially mapped to Xq25 by linkage analysis (14, 15). This was confirmed by the discovery of three unrelated patients with large, cytogenetically visible deletions at this locus (16–19). The fact that these large hemizygous deletions, spanning 2–4 Mb, were not associated with detectable genetic abnormalities other than XLP suggested that few other critical genes reside within the XLP locus. We have previously reported the presence of a 150-kb genomic deletion in a fourth XLP patient (20), although concerted efforts were unsuccessful at identifying a coding sequence within or flanking this small deletion. We now describe the presence of additional small deletions, as well as specific intragenic mutations, that target an SH2 domain-encoding gene mapping to Xq25. In vitro and in vivo analysis of gene expression suggests that this gene is an intrinsic component of lymphocyte activation pathways.
MATERIALS AND METHODS
Clinical Specimens.
Eleven patients meeting criteria for XLP were included for analysis in this study. These criteria included clinical characteristics such as fulminant IM, EBV-associated lymphoma, or hypogammaglobulinemia, and a family history of one or more maternally related affected males. EBV-immortalized cell lines were established from patients AD, R, and T by using standard procedures. Cell lines from patients D, M, 724, and 1720 were kindly provided by B. Sylla (International Agency for Research on Cancer, Lyon, France), patients 8001, 8002, and 8005 by G. Romeo (Institute Gaslini, Genoa, Italy), and patient A by J. Skare (Boston University).
Genomic Mapping and Analysis for Expressed Sequences.
Yeast artificial chromosomes (YACs) mapping to the XLP locus were identified from the CEPH [Centre d’Étude du Polymorphisme Humain (Paris)] MegaYAC library by using probes generated from genomic sequences at Xq25. YACs spanning and flanking the 150-kb deletion in XLP patient D were subcloned into a cosmid library that was used to establish a 350-kb contig across the deletion. Potential coding sequences were identified by using exon amplification of pooled and individual cosmids (21, 22), cDNA selection using pooled cosmids or YACs to screen a thymus cDNA library (23, 24), and direct genomic sequencing of cosmids followed by analysis for ORFs using the grail and gene finder programs. Potential coding sequences were used to screen tonsil, thymus, Jurkat, fetal brain, and testis cDNA libraries by using standard protocols. cDNA clones were mapped to the XLP locus and sequenced. For analysis of regions centromeric and telomeric to the 150-kb deletion in patient D, genomic sequences centered around known markers delineating the XLP locus (available in GenBank) were analyzed for homology with published expressed sequence tags. Genomic sequences of Xqter-q24 are available through the X chromosome mapping project at the Sanger Genome Center (http://www.sanger.ac.uk). Sequences contained in contig dJ1052M9 (GenBank accession no. HS1052M9) were found to have identity with several expressed sequence tags and a short cDNA clone (GenBank accession no. AL023657), both of which were deleted in patient A. Screening of a tonsil cDNA library and reverse transcription–PCR (RT-PCR) analyses were used to confirm that the cDNA called DSHP was full-length.
Mutational Analysis.
Lymphoblast DNA from XLP patients was screened for gross deletions by using Southern blot analysis. The genomic structure of DSHP was deduced from comparison of cDNA and genomic sequences, and each of the four exons and flanking introns were PCR-amplified, followed by automated sequence analysis (Applied Biosystems ABI 373). RT-PCR amplification of RNA from EBV-immortalized lymphoblasts was used to examine the DSHP transcript.
Expression Studies.
Northern blot analysis was performed by using RNA derived from multiple normal human tissues (CLONTECH) and a panel of cell lines generated from lymphoid and solid tumors according to standard procedures. For in vitro T cell activation, PBLs were isolated by using Leukoprep gradients (Becton Dickinson), washed twice with PBS, and either treated with Con A (2 μg/ml, Sigma) or plated onto flasks coated with anti-CD3 antibody (American Type Culture Collection), after which cells were harvested at various intervals. In B cell activation experiments, PBLs were cultured in the presence of goat anti-human IgM antibody (10 μg/ml, Zymed). For in vitro EBV infection, PBLs were treated with virus from the producer cell line B95–8 (American Type Culture Collection) either in the presence or absence of cyclosporine. For in situ RNA hybridization experiments, a riboprobe corresponding to 400 nt of the DSHP 3′ untranslated region was generated and used to analyze formalin-fixed, paraffin-embedded tissue. Sense and antisense probes were labeled with digoxigenin-UTP (Boehringer Mannheim) and hybridized to sections. Signal amplification was accomplished by using the TSA-Indirect Kit (NEN). Immunohistochemistry was performed by using antibodies to CD20 (1:100), CD3 (1:500), CD45RO (1:4000), Ki-1 (1:100), and Ki67 (1:50), all of which were obtained from Dako. Known immunoreactive and nonreactive tissues were used as controls.
RESULTS
We initiated our search for the XLP gene by analyzing the smallest Xq25 genomic deletion, identified in patient D. Interphase fluorescence in situ hybridization mapping and pulse-field gel electrophoresis confirmed a deletion measuring approximately 150 kb nested within a larger deletion of ≈3 Mb in patient A (see Fig. 1A). YACs from the XLP locus were used to generate a cosmid contig spanning the small deletion and flanking genomic sequences. However, analysis for coding sequences using exon trapping, cDNA selection, and direct genomic sequence analysis was unrevealing. Evaluation of sequence information derived from cosmid clones spanning the deletion and flanking regions demonstrated that it is rich in LINE repeats and that its centromeric breakpoint is the center of a large inverted duplication spanning approximately 100 kb whose outer borders are delineated by a duplicated fibulin pseudogene (data not shown). Because extensive analysis of 300 kb of genomic sequence centered around this deletion did not identify genuine transcription units, we considered the possibility that this deletion may affect regulatory sequences for the XLP gene, or that it may result from a secondary, incidental chromosomal recombination event. We therefore extended our analysis to the 3-Mb deletion in patient A, a task facilitated by the availability of genomic sequences from Xqter to Xq24 provided by the X chromosome sequencing project. Analysis of genomic sequences approximately 1 Mb centromeric to the deletion in patient D revealed identity to six expressed sequence tags and to a cDNA called DSHP, of unreported function.
Figure 1.

Physical map of the XLP locus and identification of new genomic deletions disrupting DSHP. (A) Schematic representation of the XLP locus. The locus has been defined by a 3-Mb genomic deletion in patient A. Two new patients, R and AD, harbor deletions of 200 kb that span the DSHP gene, and are approximately 1 Mb centromeric to the previously reported 150 kb deletion in patient D. (B) Delineation of the borders of genomic deletions in patients R and AD, using PCR markers for centromeric and telomeric borders, and Southern blot analysis using probes from the 5′ and 3′ regions of DSHP. DSHP is entirely deleted in patient AD, whereas the centromeric breakpoint in patient R is between exons 1 and 2. (C) Comparison of the amino acid sequence of DSHP with the SH2 domain of SHIP. Identical amino acids are in black boxes, and conserved residues are in gray boxes. DSHP exon boundaries are indicated by arrowheads.
DSHP encodes a predicted polypeptide of only 128 amino acids comprising a single SH2 domain (Fig. 1C). To determine whether it may represent a truncated gene product, we screened a human tonsil cDNA library and undertook RT-PCR analysis of multiple human tissues. These experiments confirmed the sequence of DSHP and the absence of alternatively spliced transcripts. Of particular interest, the SH2 domain of DSHP has 45% amino acid identity (63% conserved residues) with the SH2 domain of SHIP, a 145-kDa inositol polyphosphate 5-phosphatase, whose recruitment to the FcγRIIB and T cell receptors is instrumental in down-regulating B and T cell activation (25–29). A polypeptide comprising only a single SH2 domain may function as a competitive inhibitor of other proteins in which a similar SH2 domain is linked to functional moieties, raising the possibility that DSHP may serve to inhibit SHIP signaling.
Sequence comparison between DSHP and the corresponding genomic sequence indicates that the gene spans 25 kb and is encoded by four exons (data not shown). We searched for gross deletions of this gene in a panel of 10 unrelated XLP patients (excluding patient A, who had a 3-Mb deletion spanning the entire XLP locus). Southern blotting revealed deletions in two new patients, AD and R. Analysis of patient AD showed loss of probes generated from both the 5′ and 3′ DSHP untranslated sequences, but retention of PCR markers flanking the gene (Fig. 1 A and B). In contrast, the centromeric breakpoint of the deletion in patient R was internal to DSHP, with retention of exon 1, loss of the 3′ DSHP probe, but presence of the flanking telomeric PCR marker. The estimated size of both deletions is <200 kb.
To search for point mutations internal to DSHP, we sequenced PCR-amplified exons from the remaining eight XLP patients. Four were found to have mutations, all within exon 2 and resulting in premature chain termination (Table 1). Patient 8001 has a deletion of 23 nucleotides, leading to a frameshift, whereas patients D, T, and 8005 have C-to-T substitutions, resulting in the introduction of stop codons. Patients T and 8005, who are unrelated and of different ethnic origin, have identical mutations at a potential site for DNA methylation. It was of considerable interest that patient D, whose 150-kb deletion had previously been thought to define the minimal XLP locus, in fact has a point mutation within DSHP, suggesting that the genomic deletion is incidental. Repeated analysis of DSHP exons and flanking intronic sequences in the remaining XLP patients did not reveal mutations. In these patients, full-length DSHP cDNA was readily amplified by RT-PCR from EBV-transformed cell lines (data not shown), suggesting that alterations in potential regulatory sequences were unlikely to result in complete absence of gene expression. No missense mutations or potential polymorphisms were identified in any of the patients. Thus, of 11 unrelated XLP patients, hemizygous inactivation of DSHP was present in 7 (64%), of whom 4 had intragenic mutations, confirming its identity as the gene responsible for XLP syndrome.
Table 1.
DSHP mutational analysis
| Patient | Exon | Codon | Nucleotide change (nt position) | Effect on protein |
|---|---|---|---|---|
| D | 2 | 55 | C-to-T (164) | Stop at codon 55 |
| T | 2 | 58 | C-to-T (173) | Stop at codon 58 |
| 8001 | 2 | 61 | Deletion 23 bp (183) | Frameshift, stop at codon 75 |
| 8005 | 2 | 58 | C-to-T (173) | Stop at codon 58 |
The uncontrolled proliferation of EBV-infected B cells in patients with XLP has been taken as evidence that the disease may result from failure of reactive T cells to respond appropriately to viral infection (8, 11, 30). As an initial step in dissecting the physiological function of the XLP gene, we examined its expression pattern in human tissues and during lymphocyte activation. Northern blots of normal human tissues demonstrated expression of a 2.5-kb transcript in peripheral blood, spleen, thymus, and tonsil (Fig. 2A). Low levels of expression were also evident in colon, lung, and liver (not shown), probably reflecting the presence of lymphoid or reticuloendothelial cells in these organs (see below). High levels of DSHP mRNA expression was noted in 4 out of 4 transformed T cell lines (HBPALL, CEM, Jurkat, and Molt 4) (Fig. 2A). Minimal levels of expression were observed by Northern blot analysis in Daudi cells, but expression was below detection in three other transformed B cell lines, EBV-immortalized lymphoblasts derived from normal controls, and in two myeloid cell lines. A smaller transcript measuring ≈1 kb was evident in cells expressing high levels of DSHP mRNA; this less abundant species was not detected with probes derived from the extreme 3′ untranslated sequence, consistent with alternative polyadenylation sites within the transcript (data not shown).
Figure 2.

Northern blot analysis of DSHP expression. (A) DSHP expression in normal human tissues and in B, T, and myeloid cell lines, using a probe generated from the 3′ untranslated region. (B) Induction of DSHP expression following activation of PBLs using Con A or antibody to CD3.
To examine expression of DSHP during lymphocyte activation, PBLs were treated with different stimuli to induce either B or T cell proliferation. T cell activation using Con A induced expression of the transcript as early as 6 hr, with maximal expression at 48 hr (Fig. 2B). Treatment with anti-CD3 antibody also induced expression of DSHP at 48 hr. B cell activation using anti-IgM antibody, in the absence of coactivators, failed to induce expression at similar time points (data not shown). DSHP was also not detectably induced up to 72 hr following infection of PBLs with EBV, either in the presence or absence of cyclosporine, which serves to abrogate the T cell response to EBV-infected proliferating B cells (data not shown). However, the cellular events that follow in vitro infection with EBV involve both cell proliferation and cell death, as well as immediate and delayed cellular reponses, making this a more complex model than the direct activation of T cell surface receptors.
To examine the expression pattern of DSHP in vivo, we used RNA in situ hybridization to detect its expression at the single-cell level. Analysis of normal lymphoid tissues, including lymph nodes, spleen, and thymus revealed prominent expression in reactive germinal centers and paracortical regions (Fig. 3a). Strongest expression was present in morphologically activated lymphocytes, those with large vesicular nuclei and nucleoli in germinal centers, and paracortical regions. A lesser degree of hybridization was detected in some smaller lymphocytes in these regions, and no expression was detected in the mature small lymphocytes of the mantle zone and medullary cords of nonreactive lymph nodes. Immunophenotypic analysis of serial tissue sections indicated that both T and B lymphocyte-rich regions expressed DSHP (Fig. 3 b and c), and the same zones showed a high proliferation rate as determined by Ki67 immunoreactivity and increased numbers of cells positive for CD30, a marker associated with activated lymphocytes (data not shown). In the colon, expression of DSHP was limited to cells within the mucosa-associated lymphoid tissue and scattered inflammatory cells in the lamina propria, explaining the low levels of transcript detectable in this tissue by Northern blotting (data not shown). Analysis of a panel of malignant lymphomas showed DSHP expression in some cases of both Hodgkin’s and non-Hodgkin’s lymphoma. Variable expression levels were detected in diffuse large-cell lymphomas of both B and T cell types (Fig. 3d). Strong expression was present in the atypical cells of both nodular and lymphocyte-predominant Hodgkin’s disease and in some of the accompanying reactive lymphocytes (data not shown). The expression pattern of DSHP in normal lymphoid tissue and malignant lymphoma, as well as during lymphocyte activation in vitro, suggests a physiological role for the XLP gene during lymphocyte activation and proliferation.
Figure 3.
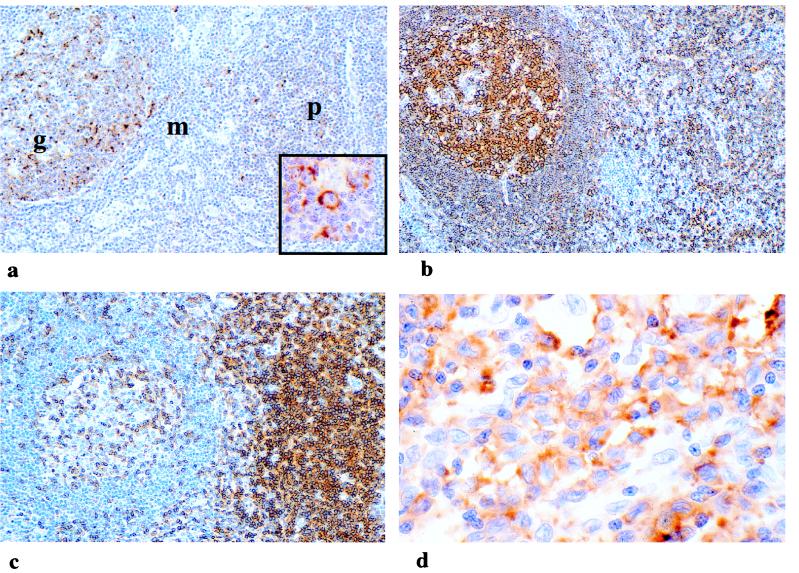
DSHP expression in normal reactive lymph node and malignant lymphoma. (a) Strong expression of DSHP in some germinal center (g) and paracortical (p) cells as demonstated by RNA in situ hybridization using antisense probe. Cells of mantle zone (m) do not express DSHP. (Inset) High magnification of strongly expressing cells with activated morphologic features of vesicular nucleus and nucleolus. Hybridization using sense probe was negative. Immunohistochemical analysis of this section demonstrates (b) the distribution of B cells using antibody to the marker CD30 and (c) the distribution of T cells using antibody to CD3. (d) Strong expression of DSHP in atypical cells of diffuse large B cell lymphoma.
DISCUSSION
The DSHP gene encodes a small protein, consisting of a single SH2 domain, expressed in proliferating cultured T cells and in the activated lymphocytes of both T and B cell areas of normal lymphoid tissues. Of the 11 XLP patients analyzed here, 7 demonstrate inactivation of DSHP, including 4 with definitive intragenic mutations, 2 with small genomic deletions that target the gene, and 1 with a large deletion of the entire XLP locus. These observations provide strong evidence supporting the specific inactivation of this gene in XLP. A number of possibilities may explain the absence of mutations in the remaining cases. First, possible mutations in regulatory sequences affecting expression of the DSHP transcript would not have been detected in our analysis. Although we were able to amplify this transcript by RT-PCR from EBV-transformed cell lines of all of the patients who did not have a chromosomal deletion, we cannot exclude subtle abnormalities in expression levels that might have been observed by more quantitative assays in more appropriate cell types. Second, the diagnosis of XLP may be questionable in some cases, because there exists significant overlap between the clinical features characteristic of this syndrome and other syndromes associated with severe IM, EBV-associated hemophagocytosis, or aberrant lymphoproliferation associated with viral infection. It is possible that mutations in other genes involved in the cellular response to EBV infection may be responsible for some cases of virus-associated lymphoproliferation.
The absence of abnormal lymphocyte proliferation in maternal XLP carriers, whose lymphocyte population is mosaic for gene inactivation as a consequence of random X chromosome inactivation (31), has been taken as evidence that loss of XLP gene function does not directly lead to malignant transformation of EBV-infected B cells. Instead, a role for the XLP gene in the regulation of reactive lymphocytes is supported by the observation that cells with an intact XLP gene in maternal carriers appear to compensate for those in which the gene has been inactivated. This postulated mechanism of action for the XLP gene is consistent with the expression pattern of DSHP. The gene is expressed at high levels in transformed T cell lines, and it is readily induced following activation of resting peripheral blood T cells using agents that are dependent as well as independent of T cell receptor signaling. Minimal expression is detectable in EBV-transformed B cell lines. RNA in situ hybridization analysis of normal lymphoid tissues indicates that DSHP expression is present in both B and T cell regions, and examination of malignant lymphomas demonstrates expression in neoplasms derived from B as well as T cells. Consistent with in vitro observations, expression of DSHP in vivo is restricted to activated lymphocytes and to high-grade lymphomas.
Together with its restricted expression profile, the sequence of DSHP indicates that it may be an important component of lymphocyte activation pathways. The presence of a single SH2 domain in this small polypeptide, without additional adaptor or functional domains, suggests that it may function as a physiological competitor of other SH2-containing proteins whose binding to cell-surface receptors triggers a signaling cascade during lymphocyte activation. It is therefore of particular interest that the SH2 domain of DSHP has extensive sequence similarity with that of SHIP, an inositol polyphosphate 5-phosphatase expressed in cells of hematopoietic origin including T and B lymphocytes, myeloid cells, and mast cells. The recruitment or phosphorylation of SHIP through activation of cell-surface receptors, including the FcγRIIB in B cells (26, 27, 32, 33), the high-affinity IgE receptor (FceRI) in mast cells, the T cell receptor in T cells (34), and the M-CSF receptor in monocytes and macrophages (25, 35), links this molecule to signal-transduction pathways activated after ligand binding and/or receptor clustering. Various mechanisms through which SHIP may repress cellular activation have been proposed, including reduction in the intracellular levels of phosphatidylinositol 3,4,5-triphosphate with blunting of the intracellular calcium response (26) and a reduction in ras activation through binding to Shc (28, 29, 33, 36, 37). By competing with SHIP, DSHP may effectively delay the termination of lymphocyte activation, functioning as a physiological enhancer of the immune response. Functional studies of the DSHP gene product will be required to test this model.
The apparent specific immunodeficiency of XLP patients to EBV infection bears reexamination in light of these insights into the physiological expression pattern and the potential function of the XLP gene. It is likely that the DSHP gene product is a general modulator of lymphocyte activation, whose inactivation may have selective consequences for different arms of the immune response. Exposure to the ubiquitous virus EBV during childhood may therefore be one of the first and most potent challenges to which a moderately compromised immune response may prove inadequate. Although the clinical diagnosis of XLP has rested on its association with EBV-induced lymphoproliferation, the availability of a genetic diagnosis is likely to result in a broader appreciation for the various manifestations of this syndrome. For instance, XLP patient AD presented with characteristic fulminant IM following a primary EBV infection; however, his two brothers, who shared the same DSHP mutation, developed diverse immunological abnormalities in the absence of EBV infection, including an atypical pulmonary infiltrate resembling Wegener’s granulomamosis in one and an atypical lymphoproliferative process with infiltration of lungs, liver, spleen, and colon in the second. Inactivation of DSHP may thus lead to aberrant lymphocytic proliferation in response to EBV, as well as other unidentified stimuli. Functional studies of the XLP gene product, together with a better understanding of the clinical syndrome resulting from its inactivation, are likely to provide important insight into the regulation of lymphocyte activation and proliferation.
Acknowledgments
We are indebted to the patients and their families who contributed blood specimens for this study and to their referring physicians, including Dr. R. Nelson (All Children’s Hospital, Miami, Florida), Dr. A. Wayne (University of Miami), and Dr. S. Shurin (University of Cleveland). We are also grateful to Drs. S. Alexander, L. Silverman, L. Lehman (Dana-Farber Cancer Institute, Boston), B. Sylla (International Agency for Research on Cancer, Lyon, France), and G. Romeo (Instituto Gaslini, Genoa, Italy) for providing additional clinical specimens. We thank D. Wahrer and J. Wissink for expert technical assistance, Drs. J. Rioux, T. Golub, and T. Hudson (Whithead Institute, Cambridge, MA) for help in identifying genomic and expressed sequence tag clones mapping to this region. This work was supported by National Cancer Institute Grants CA64088 (to D.A.H.) and CA71907 (to A.T.L.) and National Institutes of Health Grant K11 AI01331-04 (to K.E.N.).
ABBREVIATIONS
- XLP
X-linked lymphoproliferative syndrome
- EBV
Epstein–Barr virus
- IM
infectious mononucleosis
- YAC
yeast artificial chromosome
- RT-PCR
reverse transcription–PCR
- PBL
peripheral blood lymphocytes
Footnotes
A Commentary on this article begins on page 13355.
References
- 1.Straus S, Cohen J, Tosato G, Meier J. Ann Intern Med. 1993;118:45–58. doi: 10.7326/0003-4819-118-1-199301010-00009. [DOI] [PubMed] [Google Scholar]
- 2.Callan M, Steven N, Krausa P, Wilson J, Moss P, Gillespie G, Bell J, Rickinson A, McMichael A. Nat Med. 1996;2:906–911. doi: 10.1038/nm0896-906. [DOI] [PubMed] [Google Scholar]
- 3.Tosato G, Magrath I, Koski I, Dooley N, Blaese M. N Engl J Med. 1979;301:1133–1137. doi: 10.1056/NEJM197911223012101. [DOI] [PubMed] [Google Scholar]
- 4.Waele M D, Theilemans C, Camp B V. N Engl J Med. 1981;304:460–462. doi: 10.1056/NEJM198102193040804. [DOI] [PubMed] [Google Scholar]
- 5.Svedmyr E, Jondal M. Proc Natl Acad Sci USA. 1975;72:1622–1626. doi: 10.1073/pnas.72.4.1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miyashita E, Yang B, Lam K, Crawford D, Thorley-Lawson D. Cell. 1995;80:593–601. doi: 10.1016/0092-8674(95)90513-8. [DOI] [PubMed] [Google Scholar]
- 7.Purtilo D, Strobach R, Okano M, Davis J. Lab Invest. 1992;67:5–23. [PubMed] [Google Scholar]
- 8.Purtilo D, Cassel C, Yang J, Harper R. Lancet. 1975;i:935–940. doi: 10.1016/s0140-6736(75)92004-8. [DOI] [PubMed] [Google Scholar]
- 9.Grierson H, Skare L, Hawk J, Pauza M, Purtilo D. Am J Med Genet. 1991;40:294–297. doi: 10.1002/ajmg.1320400309. [DOI] [PubMed] [Google Scholar]
- 10.Grierson H, Purtilo D. Ann Int Med. 1987;106:538–545. doi: 10.7326/0003-4819-106-4-538. [DOI] [PubMed] [Google Scholar]
- 11.Seemayer T, Gross T, Egeler R, Pirruccello S, Davis J, Kelley C, Okano M, Lanyi A, Sumegi J. Pediatr Res. 1995;38:471–478. doi: 10.1203/00006450-199510000-00001. [DOI] [PubMed] [Google Scholar]
- 12.Harrington D, Weisenburger D, Purtilo D. Cancer. 1987;59:1419–1429. doi: 10.1002/1097-0142(19870415)59:8<1419::aid-cncr2820590807>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 13.Purtilo D, Grierson H, Davis J, Okano M. Pediatr Pathol. 1991;11:685–710. doi: 10.3109/15513819109065466. [DOI] [PubMed] [Google Scholar]
- 14.Skare J, Milunsky A, Byron K, Sullivan J. Proc Natl Acad Sci USA. 1987;84:2015–2018. doi: 10.1073/pnas.84.7.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skare J, Sullivan J, Milunsky A. Hum Genet. 1989;82:349–353. doi: 10.1007/BF00273996. [DOI] [PubMed] [Google Scholar]
- 16.Sanger W, Grierson H, Skare J, Wyandt H, Pirruccello S, Fordyce R, Purtilo D. Cancer Genet Cytogenet. 1990;47:163–169. doi: 10.1016/0165-4608(90)90026-7. [DOI] [PubMed] [Google Scholar]
- 17.Wyandt H, Grierson H, Sanger W, Skare J, Milunsky A, Purtilo D. Am J Med Gen. 1989;33:426–430. doi: 10.1002/ajmg.1320330331. [DOI] [PubMed] [Google Scholar]
- 18.Skare J, Wu B, Madan S, Pulijaal V, Purtilo D, Haber D, Nelson D, Sylla B, Grierson H, Nitowsky H, et al. Genomics. 1993;16:254–255. doi: 10.1006/geno.1993.1169. [DOI] [PubMed] [Google Scholar]
- 19.Wu B, Milunsky A, Nelson D, Schmeckpeper B, Porta G, Schlessinger D, Skare J. Genomics. 1993;17:163–170. doi: 10.1006/geno.1993.1298. [DOI] [PubMed] [Google Scholar]
- 20.Lamartine J, Nichols K, Yin L, Krainer M, Heitzmann F, Bernard A, Gaudi S, Lenoir G, Sullivan J, Ikeda J, et al. Eur J Hum Genet. 1996;4:342–351. doi: 10.1159/000472230. [DOI] [PubMed] [Google Scholar]
- 21.Buckler A, Chang D, Graw S, Brook J, Haber D, Sharp P, Housman D. Proc Natl Acad Sci USA. 1991;88:4005–4009. doi: 10.1073/pnas.88.9.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Church D, Stotler C, Rutter J, Murrell J, Trofatter J, Buckler A. Nat Genet. 1994;6:98–105. doi: 10.1038/ng0194-98. [DOI] [PubMed] [Google Scholar]
- 23.Tagle D, Swaroop M, Lovett M, Collins F. Nature (London) 1993;61:751–753. doi: 10.1038/361751a0. [DOI] [PubMed] [Google Scholar]
- 24.Lovett M, Kere J, Hinton L. Proc Natl Acad Sci USA. 1991;88:9628–9632. doi: 10.1073/pnas.88.21.9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lioubin M, Algate P, Tsai S, Carlberg K, Aebersold A, Rohrschneider L. Genes Dev. 1996;10:1084–1095. doi: 10.1101/gad.10.9.1084. [DOI] [PubMed] [Google Scholar]
- 26.Ono M, Bolland S, Tempst P, Ravetch J. Nature (London) 1996;383:263–266. doi: 10.1038/383263a0. [DOI] [PubMed] [Google Scholar]
- 27.Chacko G, Tridandapani S, Damen J, Liu L, Krystal G, Coggeshall K. J Immunol. 1996;157:2234–2238. [PubMed] [Google Scholar]
- 28.Ware M, Rosten P, Damen J, Liu L, Humphries R, Krystal G. Blood. 1996;88:2833–2840. [PubMed] [Google Scholar]
- 29.Liu L, Damen J, Ware M, Hughes M, Krystal G. Leukemia. 1997;11:181–184. doi: 10.1038/sj.leu.2400559. [DOI] [PubMed] [Google Scholar]
- 30.Lindsten T, Seeley J, Ballow M, Sakamoto K, Onge S S, Yetz J, Aman P, Purtilo D. J Immunol. 1982;129:2536–2540. [PubMed] [Google Scholar]
- 31.Conley M, Sullivan J, Neidich J, Puck J. Clin Immunol Immunopathol. 1990;55:486–491. doi: 10.1016/0090-1229(90)90133-b. [DOI] [PubMed] [Google Scholar]
- 32.Ono M, Bolland S, Yanagi S, Kurosaki T, Ravetch J. Cell. 1997;90:293–301. doi: 10.1016/s0092-8674(00)80337-2. [DOI] [PubMed] [Google Scholar]
- 33.Tridandapani S, Kelley T, Pradhan M, Cooney D, Justement L, Coggeshall K. Mol Cell Biol. 1997;17:4305–4311. doi: 10.1128/mcb.17.8.4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kavanaugh W, Pot D, Chin S, Deuter-Reinhard M, Jefferson A, Norris F, Masiarz F, Cousens L, Majerus P, Williams L. Curr Biol. 1996;6:438–445. doi: 10.1016/s0960-9822(02)00511-0. [DOI] [PubMed] [Google Scholar]
- 35.Odai H, Sasaki K, Iwamatsu A, Nakamoto T, Ueno H, Yamagata T, Yazaki Y, Hirai H. Blood. 1997;89:2745–2756. [PubMed] [Google Scholar]
- 36.Mitchell C, Brown S, Campbell J, Munday A, Speed C. Biochem Soc Trans. 1996;24:994–1000. doi: 10.1042/bst0240994. [DOI] [PubMed] [Google Scholar]
- 37.Lamkin T, Walk S, Liu L, Damen J, Krystal G, Ravichandran K. J Biol Chem. 1997;272:10396–10401. doi: 10.1074/jbc.272.16.10396. [DOI] [PubMed] [Google Scholar]