

Abstract
Our overall goal is to understand how viral envelope proteins mediate membrane fusion and pathogenesis. Membrane fusion is a crucial step in the delivery of the viral genome into the cell resulting in infection. On the other hand, fusion activity of viral envelope glycoproteins expressed in infected cells may cause the demise of uninfected bystander cells by apoptosis. Our general approach is to kinetically resolve steps in the pathway of viral envelope glycoprotein-mediated membrane fusion and to uncover physical parameters underlying those steps using a variety of biochemical, biophysical, virological, and molecular and cell biological techniques. Since HIV fusion involves a complex cascade of interactions of the envelope glycoprotein with two receptors, membrane organization plays an important role and interfering with it may modulate entry. To study this phenomenon, we have either examined cell lines with differential expression of sphingolipids (such as GM3), or altered membrane organization by modifying levels of cholesterol, ceramides, or glycosphingolipids. We show that the localized plasma membrane lipid microenvironment (and not the specific membrane lipids) in the vicinity of CD4 controls receptor mobility and HIV-1 fusion. The complex cascade of conformational changes that must occur to allow virus entry is also a very important target for therapy and vaccine development. We have recently designed and tested peptide analogs composed of chemical spacers and reactive moieties positioned strategically to promote permanent attachment. Using a temperature-arrested state in vitro assay we show evidence for the trapping of a pre-six helix bundle fusion intermediate by a covalent reaction with the inhibitory reactive peptide. Also, using photo-reactive hydrophobic probes we have found ways to inactivate viral envelope glycoproteins while leaving their overall structures intact. Finally, in order to study the envelope glycoprotein effects on pathogenesis, we have used an in vitro model of co-culture of envelope-expressing cells as effectors and CD4+ T cells as targets. We delineated that apoptosis mediated by envelope glycoprotein in bystander cells correlates with transmembrane subunit (gp41)-induced hemifusion. The apoptotic pathway initiated by this interaction involves caspase-3-dependent mitochondrial depolarization and reactive oxygen species production, which depends on the phenotype of the envelope glycoprotein associated with the virus. Taken as a whole, our studies have many different important implications for anti-viral therapies and vaccine development.
Introduction
Viral entry involves a complex cell biological process the elucidation of which requires a fundamental understanding of membrane trafficking, membrane receptors, conformational changes of proteins, protein-lipid interactions, and the chemistry and physics of lipids and membrane micro-domains. The studies our group has pursued on the control of mobility and disposition of the HIV receptors, CD4, CXCR4 and CCR5, by membrane composition provide important new insights to the emerging concepts of membrane dynamics and architecture. The methodologies and concepts developed in the studies on the conformational changes of HIV-1 gp41 in the course of membrane fusion, open new lines of inquiry in the area of protein folding and refolding in biology. The methodologies we have developed for the kinetic and biophysical analysis of viral fusion/entry provide substantial improvements in understanding of the mode of action of inhibitors and antibodies that block viral entry. During recent years we have continued to invest in developing photosensitized labeling techniques to gain insight into membrane protein structure and function, specifically of the viral fusion proteins. An important outcome of these labeling studies is our discovery that labeling hydrophobic domains of viral proteins results in the inactivation of enveloped viruses without affecting their conformational integrity. This important new technological advance has broad implications for the development of cancer and viral vaccines. Another important area of investigation has been the development of methods to trap intermediate conformations of the envelope glycoproteins during the fusion process. We have designed and tested peptides analogs that form a covalent bond with the transmembrane subunit (gp41) of the envelope glycoprotein. These effectively trap an intermediate conformation and will be important tools in the study of the membrane fusion process.
1. Membrane Structure and Function
The plethora of new data on membrane proteins, lipids and glycoconjugates is changing our general view of membrane structure and function [1]. Some of the emerging concepts are that membranes are patchy, with segregated regions of structure and function, that lipid regions vary in thickness and composition, and that crowding and ectodomains affect lateral mobility, and the cytoskeleton is intimately involved in membrane dynamics. Since HIV fusion involves a complex cascade of interactions of the envelope glycoprotein with two receptors, we have long argued that membrane organization plays an important role and that interfering with this may modulate entry [2;3]. We previously reported that the upregulation of cellular ceramide levels following fenretinide treatment inhibits HIV fusion [4]. As ceramide facilitates the internalization of a variety of microbes, we hypothesized that it may promote the engulfment of HIV-1 leading to its destination in intracellular compartments where it becomes inactive.
The lipid content of the cell membrane is primarily composed of glycerophospholipids (GSLs), sphingolipids and cholesterol [5]. Sphingolipids tend to self associate due to head group interactions and due to hydrophobic interactions between the saturated side chains. Cholesterol fills the void between sphingolipids and associates with sphingomyelin through hydrogen bonding. These interactions result in the segregation of sphingolipids and cholesterol from glycerophospholipids creating a more ordered phase in the cell membrane termed “rafts” [5]. The lipid composition of the target cell plays an important role in the HIV fusion process. Receptor recruitment, a prerequisite for fusion, has been shown to be sensitive to lipid modulation. In primary cells where receptor molecules are expressed in low numbers cholesterol depletion inhibits fusion and infection. However, overexpression of envelope and receptors in many model fusion systems obscures this requirement of receptor recruitment [6]. It has been demonstrated that cholesterol depletion inhibits receptor recruitment by decreasing the diffusion rate of HIV receptors, implicating receptor restriction as one possible mechanism by which modulation of cellular lipids can inhibit HIV fusion [7]. We have used a number of strategies to alter glycosphingolipid content of target cells leading to a reduction in susceptibility to HIV-1 entry. These include treatment with inhibitors of sphingolipid metabolism such as 1-phenyl-2-hexadecanoylamino-3-morpholino-1-propanol (PPMP) [8] and fenretinide [4], cells expressing various levels of GSLs [9], and treatment with sphingomyelinase (Smase) [10].
The application of Smase to cells alters the lipid content of the plasma membrane by generating ceramide upon cleaving sphingomyelin. As sphingomyelin is found primarily in rafts, this is the site where ceramide is collected. Ceramide is extremely hydrophobic and upon formation, promotes the coalescence of raft domains into what have been termed “membrane platforms” [11]. We showed previously that Smase activity, which alters the lipid composition of cells by increasing ceramide, can have adverse effects on HIV infection [4]. We have further probed the mechanistic details of how ceramide modulation inhibits HIV fusion. We have determined that Smase activity restricts the lateral diffusion of CD4 in the membrane. In contrast, the lateral diffusion of the coreceptor is unaltered. We show that restricting CD4 diffusion by antibody cross-linking also inhibits HIV infection, confirming this approach as a general mechanism to inhibit fusion. As HIV entry is a highly orchestrated event requiring the sequential interaction of CD4 and coreceptor, restricting receptor mobility would be expected to have severe consequences for viral fusion.
Viral entry may be affected by altering membrane trafficking. For instance, we have shown that treatment of target cells with fenretinide increases their cellular ceramide levels and reduces their susceptibility to HIV-1 entry [4]. However it appears that the mechanism of inhibition is different from that of other reagents like Smase which alter ceramide levels. Although HIV-1 binding to cells is not altered by fenretinide treatment and the distribution of HIV receptors was unchanged, we observed an increase in HIV-1 uptake into intracellular compartments We determined that fenretinide treatment promotes the internalization of virions from the plasma membrane and the accumulation of virus in the endocytic fraction of HeLa cells [12]. This effect of fenretinide appears to be specific for virus as the endosomal accumulation of HIV gp120, transferrin and horseradish peroxidase is not increased. Notably, fenretinide increased the infectivity of influenza, which fuses in the endosomal compartment upon low pH activation. Our data suggests that fenretinide treatment effectively inhibits HIV infection by redirecting the virus to the endocytic pathway, resulting in HIV inactivation.
We had reported that CD4 and co-receptor bearing mouse melanoma cells (B16) that express exceptionally high levels of monoganglioside GM3, a raft-associated ganglioside that interacts with CD4 were resistant to gp120-gp41-mediated membrane fusion and that the fusion activity is restored by pre-treatment of the B16 targets with PPMP [9]. On the other hand, a glycosphingolipid-deficient mouse melanoma cell line (GM95) was perfectly capable of supporting HIV-1 Env-mediated fusion following the expression of CD4 and co-receptors on the cell surface [13]. We hypothesized that the block in fusion was due to immobilization and/or segregation of the CD4 receptor in the plasma membrane of these cells. We investigated GM3-mediated modulation of CD4 localization and diffusion in the plasma membrane of B16 cells by utilizing a CD4 mutant (RA5) that supports HIV-1 entry despite its preferential localization into non-raft fraction [14]. In contrast to wild type CD4 (wt-CD4), B16 cells expressing the RA5 mutant and the cognate coreceptors (CXCR4 or CCR5) readily fused with cells expressing the corresponding HIV-1 Envs. Although the lateral diffusion of plasma membrane lipids and CCR5 was similar in B16 cells and GM95 cells, wt-CD4 mobility was significantly restricted in B16 cells (1.04±0.08×10–11cm2/sec) when compared with the RA5 mutant (3.23±2.77×10–11cm2/sec) with a p value of 0.0112 according to a student t test. In contrast, wt-CD4 and RA5 mutant when expressed in GM95 cells showed little difference in CD4 diffusion [15]. Our findings indicate that the lateral mobility of CD4 is an important determinant for HIV-1 env-mediated membrane fusion (see Fig. 1) and also provide a novel mechanism of interplay between membrane lipids and receptors by which host cells can escape viral infections. We are further investigating whether the restricted mobility of wt-CD4 in B16 cells is due to interactions with GM3 or with other host cell factors.
Figure 1.
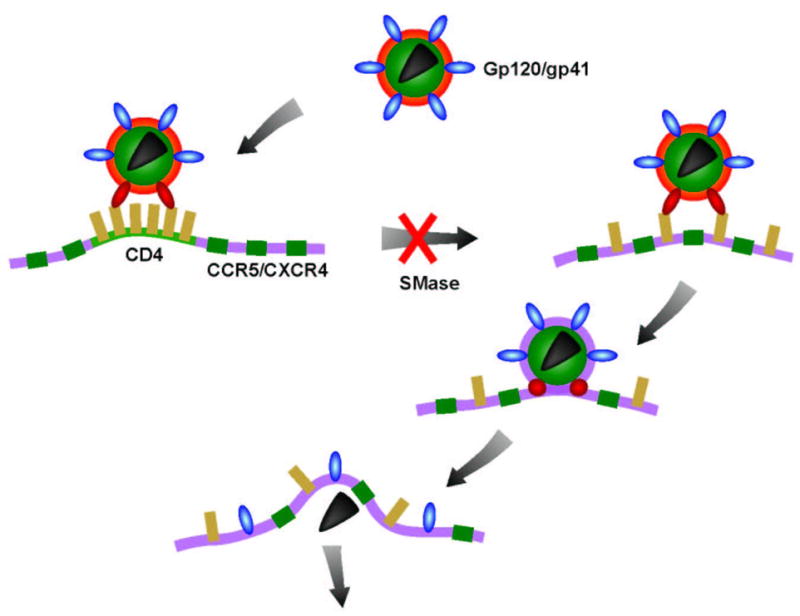
HIV-1 delivers its genetic material into the cell by direct fusion of the viral membrane with the plasma membrane of the host cells mediated by trimeric gp120/gp41 molecules (blue). Interactions between gp120 and a cluster of CD4 molecules (yellow) result in conformational changes in gp120/gp41 (red). The CD4 cluster then disperses allowing the intermingling between CD4 and CCR5/CXCR4 molecules (green) and enabling interactions of gp120 with co-receptors. A further barrage of conformational changes then leads to membrane fusion. Treatment of target cells with sphingomyelinase (Smase) inhibits the dispersion of CD4 clusters, which disables engagement with co-receptors and the subsequent fusion reaction.
2. Protein Intermediates in Membrane Fusion
Host cell surface CD4 interactions with HIV gp120-gp41 elicit conformational changes in gp120, exposing co-receptor [16;17] (CCR5 or CXCR4) binding sites, and in gp41, exposing the C-terminal heptad repeat region (CHR) and the leucine/isoleucine zipper region (NHR) (Figure 2) [18]. Co-receptor engagement by gp120 triggers a battery of additional conformational changes in HIV gp41 eventually resulting in the formation of the thermo-stable 6-helix bundle (viral hairpin) [19;20], which drives the membrane merger and eventual fusion [21]. Synthetic peptides corresponding to sequences from the NHR and CHR of the HIV transmembrane subunit (gp41) potently inhibit membrane fusion by binding to the partner domain thereby interfering with the formation of the viral hairpin [22]. Studies on the kinetics of interference with HIV fusion by a variety of inhibitors provide a way to study intermediates of the fusion process [23]. The HIV pre-hairpin conformation can also be accessed by priming HIV envelope-expressing cells with soluble CD4 (sCD4) and testing the effects of inhibitors [21;24].
Figure 2.
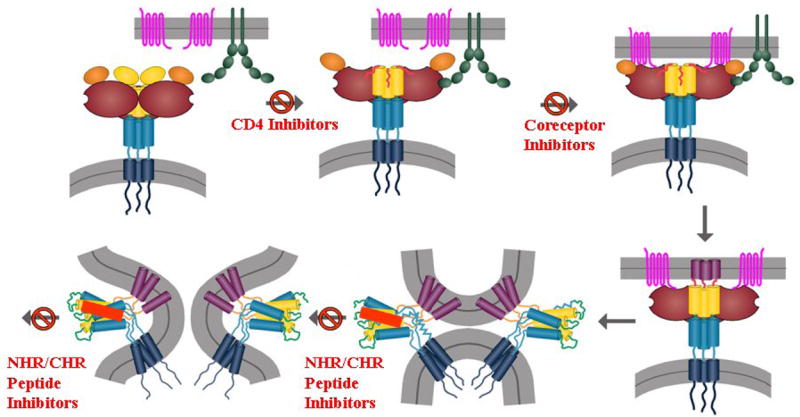
Model of the HIV envelope fusion process. The HIV envelope glycoprotein is depicted as a trimer of heterodimers with the gp120 subunit in brown/yellow and the gp41 subunit in blue/dark blue. The target membrane contains CD4 in green and the co-receptor (CXCR4 or CCR5) in pink. The CD4 and coreceptor inhibitors function upstream of the NHR/CHR gp41 peptide inhibitors in the fusion process.
We have studied the mode of action of small molecule antagonists of the viral coreceptors CCR5 and CXCR4 and peptidic fusion inhibitors such as T20 (Fuseon®/enfuvirtide) which has been approved for the treatment of AIDS. By examining fusion kinetics of various HIV strains we found that the sensitivity of HIV to entry inhibitors correlates with envelope:coreceptor affinity, receptor density and fusion kinetics [25]. The kinetic studies allowed us to determine parameters that govern fusion and inhibition. We found a window of opportunity of about 15 min between gp120-CD4 engagement and 6-helix bundle formation followed by fusion [23]. Furthermore, we have monitored the temporal sequence of conformational states of HIV-1 gp41 during the course of HIV-1 mediated cell-cell fusion by quantitative video microscopy using reagents that bind to NHR and CHR [26]. Our sCD4 prime-wash methodology [24] has proven to be very useful in revealing the mode of action of entry inhibitors. A case in point is our elucidation of the mode of action of Retrocyclin-1, a θ-defensin [27]. We found that Retrocyclin-1 completely blocked fusion mediated by HIV-1 envelope that used CXCR4 or CCR5, but had little effect on cell fusion mediated by HIV-2 and SIV envelope glycoproteins [27]. Previously it had been thought that Retrocyclin-1 inhibited HIV-1 Env-mediated fusion due to its lectin-like properties [28]. However, our kinetic studies indicated that Retrocyclin-1 acted late in the fusion cascade with kinetics that were similar to peptidic entry inhibitors such as C34. Moreover, prime-wash experiments indicated that retrocyclin targeted the HIV-1 gp41 pre-hairpin state. Surface plasmon resonance experiments showed that retrocyclin bound the ectodomain of gp41 with high affinity in a glycan-independent manner, and that it bound selectively to the gp41 C-terminal heptad repeat (CHR). Native-PAGE, ELISA, and CD spectroscopic analyses all revealed that retrocyclin-1 prevented 6 helix bundle formation. We thus identified a novel mode of action for an innate effector molecule, which resembles that of HIV-1 gp41entry inhibitors.
2.1 Trapping intermediate envelope glycoprotein conformations
In the early 1990s, researchers discovered that they could add sequences from the NHR and CHR of HIV gp41 and inhibit viral entry (Fig. 3) [29–32]. The peptide inhibitors bind to regions in HIV gp41 that are transiently exposed during the fusion process. For biochemical reasons, peptides derived from the CHR have been the most efficacious at inhibiting viral entry and one of these enfuvirtide (T20, Trimeris/Roche) is already in the clinic. However, enfuvirtide is currently reserved as a salvage therapy for treatment-experienced patients. It requires two injections daily making the regimen very difficult for patients to maintain. The problems with enfuvirtide therapy are those that are inherent to peptide therapy in general: relatively short half-lives, poor distribution in the body, susceptibility to enzymatic degradation and rapid renal clearance. The mechanism of inhibition of peptide inhibitors is based upon a protein-peptide interaction which is subject to binding equilibria. We postulated that we could permanently trap an HIV gp41 intermediate by making this interaction covalent. To achieve this, we designed and tested various peptide analogs composed of chemical spacers and reactive moieties to facilitate covalent attachment [33].
Figure 3.
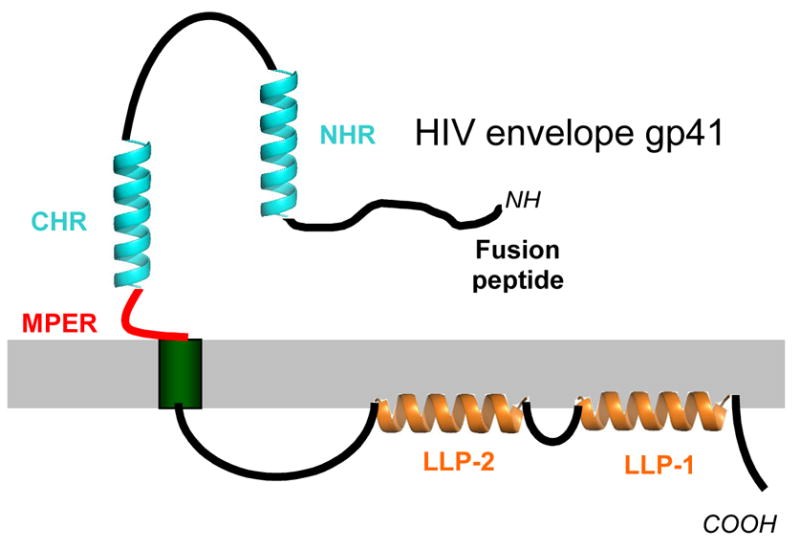
Diagram depicting the regions of HIV gp41 (the transmembrane subunit of the HIV envelope glycoprotein). The surface subunit HIV gp120 which is non-covalently associated with gp41, is not shown. The N-heptad repeat (NHR) and the C-heptad repeat (CHR) are shown in aqua. The MPER is diagrammed in red and the lentivirus lytic peptides (LLP-1 and LLP-2) are in royal blue. The fusion peptide is seen at the N-terminus (NH) of the molecule and the cytoplasmic tail is at the C-terminus (COOH). The molecule is shown as a monomer for simplicity but is most likely trimeric.
We tested the permanent inhibition by arresting the HIV entry process using low temperature, adding the covalent inhibitor and washing extensively before allowing fusion to proceed. Using this “prime-wash” method, we were able to show that we have permanently trapped an intermediate of gp41. Two of the covalent peptide inhibitors produced distinct dose dependence and the IC50 values were measured to be 680 nM and 470 nM. The covalent reaction targeted for a specific lysine residue was shown by mutagenesis to be preferentially targeted up to a concentration of 1 μM at which point we hypothesize that the binding sites are saturated and the peptide more readily available to react with other species. The full permanent inhibition time point was between 5 and 10 minutes for both compounds. The half-lives of the compounds in serum were also measured as an indication of other reactivity and were 29 and 55 minutes with serum and 61 and 79 minutes in the base medium without serum. Although there is reactivity with other species, the rapid specificity of the reaction for the specific amino acid was encouraging and this development in peptide inhibitor function will lead to new tools to better delineate the details of the fusion process and will hopefully contribute to the development of better therapies. This is also the first demonstration in live cells of the trapping of a viral envelope glycoprotein fusion intermediate.
2.2 The role of diverse regions of gp41 in HIV-1 kinetics and inhibition
Although the majority of the attention has been devoted to the role NHR, CHR and 6-helix bundle formation play in the fusion reaction, other regions appear to be of importance (Fig. 3). These include: the cytoplasmic tail (CT), the membrane proximal external domain (MPER), the loop, the membrane-spanning domain (MSD) and the fusion peptide. Mutational analysis coupled with kinetic studies revealed the fascinating interplay between these regions in the refolding of gp41 that leads to fusion.
2.2.1 The cytoplasmic tail
Using a cell-cell fusion assay and a panel of HIV Envs with stop codons at various positions in the CT, we showed that truncations of gp41 proximal to the most N-terminal alpha helix, LLP2 (lentivirus lytic peptide), increase fusion efficiency and expose CD4-induced epitopes in the envelope ectodomains [34]. These effects were not seen with a truncation distal to this domain and before LLP1. Using a dye transfer assay to quantitate fusion kinetics, we found that these truncations produced a 2–4-fold increase in the rate of fusion. These results were observed for X4, R5, and dual-tropic envelope proteins on CXCR4- and CCR5-expressing target cells and could not be explained by differences in envelope protein surface expression. These findings suggest that distal to the membrane-spanning domain an interaction of the gp41 LLP2 domain with the cell membrane restricts envelope fusogenicity during envelope protein processing. As with murine leukemia viruses where cleavage of a membrane-interactive R-peptide at the C-terminus is required for envelope protein to become fusogenic, this restriction on envelope function may serve to protect virus-producing cells from the membrane disruptive effects of the ectodomain.
2.2.2. The membrane-proximal external region
We have further investigated the role of the tryptophan-rich region immediately adjacent to the membrane-spanning domain, termed the membrane proximal external region (MPER), which is crucial for the proper functioning of gp41 as a fusion protein [35;36]. This region is conserved in the vast majority of otherwise highly variable HIV-1 isolates. Deletion of the entire stretch of 17 amino acids abrogated the ability of the envelope glycoprotein to mediate both cell-cell fusion and virus entry without affecting the normal maturation, transport, or CD4-binding ability of the protein. However, the MPER-deleted mutant gp41 is fully competent to form six-helix bundle structures following activation by CD4 and CXCR4 [37]. We had shown that alanine substitution of the five conserved tryptophan residues produces a phenotype in which the envelope glycoprotein does not induce syncytia but does permit redistribution of small aqueous dyes between host and target cells, indicating a failure in fusion-pore expansion [38]. We probed the temporal sequence of exposure of the membrane-proximal domain of HIV-1 gp41 during the course of HIV-1 mediated fusion using the broadly neutralizing monoclonal antibodies, 2F5 and 4E10 [36]. The time course for escape of HIV-1 inhibition by 2F5 and 4E10 was similar to that of inhibition by the CHR peptide, C34, which blocks 6-helix bundle formation. Immunofluorescence and a high throughput In-Cell Western assay indicated that the reactivity of antibodies 2F5 and 4E10 against this region decreased at the onset of fusion. However, addition of C34 did not counteract the loss of binding. These data suggested that conformational changes in the membrane-proximal domain occur independently of six-helix bundle formation.
2.2.3. Fusion Peptide
The envelope proteins of HIV, gp41 and gp120, make up a complex that undergoes a series of conformational changes and rearrangements during the course of the fusion process. These conformational changes lead to the exposure of the N-terminal fusion peptide of gp41. Insertion of the fusion peptide into the cellular membrane allows the gp41 protein to span both the viral and the cellular membranes leading to lipid mixing and subsequent entry of the viral core into the cell. There is no high-resolution structural information for the pre-fusion state of gp41. However, there have been numerous studies of the fusion peptide including an NMR structural determination in a micelle environment [39]. We investigated the backbone structure and dynamics of the 30 N-terminal residues of HIV-1 gp41 in membrane-mimicking environments and the effects of this peptide on lipid mixing. The ensemble of NMR structures revealed an uninterrupted alpha-helix with residues outside of the micelle showing enhanced mobility relative to those that are buried. These high-resolution structures of the post-fusion HIV gp41 ectodomain would place the fusion peptide into close proximity to the transmembrane region in the fusion-active and/or post-fusion stages. It has been hypothesized that there are necessary interactions between the fusion peptide and the transmembrane region during the fusion process [40]. However, structural studies of the transmembrane region have been difficult to pursue due to insolubility. This makes in silico studies of the transmembrane region and of its possible association with the fusion peptide highly warranted. Using computational methods it may be possible to gain insight into a membrane fusion system that is extremely difficult to study experimentally.
3. Development of Site-Directed Photosensitized Labeling to Study Membrane
Protein Interactions and for Vaccine Development
We have developed a photosensitized labeling methodology that involves binding of photoactivatable probes to membrane proteins and lipids following activation of these probes in situ by energy transfer from a variety of donor chromophores [41]. In the current studies we have used the membrane bilayer specific probe iodonaphthylazide (INA). We have successfully applied this technique on orthomyxo-, rabdo- and lentiviruses, showing that we could measure and follow the insertion and redistribution of viral envelope proteins into the target cell membrane in the course of fusion [42–44]. This method can also be used to establish which proteins of the viral envelope penetrate the target cell membrane in the course of infection and thus identify proteins and membrane compartments that participate in fusion.
Considering the high specificity and efficiency of labeling obtained with these hydrophobic photoactivable probes, we have investigated their ability to inactivate pathogens [45]. The rationale of our approach is based upon the observation that hydrophobic compounds such as INA are very highly specific towards the lipidic environment and can be activated selectively in this milieu (Fig. 4). We hypothesize that this could provide a unique way to alter the pathogenicity of any membrane containing organism while preserving its immunogenicity towards its use as a research tool or as an immunogen in a vaccine formulation.
Figure 4.
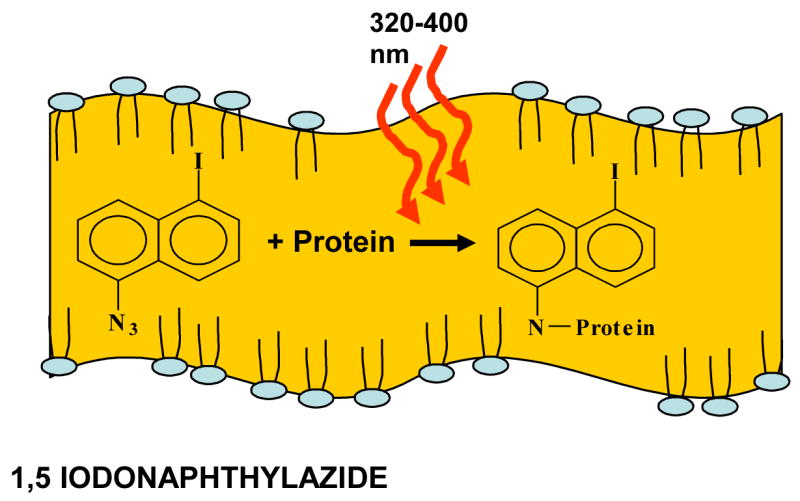
Activation of INA in biological membranes. Upon addition, INA partitions exclusively in the lipidic domain due to its very high partition coefficient. When activated by long UV (320–400 nm), the azido moiety forms a highly reactive nitrene radical that induces the covalent binding of INA to the proteins in its surroundings.
We performed experiments on HIV and SIV and showed that we could completely abolish infectivity while conserving integrity as shown by microscopy and the measurement of binding by broadly neutralizing antibodies. Through collaboration with USAMRIID, we have studied the inactivation of biothreat viruses such as Ebola and Marburg. We have determined that INA treated Ebola virus was unable to propagate in Vero cells. It was also proved to be safe in animals as all of the mice injected with the equivalent of 50,000 pfu INA-treated Ebola survived. Furthermore, this vaccination conferred a 90% protection towards challenge with 1000 pfu after three weeks and full short-term protection after 3 days. These results indicate that the technology we have developed would be efficient in inactivating highly pathogenic viruses such as Ebola and that the inactivated pathogens were able to activate both arms of the immune system.
Influenza virus is a pathogen of considerable interest. We have been able to use INA to completely inactivate influenza virus while preserving its integrity. Even though fully inactivated, the virus was still capable of undergoing the first stage of the fusion process that corresponds to the hemifusion [46] of the outer layers without delivery of the genome to the target cells. Another unique feature of this inactivation technique was shown through the selectivity of its target. Influenza virus contains on its surface an enzyme, neuraminidase, whose active site is spatially separated from the transmembrane domain targeted by our technology. While we get complete inactivation of the virus, the neuraminidase activity is conserved. Our technology therefore provides a way to inactivate influenza virus while conserving intact epitopes in a much more efficient way than is done with current inactivation techniques. We engaged in collaboration with the University of Georgia to assess the immunological potential of our inactivated virus in a mouse model. The studies showed that all of the mice subjected to INA inactivated virus were protected against heterologous challenge. Such a protection is normally only observed with live attenuated virus.
Taken together with recent studies on equine encephalitis virus [47] we established that this novel method of inactivation based on the use of hydrophobic photoinduceable alkylating probes is widely applicable to enveloped pathogens. Furthermore, the preservation of the antigenic epitopes through the targeting of the lipid domain offers new avenues for the development of whole inactivated vaccines.
As a possible mechanism of the photoactivation-mediated virus inactivation we propose that the photo-induced reaction of the membrane-spanning domain of the env Glycoprotein with INA has a similar effect on its fusion potential as truncation of the env glycoprotein into the membrane-spanning domain. Indeed, in the case of influenza virus, Judy White and coworkers have shown that the membrane-spanning domain exhibits a stringent length requirement to support the hemifusion to fusion transition [48]. Moreover, in the case of SIV, Eric Hunter and coworkers observed that progressive truncations C-terminal to the membrane-spanning domain of simian immunodeficiency virus Env reduce fusogenicity and increase concentration dependence of Env for fusion. [49]. Thus, Envs truncated in their membrane-spanning domains exhibit all of the extracytoplasmic functions required for mediating membrane fusion, but fail to mediate the dilation of the fusion pore that is required for delivering the viral genes into the cells to potentiate replication. We propose that INA-modified Envs behave in exactly the same way.
4. Mechanisms of HIV-1 gp41 Induced Pathogenesis
HIV infections cause a progressive and irreversible depletion of CD4+ T cells by a mechanism that has been suggested to rely on apoptosis of uninfected bystander cells [50]. While HIV envelope glycoprotein has been implicated in the loss of bystander cells by apoptosis, the mechanism by which envelope mediates this process remains highly debated [51]. Early studies indicated that binding of cell surface associated gp120 subunit of envelope on infected cells with uninfected cells leads to apoptosis [52]. However, later studies using the fusion inhibitor T20 (Fuseon®/enfuvirtide) suggested that the transmembrane subunit, gp41, may play a more important role in apoptosis induction than previously thought. Numerous studies have confirmed that apoptosis mediated by HIV envelope expressing cells in bystander cells can be inhibited by gp41 fusion inhibitor T20 suggesting a direct role of the fusion process [53;54]. Further studies by Blanco, et al, suggested the death of single uninfected cells by gp41 may be mediated by a process of hemifusion [55]. We have confirmed these findings and have shown that in fact gp41-mediated hemifusion triggers an apoptosis cascade that involves caspase 3-mediated mitochondrial depolarization leading to apoptosis [56]. This process was inhibited by the HIV protease inhibitor nelfinavir (Viracept®) which supports the findings that nelfinavir may have beneficial effects beyond virus suppression [57].
While these studies relied largely on the ability of gp41 inhibitor T20 to inhibit both fusion and apoptosis, direct evidence that gp41 can cause apoptosis in the absence of cell to cell fusion was missing. We undertook a mutational approach to address this issue whereby several mutations were introduced into gp41 so as to alter the fusion activity and asked what effect this had on bystander apoptosis [58]. Using a fusion defective mutant (V513E) we demonstrated that clearly, in the context of envelope-expressing cell-mediating apoptosis in bystander cells, the function of gp41 is critical. Furthermore using a mutant with either enhanced activity (CtDel) or decreased activity (G547D), we were able to show that fusion activity correlates with apoptosis induction. However our most important finding was that a hemifusion restricted mutant (D589L) that failed to induce full fusion (syncytia formation) also caused apoptosis in bystander cells in a mechanism similar to fusion competent wild type envelope. This demonstrates that hemifusion and not full fusion is both required and sufficient for induction of apoptosis. Finally in a virus replication assay we showed that the kinetics of WT but not the non-syncytia inducing (W596M) virus can be enhanced by adding caspase 3 inhibitor that prevents hemifusion induced bystander apoptosis allowing for more cells to be infected. This paradigm whereby replication rates of virus depend on the extent to which they cause bystander apoptosis in cells (Figure 5) may be a consequence of selection of virus to adapt to lower levels of CD4 and coreceptor in patients over time. The implications of our findings go beyond explanation of HIV pathogenesis. Based on our demonstration that less fusogenic viruses may be less pathogenic, we hypothesize that it may be possible to select nonpathogenic viruses in vivo by drugs targeting envelope glycoprotein. This is especially true for T20 (Enfuvirtide), that in certain cases selects for less fusogenic viruses which may also be less pathogenic as reported by Aquaro et al [59].
Figure 5.

Model of regulation of virus replication by Env glycoprotein phentoype. Transmission of virus in vivo occurs largely across the virological synapse formed between infected cells and naive cells. This interaction could result in 3 outcomes: 1) Successful transmission of virus to the target cell. 2) The target cell could fuse with the infected cell to form syncytia. 3) The target cell undergoes hemifusion-mediated apoptosis. The phenotype of the Env glycoprotein (apoptosis inducing or not) virus determines whether there is successful transmission or cell death which in turn effects the kinetics of replication.
Summary
Using a wide array of techniques from biophysical and structural techniques to novel chemistry to cell culture and virological assays, our laboratory has elucidated many details of the viral fusion process. The local membrane microenvironment has been determined to control CD4 mobility and hence HIV fusion. We have determined that sensitivity to entry inhibitors correlates with the envelope:coreceptor affinity, receptor density and fusion kinetics. The roles of various regions of gp41 including the cytoplasmic tail (Ct), the membrane-proximal external region (MPER) and the fusion peptide have been revealed as important to entry and are therefore emerging as alternate targets for inhibition. We have expanded the applicability of peptide inhibitors by proving that a permanent covalent reaction can be targeted to a specific amino acid on the HIV gp41 glycoprotein. The development of photosensitized labeling has proved to be important not only for studying the details of the mechanisms of membrane fusion but also for developing novel methods of inactivating infectious agents. The pathogenesis of HIV bystander cells has now been linked directly to the activity of the gp41 envelope glycoprotein in the hemifusion state. We hope that our studies on HIV envelope structure and function will address important issues in AIDS research including development of new and novel anti-HIV therapies, vaccine development and understanding and altering the pathogenesis of HIV.
Acknowledgments
This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. Further funding was provided by a grant from the NIH Intramural AIDS Targeted Antiviral Program (IATAP) and by a grant from the NIAID Intramural Biodefense Research Program. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract NO1-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the United States Government.
Abbreviations
- HIV
Human Immunodeficiency Virus
- SIV
Simian Immunodeficiency Virus
- gp
glycoprotein
- PPMP
1-phenyl-2-hexadecanoylamino-3-morpholino-1-propanol
- GSLs
glycosphingolipids
- Wt
wildtype
- CHR
carboxy-terminal heptad repeat of gp41
- NHR
amino-terminal heptad repeat of gp41
- sCD4
soluble CD4 receptor
- IC50
inhibitory concentration 50%
- PAGE
polyacrylamide gel electrophoresis
- ELISA
enzyme-linked immunosorbent assay
- CD
circular dichroism
- CT
cytoplasmic tail
- MPER
membrane proximal external region
- MSD
membrane spanning domain
- LLP
lentivirus lytic peptide
- NMR
nuclear magnetic resonance
- INA
iodonaphthylazide
- USAMRIID
United States Army Medical Research Institute for Infectious Diseases
- pfu
plaque forming units
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Engelman DM. Membranes are more mosaic than fluid. Nature. 2005 Dec 1;438(7068):578–80. doi: 10.1038/nature04394. [DOI] [PubMed] [Google Scholar]
- 2.Rawat SS, Viard M, Gallo SA, Rein A, Blumenthal R, Puri A. Modulation of entry of enveloped viruses by cholesterol and sphingolipids (Review) Mol Membr Biol. 2003 Jul;20(3):243–54. doi: 10.1080/0968768031000104944. [DOI] [PubMed] [Google Scholar]
- 3.Rawat SS, Johnson BT, Puri A. Sphingolipids: modulators of HIV-1 infection and pathogenesis. Biosci Rep. 2005 Oct;25(5–6):329–43. doi: 10.1007/s10540-005-2894-5. [DOI] [PubMed] [Google Scholar]
- 4.Finnegan CM, Rawat SS, Puri A, Wang JM, Ruscetti FW, Blumenthal R. Ceramide, a target for antiretroviral therapy. Proc Natl Acad Sci U S A. 2004 Oct 26;101(43):15452–7. doi: 10.1073/pnas.0402874101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simons K, Vaz WL. Model systems, lipid rafts, and cell membranes. Annu Rev Biophys Biomol Struct. 2004;33:269–95. 269–95. doi: 10.1146/annurev.biophys.32.110601.141803. [DOI] [PubMed] [Google Scholar]
- 6.Viard M, Parolini I, Sargiacomo M, et al. Role of cholesterol in human immunodeficiency virus type 1 envelope protein-mediated fusion with host cells. J Virol. 2002 Nov;76(22):11584–95. doi: 10.1128/JVI.76.22.11584-11595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steffens CM, Hope TJ. Mobility of the human immunodeficiency virus (HIV) receptor CD4 and coreceptor CCR5 in living cells: implications for HIV fusion and entry events. J Virol. 2004 Sep;78(17):9573–8. doi: 10.1128/JVI.78.17.9573-9578.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puri A, Rawat SS, Lin HM, et al. An inhibitor of glycosphingolipid metabolism blocks HIV-1 infection of primary T-cells. AIDS. 2004 Apr 9;18(6):849–58. doi: 10.1097/00002030-200404090-00002. [DOI] [PubMed] [Google Scholar]
- 9.Rawat SS, Gallo SA, Eaton J, et al. Elevated expression of GM3 in receptor-bearing targets confers resistance to human immunodeficiency virus type 1 fusion. J Virol. 2004 Jul;78(14):7360–8. doi: 10.1128/JVI.78.14.7360-7368.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finnegan CM, Rawat SS, Cho EH, et al. Sphingomyelinase restricts the lateral diffusion of CD4 and inhibits human immunodeficiency virus fusion. J Virol. 2007 May;81(10):5294–304. doi: 10.1128/JVI.02553-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gulbins E, Dreschers S, Wilker B, Grassme H. Ceramide, membrane rafts and infections. J Mol Med. 2004 Jun;82(6):357–63. doi: 10.1007/s00109-004-0539-y. [DOI] [PubMed] [Google Scholar]
- 12.Finnegan CM, Blumenthal R. Fenretinide inhibits HIV infection by promoting viral endocytosis. Antiviral Res. 2006 Feb;69(2):116–23. doi: 10.1016/j.antiviral.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 13.Rawat SS, Eaton J, Gallo SA, et al. Functional expression of CD4, CXCR4, and CCR5 in glycosphingolipid-deficient mouse melanoma GM95 cells and susceptibility to HIV-1 envelope glycoprotein-triggered membrane fusion. Virology. 2004 Jan 5;318(1):55–65. doi: 10.1016/j.virol.2003.08.042. [DOI] [PubMed] [Google Scholar]
- 14.Popik W, Alce TM. CD4 receptor localized to non-raft membrane microdomains supports HIV-1 entry. Identification of a novel raft localization marker in CD4. J Biol Chem. 2004;279:704–12. doi: 10.1074/jbc.M306380200. [DOI] [PubMed] [Google Scholar]
- 15.Rawat S, Zimmerman C, Johnson BT, et al. Restricted lateral mobility of plasma membrane CD4 impairs HIV-1 envelope glycoprotein mediated fusion. Molecular Membrane Biology. 2008 doi: 10.1080/09687680701613713. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu L, Gerard NP, Wyatt R, et al. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature. 1996;384:179–83. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- 17.Salzwedel K, Smith ED, Dey B, Berger EA. Sequential CD4-coreceptor interactions in human immunodeficiency virus type 1 Env function: soluble CD4 activates Env for coreceptor-dependent fusion and reveals blocking activities of antibodies against cryptic conserved epitopes on gp120. J Virol. 2000;74(1):326–33. doi: 10.1128/jvi.74.1.326-333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallo SA, Finnegan CM, Viard M, et al. The HIV Env-mediated fusion reaction. Biochim Biophys Acta. 2003 Jul 11;1614(1):36–50. doi: 10.1016/s0005-2736(03)00161-5. [DOI] [PubMed] [Google Scholar]
- 19.Weissenhorn W, Dessen A, Calder LJ, Harrison SC, Skehel JJ, Wiley DC. Structural basis for membrane fusion by enveloped viruses. Mol Membr Biol. 1999 Jan;16(1):3–9. doi: 10.1080/096876899294706. [DOI] [PubMed] [Google Scholar]
- 20.Jacobs A, Simon C, Caffrey M. Thermostability of the HIV gp41 wild-type and loop mutations. Protein Pept Lett. 2006;13(5):477–80. doi: 10.2174/092986606776819510. [DOI] [PubMed] [Google Scholar]
- 21.Melikyan GB, Markosyan RM, Hemmati H, Delmedico MK, Lambert DM, Cohen FS. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J Cell Biol. 2000 Oct 16;151(2):413–23. doi: 10.1083/jcb.151.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang S, Zhao Q, Debnath AK. Peptide and non-peptide HIV fusion inhibitors. Curr Pharm Des. 2002;8(8):563–80. doi: 10.2174/1381612024607180. [DOI] [PubMed] [Google Scholar]
- 23.Gallo SA, Puri A, Blumenthal R. HIV-1 gp41 Six-Helix Bundle Formation Occurs Rapidly after the Engagement of gp120 by CXCR4 in the HIV-1 Env-Mediated Fusion Process. Biochemistry. 2001 Oct 16;40(41):12231–6. doi: 10.1021/bi0155596. [DOI] [PubMed] [Google Scholar]
- 24.Gallo SA, Clore GM, Louis JM, Bewley CA, Blumenthal R. Temperature-dependent intermediates in HIV-1 envelope glycoprotein-mediated fusion revealed by inhibitors that target N- and C-terminal helical regions of HIV-1 gp41. Biochemistry. 2004 Jun 29;43(25):8230–3. doi: 10.1021/bi049957v. [DOI] [PubMed] [Google Scholar]
- 25.Reeves JD, Gallo SA, Ahmad N, et al. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc Natl Acad Sci U S A. 2002 Dec 10;99(25):16249–54. doi: 10.1073/pnas.252469399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dimitrov AS, Louis JM, Bewley CA, Clore GM, Blumenthal R. Conformational Changes in HIV-1 gp41 in the Course of HIV-1 Envelope Glycoprotein-Mediated Fusion and Inactivation. Biochemistry. 2005 Sep;44(37):12471–9. doi: 10.1021/bi051092d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gallo SA, Wang W, Rawat SS, et al. Theta-defensins prevent HIV-1 Env-mediated fusion by binding gp41 and blocking 6-helix bundle formation. J Biol Chem. 2006 Jul 7;281(27):18787–92. doi: 10.1074/jbc.M602422200. [DOI] [PubMed] [Google Scholar]
- 28.Wang W, Cole AM, Hong T, Waring AJ, Lehrer RI. Retrocyclin, an Antiretroviral theta-Defensin, Is a Lectin. J Immunol. 2003 May 1;170(9):4708–16. doi: 10.4049/jimmunol.170.9.4708. [DOI] [PubMed] [Google Scholar]
- 29.Wild C, Oas T, McDanal C, Bolognesi D, Matthews T. A synthetic peptide inhibitor of human immunodeficiency virus replication: correlation between solution structure and viral inhibition. Proc Natl Acad Sci U S A. 1992;89(21):10537–41. doi: 10.1073/pnas.89.21.10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang S, Lin K, Strick N, Neurath AR. HIV-1 inhibition by a peptide. Nature. 1993 Sep 9;365(6442):113. doi: 10.1038/365113a0. [DOI] [PubMed] [Google Scholar]
- 31.Jiang S, Lin K, Strick N, Neurath AR. Inhibition of HIV-1 infection by a fusion domain binding peptide from the HIV-1 envelope glycoprotein GP41. Biochem Biophys Res Commun. 1993;195(2):533–8. doi: 10.1006/bbrc.1993.2078. [DOI] [PubMed] [Google Scholar]
- 32.Wild C, Greenwell T, Matthews T. A synthetic peptide from HIV-1 gp41 is a potent inhibitor of virus-mediated cell-cell fusion. AIDS Res Human Retroviruses. 1993;9(11):1051–3. doi: 10.1089/aid.1993.9.1051. [DOI] [PubMed] [Google Scholar]
- 33.Jacobs A, Quraishi O, Huang X, et al. A covalent inhibitor targeting an intermediate conformation of the fusogenic subunit of the HIV-1 envelope complex. J Biol Chem. 2007 Nov 2;282(44):32406–13. doi: 10.1074/jbc.M705577200. [DOI] [PubMed] [Google Scholar]
- 34.Wyss S, Dimitrov AS, Baribaud F, Edwards TG, Blumenthal R, Hoxie JA. Regulation of Human Immunodeficiency Virus Type 1 Envelope Glycoprotein Fusion by a Membrane-Interactive Domain in the gp41 Cytoplasmic Tail. J Virol. 2005 Oct;79(19):12231–41. doi: 10.1128/JVI.79.19.12231-12241.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salzwedel K, West JT, Hunter E. A Conserved Tryptophan-Rich Motif in the Membrane-Proximal Region of the Human Immunodeficiency Virus Type 1 gp41 Ectodomain Is Important for Env-Mediated Fusion and Virus Infectivity. The Journal of Virology. 1999 Mar 1;73(3):2469–80. doi: 10.1128/jvi.73.3.2469-2480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dimitrov AS, Jacobs A, Finnegan CM, Stiegler G, Katinger H, Blumenthal R. Exposure of the membrane-proximal external region of HIV-1 gp41 in the course of HIV-1 envelope glycoprotein-mediated fusion. Biochemistry. 2007 Feb 6;46(5):1398–401. doi: 10.1021/bi062245f. [DOI] [PubMed] [Google Scholar]
- 37.Dimitrov AS, Rawat SS, Jiang S, Blumenthal R. Role of the fusion peptide and membrane-proximal domain in HIV-1 envelope glycoprotein-mediated membrane fusion. Biochemistry. 2003 Dec 9;42(48):14150–8. doi: 10.1021/bi035154g. [DOI] [PubMed] [Google Scholar]
- 38.Munoz-Barroso I, Salzwedel K, Hunter E, Blumenthal R. Role of the membrane-proximal domain in the initial stages of human immunodeficiency virus type 1 envelope glycoprotein-mediated membrane fusion. J Virol. 1999 Jul;73(7):6089–92. doi: 10.1128/jvi.73.7.6089-6092.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaroniec CP, Kaufman JD, Stahl SJ, et al. Structure and dynamics of micelle-associated human immunodeficiency virus gp41 fusion domain. Biochemistry. 2005 Dec 13;44(49):16167–80. doi: 10.1021/bi051672a. [DOI] [PubMed] [Google Scholar]
- 40.Cohen FS, Melikyan GB. The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J Membr Biol. 2004 May 1;199(1):1–14. doi: 10.1007/s00232-004-0669-8. [DOI] [PubMed] [Google Scholar]
- 41.Raviv Y, Salomon Y, Gitler C, Bercovici T. Selective labeling of proteins in biological systems by photosensitization of 5-iodonaphthalene-1-azide. Proc Natl Acad Sci U S A. 1987 Sep;84(17):6103–7. doi: 10.1073/pnas.84.17.6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pak CC, Krumbiegel M, Blumenthal R, Raviv Y. Detection of influenza hemagglutinin interaction with biological membranes by photosensitized activation of [125I]iodonaphthylazide. J Biol Chem. 1994 May 20;269(20):14614–9. [PubMed] [Google Scholar]
- 43.Raviv Y, Viard M, Bess J, Jr, Blumenthal R. Quantitative Measurement of Fusion of HIV-1 and SIV with Cultured Cells Using Photosensitized Labeling. Virology. 2002 Feb 15;293(2):243–51. doi: 10.1006/viro.2001.1237. [DOI] [PubMed] [Google Scholar]
- 44.Pak CC, Puri A, Blumenthal R. Conformational Changes and Fusion Activity of Vesicular Stomatitis Virus Glycoprotein: 125Iodonaphthylazide Photo Labeling Studies in Biological Membranes. Biochemistry. 1997;36:8890–6. doi: 10.1021/bi9702851. [DOI] [PubMed] [Google Scholar]
- 45.Raviv Y, Viard M, Bess JW, Jr, Chertova E, Blumenthal R. Inactivation of retroviruses with preservation of structural integrity by targeting the hydrophobic domain of the viral envelope. J Virol. 2005 Oct;79(19):12394–400. doi: 10.1128/JVI.79.19.12394-12400.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kemble GW, Danieli T, White JM. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell. 1994;76(2):383–91. doi: 10.1016/0092-8674(94)90344-1. [DOI] [PubMed] [Google Scholar]
- 47.Sharma A, Raviv Y, Puri A, Viard M, Blumenthal R, Maheshwari RK. Complete inactivation of Venezuelan equine encephalitis virus by 1,5-iodonaphthylazide. Biochem Biophys Res Commun. 2007 Jun 29;358(2):392–8. doi: 10.1016/j.bbrc.2007.04.115. [DOI] [PubMed] [Google Scholar]
- 48.Armstrong RT, Kushnir AS, White JM. The transmembrane domain of influenza hemagglutinin exhibits a stringent length requirement to support the hemifusion to fusion transition. J Cell Biol. 2000 Oct 16;151(2):425–37. doi: 10.1083/jcb.151.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin X, Derdeyn CA, Blumenthal R, West J, Hunter E. Progressive Truncations C Terminal to the Membrane-Spanning Domain of Simian Immunodeficiency Virus Env Reduce Fusogenicity and Increase Concentration Dependence of Env for Fusion. J Virol. 2003 Jun 15;77(12):7067–77. doi: 10.1128/JVI.77.12.7067-7077.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gougeon ML, Montagnier L. Programmed cell death as a mechanism of CD4 and CD8 T cell deletion in AIDS. Molecular control and effect of highly active anti-retroviral therapy. Ann N Y Acad Sci. 1999;887:199–212. 199–212. doi: 10.1111/j.1749-6632.1999.tb07934.x. [DOI] [PubMed] [Google Scholar]
- 51.Perfettini JL, Castedo M, Roumier T, et al. Mechanisms of apoptosis induction by the HIV-1 envelope. Cell Death Differ. 2005 Aug;12( Suppl 1):916–23. doi: 10.1038/sj.cdd.4401584. [DOI] [PubMed] [Google Scholar]
- 52.Biard-Piechaczyk M, Robert-Hebmann V, Richard V, Roland J, Hipskind RA, Devaux C. Caspase-dependent apoptosis of cells expressing the chemokine receptor CXCR4 is induced by cell membrane-associated human immunodeficiency virus type 1 envelope glycoprotein (gp120) Virology. 2000 Mar 15;268(2):329–44. doi: 10.1006/viro.1999.0151. [DOI] [PubMed] [Google Scholar]
- 53.Scheller C, Jassoy C. Syncytium formation amplifies apoptotic signals: a new view on apoptosis in HIV infection in vitro. Virology. 2001 Mar 30;282(1):48–55. doi: 10.1006/viro.2000.0811. [DOI] [PubMed] [Google Scholar]
- 54.Meissner EG, Zhang L, Jiang S, Su L. Fusion-induced apoptosis contributes to thymocyte depletion by a pathogenic human immunodeficiency virus type 1 envelope in the human thymus. J Virol. 2006 Nov;80(22):11019–30. doi: 10.1128/JVI.01382-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blanco J, Barretina J, Ferri KF, et al. Cell-surface-expressed HIV-1 envelope induces the death of CD4 T cells during GP41-mediated hemifusion-like events. Virology. 2003 Jan 20;305(2):318–29. doi: 10.1006/viro.2002.1764. [DOI] [PubMed] [Google Scholar]
- 56.Garg H, Blumenthal R. HIV gp41-induced apoptosis is mediated by caspase-3-dependent mitochondrial depolarization, which is inhibited by HIV protease inhibitor nelfinavir. J Leukoc Biol. 2006 Feb;79(2):351–62. doi: 10.1189/jlb.0805430. [DOI] [PubMed] [Google Scholar]
- 57.Miro O, Villarroya J, Garrabou G, et al. In vivo effects of highly active antiretroviral therapies containing the protease inhibitor nelfinavir on mitochondrially driven apoptosis. Antivir Ther. 2005;10(8):945–51. [PubMed] [Google Scholar]
- 58.Garg H, Joshi A, Freed EO, Blumenthal R. Site-specific mutations in HIV-1 gp41 reveal a correlation between HIV-1-mediated bystander apoptosis and fusion/hemifusion. J Biol Chem. 2007 Jun 8;282(23):16899–906. doi: 10.1074/jbc.M701701200. [DOI] [PubMed] [Google Scholar]
- 59.Aquaro S, D’Arrigo R, Svicher V, et al. Specific mutations in HIV-1 gp41 are associated with immunological success in HIV-1-infected patients receiving enfuvirtide treatment. J Antimicrob Chemother. 2006 Oct;58(4):714–22. doi: 10.1093/jac/dkl306. [DOI] [PubMed] [Google Scholar]