

Abstract
Background
Medicago truncatula is a model legume species that is currently the focus of an international genome sequencing effort. Although several different oligonucleotide and cDNA arrays have been produced for genome-wide transcript analysis of this species, intrinsic limitations in the sensitivity of hybridization-based technologies mean that transcripts of genes expressed at low-levels cannot be measured accurately with these tools. Amongst such genes are many encoding transcription factors (TFs), which are arguably the most important class of regulatory proteins. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) is the most sensitive method currently available for transcript quantification, and one that can be scaled up to analyze transcripts of thousands of genes in parallel. Thus, qRT-PCR is an ideal method to tackle the problem of TF transcript quantification in Medicago and other plants.
Results
We established a bioinformatics pipeline to identify putative TF genes in Medicago truncatula and to design gene-specific oligonucleotide primers for qRT-PCR analysis of TF transcripts. We validated the efficacy and gene-specificity of over 1000 TF primer pairs and utilized these to identify sets of organ-enhanced TF genes that may play important roles in organ development or differentiation in this species. This community resource will be developed further as more genome sequence becomes available, with the ultimate goal of producing validated, gene-specific primers for all Medicago TF genes.
Conclusion
High-throughput qRT-PCR using a 384-well plate format enables rapid, flexible, and sensitive quantification of all predicted Medicago transcription factor mRNAs. This resource has been utilized recently by several groups in Europe, Australia, and the USA, and we expect that it will become the 'gold-standard' for TF transcript profiling in Medicago truncatula.
Background
Legumes are second only to grasses in agricultural importance [1]. They are a mainstay of sustainable agricultural systems because of their ability to reduce atmospheric nitrogen (N2) to ammonia via a symbiosis with bacteria called rhizobia. This provides legumes and subsequent crops with a free and renewable source of nitrogen in lieu of expensive, environmentally-unfriendly fertilizers. Development and differentiation of root nodules, the organ that accommodates nitrogen-fixing rhizobia in legumes, is orchestrated by transcription factors [2-9]. Transcription factors are DNA-binding proteins that regulate the transcription of most, if not all genes [10]. As a result, TFs play central roles in all aspects of plant biology, including development and differentiation of organs and adaptive responses to changes in the environment [11]. Transcription factors as a whole are an important target of plant research because they are a key to understanding the regulation of important plant processes as well as potential tools to optimize these processes for agriculture.
The importance of TFs in plant biology is reflected by the fact that approximately 5% of all plant genes encode such proteins [10]. Thus, even species with relatively small genomes, such as Arabidopsis thaliana contain thousands of TF genes [10]. This presents a real challenge for systematic approaches to decipher the function of TF genes in plants. Classical, 'forward' genetics has uncovered the roles of perhaps a hundred TF genes in Arabidopsis [12] and far fewer in other species [11]. Reverse-genetic approaches, using T-DNA insertion mutants for instance [13], provide a means to decipher in a systematic and relatively rapid manner the function of TF genes/proteins, although gene-redundancy often stymies this enterprise [12]. Another stumbling-block is that phenotypes associated with non-redundant TFs may be subtle in nature.
Transcript profiling can help to uncover the functions of TF genes/proteins by revealing where and when in a plant TF genes are expressed. This information can help direct our attention to particular organs, developmental stages, or conditions under which aberrant phenotypes might become apparent in a TF mutant of interest.
Medicago truncatula is a model legume species that is currently the focus of an international genome sequencing effort [14]. Several generations of cDNA [15] and oligonucleotide arrays [16] have been developed for transcriptome analysis of Medicago truncatula, including most-recently an Affymetrix GeneChip that contains 51,000 probe-sets representing a large proportion of all the genes in this species [17]. While these tools now provide a means to measure the transcriptional output of a large proportion of genes in Medicago, inherent limitations in the sensitivity of hybridization-based technologies [18] mean that transcripts of a substantial number of genes cannot be detected even when probes for these transcripts are present on the array/chip. Furthermore, expansion of arrays to encompass novel genes uncovered by genome sequencing is not a trivial task. An alternative to arrays that is 2–3 orders of magnitude more sensitive and more flexible in terms of expansion to encompass novel genes is quantitative reverse transcription-polymerase chain reaction (qRT-PCR). Platforms for qRT-PCR analysis of thousands of Arabidopsis and rice TF genes have been developed by us and others [19,20], and utilized to identify TF genes involved in Arabidopsis responses to nutrient stress and pathogen attack [21-24]. Here we describe a bioinformatics pipeline to identify putative TF genes in Medicago truncatula and to design gene-specific oligonucleotide primers for qRT-PCR analysis of all predicted TF transcripts. Over 1000 TF primer pairs were tested and used to identify sets of organ-enhanced TF genes that may play important roles in organ development or differentiation in this species.
Results and Discussion
Identification of putative transcription factors
TF protein families are generally defined by the type(s) of DNA-binding domain they contain and putative TF genes are often identified on the basis of DNA sequences that encode known DNA-binding domains [10,11,25,26]. We utilized this approach to identify putative TFs of Medicago amongst the set of proteins predicted from genomic sequence by the International Medicago Genome Annotation Group (IMGAG). Proteins of IMGAG release 1, which contained over 40,000 predicted proteins, were screened for the presence of known or presumed DNA-binding domains (Table 1), using INTERPRO [27]. Medicago proteins containing putative DNA binding domains and other domains associated with TFs were then used as query sequences in WU-BLASTX [28] which included searches of both the non-redundant DNA database of NCBI [29] and the well-curated protein database, UniProt [30] to check annotations of related proteins in support of tentative Medicago TF assignments. This process resulted in a list of 1045 putative TF genes (see Additional file 1). We utilized genomic sequences rather than the large collection of partial cDNA sequences present in Expressed Sequence Tag (EST) databases for Medicago as the starting point for TF gene discovery because protein sequences derived from genomic sequence are more complete and the set of IMGAG proteins essentially contains no redundancy. Although identification of the 'complete' set of Medicago TFs from IMGAG-annotated proteins will only be possible upon completion of genome sequencing, we expect little or no redundancy in the protein set targeted by our primer set. This approach avoids wasting money on redundant primer sets and re-organization of primers when redundancy is detected, both of which would have been inevitable if we chose to use ESTs in addition to genomic sequences to identify Medicago TFs.
Table 1.
Classification of putative transcription factors of Medicago into families and sub-families
| TF Family |
No. of Genes |
Characteristic Domain (InterPro No.) |
Domain Function |
Domain Description |
| MYB/HD-like | 76 | IPR001005, IPR009057 | D | Myb, DNA-binding; homeodomain like |
| MYB | 58 | IPR001005 | D | Myb, DNA-binding |
| C2H2 (Zn) | 64 | IPR007087 | NA | Zn-finger, C2H2 type |
| AP2/EREBP | 55 | IPR001471 | D | Pathogenesis-related transcriptional factor and ethylene response factor |
| BHLH | 49 | IPR001092 | D | Basic helix-loop-helix dimerisation region bHLH |
| HD-like | 50 | IPR009057 | D | Homeodomain like |
| HD family | IPR001356 | D | Homeobox | |
| HD | 25 | |||
| HD-ZIP | 5 | IPR006712 | P | HD-ZIP protein, N terminus |
| HD-PHD finger | 2 | IPR001965 | P | Zn-finger like, PHD-finger |
| MADS | 48 | IPR002100 | D | TF, MADS-box |
| BZIP | 41 | IPR004827 | D | Basic Leu zipper (bZIP) TF |
| PHD | 34 | IPR001965 | P | Zn-finger like, PHD-finger |
| WRKY family | IPR003657 | D | DNA-binding WRKY | |
| WRKY | 29 | |||
| LLR WRKY | 1 | IPR001611 | Leu-rich repeat | |
| ABI3/VP1 | 29 | IPR003340 | D | TF B3 |
| NAC | 29 | IPR003441 | D | No apical meristem (NAM) protein |
| C3H-type1(Zn) | 27 | IPR000571 | D | Zn-finger, C-x8-C-x5-C-x3-H type |
| ARF | 23 | IPR003340, IPR010525, IPR011525 | D | |
| JUMONJI | 20 | IPR003347 | D | TF jumonji, jmjC |
| GRAS | 19 | IPR005202 | P | GRAS TF |
| HMG | 15 | IPR000637 | D | HMG-I and HMG-Y, DNA binding |
| AS2 | 14 | IPR004883 | P | Lateral organ boundaries |
| C2C2 (Zn) | ||||
| Dof | 13 | IPR003851 | D | Zn-finger, Dof type |
| GATA | 7 | IPR000679 | D | Zn-finger, GATA type |
| CO-like | 6 | IPR000315 | D | Zn-finger, B-box |
| YABBY | 5 | IPR006780 | D | YABBY protein |
| CCAAT-HAP3 type | 12 | IPR003958 | D | TF CBF/NF-Y/archaeal histone |
| GRF | 8 | IPR010666 | D | Zn-finger, GRF type |
| SBP | 8 | IPR004333 | D | SBP |
| EIL | 7 | IPR006957 | D | Ethylene insensitive 3 |
| LIM | 7 | IPR001781 | P | Zn-binding protein, LIM |
| SNF2 | 6 | IPR000330 | D | SNF2 family N-terminal domain |
| E2F/DP | 5 | IPR003316 | D | TF E2F/dimerisation partner (TDP) |
| TCP | 5 | IPR005333 | D | TCP TF |
| FHA | 5 | IPR000253 | D | Forkhead-associated |
| ARID | 4 | IPR001606 | D | AT-rich interaction region |
| HSF | 4 | IPR000232 | D | Heat shock factor (HSF)-type, DNA binding |
| AUX/IAA | 3 | IPR003311 | D | AUX/IAA protein |
| SRS | 3 | IPR006510 | D | Zn-finger, LRP1 type |
| TUB | 3 | IPR000007 | D | Tubby |
| ZIM | 3 | IPR010399 | D | ZIM |
| DDT | 3 | IPR004022 | D | DDT |
| ZF-HD | 2 | IPR006455 | D | Homeobox domain, ZF-HD class |
| MBF1 | 2 | IPR001387 | D | Helix-turn-helix type 3 |
| S1Fa-like | 2 | IPR006779 | D | DNA binding protein S1FA |
| CAMTA | 2 | IPR005559 | D | CG-1 |
| LFY | 1 | IPR002910 | D | Floricaula/leafy protein |
| Nin-like | 1 | IPR003035 | D | Plant regulator RWP-RK |
| TAZ | 1 | IPR000197 | P | Zn-finger, TAZ-type |
| Potentially novel plant TFs and transcriptional regulators | ||||
| CCHC (Zn) | 112 | IPR001878 | NA | Zn-finger, CCHC-type |
| RR | 16 | IPR001789, IPR011006 | RD | Response regulator receiver |
| DHHC (Zn) | 14 | IPR001594 | D or P | Zn-finger, DHHC-type |
| HTH | ||||
| FIS | 11 | IPR002197 | D | Helix-turn-helix, Fis-type |
| AraC | 2 | IPR000005 | D | Helix-turn-helix, AraC type |
| BTB/POZ | 7 | IPR000210 | P | BTB |
| TTF-type (Zn) | 6 | IPR006580 | D | Zn-finger, TTF-type |
| BD | 6 | IPR001487 | P | Bromodomain |
| Lambda-DB | 3 | IPR010982 | D | Lambda_DNA_bd |
| TrpR | 3 | IPR010921 | D | Trp repressor/replication initiator |
| TPR | 3 | IPR001440 | P | Tetratricopeptide TPR_1 |
| KRAB-box | 2 | IPR001909 | P | KRAB box |
| NRs | 2 | IPR008946 | LBD | Steroid nuclear receptor, ligand binding |
| R3H | 2 | IPR001374 | NA | Single-stranded nucleic acid binding R3H |
| YEATS | 2 | IPR005033 | TA | YEATS |
| U1-type (Zn) | 2 | IPR003604 | NA | Zn-finger, U1-type |
| A20-like | 2 | IPR002653 | P | Zn-finger, A20-type |
| Euk_TF | 1 | IPR008917 | D | Euk_TF, DNA binding |
| NGN | 1 | IPR006645 | D | NGN |
| p53-like | 1 | IPR008967 | D | p53-like TF, DNA binding |
| SSB protein | 1 | IPR011344 | D | Single-strand binding protein |
| ssDB TR | 1 | IPR009044 | D | Single-strand DNA binding transcriptional regulator |
| TCoAp15 | 1 | IPR003173 | D | Transcriptional coactivator p15 |
| BED-type (Zn) | 1 | IPR003656 | D | Zn-finger, BED-type predicted |
| TCoA | 1 | IPR009255 | TA | Transcriptional coactivation |
| Tc/PD | 1 | IPR001533 | TA | Transcriptional coactivator |
IMGAG (International Medicago Gene Annotation Group)-proteins were classified as putative TFs if they contained characteristic DNA-binding or other characteristic TF domains and if annotations of matching proteins obtained by BLAST searches were consistent with such a classification. TF families previously identified in plants are presented in the first part of the table while potentially novel plant TF families, which were identified by the presence of domains associated with TFs and other transcriptional regulators outside the plant kingdom, are presented in the latter part of the table. D = DNA binding domain; P = protein-protein interaction domain; NA = nucleic acid (DNA and RNA) binding domain; RD = receiver domain; LBD = ligand binding; transcriptional co-activator. Plant-specific TF families and sub-families are indicated in bold (according to [12])
PCR primer design
To ensure maximum specificity and efficiency during PCR amplification of TF cDNA under a standard set of reaction conditions, a stringent set of criteria was used for primer design. This included predicted melting temperatures (Tm) of 58°C to 61°C, limited self-complementarity and poly-X, and PCR amplicon lengths of 100–150 base pairs (bp). Secondary hits were minimized by aligning primer candidates to all known Medicago sequences via WU-BLAST [28] and eliminating primer pairs with multiple potential hits.
PCR primer testing: gene-specificity and amplification efficiency
PCR primers were tested on Medicago cDNA free of genomic DNA contamination as follows. First, total RNA was extracted from various organs using Trizol reagent (Invitrogen GmbH, Karlsruhe, Germany), which yielded high quality RNA as judged by gel electrophoresis and by Agilent 2100 BioAnalyser using RNA 6000 Nano Chips (Agilient Technologies, Waldbronn, Germany). Typical RNA yields ranged from 0.5–1.0 μg RNA/mg fresh mass for nodules and leaves, respectively. Isolated RNA was treated with DNAse I (Ambion, product number 1907) to remove all contaminating genomic DNA, and this was always confirmed by PCR using primers to non-coding regions of the Ubiquitin gene (TC102473; AC137828-19.4). After inactivation of DNAse I, RNA was reverse transcribed using SuperScript III reverse transcriptase (Invitrogen GmbH, Karlsruhe, Germany) and oligo-dT12–18 to prime the reaction.
Specificity of PCR primers was assessed in three ways: by melting curve analysis of PCR reaction products; by separating the products of all reactions via electrophoresis in 3% agarose gels; and by sequencing a sub-set of PCR reaction products (Figure 1). 94.5% (998/1045) of primer pairs gave unique PCR products of the expected size. Only 3.3% (34/1045) of primer pairs yielded no product and 2.2% (23/1045) gave non-specific products. (see Additional file 1). Sequencing was performed on 178 randomly-chosen PCR products amplified from a 1:1 mixture of leaf and root cDNA. In the vast majority of cases (92.7% or 165/178), the sequence of the PCR product was identical to that of the intended target gene. In 5.1% of cases, the amplicon sequence matched multiple related genes, including the target gene, while in only 2.2% of cases the amplicon sequence did not match the target gene sequence.
Figure 1.
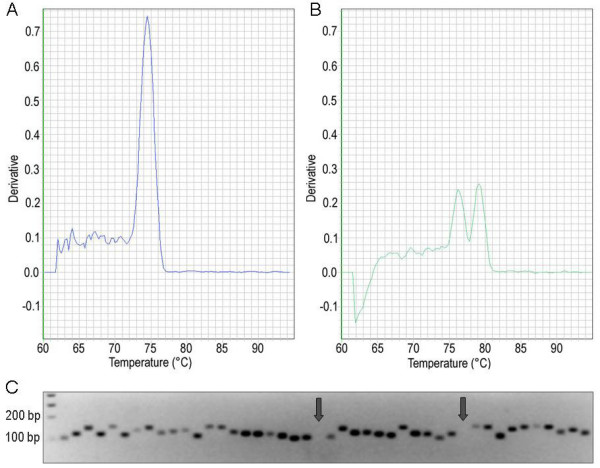
Specificity of transcription factor PCR primers. Specificity was confirmed by dissociation curves with a single peak (A) while double peaks (B) indicated off-target ampification. The derivative of fluorescence intensity is shown on the y-axis. Separation of PCR products on 3% (v/w) agarose gels following electrophoresis (C) confirmed the presence of unique amplicons of the expected size for most reactions. Few reactions yielded no products (indicated by arrow).
Ideally, PCR results in an exact doubling of the amount of dsDNA after each temperature cycle. In practice, however, this is generally not the case because the reactions are less than 100% efficient. Primer sequences can affect PCR efficiency, so we determined the efficiency of each TF primer pair from amplification plots, using LinRegPCR software [31]. First, the correlation coefficient derived from linear regression analysis of each amplification plot (e.g. see Figure 2) was used to assess the 'quality' of each reaction, and all reactions with an R2 < 0.990 were excluded from further analysis (10.6% of reactions). Next, average PCR efficiencies (E) were computed for each individual primer pair across all analyzed samples. 53.4% (558 TF genes) displayed PCR efficiencies greater than 0.80, while 39.7% (415 TF genes) had efficiencies between 0.51–0.80. Only 2.6% (27 TF genes) had mean E values below 0.4; these all yielded R2 < 0.99 in LinRegPCR analysis and mostly represented reactions that lacked detectable fragment amplification (CT > 40) or that generated unspecific PCR products (Figure 2; see Additional file 1). A similar range of PCR efficiencies were determined for Arabidopsis and rice TF primers previously [19,20].
Figure 2.
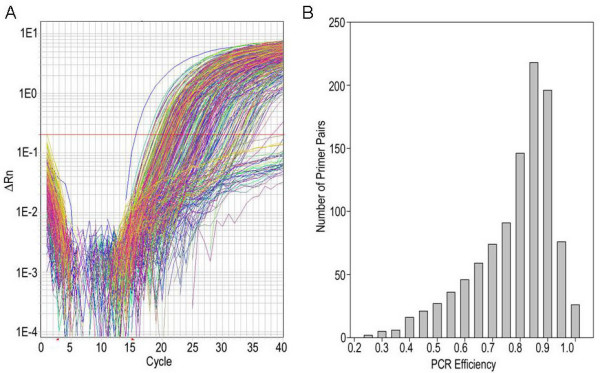
Amplification efficiency of transcription factor-specific primer pairs. Typical real-time RT-PCR amplification plots of 384 TF genes (left) and distribution of PCR efficiencies for all 1045 TF primer pairs (right).
Selection of reference genes
Reference genes with stable expression/transcript levels throughout development and in the face of environmental challenge are crucial for the normalization of expression data of other genes. Potentially useful reference genes were chosen based on published data for Medicago (e.g. Msc27 [32]) and Arabidopsis thaliana [33]. The closest Medicago homologues of Arabidopsis genes were identified by BlastN [29]. Gene-specific primer pairs for the genes encoding elongation factor 1α (EST317575), glyceralaldehyde-3-phosphate dehydrogenase (MtC00030_GC; CT573421_3.4), β-tubulin (TC106341), Pentatricopeptide repeat protein (TC96273), actin2 (TC107326; AC137836_27.5), Ubiquitin (TC102473; AC137828-19.4), Helicase (CB892427), and the genes PDF2 (TC107161), UPL7 (TC111218), PTB (TC111751), UBC (AW686873), bHLH (CX538576), and UBC9 (TC106312) were designed using the criteria described above (Table 2). The specificity of PCR primers was tested using 18 first-strand cDNAs from six different organs of Medicago (three biological replicates each). All primer pairs produced a single PCR product of the expected size, as shown by gel electrophoresis and unique dissociation curves generated by the PCR machine after 40 cycles (Figure 3). To determine which reference genes were best suited for transcript normalisation, we used the software geNORM [34], which uses pair-wise comparison and geometric averaging across a matrix of biological samples to determine gene expression stability (M; [35]). The genes PDF2, PPRep, Ubiquitin, and PTB had the lowest M (greatest transcript stability) and, therefore, were judged to be the best reference genes for this diverse set of developmental samples (Figure 4; Table 2).
Table 2.
Medicago reference genes and primers for qRT-PCR
| Gene Name | TC |
Accession Number |
Forward/Reverse Primer (5'-3') |
PCR Product Size (bp) |
PCR efficiency (E) |
R2 |
| β Tubulin | TC106341 | N | TTTGCTCCTCTTACATCCCGTG / GCAGCACACATCATGTTTTTGG | 100 | 1.08 | 1.00 |
| PPRrep | TC96273 | N | GGAAAACTGGAGGATGCACGTA / ACAAGCCCTCGACACAAAACC | 100 | 0.93 | 1.00 |
| PDF 2 | TC107161 | N | GTGTTTTGCTTCCGCCGTT / CCAAATCTTGCTCCCTCATCTG | 100 | 0.99 | 1.00 |
| bHLH | CX538576 | N | TAGCGAGTACCATGATGCCAGA / GCGCCTCTTTTGTTTTCAGC | 100 | 0.89 | 1.00 |
| UBC | AW686873 | N | CTGACAGCCCACTGAATTGTGA / TTTTGGCATTGCTGCAAGC | 100 | 0.96 | 1.00 |
| PTB | TC111751 | N | CGCCTTGTCAGCATTGATGTC / TGAACCAGTGCCTGGAATCCT | 100 | 0.85 | 1.00 |
| Ubiquitin | TC102473 | AC137828_19.4 | GCAGATAGACACGCTGGGA / AACTCTTGGGCAGGCAATAA | 100 | 0.95 | 1.00 |
| UBC9 | TC106312 | AC137602_2.4 | GGTTGATTGCTCTTCTCTCCCC / AAGTGATTGCTCGTCCAACCC | 100 | 1.13 | 0.99 |
| Helicase | CB892427 | N | GTACGAGGTCGGTGCTCTTGAA / GCAACCGAAAATTGCACCATAC | 100 | 0.91 | 1.00 |
| ELF1α | EST317575 | N | GACAAGCGTGTGATCGAGAGATT / TTTCACGCTCAGCCTTAAGCT | 100 | 0.68 | 0.98 |
| UPL7 | TC111218 | N | CCAGTTGTTCTCGTGGTCCATT / CCTCCAATTGTCGCCCAAA | 100 | 0.93 | 1.00 |
| GAPDH | MtC00030_GC | CT573421_3.4 | TGCCTACCGTCGATGTTTCAGT / TTGCCCTCTGATTCCTCCTTG | 100 | 1.04 | 0.99 |
| Actin2 | TC107326 | AC137836_27.5 | TCAATGTGCCTGCCATGTATGT / ACTCACACCGTCACCAGAATCC | 100 | 1.12 | 0.99 |
| MSC27 | X63872 (M. sativa) | N | GTTGAAGTAGACATTGGTGCTAACG / AGCTGAGTCATCAACACCCTCAT | 100 | 0.76 | 0.99 |
Mean PCR efficiency (E) was determined from three biological replicates of each of six organs, using LinRegPCR [31], which also yielded mean R2. N, no corresponding GenBank accession number.
Figure 3.

Specificity, efficiency, and reproducibility of PCR primers designed to amplify reference gene transcripts. Specificity of primers was confirmed by the presence of unique amplicons of the expected size following electrophoresis on 3% (v/w) agarose gels (A) and by dissociation curves with a single peak (B to D). Typical real-time RT-PCR amplification plots of three reference gene transcripts (E to G).
Figure 4.

Ranking of 8 reference genes in M. truncatula. Transcript levels of all 8 genes were measured by qRT-PCR, using 18 independent cDNA preparations from six different organs with three replicate measurements of each cDNA preparation. A low value for the average expression stability M, as calculated by geNORM software, indicates more stable expression throughout the various organs.
Identification of organ-enhanced TFs of Medicago
To get an overview of TF gene expression in Medicago truncatula and to identify TFs induced in specific organs, we used the real-time RT-PCR platform described above. Transcript profiling was performed on six different organs of Medicago (leaves, stems, flowers, pods, roots, and nodules) with three independent biological replicates for each (see Additional file 2). The fraction of genes for which transcripts were detected within 40 cycles ranged from 77.2% in leaves to 90.8% in pods. Transcripts from nearly all putative TF genes (96.8% or 1011/1045) were detected in at least one organ. Genes were called detected if they were expressed in at least two biological replicates with a CT < 40. Approximately half of all TF genes exhibited differential expression during plant development, based on significant differences (p ≤ 0.05) in transcript levels between organs. Few TF genes (1.19% or 12/1011) were expressed exclusively in vegetative organs (leaves, stems, roots or nodules), and even fewer (0.5% or 5/1011) were expressed only in reproductive organs (flowers or pods). A relatively small number of TF genes exhibited greater than ten-fold ratios in expression level in one organ compared to any other organ (Table 3). For comparison, we have included gene expression ratios derived from Affymetrix array data from the same RNA samples. While there is reasonable qualitative agreement between gene expression ratios obtained using the two methods, the lack of quantitative agreement is likely due to the limited sensitivity and low signal to noise ratio near the detection limit of Affymetrix arrays [19]. The genes listed in Table 3 may control development and/or differentiation in Medicago and are interesting targets for future research.
Table 3.
Organ-enhanced TF genes
| A Root-enhanced TF genes identified by real-time RT-PCR | |||||||||||
| Real-time RT-PCR Expression Ratio | Affymetrix Expression Ratio | ||||||||||
| Accession Number | R/L | R/S | R/F | R/P | R/N | R/L | R/S | R/F | R/P | R/N | Affymetrix Chip ID |
| AC140721_12.1 | 121.5 | 320.1 | 1623.4 | 2676.5 | 64.4 | 161.2b | 144.5b | 219.2b | 158.1b | 99.9 | Mtr.50075.1.S1_s_at |
| AC135101_25.1 | 312.2 | 438.0 | 291.6 | 787.0 | 61.3 | n | n | n | n | n | n |
| AC140721_14.1 | 1204.9 | 751.2 | 956.6 | 6296.6 | 32.6 | n | n | n | n | n | n |
| AC140031_3.1 | 275.4 | 1157.3 | 923.4 | 258916.9a | 31.1 | 0.4 | 0.7 | 0.8 | 0.9 | 2.2 | Mtr.47227.1.S1_s_at |
| AC140721_13.1 | 39.9a | 179.8 | 614.8 | 935.1a | 26.5 | 35.9 | 48.1b | 41.2 | 44.2b | 39.5 | Mtr.50074.1.S1_at |
| AC140031_7.1 | 360.2a | 830.6 | 1542a | 6868.8 | 19.7 | 0.7b | 1b | 1.1b | 1b | 0.9b | Mtr.47229.1.S1_at |
| AC146574_6.1 | 1095.7 | 11.9a | 48.8 | 51.8 | 88.8 | 1.2b | 1.1b | 1.3b | 1.1b | 1.2b | Mtr.40781.1.S1_s_at |
| AC125478_13.7 | 1117.4 | 7008.0 | 3264.9a | 7256.6 | 11.6 | 211.5b | 248.8b | 296.4b | 228.7b | 45.3 | Mtr.15416.1.S1_at |
| AC125478_7.2 | 1052.3 | 503.3 | 938.4a | 2754.5 | 11.2 | n | n | n | n | n | n |
| AC122726_21.111 | 50.0 | 48.0 | 20943.9 | 2658.6 | 10.4 | 74.3b | 76.4b | 72.8b | 82.8b | 26.6 | Mtr.15568.1.S1_s_at |
| B Nodule-enhanced TF genes identified by real-time RT-PCR | |||||||||||
| Real-time RT-PCR Expression Ratio | Affymetrix Expression Ratio | ||||||||||
| Accession Number | N/L | N/S | N/F | N/P | N/R | N/L | N/S | N/F | N/P | N/R | Affymetrix Chip ID |
| AC148816_3.2 | 9966.7 | 1826.2 | 6262.9 | 21709.1 | 662.7 | 434.6b | 516.8b | 455b | 521.7b | 551.4b | Mtr.14503.1.S1_at |
| AC147774_3.2 | 55.2a | 466.7 | 726.3 | 881.2 | 498.5 | 17.7b | 16b | 17.8b | 19.1b | 16.1b | Mtr.19554.1.S1_at |
| AC138056_33.241 | 43.9 | 93.2 | 18.3 | 17.3 | 13.1 | 0.9b | 0.8b | 0.8b | 1.1b | 1.1b | Mtr.17993.1.S1_at |
| AC124214_39.2 | 121.8 | 92.1 | 44.8 | 64.9 | 10.5 | n | n | n | n | n | n |
| C Pod-enhanced TF genes identified by real-time RT-PCR | |||||||||||
| Real-time RT-PCR Expression Ratio | Affymetrix Expression Ratio | ||||||||||
| Accession Number | P/L | P/S | P/F | P/N | P/R | P/L | P/S | P/F | P/N | P/R | Affymetrix Chip ID |
| AC143340_4.7 | 154.1 | 44.8 | 173.3 | 180.3a | 407.3 | 1.6b | 1.8b | 1.7b | 1.7b | 1.7b | Mtr.17931.1.S1_at |
| D Flower-enhanced TF genes identified by real-time RT-PCR | |||||||||||
| Real-time RT-PCR Expression Ratio | Affymetrix Expression Ratio | ||||||||||
| Accession Number | F/L | F/S | F/P | F/N | F/R | F/L | F/S | F/P | F/N | F/R | Affymetrix Chip ID |
| AC129092_13.1 | 40.3 | 383.3 | 65.1 | 1683.9 | 318.1 | 24.2 | 47.1b | 38.2 | 45.4b | 55.1b | Mtr.16432.1.S1_at |
| AC148485_10.1 | 476.2 | 203.7 | 27.2 | 174.6 | 44.5 | 36.5b | 32.9b | 22.0 | 34.7b | 29.7b | Mtr.20392.1.S1_at |
| AC140915_20.1 | 169.5 | 101.1 | 18.6 | 481.0 | 178.7 | 1.4b | 1.4b | 1.3b | 1.3b | 1.6b | Mtr.51688.1.S1_at |
| AC141107_50.2 | 38.2 | 33.0 | 17.9 | 114.0 | 97.4 | n | n | n | n | n | n |
| AC144731_15.21 | 64.4 | 44.3 | 33.6 | 61.5 | 16.3 | 0.6b | 0.6b | 0.8b | 0.8b | 0.8b | Mtr.19093.1.S1_at |
| AC150978_12.1 | 235.3 | 198.5 | 48.4 | 14.0 | 16.9 | n | n | n | n | n | n |
| AC141107.5.61 | 261.1 | 127.8 | 13.3 | 96.4 | 71.5 | 0.9b | 0.9b | 0.8b | 0.1b | 1.1b | Mtr.51651.1.S1_at |
| AC157472_19.1 | 60.2a | 92.1 | 13.2 | 61.5a | 113.6 | n | n | n | n | n | n |
| AC148527_19.141 | 188.4 | 42.1 | 17.3 | 26.5 | 13.0 | n | n | n | n | n | n |
| AC144726_6.1 | 254.7 | 55.2 | 10.9 | 112.7a | 754.7 | 59.2 | 73.7b | 7.6 | 89.2 | 97.9b | Mtr.19024.1.S1_at |
| AC157488_16.1 | 7053.2a | 141.1 | 18.7 | 81.8 | 10.7 | n | n | n | n | n | n |
| AC123899_15.181 | 10.2 | 92.2 | 19.1 | 62.8 | 24.9 | 0.9b | 0.5b | 0.8b | 0.1b | 0.8b | Mtr.52015.1.S1_at |
A TF gene was considered organ-enhanced if transcript levels for that gene were more than 10-fold higher in one organ than in any other organ. Transcript ratios were calculated using the mean of three biological replicates for each organ. Data from qRT-PCR are compared with data for the same RNA samples obtained from Affymetrix Gene chips [38]. Affymetrix data were normalized using the Robust Multiarray Average (RMA) method, as described by [38], prior to calculation of ratios. Data in bold represent the lowest transcript ratio of the corresponding gene across all organs. n = not present on Affymetrix chip; a = Ct > 40 in two or three biological replicates of denominator organ; b = transcript called 'absent' by Affymetrix software in two or three biological replicates of denominator organ. L = leaf; S = stem; P = pod; F = flower; R = root; N = nodule.
Conclusion
We have established a flexible platform for high-throughput qRT-PCR analysis of Medicago TF gene expression that is based on gene-specific primers arrayed in 384-well plates and SYBR® Green detection of gene-specific PCR amplicons. Currently, the platform has primer pairs for 1045 TF genes and we have plans to extend this to all predicted Medicago TF genes as genome sequencing progresses. At this stage, the resource has been utilized by several groups in Europe, Australia, and the USA, and we expect it will become the 'gold-standard' for TF transcript profiling in Medicago truncatula.
Methods
Plant material and growth conditions
Medicago truncatula cv. Jemalong, line A17 wild type plants were vernalized for 3 days in the dark at 4°C on sterile, wet filter paper. Germinated seedlings were transferred to pots containing Turface (BWI Texarcana, Texarcana, TX). Plants were grown in growth chambers under a 16 h day and 8 h night regime, at 200 μE light intensity, 24°C and 40% relative humidity.
Vegetative organs (leaves, stems, roots, and nodules) were harvested 28 days after planting. Leaf material did not include petioles and stems did not include buds. Roots consisted of the entire root system with laterals. Several plants grown at the same time were pooled for each of the three biological replicates. Biological replicates were planted on separate days. Nodules were harvested from plants inoculated with Sinorhizobium meliloti strain 1021 one and seven days after sowing. Reproductive organs were harvested from plants that were vernalized for two weeks to decrease the time to flowering. Flowers were harvested on the day of opening. Pods were harvested from 1 to 21 days after the appearance of the floral bud to cover a wide range of developmental stages. Harvested plant material was frozen in liquid nitrogen before storage at -80°C.
RNA isolation and cDNA synthesis
Total RNA was extracted using Trizol reagent [36], following the manufacturer's instructions (Invitrogen GmbH, Karlsruhe, Germany). RNA was quantified using a Nanodrop Spectrophotometer ND-100 (NanoDrop Technologies, Wilington, DE). Sixty μg of total RNA were digested with RNase free DNase1 (Ambion Inc., Houston, TX), according to manufacturer's protocol. RNA integrity was checked using an Agilent 2100 BioAnalyser and RNA 6000 Nano Chips (Agilient Technologies, Waldbronn, Germany), and by electrophoresis on a 3% (v/w) agarose gel before and after DNase I treatment. The absence of contaminating genomic DNA after DNase I treatment was verified by PCR analysis, using primer pairs designed to amplify a 107 bp genomic fragment of the control gene, Ubiquitin (TC102473intronF, 5'-GTCCTCTAAGGTTTAATGAACCGG-3'; TC102473intronR, 5'-GAAAGACACAGCCAAGTTGCAC-3').
First-strand complementary DNA was synthesized by priming with oligo-dT12–18 (Qiagen, Hilden, Germany), using SuperScript III reverse transcriptase (Invitrogen GmbH, Karlsruhe, Germany) following the instructions of the provider. To assess cDNA synthesis efficiency, qPCR was used to amplify segments in the 5' and 3' regions of Ubiquitin cDNA approx. 1600 and 400 bp from the 3'-end, respectively (primers: TC102473_5'F, 5'-TTGGAGACGGATTCCATTGCT-3'; TC102473_5'R, 5'-GCCAATTCCTTCCCTTCGAA-3; TC102473_3'F, 5'-GGCCCTAGAACATTTCCTGTGG-3'; and TC102473_3'R, 5'-TTGGCAACCAAAATGTTCCC-3'). If ΔCt (Ct3'-Ct5') < 2, then cDNA synthesis efficiency was judged to be satisfactory, and the cDNA was considered suitable for qRT-PCR analysis.
PCR primer design
The primer design pipeline was implemented in object-oriented PERL modules supported by a MySQL relational database. Primers iterated through three phases before approval: design, specificity, and selection.
The design phase interrogated TF genes with a sliding window 250 bp across that stepped 50 bp along the entire target sequence, generating primer candidates at each window. Experimental conditions, as outlined in the Results section above, were enforced by the following MIT Primer3 parameters: PRIMER_MIN_TM 58, PRIMER_OPT_TM 60, PRIMER_MAX_TM 61, PRIMER_SELF_ANY 6, PRIMER_SELF_END 2, PRIMER_MAX_POLY_X 3, and PRIMER_PRODUCT_SIZE_RANGE '100–150' [37]. The specificity phase aligned primer candidates via WU-Blast to a database of all known Medicago sequences. The selection phase sorted primer candidates by the number of possible secondary hits, self-complementarity, and poly-X characteristics. Secondary hits were defined as specificity alignments that contained at least one of the terminal ends of the primer and achieved 80% or greater identity over the length of the primer. The sequences of each primer pair are given in Supplementary Material (see Additional file 1).
Real-time PCR conditions and analysis
PCR reactions were carried out in an ABI PRISM® 7900 HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA). SYBR® Green was used to quantify dsDNA synthesis. Reactions (5 μl total volume) were performed in an optical 384-well plate containing 2.5 μl 2 × SYBR® Green Power Master Mix reagent (Applied Biosystems, Warringen, UK), 5 ng cDNA and 200 nM of each gene-specific primer. Primer pairs were aliquoted using a pipetting robot (Evolution P3 liquid handling system, Perkin Elmer, MA, USA) to minimize pipetting errors. cDNA was aliquoted as a master mix of cDNA and 2 × SYBR® Green reagent, using an electronic Eppendorf multipipette. Reaction plates were sealed with a transparent adhesive cover before proceeding (Applied Biosystems, Foster City, CA, USA). All templates were amplified using the following standard PCR protocol: 50°C for 2 min; 95°C for 10 min; 40 cycles of 95°C for 15 sec and 60°C for 1 min, and SYBR® Green fluorescence was measured continuously. Melting curves were generated after 40 cycles by heating the sample up to 95°C for 15 sec followed by cooling down to 60°C for 15 s and heating the samples to 95°C for 15 sec.
Data analysis was performed with the SDS 2.2.1 software (Applied Biosystems). To determine the threshold cycle value (CT) for each PCR reaction, the threshold (ΔRn) was set within the logarithmic amplification phase. All amplification plots were analyzed with an ΔRn of 0.2. PCR efficiency (E) was estimated using LinReg software with data obtained from the exponential phase of each individual amplification plot and the equation (1+E) = 10slope [31]. To compare data from different PCR runs and different cDNA samples, CT values were normalized against the geometric mean of four reference genes (Ubquitin, PPRep, PDF2, and PTB), whose transcript levels were most stable across the biological samples analyzed. The average of the geometric mean of these four genes for all 18 samples was CT 21.23 ± SD1.15. For normalization, the mean reference gene CT value was substracted from the CT value of the TF gene of interest, yielding a ΔCT value. The expression ratios for the identification of organ-enhanced genes were obtained using the following formula on all 30 organ combinations: , where ΔΔCT was calculated by ΔCTA minus ΔCTb, A and B are averages of three biological replicates of the two organs being compared, and E is the PCR efficiency. Dissociation curves were analysed using SDS 2.2.1 software (Applied Biosystems). RT-PCR products were resolved on 3% (w/v) agarose gels (LE Agarose, Biozym, Oldendorf, Germany) run at 4 V cm-1 in TAE Tris-Acetate-EDTA buffer, along with a 200-bp DNA-standard ladder (Promega GmbH). A subset of 178 RT-PCR products was sequenced at the JC Venter Institute (Rockville, MD, USA).
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
KK performed the experimental work and helped draft the manuscript. W-RS, TC, and MS helped to conceive the project and provided practical advice. MW, FC, and CDT carried out bioinformatic analysis of Medicago TFs, and HW designed gene-specific primers. JCR and YX were responsible for the sequencing and analysis of the PCR products. IT carried out qRT-PCR and TG performed statistical analyses. MU designed and coordinated the project and wrote the manuscript.
Supplementary Material
Complete list of TF genes, primer sequences and corresponding PCR efficiencies. The TF primer platform was established based on the data for the gene models according to SA, specific amplification; NA, no amplification; NS, non-specific amplification. RT-PCR products of primer sequences indicated in bold were sequenced.
Complete list of TF genes, gene families and experimental data. Shown are the Medicago gene Accession Numbers (TIGR) in ascending order, the TC number if available, as well as the transcription factor family and the subfamily names. The next columns show experimental results for the real-time RT-PCR reactions performed on six different organs of Medicago in three indipendent biological replicates, as explained in the manuscript. CT = not normalized CT value, ΔCT = CT value normalized against the geometric mean of 4 house keeping genes; ΔΔCT = power(PCReff;-ΔCT); log2 ΔΔCT = logarithmus of ΔCT.
Acknowledgments
Acknowledgements
This work was supported in part by the European Commission FP6 Framework Programme Grain Legume Integrated Project (FOOD-CT-2004-506223), the Max Planck Society, the National Research Initiative (NRI) Plant Genome Program of the USDA Cooperative State Research, Education and Extension Service (CSREES), and the Samuel Roberts Noble Foundation.
Contributor Information
Klementina Kakar, Email: kakar@mpimp-golm.mpg.de.
Maren Wandrey, Email: wandrey@gfz-potsdam.de.
Tomasz Czechowski, Email: tc519@york.ac.uk.
Tanja Gaertner, Email: gaertner@mpimp-golm.mpg.de.
Wolf-Rüdiger Scheible, Email: scheible@mpimp-golm.mpg.de.
Mark Stitt, Email: stitt@mpimp-golm.mpg.de.
Ivone Torres-Jerez, Email: itjerez@noble.org.
Yongli Xiao, Email: yxiao@jcvi.org.
Julia C Redman, Email: jredman@jcvi.org.
Hank C Wu, Email: hwu@jcvi.org.
Foo Cheung, Email: FCheung@jcvi.org.
Christopher D Town, Email: cdtown@jcvi.org.
Michael K Udvardi, Email: mudvardi@noble.org.
References
- Graham PH, Vance CP. Legumes: importance and constraints to greater use. Plant Physiology. 2003;131:872–877. doi: 10.1104/pp.017004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauser L, Roussis A, Stiller J, Stougaard J. A plant regulator controlling development of symbiotic root nodules. Nature. 1999;402:191–195. doi: 10.1038/46058. [DOI] [PubMed] [Google Scholar]
- Frugier F, Poirier S, Satiat-Jeunemaitre B, Kondorosi A, Crespi M. A Kruppel-like zinc finger protein is involved in nitrogen-fixing root nodule organogenesis. Genes Dev. 2000;14:475–482. [PMC free article] [PubMed] [Google Scholar]
- Nishimura R, Ohmori M, Fujita H, Kawaguchi M. A Lotus basic leucine zipper protein with a RING-finger motif negatively regulates the developmental program of nodulation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:15206–15210. doi: 10.1073/pnas.222302699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronlund M, Gustafsen C, Roussis A, Jensen D, Nielsen LP, Marcker KA, Jensen EO. The Lotus japonicus ndx gene family is involved in nodule function and maintenance. Plant Molecular Biology. 2003;52:303–316. doi: 10.1023/A:1023967214199. [DOI] [PubMed] [Google Scholar]
- Kaló P, Gleason C, Edwards A, Marsh J, Mitra RM, Hirsch S, Jakab J, Sims S, Long SR, Rogers J, Kiss GB, Downie JA, Oldroyd GED. Nodulation signaling in legumes requires NSP2, a member of the GRAS family of transcriptional regulators. Science. 2005;308:1786–1789. doi: 10.1126/science.1110951. [DOI] [PubMed] [Google Scholar]
- Smit P, Raedts J, Portyanko V, Debelle F, Gough C, Bisseling T, Geurts R. NSP1 of the GRAS protein family is essential for rhizobial Nod factor-induced transcription. Science. 2005;308:1789–1791. doi: 10.1126/science.1111025. [DOI] [PubMed] [Google Scholar]
- Combier JP, Frugier F, de Billy F, Boualem A, El-Yahyaoui F, Moreau S, Vernie T, Ott T, Gamas P, Crespi M, Niebel A. MtHAP2-1 is a key transcriptional regulator of symbiotic nodule development regulated by microRNA169 in Medicago truncatula. Genes & Development. 2006;20:3084–3088. doi: 10.1101/gad.402806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton PH, Jakab J, Penmetsa RV, Starker CG, Doll J, Kaló P, Prabhu R, Marsh JF, Mitra RM, Kereszt A, Dudas B, VandenBosch K, Long SR, Cook DR, Kiss GB, Oldroyd GE. An ERF transcription factor in Medicago truncatula that is essential for Nod factor signal transduction. Plant Cell. 2007;19:1221–1234. doi: 10.1105/tpc.106.048264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann JL, Heard J, Martin G, Reuber L, Jiang CZ, Keddie J, Adam L, Pineda O, Ratcliffe OJ, Samaha RR, Creelman R, Pilgrim M, Broun P, Zhang JZ, Ghandehari D, Sherman BK, Yu GL. Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes. Science. 2000;290:2105–2110. doi: 10.1126/science.290.5499.2105. [DOI] [PubMed] [Google Scholar]
- Udvardi MK, Kakar K, Wandrey M, Montanari O, Murray J, Andriankaja A, Zhang JY, Benedito V, Hofer JMI, Chueng F, Town CD. Legume Transcription Factors: Global Regulators of Plant Development and Response to the Environment. Plant Physiol. 2007;144:538–549. doi: 10.1104/pp.107.098061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann JL. Transcriptional Regulation: a Genomic Overview. In: Somerville CR, Meyerowitz EM, editor. The Arabidopsis Book. The American Society of Plant Biologists, Rockville; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso JM, Ecker JR. Moving forward in reverse: genetic technologies to enable genome-wide phenomic screens in Arabidopsis. Nat Rev Genet. 2006;7:524–536. doi: 10.1038/nrg1893. [DOI] [PubMed] [Google Scholar]
- Young ND, Cannon SB, Sato S, Kim D, Cook DR, Town CD, Roe BA, Tabata S. Sequencing the genespaces of Medicago truncatula and Lotus japonicus. Plant Physiol. 2005;137:1174–1181. doi: 10.1104/pp.104.057034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Yahyaoui F, Küster H, Amor BB, Hohnjec N, Pühler A, Becker A, Gouzy J, Vernié T, Gough C, Niebel A, Godiard L, Gamas P. Expression profiling in Medicago truncatula identifies more than 750 genes differentially expressed during nodulation, including many potential regulators of the symbiotic program. Plant Physiology. 2004;136:3159–3176. doi: 10.1104/pp.104.043612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohnjec N, Vieweg MF, Pühler A, Becker A, Küster H. Overlaps in the transcriptional profiles of Medicago truncatula roots inoculated with two different Glomus fungi provide insights into the genetic program activated during arbuscular mycorrhiza. Plant Physiol. 2005;137:1283–1301. doi: 10.1104/pp.104.056572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes P, Goffard N, Weiller GF, Rolfe BG, Imin N. Transcriptional profiling of Medicago truncatula meristematic root cells. BMC Plant Biol. 2008;8:21. doi: 10.1186/1471-2229-8-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak CE, Snyder M. Global analysis of gene expression in yeast. Funct Integr Genomics. 2002;2:171– 180. doi: 10.1007/s10142-002-0065-3. [DOI] [PubMed] [Google Scholar]
- Czechowski T, Bari RP, Stitt M, Scheible WR, Udvardi MK. Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: unprecedented sensitivity reveals novel root- and shoot-specific genes. Plant Journal. 2004;38:366–379. doi: 10.1111/j.1365-313X.2004.02051.x. [DOI] [PubMed] [Google Scholar]
- Caldana C, Scheible WR, Mueller-Roeber B, Ruzicic S. A quantitative RT-PCR platform for high-throughput expression profiling of 2500 rice transcription factors. Plant Methods. 2007;3:7. doi: 10.1186/1746-4811-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheible WR, Morcuende R, Czechowski T, Fritz C, Osuna D, Palacios-Rojas N, Schindelasch D, Thimm O, Udvardi MK, Stitt M. Genome-wide reprogramming of primary and secondary metabolism, protein synthesis, cellular growth processes, and the regulatory infrastructure of Arabidopsis in response to nitrogen. Plant Physiology. 2004;136:2483–2499. doi: 10.1104/pp.104.047019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath KC, Dombrecht B, Manners JM, Schenk PM, Edgar CI, Maclean DJ, Scheible WR, Udvardi MK, Kazan K. Repressor- and activator-type ethylene response factors functioning in jasmonate signaling and disease resistance identified via a genome-wide screen of Arabidopsis transcription factor gene expression. Plant Physiol. 2005;139:949–959. doi: 10.1104/pp.105.068544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morcuende R, Bari R, Gibon Y, Zheng W, Pant BD, Bläsing O, Usadel B, Czechowski T, Udvardi MK, Stitt M, Scheible WR. Genome-wide reprogramming of metabolism and regulatory networks of Arabidopsis in response to phosphorus. Plant Cell Environ. 2007;30:85–112. doi: 10.1111/j.1365-3040.2006.01608.x. [DOI] [PubMed] [Google Scholar]
- Libault M, Wan J, Czechowski T, Udvardi M, Stacey G. Identification of 118 Arabidopsis transcription factor and 30 ubiquitin-ligase genes responding to chitin, a plant-defense elicitor. Mol Plant Microbe Interact. 2007;20:900–911. doi: 10.1094/MPMI-20-8-0900. [DOI] [PubMed] [Google Scholar]
- Guo A, He K, Liu D, Bai S, Gu X, Wei L, Luo J. DATF: a database of Arabidopsis transcription factors. Bioinformatics. 2005;21:2568–2569. doi: 10.1093/bioinformatics/bti334. [DOI] [PubMed] [Google Scholar]
- Iida K, Seki M, Sakurai T, Satou M, Akiyama K, Toyoda T, Konagaya A, Shinozaki K. RARTF: Database and tools for complete sets of Arabidopsis transcription factors. DNA Res. 2005;12:247–256. doi: 10.1093/dnares/dsi011. [DOI] [PubMed] [Google Scholar]
- InterPro: Database of protein families, domains and sites http://www.ebi.ac.uk/interpro/
- WU-BLAST: Washington University BLAST Archives http://blast.wustl.edu
- NCBI: The National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/blast
- UniProt: The Universal Protein Resource http://www.uniprot.org/
- Ramakers C, Ruijter JM, Deprez RH, Moorman AF. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett. 2003;339:62–66. doi: 10.1016/S0304-3940(02)01423-4. [DOI] [PubMed] [Google Scholar]
- Pay A, Heberle-Bors E, Hirt H. An alfalfa cDNA encodes a protein with homology to translationally controlled human tumor protein. Plant Molecular Biology. 1992;19:501–503. doi: 10.1007/BF00023399. [DOI] [PubMed] [Google Scholar]
- Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible WR. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiology. 2005;139:5–17. doi: 10.1104/pp.105.063743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- geNORM software http://medgen.ugent.be/%7Ejvdesomp/genorm
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology. 2002;3:research0034.0031–research0034.0011. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Mackey K. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. Biotechniques. 1995;19:942–945. [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer 3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editor. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Totowa, NJ , Humana Press; 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- Benedito VA, Torres-Jerez I, Murray JD, Andriankaja A, Allen S, Kakar K, Wandrey M, Verdier J, Zuber H, Ott T, Moreau S, Niebel A, Frickey T, Weiller G, He J, Dai X, Zhao PX, Tang Y, Udvard MK. A gene expression atlas of the model legume Medicago truncatula. Plant Journal. 2008;(in press) doi: 10.1111/j.1365-313X.2008.03519.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete list of TF genes, primer sequences and corresponding PCR efficiencies. The TF primer platform was established based on the data for the gene models according to SA, specific amplification; NA, no amplification; NS, non-specific amplification. RT-PCR products of primer sequences indicated in bold were sequenced.
Complete list of TF genes, gene families and experimental data. Shown are the Medicago gene Accession Numbers (TIGR) in ascending order, the TC number if available, as well as the transcription factor family and the subfamily names. The next columns show experimental results for the real-time RT-PCR reactions performed on six different organs of Medicago in three indipendent biological replicates, as explained in the manuscript. CT = not normalized CT value, ΔCT = CT value normalized against the geometric mean of 4 house keeping genes; ΔΔCT = power(PCReff;-ΔCT); log2 ΔΔCT = logarithmus of ΔCT.