

Abstract
A series of bis-(arylsulfonamide) hydroxamate inhibitors were synthesized. These compounds exhibit good potency against MMP-7 and MMP-9 depending on the nature, steric bulk and substitution pattern of the substituents in the benzene ring. In general, the preliminary structure-activity relationships (SAR) suggest that among the DAPA hydroxamates (i) electron-rich benzene rings of the sulfonamides may produce better inhibitors than electron-poor analogs. However, potential H-bond acceptors can reverse the trend depending on the isozyme; (ii) isozyme-selectivity between MMP-7 and -9 can be conferred through steric bulk and substitution pattern of the substituents in the benzene ring and (iii) the MMP-10 inhibition pattern of the compounds paralleled that for MMP-9.
Keywords: MMP inhibitors, bis(arylsulfonamide), hydroxamate
Matrix metalloproteinases (MMPs) are a family of more than 26 zinc- and calcium-dependent endopeptidases that mediate several physiological processes like tissue remodeling, angiogenesis and cell signaling, among others.1 Abnormal MMP expression has been implicated in various pathological conditions including cardiovascular diseases, cancer progression and metastasis, rheumatoid arthritis and multiple sclerosis.2
The MMP inhibitors in cancer clinical trials yielded disappointing results.3 Part of the failure have been attributed to the lack of isozyme selectivity of the inhibitors resulting in poor or indifferent clinical outcomes.3 Matrilysin (MMP-7) often is overexpressed by cancer cells4 and has been implicated to facilitate cell transformation5 and resist apoptosis.6 MMP-9 has been shown to promote tumor invasiveness and metastasis7 apart from occasional tumor-preventive roles.4,8 Isozyme-selective inhibition of MMPs can therefore potentially result in better clinical outcomes.3
Succinyl hydroxamate derivatives have been shown to be potent inhibitors of MMPs (e.g. marimastat and batimastat, Figure 1).9,10 We initiated our efforts of MMP inhibitor synthesis based on the 2,3-diaminopropanoic acid (DAPA) scaffold for the following reasons: (i) the DAPA scaffold is of comparable length to the succinate scaffold, and thus the β-substituent can potentially orient into the S1’ pocket, commonly referred to as the “selectivity pocket” of MMPs; (ii) simple synthesis protocols; (iii) a sulfonamide derivative can provide crucial H-bonding interactions to the enzyme backbone either as a donor (NH) or as an acceptor (S=O),10 either in the P1 or P1’ sites (Figure 2) and (iv) the α-substituent may contribute to increase the potency of the inhibitors.
Figure 1.
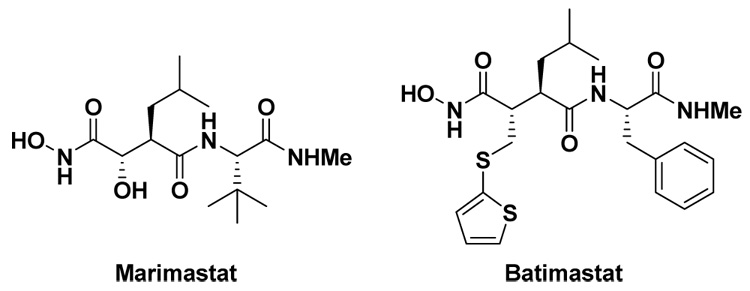
Succinyl hydroxamate derivatives.
Figure 2.
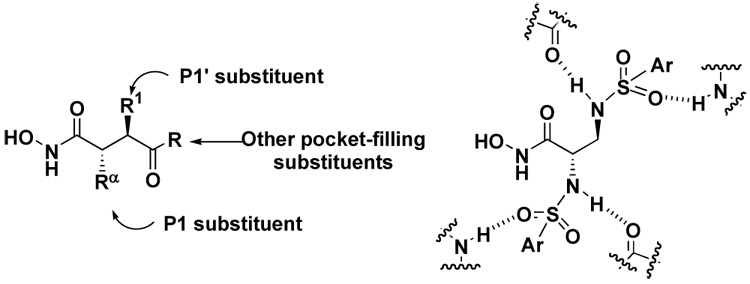
Succinyl hydroxamate derivatives versus DAPA sulfonamide hydroxamate derivatives.
The synthesis of the DAPA hydroxamates is shown in Scheme 1. Racemic 2,3-DAPA was converted to the bis-(arylsulfonamide) derivative under alkaline conditions in one step by treating with the respective aryl sulfonylchloride. The bis-(arylsulfonamide) carboxylic acid derivatives were converted to the corresponding O-benzyl hydroxamates under standard peptide coupling conditions. Removal of the benzyl group yielded the desired hydroxamates in moderate to excellent yields (Scheme 1).
Scheme 1.
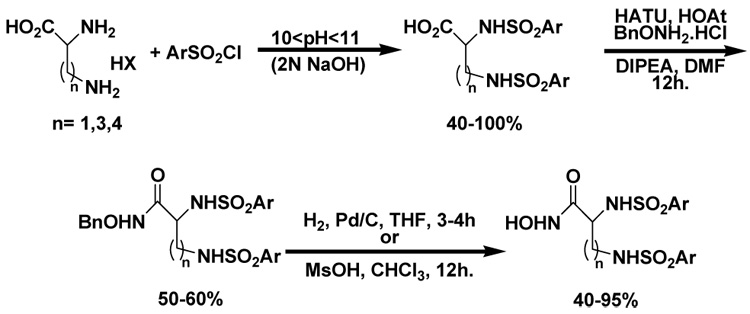
Synthesis of the bis-(arylsulfonamide) hydroxamate inhibitors.
Sulfonamide derivatives of α-amino hydroxamates have been shown to be potent inhibitors of MMPs. One of the key interactions for the potency was the α-sulfonamide interaction with the “MMP selectivity” P1’ pocket by a H-bond.10,11 Hence, bis-(arylsulfonamide) hydroxamate derivatives of the L-ornithine and L-lysine were also prepared (Scheme 1) to explore the ω-substituent interaction to the other pockets of the MMPs (P2’, P3’ etc.) while the α-substituent was expected to act as the P1’ substituent (Figure 3).
Figure 3.
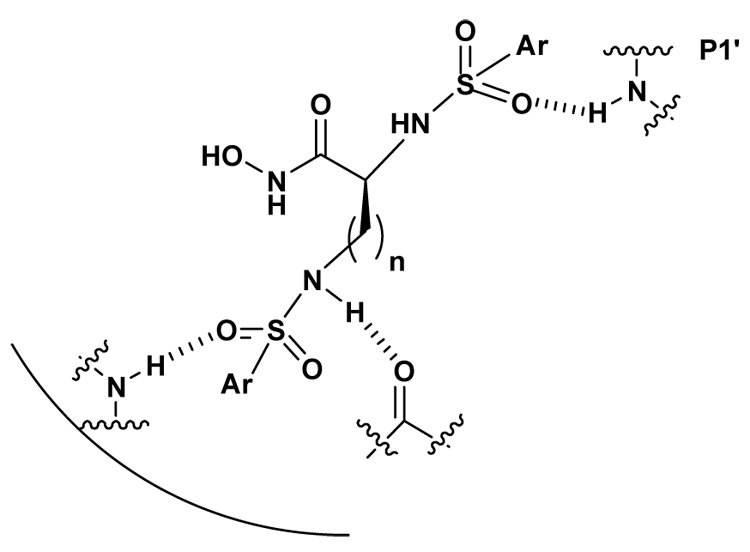
α-Sulfonamide interaction in P1'
The synthesized compounds were tested in vitro against recombinant human MMP-7, -9 and -10. The results for the DAPA derivatives are shown in Table 1. The effect of the electron withdrawing and releasing properties of the substituents on the arylsulfonamides was first studied (Compounds 1–4). It was observed that for all the MMPs studied, inhibitors with electron-rich aromatic rings (p-OMe or 2,4-di-OMe) were more potent inhibitors than electron-poor analogs (p-NO2, p-CF3 or C6F5). However, this pattern could be reversed by the presence of strong H-bonding interactions as was seen with halogenated derivatives in the case of MMP-7 (see below).
Table 1.
Inhibition constants for various DAPA bis(arylsulfonamide) hydroxamate derivatives.
| Ki Value (µM)a | |||||
|---|---|---|---|---|---|
| Compound # | Ar- | ||||
| Yield (%) | MMP-7 | MMP-9 | MMP-10 | ||
| 1 | 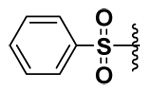 |
70 | 2.8 | 0.227 | 0.356 |
| 2 | 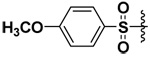 |
44 | 0.605 | 0.039 | 0.335 |
| 3 | 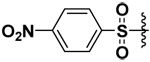 |
93 | 6.6 | 1.7 | 22 |
| 4 | 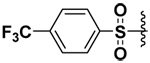 |
72 | 10 | 7.9 | 23 |
| 5 | 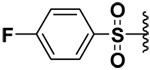 |
50 | 0.625 | 2.75 | 4.57 |
| 6 | 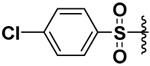 |
72 | 0.237 | 0.23 | ND |
| 7 | 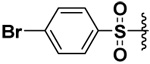 |
70 | 5.5 | 0.15 | ND |
| 8 | 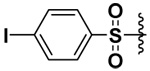 |
75 | 1.8 | 0.0026 | 1.6 |
| 9 | 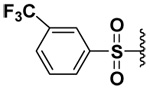 |
72 | 0.602 | NI | NI |
| 10 | 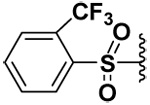 |
92 | 6.0 | NI | NI |
| 11 | 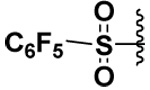 |
40 | 8.6 | 212 | NI |
| 12 | 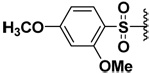 |
64 | 0.289 | 124 | 372 |
| 13 | 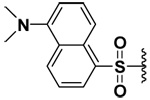 |
46 | 4.8 | 150 | ND |
NI= No inhibition ND= Not Determined.
In order to be able to make a direct comparison, the halogenated bis-(arylsulfonamide) hydroxamates were prepared (Compounds 5–8). It was observed that as the size of the halogen increased, the inhibitory potency increased for MMP-9, with the iodo derivative (compound 8) having single digit nanomolar inhibitory potency (Ki = 2.6 nM, Table 1). The opposite behavior was observed for MMP-7. The inhibitory potency of the chloro derivative 6 was similar for both the MMP-7 and -9. Thus the iodo derivative 8 exhibited a higher selectivity by at least three orders of magnitude toward MMP-9, while the fluoro derivative 5 exhibited four times selectivity for MMP-7 versus MMP-9 (Table 1).
Surprisingly compound 9 exhibited an inhibitory potency that was similar to compound 5 (sub-micromolar) for MMP-7, but did not inhibit MMP-9 or -10. Usually longer alkyl or phenylalkyl groups have been employed to achieve isozyme selectivity for MMP-9 over MMP-1 or -7.9 To the best of our knowledge, this is the first report of achieving MMP-7 selective inhibition by employing small P1’ substituents. That the inhibitory potencies of the compounds 5 and 9 were similar indicated that P1’ pocket of MMP-7 probably contained an H-bond donor with which fluorine could have favorable interaction. In compound 4, the fluorine atoms were probably forced against the wall of the P1’pocket that the fluorines are not having a favorable orientation to act as H-bond acceptors.
The presence of H-bond donors elsewhere in the P1’ MMP-7 pocket is indicated by the fact that compound 10 was a selective inhibitor for MMP-7 with low micromolar inhibitory potency, while sparing MMP-9 and -10. Also compound 11 exhibited Ki values that were comparable to compound 10 toward MMP-7 and had a better inhibitory potency against MMP-7 by at least two orders of magnitude compared to MMP-9. Compound 11 did not inhibit MMP-10.
The selectivity of an inhibitor toward a MMP could be inversed by introducing another substituent in the aromatic ring. As shown in Table 1, compound 2 had an inhibitory potency that was at least a magnitude higher for MMP-9 compared to MMP-7. Introduction of another methoxy group in the aromatic ring yielded compound 12 that had at least three orders of magnitude higher inhibitory potency toward MMP-7 as opposed to MMP-9 and -10. This selectivity is probably conferred by the H-bond donor present in the MMP-7 P1’ pocket, that interacts favorably with the o-methoxy group in the aromatic ring.
Increasing steric bulk also conferred selectivity for MMP-7. As shown in Table 1, the bis-(dansylsulfonamide) derivative had low micromolar inhibitory potency against MMP-7 compared to MMP-9.
At this point it should be noted that the synthesized bis- (arylsulfonamide) hydroxamate derivatives of DAPA were racemic compounds. It has been shown that while chirality with small α-substituents does not make any significant difference to the inhibitory potency, larger groups can cause at least one to two orders of magnitude higher inhibitory potency to one enantiomer versus the other.11 Currently, we are synthesizing the bis(arylsulfonamide) derivatives of both enantiomers of DAPA and the inhibitory potencies of these compounds will be reported in the future.
The structure-activity relationships of the L-ornithine and L-lysine derivatives were also explored toward MMP-7, -9 and -10. Based on literature reports, it was anticipated that the sulfonamide group on the α-carbon could potentially occupy the P1’ pocket10,11 and the longer methylene spacer in these derivatives could provide hydrophobic interactions with the enzyme backbone while the sulfonamide on the ω-carbon provides interaction in the other pockets (P2’, P3’ etc.) of the targeted MMPs.
However, this was not found to be the case (Table 2 and Table 3). With longer methylene spacers in the bis(arylsulfonamide) hydroxamate derivatives, the sulfonamide on the α-carbon still seemed to orient to the P1 pocket as opposed to the P1’ pocket, while the ω-sulfonamide interacted elsewhere or was oriented toward the solvent. Among the ornithine derivatives, compounds 14, 16 and 20 appeared to be selective for MMP-7 and inhibit the enzyme with low micromolar potency (Table 2). Compound 14 was particularly selective for MMP-7 while not inhibiting MMP-9 and -10. Compound 16 exhibited about 50 fold selectivity for MMP-7 compared to MMP-9 and -10. Compound 20 was more selective for MMP-7 (44x) than MMP-9 while not inhibiting MMP-10 at all. On the other hand, compound 15 did not inhibit MMP-7 and was about four times more selective toward MMP-9 than MMP-10.
Table 2.
Inhibition constants for various bis(arylsulfonamide) hydroxamate derivatives of (L)-ornithine.
| Ki Value (µM)a,b | |||||
|---|---|---|---|---|---|
| Compound # | Ar- | ||||
| Yield (%) | MMP-7 | MMP-9 | MMP-10 | ||
| 14 | 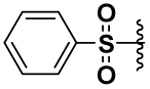 |
64 | 3 | NI | NI |
| 15 | 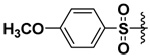 |
50 | NI | 4.2 | 16 |
| 16 | 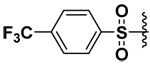 |
46 | 7 | 329 | 337 |
| 17 | 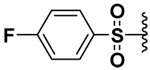 |
58 | 17%b | NI | 20%b |
| 18 | 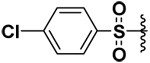 |
63 | 31%b | NI | 34.5%b |
| 19 | 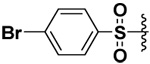 |
60 | 32%b | 9.3%b | 16.4%b |
| 20 | 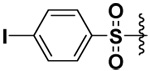 |
79 | 5 | 221 | NI |
NI= No inhibition.
% inhibition at 10 µM inhibitor concentration.
Table 3.
Inhibition constants for various bis(arylsulfonamide) hydroxamate derivatives of (L)-lysine.
| Ki Value (µM)a,b | |||||
|---|---|---|---|---|---|
| Compound # | Ar- | ||||
| Yield (%) | MMP-7 | MMP-9 | MMP-10 | ||
| 21 | 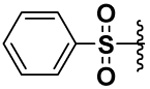 |
41 | 44b | 7.5b | 34.5b |
| 22 | 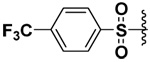 |
71 | 24b | NI | 1.8b |
| 23 | 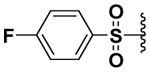 |
69 | 3.1 | NI | 27.4b |
| 24 | 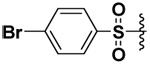 |
42 | 2.2 | NI | 34.5b |
| 25 | 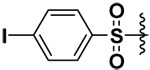 |
45 | 46%b | NI | 30.9b |
NI= No inhibition.
% inhibition at 10 μM inhibitor concentration.
The corresponding carboxylic acids were also tested for their inhibitory potency. Surprisingly, except for the DAPA derivative of bis-(4-iodobenzene sulfonamide) none of the carboxylate derivatives showed any appreciable inhibition of the MMPs studied. In fact, some of the carboxylate derivatives increased the activity for MMP-9 and/or MMP-10. The bis-(4-iodobenzene sulfonamide) DAPA derivative, however, was a potent inhibitor with submicromolar Ki values (0.327 µM, 0.936 µM and 65 µM for MMP-7, MMP-9 and MMP-10, respectively). Studies are underway to understand this inhibition pattern.
Among the lysine derivatives (Table 3), only compounds 23 and 24 inhibited MMP-7 with low micromolar Ki values while not inhibiting MMP-10 appreciably and MMP-9 only sparingly. We do not yet understand the reason for these observations and mechanistic studies are underway to gain a better understanding of the inhibition.
In conclusion, we have developed a series of bis-(arylsulfonamide) hydroxamates as novel MMP inhibitors. Preliminary data suggest that among the DAPA hydroxamates (i) electron-rich benzene rings of the sulfonamides may provide better inhibitors than electron-poor analogs. However, potential H-bond acceptors can reverse the trend depending on the isozyme; (ii) isozyme-selectivity between MMP-7 and -9 can be conferred through steric bulk and substitution pattern of the substituents in the benzene ring and (iii) the MMP-10 inhibition pattern of the compounds parallels that for MMP-9. Detailed structure-activity relationship studies involving these inhibitors and synthesis of bis-(arylsulfonamide) hydroxamate derivatives of DAPA with other groups appended on the β-carbon to access other MMP pockets are currently in progress to make more potent and isozyme-selective inhibitors for MMPs.
Supplementary Material
Supplementary data Experimental details and characterization data for the synthesis of the compounds reported in Table 1–Table 3 are available as Supplementary data. Supplementary data associated with this article can be found, in online version, at doi:
Acknowledgments
This research was supported by the NIH grant 1R01 CA113746 and NSF DMR-0705767 to SM and DKS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Recent reviews: Ra H-J, Parks WC. Matrix Biol. 2007;26:587. doi: 10.1016/j.matbio.2007.07.001.Ethell IM, Ethell DW. J. Neurosci. Res. 2007;85:2813. doi: 10.1002/jnr.21273.Ganea E, Trifan M, Laslo AC, Putina G, Cristescu C. Biochem. Soc. Trans. 2007;35:689. doi: 10.1042/BST0350689.Vincenti MP, Brinckerhoff CE. J. Cell. Physiol. 2007;213:355. doi: 10.1002/jcp.21208.Bénédicte C, Van den Steen PE, Opdenakker G. Critical Rev. Biochem. Mol. Biol. 2007;42:113. doi: 10.1080/10409230701340019.
- 2.Recent reviews: Raffett JD, Khalil RA. Biochem. Pharmacol. 2008;75:346. doi: 10.1016/j.bcp.2007.07.004.Muroski ME, Roycik MD, Newcomer RG, Van den Steen PE, Opdenakker G, Monroe HR, Sahab ZJ, Sang Q-X. Curr. Pharm. Biotech. 2008;9:34. doi: 10.2174/138920108783497631.Dorman G, Kocsis-Szommer K, Spadoni C, Ferdinandy P. Rec. Patents Cardiovascular Drug Disc. 2007;2:186. doi: 10.2174/157489007782418964.Fingleton B. Current Pharm. Design. 2007;13:333. doi: 10.2174/138161207779313551.Verma RP, Hansch C. Bioorg. Med. Chem. 2007;15:2223. doi: 10.1016/j.bmc.2007.01.011.Fingleton B. Frontiers Biosc. 2006;11:479. doi: 10.2741/1811.
- 3.Recent reviews: Hu J, Van den Steen PE, Sang Q-XA, Opdenakker G. Nat. Rev. Drug Disc. 2007;6:480. doi: 10.1038/nrd2308.Fisher JF, Mobashery S. Cancer Metastasis Rev. 2006;25:115. doi: 10.1007/s10555-006-7894-9.Overall CM, Kleifeld O. Br. J. Cancer. 2006;94:941. doi: 10.1038/sj.bjc.6603043.
- 4.Overall CM, Kleifeld O. Nat. Rev. Cancer. 2006;6:227. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- 5.Lynch CC, Vargo-Gogola T, Martin MD, Fingleton B, Crawford HC, Matrisian LM. Cancer Res. 2007;67:6760. doi: 10.1158/0008-5472.CAN-07-0026. [DOI] [PubMed] [Google Scholar]
- 6.Fingleton B, Vargo-Gogola T, Crawford HC, Matrisian LM. Neoplasia. 2001;3:459. doi: 10.1038/sj.neo.7900190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egebald M, Werb Z. Nat. Rev. Cancer. 2002;2:161. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 8.Martin MD, Matrisian LM.Cancer Metastasis Rev 200726717 and references therein. [DOI] [PubMed] [Google Scholar]
- 9.Whittaker M, Floyd CD, Brown P, Gearing AJH. Chem. Rev. 1999;99:2735. doi: 10.1021/cr0100345. [DOI] [PubMed] [Google Scholar]
- 10.(a) Winum J-Y, Scozzafava A, Montero J-L, Supuran CT. Med. Res. Rev. 2006;26:767. doi: 10.1002/med.20068. [DOI] [PubMed] [Google Scholar]; (b) Supuran CT, Casini A, Scozzafava A. Med. Res. Rev. 2003;23:535. doi: 10.1002/med.10047. [DOI] [PubMed] [Google Scholar]
- 11.(a) Yang S-M, Scannevin RH, Wang B, Burke SL, Wilson LJ, Karnachi P, Rhodes KJ, Lagu B, Murray WV. Bioorg. Med. Chem. Lett. 2008;18:1135. doi: 10.1016/j.bmcl.2007.11.119. [DOI] [PubMed] [Google Scholar]; (b) Yang S-M, Scannevin RH, Wang B, Burke SL, Huang Z, Karnachi P, Wilson LJ, Rhodes KJ, Lagu B, Murray WV. Bioorg. Med. Chem. Lett. 2008;18:1140. doi: 10.1016/j.bmcl.2007.11.129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data Experimental details and characterization data for the synthesis of the compounds reported in Table 1–Table 3 are available as Supplementary data. Supplementary data associated with this article can be found, in online version, at doi: