

Abstract
Calcium permeability of l-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors (AMPARs) in excitatory neurons of the mammalian brain is prevented by coassembly of the GluR-B subunit, which carries an arginine (R) residue at a critical site of the channel pore. The codon for this arginine is created by site-selective adenosine deamination of an exonic glutamine (Q) codon at the pre-mRNA level. Thus, central neurons can potentially control the calcium permeability of AMPARs by the level of GluR-B gene expression as well as by the extent of Q/R-site editing, which in postnatal brain, positions the R codon into >99% of GluR-B mRNA. To study whether the small amount of unedited GluR-B is of functional relevance, we have generated mice carrying GluR-B alleles with an exonic arginine codon. We report that these mutants manifest no obvious deficiencies, indicating that AMPAR-mediated calcium influx into central neurons can be solely regulated by the levels of Q/R site-edited GluR-B relative to other AMPAR subunits. Notably, a targeted GluR-B gene mutant with 30% reduced GluR-B levels had 2-fold higher AMPAR-mediated calcium permeability in hippocampal pyramidal cells with no sign of cytotoxicity. This constitutes proof in vivo that elevated calcium influx through AMPARs need not generate pathophysiological consequences.
l-α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors (AMPARs) mediate fast excitatory neurotransmission in central neurons (1). AMPARs in principal excitatory neurons possess low Ca2+ permeability but display elevated Ca2+ permeability in interneurons, a functional distinction that was traced to different levels of GluR-B subunit expression (2). GluR-B is unique among four AMPAR subunits (GluR-A to GluR-D) (3) in that an exonic glutamine (Q) codon for a critical channel position is changed to an arginine (R) codon by a process of RNA editing (4). The presence of R instead of Q in the critical channel position of GluR-B prevents Ca2+ conductance through AMPARs containing this subunit, whereas AMPARs without GluR-B are Ca2+-permeable (5–8).
Thus, in principle, neurons can regulate the Ca2+ conductance of AMPARs by the extent of GluR-B gene transcription as well as by the extent of GluR-B Q/R-site editing. Although regulation by GluR-B gene expression has been established, it is unknown whether Q/R-site editing is regulated. RNA editing ensures that nearly 100% of the GluR-B subunits in postnatal brain contain the R residue in the channel pore (4, 9). This mechanism operates at the pre-mRNA level and requires the formation of a double-stranded RNA structure of exonic and intronic sequences, directed by a cis-acting, exon-complementary intronic sequence (ECS) (9). A reduction by approximately 25% in the efficiency of Q/R-site editing led to severe neurological dysfunctions and premature death of mice carrying the editing-deficient GluR-BΔECS allele, created by deleting the intronic cis-acting ECS element (10). Although these mice demonstrated that a high preponderance of the edited over the unedited GluR-B subunit is essential for normal brain physiology, the potential role of the unedited GluR-B subunit in brain remained unaddressed. A functional role may be assumed from the findings that in the mammalian brain the Q/R site of GluR-B is edited to less than 100% (4, 9) and hence, distinct cell populations may express only unedited GluR-B. In certain interneurons, for example, the Q/R-site editing of GluR-B may be down-regulated to contribute to the high Ca2+ permeability of AMPARs. Moreover, at early embryonal stages (E12 in rat), the unedited form can constitute up to 20% of GluR-B mRNA (11). Furthermore, Q/R-site editing may be regulated, for instance by neuronal activity, to increase transiently the Ca2+ permeability of AMPAR channels.
To ascertain whether the Q/R site-unedited GluR-B subunit has a physiological function, we substituted into the GluR-B gene the exonic Q (CAG) codon for the Q/R site by an R (CGG) codon. We found that absence of Q/R site-unedited GluR-B did not noticeably perturb development and function of the mouse brain. This result indicates that the Ca2+ conductance of AMPARs can be regulated solely by relative expression levels of the Q/R site-edited GluR-B subunit. One mouse mutant that carries the phosphoglycerate kinase–neomycin phosphotransferase (PGK-neo) gene inserted in GluR-B gene intron 11 showed moderate reduction in GluR-B expression and afforded experimental proof that the concomitant 2-fold increase in the Ca2+ permeability of hippocampal pyramidal cell AMPARs in vivo is tolerated without phenotypic consequences.
MATERIALS AND METHODS
Generation of GluR-BRneo/Rneo and GluR-BR/R Gene-Targeted Mice.
A mouse 129/sv genomic BsrGI-SalI fragment in pBluescript SKII(−) containing exons 10–12 of the GluR-B locus (10, 12) was used for the construction of the targeting vector pGRBRNEO. A codon for arginine at the Q/R site was introduced by PCR mutagenesis along with four silent mutations in exon 11(see Fig. 1). For selection of embryonic stem (ES) cells, a loxP-PGK-neo-loxP sequence from vector pLOXPNEO3 was introduced into intron 11 such that it replaced a 56-bp sequence between NcoI and KpnI sites. pLOXPNEO3 is a derivative of pLOXPNEO1 (13) with changes in the multiple cloning sites flanking the loxP-PGK-neo-loxP module. R1 ES cells (14) were electroporated (BioRad gene pulser set at 240 V, 500 μF) with 70 μg of the SalI linearized targeting vector. Cell clones resistant to G418 (250 μg/ml) were isolated after 10 days. Four targeted clones were identified by nested PCR analysis with rsp27 (5′-AGGACGCGCGGCAAACGAGGGCACCCG-3′) and rsp28 (5′-GCAGGCAAGAGCCGAGGCGAGGCCAAG-3′) as sense primers in intron 9 sequences outside the targeting vector and pgkprom1 (5′-GAATGTGTGCGAGGCCAGAGG-3′) and pgkprom2 (5′-CAGACTGCCTTGGGAAAAGCG-3′) as antisense primers in the loxP-PGK-neo-loxP sequence. The correct targeting in these clones was confirmed by Southern blot analysis (data not shown). One clone, GRB/R44, was injected into C57/Bl6 blastocysts. A highly chimeric male transmitted to offspring the targeted GluR-BRneo allele, yielding the line GluR-B+/Rneo. The same chimeric male was bred with females of the “deleter” line (15) for germ-line transmission of the GluR-BRneo allele and simultaneous removal from this allele of the floxed PGK-neo marker in intron 11, generating the GluR-B+/R line. The GluR-BR allele still contains 105 bp of exogenous sequence (one loxP site and part of a multiple cloning site) in the proximal part of intron 11. Genotyping was carried out by PCR with MH53 as sense primer and rsp36 as antisense primer (10). Primers rspneo4 (5′-GGCTATTCGGCTATGACTGGGC-3′) and rspneo5 (5′-GGGTAGCCAACGCTATGTCCTG-3′) were used to detect the presence of the PGK-neo cassette in GluR-B+/Rneo mice. PCR on genomic DNA from brains confirmed the complete removal of PGK-neo sequences in GluR-B+/R mice (data not shown).
Figure 1.

GluR-B alleles having an exonic arginine codon for the inner pore segment of AMPAR channels. (A) Schematic representation of the GluR-B subunit and of GluR-B gene segments corresponding to different alleles: GluR-B+ (wild-type allele), GluR-BRneo (targeted allele), and GluR-BR (targeted allele after Cre-mediated removal of the floxed neo cassette). In the GluR-B protein (hatched), the N and C termini and the Q/R site are indicated; black boxes mark membrane insertion domains M1–M4. The GluR-B gene segments show numbered exons (boxes), the exon-complementary, cis-acting ECS element in intron 11, the position of the Q/R site, the location of the exonic arginine (R) codon for the Q/R site, as well as the locations of primers MH53 (a) and RSP36 (b) used for genotyping. The loxP sites are shown as filled arrowheads. The floxed PGK-neo marker in the GluR-BRneo allele was inserted between intronic NcoI (No) and KpnI (Kp) sites. • in the GluR-BRneo allele border the targeting vector (BsrGI-SalI fragment). (B) Genotyping by PCR. The expected sizes of amplicons for wild-type and GluR-BR alleles are 494 and 599 bp, respectively. Genotypes are indicated on top of gel, size markers are given in kbp on left of gel.
Gene Expression Analysis.
Reverse–transcription (RT)-PCR amplification of GluR-B mRNA from brains of GluR-B+/R and GluR-B+/Rneo mice was carried out with sense primer BG PCR1 (5′-GCGAATTCAGAAGTCCAAACCAGGAG-3′) and antisense primer BG PCR2 (5′-GCGGTACCTTGGCGAAATATCGCATC-3′) in exon 11. The amplicons were directionally cloned in M13 mp18 replicative-form DNA for recombinant plaque analysis by differential oligonucleotide hybridization (4, 10). In addition, amplicons were sequenced on an ABI377 automated sequencer (Perkin–Elmer), and relative expression levels from different alleles were derived from visual comparison of peak areas corresponding to nucleotides at exon 11 positions where silent mutations were introduced.
Immunoblotting.
To compare the overall levels of GluR-B protein in wild-type and mutant mice, forebrains of adult homozygous mice were dissected, homogenized in 0.32 M sucrose in 5 mM Hepes (pH 7.4) containing a mixture of protease inhibitors (Complete; Boehringer Mannheim), and centrifuged for 15 min at 3,000 × g. The supernatant was subsequently centrifuged for 30 min at 10,000 × g. The pellet was resuspended in 1% SDS, and the amount of total protein was determined (BCA protein assay reagent, Pierce). For each sample, 20 μg of total membrane protein was resolved on an 8% SDS/polyacrylamide gel, and the separated proteins were transferred to nitrocellulose membranes. Blots were probed with anti-GluR-B antisera T62–3B (16) at 1 μg/ml, followed by horseradish peroxidase-linked anti-rabbit secondary antibodies. The enhanced chemiluminescence method (Amersham) was used to detect GluR-B. To verify that equal amounts of protein were loaded into each lane, the lower part of the SDS/polyacrylamide gel was cut off before electroblotting and Coomassie-stained, and the image was densitometrically analyzed (National Institutes of Health image 1.56, data not shown).
Electrophysiological Recordings.
Transverse slices of 250 μm were prepared from the brains of 14-day-old mice. Cells were visually identified by infrared differential-contrast video microscopy (17) and by their firing pattern after current injection (18). Glutamate (1 mM) was applied to nucleated patches (19) with a piezo-controlled (P 245.70, Physik Instruments, Waldbronn, Germany) fast application system with a double-barrel application pipette (20). Duration of the glutamate pulse was 2 ms. Currents were recorded on an EPC-7 amplifier with pulse software (HEKA Elektronics, Lambrecht/Pfalz, Germany), filtered at 3–5-kHz bandwidth (−3 dB) with an 8-pole low-pass Bessel filter, and digitized at 10–20 kHz. Analysis was done offline with igor pro software package (WaveMetrics, Lake Oswego, OR). Averaged data are given as mean ± SD. All recordings were at room temperature (22–24°C). Relative calcium to monovalent ion ratios (PCa2+/PNa+) were calculated as in ref. 10. The control extracellular solution (Normal Rat Ringer) consisted of 135 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, and 10 mM Hepes (pH 7.2, adjusted with NaOH). High-Ca2+ extracellular solution contained 105 mM N-methyl-d-glucamine (NMDG), 30 mM CaCl2, 5 mM Hepes (pH 7.2, adjusted with HCl). Intracellular solution contained 130 mM CsCl, 10 mM Hepes, 0.5 mM EGTA, 4 mM ATP-Mg2+ (pH 7.2, adjusted with CsOH). All recordings were in the presence of 50 μM d-2-amino-5-phosphonopentanoic acid (D-AP5; Tocris Neuramin, Bristol, U.K.).
Immunohistochemical Staining.
Adult mice were anesthetized with halothane (Hoechst–Roussel) and perfused intracardially with 0.1 M sodium phosphate buffer (pH 7.4) at room temperature followed by ice-cold 4% paraformaldehyde in PBS. Brains were removed and postfixed overnight in PBS containing 4% paraformaldehyde at 4°C. The next day, brains were rinsed in PBS, embedded in 2% agarose in PBS as a supportive material, and cut coronally into 50-μm sections on a vibratome (Campden Instruments, Loughborough, U.K.). The sections were then transferred into 50 mM Tris⋅HCl (pH 7.4) containing 1.5% NaCl (Tris buffered saline, TBS) and permeabilized in TBS containing 0.4% Triton X-100 for 30 min. Preincubation was done in 4% normal goat serum (NGS) containing 0.4% Triton X-100 for another 30 min at room temperature. Sections were incubated in 2% NGS/0.4% Triton X-100 at 4°C overnight with polyclonal anti-glutamate receptor 1 (Chemicon) at a final concentration of 1–3 μg/ml. The monoclonal primary antibodies anti-calbindin-D (1:200, clone CL-300), anti-parvalbumin (1:200, clone PARV-19), and anti-MAP2 (microtubule-associated protein 2) (1:500, clone HM-2) were obtained from Sigma. The next day, sections were washed three times for 10 min with cold TBS and incubated for 2.5 hr in the dark at room temperature with goat anti-rabbit or goat anti-mouse fluorescein-isothiocyanate (FITC)-conjugated secondary antibodies (1:200; Jackson ImmunoResearch). The sections were then washed twice in TBS supplemented with 1% NGS and twice in TBS (10 min each). After a brief rinse in 10mM Tris⋅HCl (pH 7.5), sections were mounted in Mowiol (Polysciences). Immunostained sections were visualized under epifluorescent illumination on Axioplan-2 microscope (Zeiss). For overall anatomical comparison, brain slices from wild-type and mutant mice were also visualized by Nissl staining.
RESULTS
Targeted GluR-B Alleles with an Arginine Codon for the Q/R Site.
The CAG codon for the Q/R site of GluR-B resides in the GluR-B gene toward the 3′ end of exon 11, which encodes the putative membrane-spanning domain M1 and the inner-channel pore lining domain M2 (12). A targeting vector was constructed in which this codon was changed into one for arginine (CGG) by site-directed mutagenesis. At the same time, five silent mutations were introduced into exon 11 to facilitate the analysis of mRNA expression levels from the modified GluR-B allele in mutant mice. The targeting construct allowed for the subsequent removal by Cre recombinase of the floxed neo selection marker placed in intron 11 (Fig. 1).
One of four correctly targeted ES cell clones was injected into C57/BL6 blastocysts. A highly chimeric male transmitted the GluR-BRneo allele successfully through the germ line with about 50% of the offspring being of the GluR-B+/Rneo genotype. GluR-B+/R mice, lacking the neo marker in intron 11 of the GluR-BRneo allele, were generated by breeding the highly chimeric male with females of the deleter strain, which expresses Cre in germ cells (14). In offspring, removal of the floxed neo marker from the GluR-BRneo allele was complete, as judged by PCR on genomic DNA from GluR-B+/R mouse tissue.
GluR-B Expression Levels from the Wild-Type and Mutant Alleles.
A differential oligonucleotide hybridization assay (9, 10) performed on RT-PCR amplicons of brain RNA from heterozygous GluR-B+/R animals demonstrated that the GluR-BR and GluR-B+ alleles contributed equally to GluR-B mRNA [50.1 ± 0.3% GluR-BR mRNA (n = 3)]. A visual estimate of expression levels from sequencing chromatograms (Fig. 2A) of these amplicons, covering all exon 11 mutations, agreed well with the hybridization assay. Hence, in GluR-B+/R mice, the expression of the GluR-BR allele is not affected by the loxP site in intron 11. Moreover, no differences in GluR-B subunit levels between wild-type and GluR-BR/R mice were apparent by immunoblots of mouse brain extracts with a GluR-B antibody (Fig. 2B).
Figure 2.
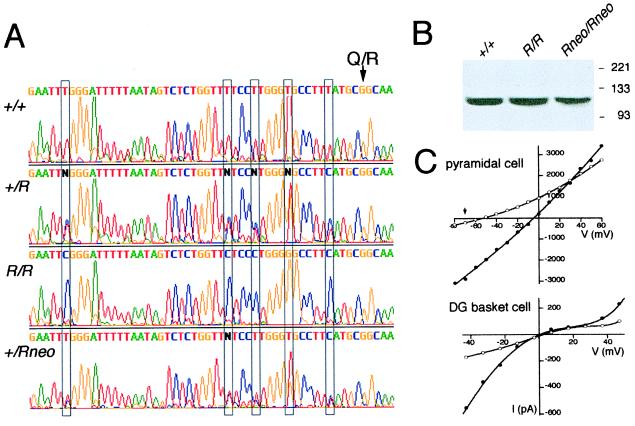
Expression of GluR-BR and GluR-BRneo alleles and increased Ca2+ permeability in hippocampal CA1 neurons of GluR-BRneo/Rneo mice. (A) Aligned sequence chromatograms of RT-PCR products encompassing exon 11 sequences of GluR-B mRNA from homozygous and heterozygous mice. The GluR-B alleles are listed on the left. The Q/R site is indicated by an arrow. The positions for the five silent mutations in the mutant alleles are boxed. The automatic sequence-analysis software attributes N to those nucleotide positions where the different allelic sequences in the RT-PCR product from GluR-B+/R mice have comparable peak heights but differ in sequence. The differential oligonucleotide-hybridization analysis shows that in GluR-B+/R mice, 50.1 ± 0.3% (n = 3) of GluR-B mRNA is from the mutant allele. In RT-PCR amplicons from heterozygous GluR-B+/Rneo mice, peak heights corresponding to the sequence of the mutant allele are significantly lower from those of the wild-type allele. (B) Immunoblot analysis of GluR-B protein in brains from wild-type and homozygous mutant mice. Genotypes are indicated on top of gel, size markers in kDa are shown on the right. The GluR-B antibody recognizes a single band corresponding to a polypeptide of about 110 kDa. (C) Current–voltage relations for glutamate-evoked AMPAR currents recorded in nucleated patches pulled from CA1 pyramidal cells and dentate gyrus (DG) basket cells of GluR-BRneo/Rneo mice in control (•, Normal Rat Ringer) and high Ca2+ (○, 30 mM Ca2+) solutions. For the pyramidal cells, the reversal potential in high-Ca2+ solution was Vrev = −54.5 ± mV and in control solution, Vrev = −4.9 ± mV. Current–voltage relations for wild-type and GluR-BR/R mice were identical, with the averaged Ca2+ reversal potential in high-Ca2+ solution (−69 ± mV, n = 5) indicated by arrow. For the DG basket cells, the reversal potentials in high Ca2+ and control extracellular solutions were Vrev = 2.7 ± mV and Vrev = 0.3 ± mV, respectively, and were indistinguishable from GluR-BR/R and wild-type mice.
The situation differed for the GluR-BRneo allele for which the neo cassette reduced expression (Fig. 2A), most likely by slowing intron 11 removal (see ref. 10) or by using splice sites in the PGK-neo marker (13). Differential hybridization analysis of cloned RT-PCR products from heterozygous GluR-B+/Rneo mice indicated that the contribution of the GluR-BRneo allele to the entire GluR-B mRNA was 70% that of the wild-type allele. In two independent experiments, the ratio of recombinant plaques from the GluR-BRneo mRNA versus the total number of plaques (i.e., those from GluR-BRneo and wild-type mRNA) was 133/322 and 138/337. Congruent with this result, immunoblot analysis showed less GluR-B protein in GluR-BRneo/Rneo than in wild-type mice (Fig. 2B).
Electrophysiological Recordings.
Two cell types, representatives of principal cells and interneurons, were characterized electrophysiologically in GluR-BR/R and in GluR-BRneo/Rneo mice. Hippocampal CA1 pyramidal neurons normally express high levels of Q/R site-edited GluR-B relative to the other AMPAR subunits and, as a consequence, AMPARs in this cell type are Ca2+ impermeable. This was not changed in GluR-BR/R mice. The AMPAR-mediated currents in nucleated patches of CA1 neurons of GluR-BR/R mice displayed current–voltage relations identical to wild type. The reversal potential for CA1 pyramidal cells from wild-type and GluR-BR/R mice in high-Ca2+ solution was (mean ± SD) −69 ± 4 mV (n = 9) (Fig. 2C), corresponding to a calculated PCa/PNa of 0.11 ± 0.02.
In GluR-BRneo/Rneo mice, however, which have a 30% reduced GluR-B gene expression as compared with wild-type mice, the reversal potential of AMPAR-mediated currents in high-Ca2+ solution in nucleated patches of CA1 pyramidal neurons shifted from about −70 mV for wild-type and GluR-BR/R mice to −54.5 mV (Fig. 2C), whereas no change was observed in standard extracellular solution (data not shown). The average reversal potential in high-Ca2+ solution was −55.2 ± 5.6 (n = 7), which corresponded to a PCa/PNa of 0.19 ± 0.04, indicating that the AMPAR-mediated Ca2+ permeability in CA1 pyramidal neurons in GluR-BRneo/Rneo mice was elevated nearly 2-fold (P < 0.05; two-tailed t test).
Inhibitory interneurons have low GluR-B levels and concomitantly high Ca2+ permeability of AMPARs. In some interneurons, the unedited GluR-B form may contribute to their characteristically high Ca2+ permeability of AMPARs. Dentate gyrus basket cells express the GluR-B subunit to about 12% of all AMPAR subunits (2). These cells of wild-type and mutant mice were electrophysiologically compared to search for a possible shift in the Ca2+ permeability of AMPARs (Fig. 2C). However, the reversal potential determined from current–voltage relations in the presence of 30 mM Ca2+ and standard extracellular solution was the same in GluR-BR/R, wild-type, and GluR-BRneo/Rneo mice. Thus, the low GluR-B expression in DG cells and the resolution of the method used did not permit a conclusion as to whether the majority of GluR-B is Q/R site-edited in this interneuron.
General Phenotype and Brain Histology of Mutant Mice.
GluR-BR/R and GluR-B+/R mice were healthy and did not differ in appearance, body weight, and average life span from their wild-type littermates. The GluR-BR/R genotype was represented in a Mendelian fashion in offsprings of GluR-B+/R breedings, indicating that embryonal development was not perturbed by the absence of the unedited GluR-B. GluR-BRneo/Rneo mice appear phenotypically normal in spite of 30% lower than wild-type GluR-B levels.
Nissl-stained brain slices from wild-type and GluR-BR/R mice showed no differences in overall brain architecture (Fig. 3A). Immunostaining of GluR-A, a prominently expressed AMPAR subunit (2, 3, 21), revealed no change in level or pattern in GluR-BR/R mice (Fig. 3B). Dendritic arborization is normally developed in GluR-BR/R mice, as visualized by immunostaining with MAP-2 antibodies (Fig. 3C). The strong labeling of brain slices by MAP-2 antibodies also permitted a good visual comparison of the brains from all genotypes. Similar to Nissl-stained sections, no detectable differences were observed. Immunostaining of two major classes of γ-aminobutyric acid (GABA)ergic interneurons, the parvalbumin- and calbindin-positive cells (22), indicated that number and distribution of these cells seemed unaffected in the GluR-BRneo/Rneo and GluR-BR/R mice (Fig. 3 D and E).
Figure 3.

Histochemistry of adult mutant and wild-type mouse brains. (A) Nissl-stained hemibrain slice. (B–E) Representative hippocampal images of immunohistochemical stainings for marker genes. (B), GluR-A; (C), MAP-2; (D), parvalbumin; (E), calbindin. Mouse genotypes are indicated below images.
DISCUSSION
We have generated GluR-BR/R mice, which carry in their GluR-B alleles a Q-to-R codon substitution for the Q/R site of the inner pore lining segment of the AMPAR GluR-B subunit. Whereas in wild-type animals the critical R codon is created by site-selective adenosine deamination of the Q (CAG) codon at the pre-mRNA level and is present in >99% of all GluR-B mRNA, the mutant animals lack all Q/R site-unedited GluR-B. The molecular and phenotypic analysis of mouse mutants carrying the GluR-BR allele and its precursor, the GluR-BRneo allele, permit the following conclusions to be drawn.
The Expression of GluR-B in Its Unedited Form, as It Occurs to a Minor Extent in Wild-Type Animals, Is of Unknown Functional Significance.
The Q form of GluR-B is expressed in adult brain at very low levels (<0.1% of R form) (4), but represents up to 20% of GluR-B mRNA during early embryonic development (11). The high level of the unedited form in the early embryonic central nervous system may suggest a role for glutamate as a trophic factor acting on neurons via a selectively increased Ca2+ influx through AMPARs. The normal development of GluR-BR/R mice demonstrates, however, that loss of the Q/R site-unedited form of GluR-B has no obvious functional consequence. Our analysis of the postnatal brain of GluR-BR/R mice, performed at a gross anatomical and immunocytochemical level, leaves the possibility that certain minor cell types may be affected in the mutant brain. If so, no untoward consequences arise from it, as evidenced by the apparently normal phenotype of GluR-BR/R mice. However, we cannot exclude that more detailed analyses such as quantitative cell counts, vulnerability to injury, and behavioral testing may reveal differences from wild-type phenotypes.
It Remains Unclear Why the Critical R Residue for the Q/R Site of GluR-B Is Generated by Site-Selective RNA Editing but Not by Exon Coding.
The lethal phenotype of mouse mutants expressing elevated levels of the unedited form of GluR-B attest to the physiological importance of the R residue at the Q/R site of GluR-B (10). The most straightforward way to position this residue into the critical channel site is to encode it exonically on the GluR-B gene. Unexpectedly, the arginine is generated by a process of site-selective adenosine deamination at the posttranscriptional level, at slightly less than 100% efficiency. The advantage RNA editing may have over exon coding is that it affords regulation such that the unedited subunit could be produced at higher levels in certain neurons under particular physiological conditions. Although we cannot exclude that such regulation exists, it is not essential.
We surmise that at some point in evolution, the posttranscriptional mechanism of RNA editing by site-selective adenosine deamination was co-opted to lower the Ca2+ permeability of GluRs. Once recruited, it was kept in operation. The recent report of a GluR-B-like gene with an exonic R codon for the Q/R site in the freshwater fish Oreochromis sp. (23) may contradict this evolutionary scheme. However, this fish species is tetraploid and hence, the particular gene may be an allele that has sequence-drifted in the presence of another allele, the transcript of which is Q/R site-edited. The evolutionary history of Q/R site editing of GluR subunits is poorly charted. In Caenorhabditis elegans and Drosophila melanogaster, all AMPAR-like subunits characterized to date (GenBank and A C. elegans Database (ACEDB) carry a Q codon for the critical channel position. In avian neurons, GluR-B cDNA specifies an R codon for the channel position but the corresponding exon has the Q codon (24), as in rodents and humans. No information is available on amphibian GluR-B. Thus, the period in evolution when the Ca2+ permeability of central AMPAR-like receptor channels came under the control of RNA editing remains to be determined.
Q/R Site Editing May Accelerate Intron 11 Splicing.
Wild-type GluR-B pre-mRNA contains a double-stranded structure composed of exon 11 and intron 11 sequences (9). This structure is the substrate for an RNA-dependent adenosine deaminase, which edits the Q/R site codon before intron 11 splicing (9). The GluR-BR allele contains in intron 11 an extended loxP element, which disrupts the double-stranded RNA structure. This mutant allele expresses as well as the wild-type GluR-B allele. The dominant negative GluR-BΔECS allele (10) is closely related to the GluR-BR allele. It carries the same extended loxP element, 56 bp 3′ of its location in the GluR-BR allele, but has the exonic Q codon for the Q/R site. However, the GluR-BΔECS allele expresses to only 50% of the GluR-B+ allele as a result of attenuated intron 11 splicing (10). It appears that the double-stranded RNA structure slows intron 11 splicing in pre-mRNA of the GluR-BΔECS allele and, very likely, the GluR-B+ allele when the Q/R site codon is not edited. Proof of this should come from ablating the gene for the enzyme that edits the Q/R site codon in GluR-B pre-mRNA.
Increased Ca2+ Influx Through AMPARs in Hippocampal Principal Neurons Is Caused by a Moderate Decline in GluR-B Expression.
The expression of GluR-B relative to other AMPAR subunits in principal excitatory cells is sufficient to ensure that most AMPARs contain this subunit and thus become impermeable to Ca2+ ions (2). Interestingly, in the CA1 pyramidal neurons, the Ca2+ permeability was elevated by nearly 2-fold in GluR-BRneo/Rneo mice, which express the modified allele by 30% less than the wild-type GluR-B allele (Fig. 4A). This indicates that the low Ca2+ permeability of AMPARs in these neurons results from merely a moderate excess of GluR-B. Hence, the Ca2+ permeability of AMPARs in hippocampal pyramidal neurons can increase with a relatively small decrease in GluR-B gene expression. It is important to note that the increased AMPAR-mediated Ca2+ influx into pyramidal cells did not induce cytotoxicity in hippocampal neurons.
Acknowledgments
We thank Dr. R. Wenthold for antisera to GluR-A and GluR-B, Dr. F. Schwenk for the deleter mouse line, Dr. R. Brusa for the plasmid containing a GluR-B gene fragment, Dr. A. Nagy for plasmid ploxPneo1, F. N. Single for help with ES cell culture, F. Zimmermann for blastocyst injections, S. Grünewald and H. Grosskurth for DNA sequencing, M. Pfeffer for animal care, and A. Herold and M. Lang for technical assistance. K.K. was the recipient of a European Molecular Biology Organization long-term fellowship. This study was funded, in part, by the German Chemical Society and Human Frontier Science Program Grant RG0003 to P.H.S.
ABBREVIATIONS
- AMPAR
l-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor
- ECS
exon-complementary intronic sequence
- DG
dentate gyrus
- ES cell
embryonic stem cell
- neo
neomycin phosphotransferase gene
- PGK
phosphoglycerate kinase
- RT-PCR
reverse transcription–PCR
References
- 1.Mayer M L, Westbrook G L. Prog Neurobiol. 1987;28:197–276. doi: 10.1016/0301-0082(87)90011-6. [DOI] [PubMed] [Google Scholar]
- 2.Geiger J R, Melcher T, Koh D-S, Sakmann B, Seeburg P H, Jonas P, Monyer H. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 3.Hollmann M, Heinemann S F. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 4.Sommer B, Köhler M, Sprengel R, Seeburg P H. Cell. 1991;67:11–19. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- 5.Hollmann M, Hartley M, Heinemann S F. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 6.Verdoorn T A, Burnashev N, Monyer H, Seeburg P H, Sakmann B. Science. 1991;252:1715–1718. doi: 10.1126/science.1710829. [DOI] [PubMed] [Google Scholar]
- 7.Hume R I, Dingledine R, Heinemann S F. Science. 1991;253:1028–1031. doi: 10.1126/science.1653450. [DOI] [PubMed] [Google Scholar]
- 8.Burnashev N, Monyer H, Seeburg P H, Sakmann B. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- 9.Higuchi M, Single F N, Köhler M, Sommer B, Sprengel R, Seeburg P H. Cell. 1993;75:1361–1370. doi: 10.1016/0092-8674(93)90622-w. [DOI] [PubMed] [Google Scholar]
- 10.Brusa R, Zimmermann F, Koh D S, Feldmeyer D, Gass P, Seeburg P H, Sprengel R. Science. 1995;270:1677–1680. doi: 10.1126/science.270.5242.1677. [DOI] [PubMed] [Google Scholar]
- 11.Melcher, T., König, N., Berger, T., Bardoul, M., Jonas, P., Seeburg, P. H. & Monyer, H. (1997) Soc. Neurosci. Abstr. 478.26.
- 12.Köhler M, Kornau H-C, Seeburg P H. J Biol Chem. 1994;269:17367–17370. [PubMed] [Google Scholar]
- 13.Nagy A, Moens C, Ivanyi E, Pawling J, Gertsenstein M, Hadjantonakis A-K, Pirity M, Rossant J. Curr Biol. 1998;8:661–664. doi: 10.1016/s0960-9822(98)70254-4. [DOI] [PubMed] [Google Scholar]
- 14.Nagy A, Rossant J, Nagy N, Abramow N W, Roder J C. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwenk F, Baron U, Rajewsky K. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petralia R S, Wang Y X, Mayat E, Wenthold R J. J Comp Neurol. 1997;385:456–476. doi: 10.1002/(sici)1096-9861(19970901)385:3<456::aid-cne9>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 17.Stuart G J, Dodt H-U, Sakmann B. Pflügers Arch. 1993;423:511–518. doi: 10.1007/BF00374949. [DOI] [PubMed] [Google Scholar]
- 18.Koh D-S, Geiger J R, Jonas P, Sakmann B. J Physiol (London) 1995;485:383–402. doi: 10.1113/jphysiol.1995.sp020737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sather W, Dieudonné S, MacDonald J F, Ascher P. J Physiol (London) 1992;450:643–672. doi: 10.1113/jphysiol.1992.sp019148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colquhoun D, Jonas P, Sakmann B. J Physiol (London) 1992;458:261–287. doi: 10.1113/jphysiol.1992.sp019417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keinänen K, Wisden W, Sommer B, Werner P, Herb A, Verdoorn T A, Sakmann B, Seeburg P H. Science. 1990;249:556–560. doi: 10.1126/science.2166337. [DOI] [PubMed] [Google Scholar]
- 22.Hendry S H, Jones E G, Emson P C, Lawson D E, Heizmann C W, Streit P. Exp Brain Res. 1989;76:467–472. doi: 10.1007/BF00247904. [DOI] [PubMed] [Google Scholar]
- 23.Kung S S, Wu J M, Chow W Y. Brain Res Mol Brain Res. 1996;35:119–130. doi: 10.1016/0169-328x(95)00193-v. [DOI] [PubMed] [Google Scholar]
- 24.Lowe D L, Jahn K, Smith D O. Mol Brain Res. 1997;48:37–44. doi: 10.1016/s0169-328x(97)00072-7. [DOI] [PubMed] [Google Scholar]