

Abstract
The activity of human TREX2-catalyzed 3′ → 5′-deoxyribonuclease has been analyzed in steady-state and single turnover kinetic assays and in equilibrium DNA binding studies. These kinetic data provide evidence for cooperative DNA binding within TREX2 and for coordinated catalysis between the TREX2 active sites supporting a model for communication between the protomers of a TREX2 dimer. Mobile loops positioned adjacent to the active sites provide the major DNA binding contribution and facilitate subsequent binding into the active sites. Mutations of three arginine residues on these loops cause decreased TREX2 activities by up to 60-fold. Steady-state kinetic assays of these arginine to alanine TREX2 variants result in increased Km values for DNA substrate with no effect on kcat values indicating contributions exclusively to DNA binding by all three of the loop arginines. TREX2 heterodimers were prepared to determine whether exonuclease activity in one protomer is communicated to the opposing protomer. Evidence for communication across the dimer interface is provided by the 7-fold lower catalytic activity measured in the TREX2WT/H188A heterodimer compared with the TREX2WT homodimer, contrasting the 2-fold lower activity measured in the TREX2WT/R163A,R165A,R167A heterodimer. The measured activity in TREX2WT/H188A heterodimer indicates that defective catalysis in one protomer reduces activity in the opposing protomer. A DNA binding analysis of TREX2 and the heterodimers indicates a cooperative binding effect within the TREX2 protomer. Finally, single turnover kinetic assays identify DNA binding as the rate-limiting step in TREX2 catalysis.
The 3′ → 5′-deoxyribonucleases play fundamental roles in a variety of DNA metabolic pathways. These enzymes process DNA 3′ ends by removing nucleotides one at a time to remodel 3′ termini for subsequent molecular events in cells (1, 2). TREX1 is the most active 3′-deoxyribonuclease purified from mammalian cells (3, 4). TREX2 has not been purified from cells, but recombinant TREX1 and TREX2 generated in Escherichia coli confirm the robust nature of these deoxyribonucleases with TREX1 exhibiting about a 10-fold greater activity than TREX2 (5–7). The high catalytic efficiencies measured for TREX1 and TREX2 can be attributed in part to the nature of the active sites. The TREX gene sequences and Mg2+ ion dependence suggested that these deoxyribonucleases might be members of the DnaQ-like exonuclease family that utilize a two Mg2+ ion-dependent mechanism for catalysis (8–12). The DnaQ-like family contains both deoxyribo- and riboexonucleases and has been divided into two subfamilies characterized by the presence of four conserved carboxylate residues and a histidine (DEDD-h) or a tyrosine (DEDD-y) positioned in the active site (13). The TREX enzymes prefer excision of deoxynucleotide polymers over ribonucleotide polymers by about 1000-fold (5)2 with specificity for unmodified 3′ termini (14, 15). The four carboxylate residue side chains in the DnaQ-like enzymes coordinate two divalent metal ions, and the histidine or tyrosine is positioned to deprotonate a water producing the nucleophile for attack on the target phosphate to affect bond cleavage (16–19). TREX1 and TREX2 are the only mammalian deoxyribonuclease members of the DEDDh subfamily. The structures of TREX1 and TREX2 and mutations in the active site carboxylates and histidine confirm their catalytic role (20, 21).
That TREX1 and TREX2 are homodimers was first proposed from solution studies (5–7) and was subsequently observed in the crystal structures (20–22). Dimerization of the TREX enzymes generates a central extended β-sheet across the entire molecule and positions the active sites toward opposite outer edges on the same face of the enzyme. Additional structural elements unique to TREX1 and TREX2 are found flanking the active sites and positioned near the dimer interface. The presence of two related TREX genes is only seen in mammalian genomes (23), whereas other metazoans, including amphibians and insects, have a single TREX gene. The presence of two related TREX genes in mammals and not in other species suggests the need in higher organisms for distinct dimeric 3′-deoxyribonucleases with modified unique elements to provide for specific cellular functions.
Mutations in the human TREX1 gene cause Aicardi-Goutieres syndrome mediated through immune dysfunction (24), and Trex1-/- mice exhibit an autoinflammatory phenotype proposed to be mediated through the failed processing of aberrant DNA replication intermediates resulting in chronic checkpoint activation and autoimmunity (25, 26). TREX1 is located in the endoplasmic reticulum and relocates into the nucleus along with the SET complex in response to DNA-damaging conditions. In the nucleus TREX1 is proposed to participate in a cell death pathway by acting with the NM23-H1 endonuclease to degrade the genomic DNA (27). Consistent with this view, TREX1 mutations have now been associated with several autoimmune disease phenotypes presumably resulting from failure to appropriately process DNA during replication, repair, or cell death (24, 28–32).
Proofreading of DNA synthesis by 3′-exonucleases is one of the major determinants of mutagenesis and genome stability. Cells lacking this ability show a high incidence of cancers (33, 34). Similarities to the proofreading exonucleases and the preference for frayed DNA ends lead to the proposal that the TREX enzymes might serve an extrinsic function to proofread errors generated by DNA polymerases that lack intrinsic exonuclease activities (5, 6). There is some genetic evidence for extrinsic proofreading by some exonucleases of DNA polymerases (35–37), but evidence that the TREX enzymes act as proofreaders is supported only by in vitro studies (1). Although direct genetic evidence for a TREX2 role in mammalian DNA maintenance is lacking, cells deleted of TREX2 exhibit increased levels of chromosomal rearrangements suggesting a role in genome stability (38). The severe consequences of 3′-deoxyribonuclease loss of function in cells highlight the need to understand the precise mechanisms of these exonucleases in DNA metabolic processes.
The nature of efficient catalysis at DNA 3′ termini was revealed in the TREX2 structure, and mutagenesis confirmed contributions of key residues on a disordered loop positioned adjacent the active sites (20). A flexible loop region containing three Arg residues is appropriately positioned to bind a nucleic acid polymer substrate and control entry into the active site. The sequence of the loop region in TREX2 is distinguished from that of TREX1. The kinetic studies presented here address the 3′-deoxyribonuclease action of the TREX2 enzyme by precisely quantifying contributions of key residues within TREX2 to catalysis of deoxyribonucleotide polymers. Mutagenesis studies demonstrate that each of the three arginines positioned on the α6–α7 loop, which is disordered in the crystal structure, all contribute to nucleic acid binding. Studies presented here provide evidence that there is direct communication between the TREX2 active sites within the dimer despite the considerable distance separating these sites. These data indicate that the dimer structure of the TREX enzymes has both catalytic and biologic implications.
EXPERIMENTAL PROCEDURES
Materials—The synthetic 30-mer oligonucleotide 5′-ATACGACGGTGACAGTGTTGTCAGACAGGT-3′ and the same oligonucleotide with 5′-fluorescein (FAM)3 were from Operon.
Enzyme Preparation—The plasmid constructs used to express the human TREX2 enzyme and variants in Escherichia coli and purification of the enzymes have been described (20, 29, 39). The TREX2 variant plasmid constructs were produced using a PCR site-directed mutagenesis strategy (40), and the constructs were confirmed by DNA sequencing.
Variant TREX2 heterodimers were prepared using a procedure similar to that described for preparation of TREX1 heterodimers (29). Briefly, a variant and a wild-type copy of TREX2 were cloned on separate plasmids for coexpression in E. coli. The TREX2R163A,R165A,R167A and TREX2H188A variant genes were cloned into the pLM303X plasmid lacking a polyhistidine sequence retaining the rhinovirus 3C protease recognition site between the maltose-binding protein and TREX2. The WT TREX2 gene was cloned as a fusion of the His-tagged NusA with TREX2 in the pCDFDuet-1 plasmid (Novagen). The pLM303X-TREX2 variants and pCDFDuet-1-TREX2-WT plasmids were co-expressed in E. coli BL21(DE3) Rosetta 2 cells (Novagen) resulting in the possible formation of WT/WT homodimers, WT/MUT heterodimers, and MUT/MUT homodimers. The WT/WT TREX2 homodimers and WT/MUT TREX2 heterodimers were recovered using a nickel-nitrilotriacetic acid resin (Qiagen). The WT/MUT TREX2 heterodimers were recovered by subsequent chromatography using an amylose resin. The His-tagged NusA and maltose-binding proteins were removed from the fusion by treatment with PreScission Protease™ (GE Biosciences), and the TREX2 enzyme was purified using a phosphocellulose column.
Exonuclease Assays-Time Course Reactions—The exonuclease time course reactions (210 μl) contained 20 mm Tris-HCl (pH 7.5), 5 mm MgCl2, 2 mm dithiothreitol, 100 μg/ml bovine serum albumin, 50 nm FAM-30-mer oligonucleotide, and 380 pm TREX2 protein. Incubations were at 25 °C. At the indicated times, the samples were removed, quenched by addition of 3 volumes of cold ethanol, and dried in vacuo. Pellets were resuspended in 6 μl of formamide, heated to 95 °C for 5 min, and separated on 23% denaturing polyacrylamide gels. Fluorescent-labeled bands were visualized and quantified using a Storm PhosphorImager (GE Healthcare). The amount of dNMP excised in a reaction was quantified as described (39).
Steady-state Reactions—The exonuclease reactions (10 μl) to determine Km and kcat values for the variant TREX2 proteins were as described above with the concentrations of FAM-30-mer oligonucleotide and TREX2 proteins indicated in the figure and table legends. The reactions to determine Km and kcat values for the wild-type TREX2 were 50 μl. Incubations were at 25 °C for 20 min. Reaction products were quantified to determine the amount of dNMP excised as described above. All enzyme dilutions were prepared on ice in 1 mg/ml bovine serum albumin. The steady-state kinetic data were fit to a hyperbolic equation to calculate the Michaelis-Menten kinetic parameters. The Km and kcat values and standard errors for TREX2 and variant proteins were determined by nonlinear regression using SigmaPlot 8.02 (SPSS Science, Inc.).
Equilibrium DNA Binding Analysis—The DNA binding reactions (30 μl) contained 20 mm Tris-HCl (pH 7.5), 2 mm CaCl2, 2 mm dithiothreitol, 5 mm NaCl, 1% glycerol, 10 nm FAM-30-mer plus unlabeled 30-mer to yield the indicated final oligonucleotide concentration and TREX2 protein. Fluorescence anisotropy measurements to quantify TREX2 binding to the FAM-30-mer were obtained at 25 °C from reaction samples (20 μl) in microtiter plates using a Tecan Safire2TM with fluorescence polarization module using excitation and emission wavelengths of 470 and 525 nm, respectively. The observed relative fluorescence anisotropy (relative Aobs) is presented as follows: relative Aobs = (An - Ao)/Ao, where An is the anisotropy at point n in the titration, and Ao is the anisotropy of the DNA in the absence of TREX2. The maximum relative Aobs in TREX2 titrations using 10 nm FAM-30-mer and Ca2+ is 15 ± 0.6.
A modification of the macromolecular competition titration method described by Bujalowski and co-workers (41, 42) was used to quantify TREX2 binding to the 30-mer DNA. A series of titrations was performed with increased amounts of TREX2 and 10 nm FAM-30-mer plus unlabeled 30-mer to yield the indicated final DNA concentrations. Binding of TREX2 to the unlabeled 30-mer competes with binding to the FAM-30-mer. The average binding density (BD) to the 30-mer and the concentration of free TREX2, TREX2F, were calculated using the mass conservation (Equations 1 and 2) (42, 43),
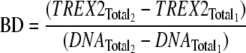 |
(Eq. 1) |
and
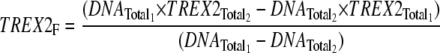 |
(Eq. 2) |
Model-independent binding isotherms were generated for TREX2 and variants by relating the dependence of the observed relative fluorescence anisotropy to the calculated BD and TREX2F from DNA binding curves measured at five different DNA concentrations between 300 and 1500 nm. The TREX2 DNA titration data were analyzed using Equations 13–15 from Jezewska et al. (44) to obtain the intrinsic binding constant K and cooperativity parameter ω for TREX2 binding to the 30-mer DNA. The binding data were fit to the indicated equations using SigmaPlot 8.02 (SPSS Science, Inc.).
Single Turnover Reactions—Single turnover kinetic assays were performed using a Biologic SFM-400 (Bio-Logic USA, LLC, Knoxville, TN) configured for quench-flow and the same reaction buffer as the steady-state experiments except the bovine serum albumin was replaced with 10% glycerol. An aliquot (75 μl) of the TREX2 solution from one syringe was mixed with the oligonucleotide substrate (75 μl) from the second syringe, and the reaction was allowed to proceed at 25 °C for time intervals ranging from 4 to 2000 ms. Reactions were quenched in 0.3 m EDTA (final concentration) by an aliquot (75 μl) from a third syringe. A portion of the quenched reaction mixture (10 μl) was dried in vacuo, resuspended in 8 μl of formamide, and processed as described above to quantify the products. The amount of 30-mer was plotted against time and fit by nonlinear regression using SigmaPlot 8.0 to the single exponential equation: [30-mer] = A·e-kobs·t + C, where A is the amplitude; t is the time; C is the final [30-mer], and kobs is the observed TREX2 exonuclease rate.
Protein and Nucleic Acid Concentrations—The proteins and oligonucleotides were quantified by A280 and A260, respectively. Protein and DNA concentrations were determined using the web tools available from ExPAsy (45) and OligoCalc, respectively. TREX2 concentrations are expressed as protomer, and oligomer concentrations are expressed as 3′ termini.
RESULTS
Catalytic Efficiency of TREX2—Three arginines located at residue 163, 165, and 167 on the α6-α7 loops positioned adjacent to the active sites contribute to TREX2 catalytic efficiency. Our previous studies demonstrate the high catalytic efficiencies of TREX1 and TREX2, with about a 10-fold lower efficiency for TREX2 relative to that of TREX1 (23). These data indicated that this 10-fold lower efficiency of TREX2 could be largely attributed to differences in DNA binding. Mutation of all three arginines to alanine results in a dramatically decreased catalytic efficiency of the TREX2 enzyme, almost exclusively mediated through an increased Km value suggesting a DNA binding contribution by one or all three of these arginines (20). To identify contributions to exonuclease activity by the Arg-163, Arg-165, and Arg-167, a series of TREX2 variant enzymes was generated in which each of these residues was changed to alanine individually or in combinations, and the variant TREX2 enzymes were expressed and purified (supplemental Fig. 1). Interestingly, we were not able to express and purify a TREX2R165A enzyme indicating considerable instability in this variant TREX2 protein. The TREX2H160A enzyme was also prepared to test possible contributions to activity by the histidine located at position 160 on the TREX2 α6-α7 loop. The TREX2 and variant proteins were tested for 3′ → 5′-deoxyribonuclease activity in time course reactions using a 30-mer oligonucleotide to confirm the presence of exonuclease activity in each of the TREX2 proteins and to establish the relative 3′-nucleotide excision activities for the variant TREX2 enzymes. In separate reactions TREX2 or variant enzyme was incubated with the ssDNA oligonucleotide; samples were removed at the indicated times, and products were quantified after separation on urea-polyacrylamide gels (Fig. 1 and supplemental Fig. 2). These data showed reduced excision activities for all of the TREX2 arginine to alanine variant proteins relative to the wild-type TREX2. The excision rates of the single Arg TREX2 variants were reduced 2–3-fold (TREX2R163A = 1.3 nm dNMP/s/nm enzyme and TREX2R167A = 0.94 nm dNMP/s/nm enzyme) relative to the TREX2WT rate of 2.8 nm dNMP/s/nm enzyme. The excision rates of the double Arg TREX2 variants were reduced further by 11–18-fold (TREX2R163A,R165A = 0.24 nm dNMP/s/nm enzyme, TREX2R163A,R167A = 0.17 nm dNMP/s/nm enzyme, and TREX2R165A,R167A = 0.16 nm dNMP/s/nm enzyme) relative to the TREX2WT. Finally, the excision rate of the triple Arg TREX2 variant was reduced 60-fold (TREX2R163A,R165A,R167A = 0.048 nm dNMP/s/nm-enzyme) relative to the TREX2WT. In contrast to the loop-arginine variants, the TREX2H160A enzyme activity of 2.6 nm dNMP/s/nm enzyme was indistinguishable from the wild-type TREX2 enzyme activity indicating that the histidine at position 160 does not participate directly in catalysis using an ssDNA oligonucleotide substrate.
FIGURE 1.

The 3′ → 5′-deoxyribonuclease activities of TREX2 and variants. Time course reactions (210 μl) were prepared containing 50 nm FAM-labeled ssDNA 30-mer oligonucleotide and 380 pm TREX2WT (•), TREX2H160A (⋄), TREX2R163A (○), TREX2R167A (▾), TREX2R163A,R165A (▿), TREX2R163A,R167A (▪), TREX2R165A,R167A (□), or TREX2R163A,R165A,R167A (♦). Samples (30 μl) were removed after incubation at 25 °C for the indicated times. Reaction products were subjected to electrophoresis on 23% polyacrylamide gels (see supplemental Fig. 2). The products were quantified, and the amounts of dNMP excised were determined as described under “Experimental Procedures.”
Steady-state Kinetics—Mutation of any one of the three Arg residues on the TREX2 α6-α7 loop reduces the nucleic acid binding capacity with little effect on catalysis of 3′-nucleotides. A steady-state kinetic analysis of the TREX2R163A, TREX2R167A, TREX2R163A,R165A, TREX2R163A,R167A, TREX2R165A,R167A, and TREX2R163A,R165A,R167A variants was performed and compared with the wild-type TREX2 protein to identify the nature of the reduced catalytic activity in the variant TREX2 enzymes (Table 1 and supplemental Fig. 3). These data show that the Km values of the variant TREX2 enzymes are increased for all of the Arg to Ala mutations. Measured Km values ranged from 23,000 nm for the TREX2R163A,R165A,R167A triple mutant using the 30-mer to Km values of ∼5000 nm for the arginine double mutants to Km values of ∼1000 nm for the arginine single mutants compared with a Km value of 97 nm for wild-type TREX2. In contrast, the kcat values of 12–20 s-1 were similar for each of the α6-α7 loop Arg to Ala variant TREX2 enzymes. Similar kinetic values were obtained using a (dT)20-mer oligonucleotide substrate demonstrating the lack of effect of nucleotide sequence context on the TREX2 excision activities (data not shown). These increased Km values for the variant TREX2 enzymes relative to the Km value of 97 nm for the wild-type TREX2 enzyme and the lack of effect on the measured kcat values indicate a reduced DNA binding affinity for the α6-α7 loop variants and provide direct evidence for the contribution of the Arg-163, Arg-165, and Arg-167 in the reaction mechanism of 3′-nucleotide excision by TREX2.
TABLE 1.
Steady-state kinetics of TREX2 and variants
| Kma | kcata | kcat/Km | Relative efficiencyb | |
|---|---|---|---|---|
| m (×10–9) | s–1 | m–1 s–1 | ||
| TREX2wt | 97 ± 18 | 12 ± 0.92 | 1.2 × 108 | 1 |
| R163A | 870 ± 91 | 16 ± 0.56 | 1.8 × 107 | 1/6.7 |
| R167A | 1400 ± 83 | 20 ± 0.46 | 1.4 × 107 | 1/8.6 |
| R163A,R165A | 3900 ± 810 | 14 ± 1.2 | 3.6 × 106 | 1/33 |
| R163A,R167A | 5200 ± 590 | 12 ± 0.64 | 2.3 × 106 | 1/52 |
| R165A,R167A | 7600 ± 1800 | 19 ± 2.6 | 2.5 × 106 | 1/48 |
| R163A,R165A,R167A | 23,000 ± 2400 | 14 ± 0.59 | 6.1 × 105 | 1/200 |
Kinetic constants were derived from reactions in supplemental Fig. 3
Relative efficiency is given as m–1 s–1 TREX2 variant/m–1 s–1 TREX2wt
Interdependence of Activities in the TREX2 Dimer—Efficient catalysis in one TREX2 protomer is dependent upon catalysis in the other protomer. The positions of the active sites on opposite sides of the dimeric molecule suggest the possible independent actions of each TREX2 protomer. However, the extensive network of conserved contacts at the dimer interface in TREX enzymes indicates the importance of the dimeric structure to biological function and the possible interdependence of protomers A and B within the TREX2 dimer on activity (20). To address the interdependence of TREX2 activities within the dimeric molecule, a strategy was developed to prepare TREX2 heterodimers containing a TREX2WT protomer and a variant TREX2 protomer within the same TREX2 dimer. Co-expression of WT and variant TREX2 plasmid constructs and subsequent purification of the TREX2 dimers containing two different affinity tags resulted in the isolation of TREX2 heterodimer enzymes. The TREX2WT/R163A,R165A,R167A heterodimer was prepared to test the effects of defective DNA binding in the α6-α7 loop in protomer A on the WT activity of TREX2 in protomer B within the same dimer. In addition, the TREX2WT/H188A heterodimer was prepared to test the effects of defective catalysis at the active site in protomer A on the WT activity of TREX2 in protomer B within the same dimer. The activity of the TREX2R163A,R165A,R167A homodimer is reduced by ∼60-fold (greater than 98% under our standard exonuclease assay conditions) relative to the TREX2WT homodimer (Fig. 1), and the activity of the TREX2H188A homodimer is reduced by ∼100% relative to the wild-type TREX2 homodimer (20). Therefore, we reasoned that if the presence of the TREX2R163A,R165A,R167A and TREX2H188A variants as protomer A does not affect the activity of the TREX2WT as protomer B, the predicted reduction of TREX2 activity would be 2-fold in the heterodimers. However, if the mutation in protomer A impacts the activity of protomer B, the overall activity of the dimer would be reduced by a factor greater than 2-fold.
Time course reactions using the TREX2 heterodimers were performed with the ssDNA oligonucleotide, and the products were quantified after separation on urea-polyacrylamide gels (Fig. 2). These data showed reduced excision activities for the TREX2WT/R163A,R165A,R167A and for the TREX2WT/H188A heterodimers. The TREX2WT/R163A,R165A,R167A heterodimer activity was reduced 2.4-fold (TREX2WT/R163A,R165A,R167A = 1.6 nm dNMP/s/nm-enzyme) relative to the TREX2WT rate of 4.0 nm dNMP/s/nm enzyme. The ∼2-fold reduction in the TREX2WT/R163A,R165A,R167A heterodimer suggests that mutations in the DNA binding loop of one TREX2 protomer have little effect on catalysis in the wild-type TREX2 protomer within the TREX2 dimer. In contrast, the TREX2WT/H188A heterodimer activity was reduced to a greater extent of 6.8-fold (TREX2WT/H188A = 0.59 nm dNMP/s/nm enzyme) relative to the TREX2WT enzyme. The greater than 2-fold reduction in overall activity in the TREX2WT/H188A heterodimer provides direct evidence for the interdependence of the TREX2 protomers within the dimer.
FIGURE 2.

Interdependence of activities in the TREX2 dimer. Time course reactions (210 μl) were prepared containing 50 nm FAM-labeled ssDNA 30-mer oligonucleotide and 380 pm TREX2WT (•), TREX2WT/R163A,R165A,R167A (○), TREX2WT/H188A (▾), or TREX2R163A,R165A,R167A/R163A,R165A,R167A (▿). Samples (30 μl) were removed after incubation at 25 °C for the indicated times. Reaction products were subjected to electrophoresis on 23% polyacrylamide gels (A). The products were quantified, and the amounts of dNMP excised (B) were determined as described under “Experimental Procedures.”
DNA Binding by TREX2—Equilibrium DNA binding analysis reveals a positive cooperativity effect in the TREX2 DNA binding mechanism with the α6-α7 loop arginines providing the principle binding contribution. A series of equilibrium binding studies was performed using the 30-mer oligonucleotide to explore the mechanism in TREX2 DNA binding. Our previous studies demonstrated the absolute requirement for an activating divalent metal in TREX2 catalysis with preference for Mg2+ over Mn2+ and the lack of exonuclease activation in the presence of Zn2+ or Ca2+ (7).3 Binding reactions containing the 30-mer DNA were performed with increased TREX2 concentrations in the absence of divalent metal to prevent degradation of the DNA, and binding was quantified using the observed increase in anisotropy of the FAM-labeled DNA (Fig. 3A). An increase in the observed relative anisotropy (Aobs) is detected in the presence of increased amounts of TREX2 using 10 and 30 nm 30-mer DNA with a maximum increase in the relative Aobs of ∼4-fold demonstrating that TREX2 binds DNA in the absence of an activating divalent cation. The TREX2 DNA binding curves generated in the absence of divalent metal are sigmoidal suggesting cooperativity in the TREX2 dimer association with DNA. Addition of the nonactivating divalent cation Ca2+ to the TREX2 DNA binding reactions results in an additional 4-fold increase in the maximum relative Aobs indicating a greater stability in the TREX2-DNA interaction. Titrations of TREX2 with the 30-mer DNA were performed in reactions containing Ca2+ over a broad range of DNA concentrations from 10 to 1000 nm to identify appropriate DNA concentrations that generate binding isotherms with well separated curves (Fig. 3B). This result shows that TREX2 titrations at DNA concentrations greater than 300 nm produce binding curves appropriate for rigorous quantification of TREX2 DNA binding. Finally, we compared TREX2WT association with 30-mer DNA in the presence of Ca2+ with TREX2H188A homodimer variant in the presence of Mg2+ to determine how closely Ca2+-mediated TREX2 DNA binding recapitulates Mg2+-mediated TREX2 DNA binding (Fig. 3C). Our previous TREX2 structural studies show that the His-188 residue is positioned to deprotonate a water molecule and does not participate directly in divalent metal ion binding (20). The Ca2+-mediated TREX2WT DNA binding is similar to that of Mg2+-mediated TREX2H188A DNA binding supporting the use of Ca2+ in these binding studies. These titrations establish the appropriate conditions and TREX2 ligand and DNA concentrations for generating model-independent DNA binding isotherms. The model-independent approach described under “Experimental Procedures” was used to quantify TREX2-DNA binding independent of assumptions relating the specific proportions of TREX2-DNA complexes to the observed anisotropy (42).
FIGURE 3.

Fluorescence anisotropy of TREX2 binding to an ssDNA 30-mer. Equilibrium DNA binding reactions (30 μl) were prepared containing the indicated concentration of FAM-labeled ssDNA 30-mer oligonucleotide (A and B) or at 300 nm (C) and the indicated TREX2WT (A–C, closed circles) or TREX2H188A (C, open circles). The reactions contained no divalent metal ions (A), 2 mm CaCl2 (B and C, closed circles), or 5 mm MgCl2 (C, open circles). The observed fluorescence anisotropy Aobs were obtained as described under “Experimental Procedures.” The best fit line in A has no theoretical basis. The lines in B and C were generated by computer fit of the data using the Hill equation. The Hill coefficient is 1.5 (B and C) indicating positive cooperativity in TREX2 DNA binding.
The equilibrium DNA binding analysis identifies a cooperativity effect in the association of TREX2 with DNA resulting from binding to the DNA within the TREX2 protomer. Our previous results (20) and the steady-state analysis above indicate that the TREX2R163A,R165A,R167A homodimer is dramatically impaired in DNA binding, and the TREX2H188A is catalytically deficient. The TREX2WT/R163A,R165A,R167A and TREX2WT/H188A heterodimers were used in equilibrium binding titrations to distinguish cooperative binding of DNA within the TREX2 protomer from possible cooperative binding effects transmitted across the dimer interface. Fluorescence anisotropy titration curves were first generated at five different DNA concentrations to quantify DNA binding in the TREX2WT homodimer (Fig. 4A). As expected, at each successively higher DNA concentration, the amount of TREX2 required to generate the same relative fluorescence anisotropy increase Aobs is also increased. The higher amount of TREX2 is required to bind the increased 30-mer DNA generating equivalent Aobs at the same calculated free TREX2 concentration, TREX2F, demonstrating the general principle of the direct relationship between protein binding density and free protein concentration in nucleic acid association (42, 43, 46).
FIGURE 4.

Fluorescence anisotropy of TREX2 and TREX2 heterodimer variants. Equilibrium DNA binding reactions (30 μl) were prepared containing the indicated concentration of FAM-labeled ssDNA 30-mer oligonucleotide and the indicated TREX2WT (A), TREX2WT/R163A,R165A,R167A (B), or TREX2WT/H188A (C) in the presence of 2 mm CaCl2. The observed fluorescence anisotropy Aobs were obtained as described under “Experimental Procedures.” The lines were generated by computer fit of the data using the Hill equation. The Hill coefficient for TREX2WT (A) and TREX2WT/H188A (C) is 1.4 and for TREX2WT/R163A,R165A,R167A (B) is 1.9 confirming positive cooperativity within the TREX2 protomer.
The equilibrium binding analysis of TREX2WT/R163A,R165A,R167A heterodimer binding to the 30-mer DNA indicates that the cooperativity observed in the TREX2 dimer results from DNA binding effects from within the protomer. As observed with TREX2WT, the TREX2WT/R163A,R165A,R167A heterodimer binding curves are sigmoidal indicating that the cooperative binding effect results from DNA contacts within the active protomer of the heterodimer (Fig. 4B). Furthermore, the maximum DNA binding, as reflected in the Aobs, is approximately half that observed using the TREX2WT enzyme supporting the principal role of the three loop arginines in TREX2 DNA binding. The results of the equilibrium binding analysis using the TREX2WT/R163A,R165A,R167A heterodimer contrast those using the TREX2WT/H188A heterodimer, which demonstrate binding properties indistinguishable from the TREX2WT homodimer (Fig. 4C). These data show that the three arginines positioned on the TREX2 α6-α7 loop are the predominant residues in the TREX2 30-mer DNA binding process.
The titration curves shown in Fig. 4 were used to calculate the binding densities and the concentrations of free TREX2 in the binding reactions. The TREX2 DNA binding isotherms relating the binding densities to the free enzyme concentrations for TREX2WT, TREX2WT/R163A,R165A,R167A, and TREX2WT/H188A are shown in Fig. 5. The anisotropy data for the TREX2WT and the TREX2WT/H188A heterodimer indicate very similar DNA binding properties, contrasting the anisotropy detected upon binding of the TREX2WT/R163A,R165A,R167A heterodimer to the 30-mer DNA (Fig. 5A). A plot of the dependence of the observed increase in anisotropy Aobs as a function of the binding density for TREX2WT and the two heterodimer variants is shown in Fig. 5B. For the TREX2WT and TREX2WT/H188A heterodimer, we calculate three TREX2 dimers bind to the 30-mer DNA at saturation of the protein-nucleic acid complex. Because TREX2 is a dimer, two 30-mer DNA oligomers bind to each TREX2 dimer to generate the maximum observed anisotropy that is approximately twice that observed for the TREX2WT/R163A,R165A,R167A heterodimer (Fig. 5A). These data indicate a DNA-binding site size of approximately 10 nucleotides for TREX2. In addition, these binding data indicate a nonlinear relationship with two binding phases. In the first binding phase the observed anisotropy increases to ∼7 corresponding to a BD of 2.3 for the TREX2WT and TREX2WT/H188A heterodimer. In the first binding phase for the TREX2WT/R163A,R165A,R167A heterodimer, the observed anisotropy increases to ∼2.5 corresponding to a BD of 1.7. In the second binding phase the observed anisotropy increases to ∼14 corresponding to a BD of 7.0 for the TREX2WT and TREX2WT/H188A heterodimer, and the observed anisotropy increases to ∼5.5 corresponding to a BD of 3.7 for the TREX2WT/R163A,R165A,R167A heterodimer. Together these data indicate that TREX2 binding in the first phase to the 30-mer DNA generates a greater change in the observed anisotropy than detected in the second binding phase. These data likely reflect the initial TREX2 DNA binding by the three loop arginines and subsequent binding of the 3′ terminus into the active site within a protomer in the first phase. Additional TREX2 dimers bind to the 30-mer by the three loop arginines in the second phase.
FIGURE 5.

Binding isotherms of TREX2 and variants. The concentrations of free TREX2 and TREX2 binding density were calculated in DNA binding reactions from the TREX2 titrations in Fig. 4 and analyzed as described under “Experimental Procedures.” Plots relating the observed anisotropy Aobs to the free TREX2 (A) are fits of the TREX2 cooperative binding to the 30-mer DNA using a binding site size n = 10 nucleotides, an intrinsic binding constant K = 1.8 × 104 m-1, and a cooperativity parameter ω = 1.0 × 103. Plots relating the observed anisotropy Aobs to the calculated binding density (B) show the two TREX2 binding phases. In the first binding phase the observed anisotropy increases to ∼7 for TREX2WT and TREX2WT/H188A heterodimer and to ∼2.5 for TREX2WT/R163A,R165A,R167A heterodimer. In the second binding phase the observed anisotropy increases to ∼14 for the TREX2WT and TREX2WT/H188A heterodimer and to ∼5.5 for the TREX2WT/R163A,R165A,R167A heterodimer.
Single Turnover Kinetic Analysis of TREX2—A single turnover kinetic analysis shows that DNA binding is rate-limiting in the TREX2-catalyzed reaction. The native TREX2 structure modeled with DNA bound indicates that Arg-152 and Asn-118 in addition to the metal ion-coordinating interactions of Asp-14, Glu-16, Asp-123, and Asp-193 provide the major electrostatic interactions with a DNA substrate within the 3′-exonuclease active site (20). Our recent TREX2 structure with bound DNA confirms these interactions.4 Together, our available structural and kinetic data support a model where the TREX2 Arg-163, Arg-165, and Arg-167 on the α6-α7 loop bind first to the DNA backbone and subsequently position the DNA 3′ end into the active site. This model predicts that DNA binding might be rate-limiting in the TREX2-catalyzed reaction, and we performed single turnover reactions comparing TREX2WT with TREX2R163A,R165A,R167A variant to test this possibility. We first determined the appropriate TREX2 and DNA concentrations that establish pseudo-first order reaction conditions by incubating a single amount of TREX2 with four different concentrations of the 30-mer DNA substrate (Fig. 6A) and by incubating four different amounts of TREX2 with a single concentration of the 30-mer DNA substrate (Fig. 6B). The TREX2 was in excess (2.5–10-fold) over DNA substrate in all reactions. The observed excision rate (kobs) for TREX2WT was determined in these time course experiments by quantifying the oligonucleotide products and plotting the disappearance of full-length 30-mer DNA substrate. The data show that disappearance of the 30-mer DNA corresponds closely to a single exponential fit. Furthermore, these results demonstrate that the TREX2WT kobs = 63 s-1 is independent of the DNA substrate concentrations that were tested (Fig. 6C) and that the kobs is directly related to the concentration of TREX2WT used in these reactions (Fig. 6D). These data support a TREX2 mechanism in which DNA binding is the rate-limiting step.
FIGURE 6.

Single turnover kinetics of TREX2 using an ssDNA 30-mer. The TREX2 was mixed with FAM-labeled ssDNA 30-mer oligonucleotide to yield the indicated final enzyme and DNA concentrations and quenched at the times indicated (A and B). The oligonucleotide products were quantified, and the amount of 30-mer remaining in the time course reaction is shown (A and B). The curves of the loss of the 30-mer in the time course reactions were generated by fitting the data to a single exponential equation as described under “Experimental Procedures.” A plot of the TREX2 kobs at different DNA concentrations (C) and using different TREX2 concentrations (D) supports a mechanism in which DNA binding is rate-limiting in TREX2 catalysis.
Single turnover analysis of TREX2R163A/R165A/R167A indicates that binding of DNA by the α6-α7 loop constitutes a binding region distinct from that of the active site, thus identifying two DNA-binding sites in each TREX2 protomer. Like TREX2WT, single turnover reactions using TREX2R163A,R165A,R167A show that the kobs = 2.5 s-1 is independent of the DNA substrate concentration tested and that the kobs is directly related to the concentration of enzyme used in the reactions (data not shown). A direct comparison of TREX2WT with TREX2R163A,R165A,R167A excision under single turnover conditions is shown in Fig. 7. The time course for TREX2R163A,R165A,R167A excision was extended relative to that of TREX2WT, and the excision products were separated and quantified for comparison. Importantly, despite elimination of all three arginines on the principal DNA binding loops, the TREX2R163A,R165A,R167A enzyme retains considerable catalytic function at a reduced rate demonstrating binding of the 30-mer DNA substrate into the active site and subsequent catalysis. These data indicate the presence of two distinct DNA-binding sites in each TREX2 protomer.
FIGURE 7.

Comparison of TREX2WT and TREX2R163A,R165A,R167A single turnover kinetics. The TREX2WT (○) or TREX2R163A,R165A,R167A (•) (2000 nm) was mixed with FAM-labeled ssDNA 30-mer oligonucleotide (200 nm) and quenched at the times indicated. The oligonucleotide products were subjected to electrophoresis on 23% polyacrylamide gels (upper panels) and quantified. The amounts of 30-mer remaining at the sampled times are plotted. The curves of the loss of the 30-mer in the time course reactions were generated by fitting the data to a single exponential equation yielding the TREX2WT kobs = 63 ± 4.7 s-1 and TREX2R163A,R165A,R167A kobs = 2.5 ± 0.30 s-1. The amplitudes for the TREX2WT and TREX2R163A,R165A,R167A reactions were 180 and 130 nm, respectively.
DISCUSSION
The TREX2 protein is a dimer consisting of two identical protomers. Our previous studies used a sensitive biochemical assay to confirm the catalytically robust activity of this dimeric enzyme (7, 39). The catalytic residues responsible for phosphodiester bond cleavage were identified (20), but how precisely TREX2 achieves the measured high catalytic efficiency has remained obscure. The studies presented here show that TREX2 combines the efficient hydrolytic chemistry of the active sites with a DNA binding mechanism that includes two distinct regions in each of the TREX2 protomers of the dimer. In addition, there is communication across the dimer interface demonstrating the dependence of the dimeric structure for coordination of exonuclease action between the distantly located active sites contributing to the high catalytically efficiency of the enzyme.
Specifically targeted variant enzymes were prepared to address functional requirements for high catalytic efficiency in the TREX2 dimer. All of the TREX2 enzymes in which Arg residues on the α6-α7 loop were changed to Ala clearly show decreased exonuclease activities (Fig. 1 and supplemental Fig. 2), and a steady-state kinetic analysis shows that the diminished catalytic efficiency in these enzymes is exhibited through altered DNA binding capacity as indicated by the increased Km values (Table 1 and supplemental Fig. 3). Thus, our data show that Arg-163, Arg-165, and Arg-167 each contribute to DNA binding and high catalytic efficiency in TREX2. These data are in conflict with a previously published report that failed to detect a contribution in TREX2 DNA binding by Arg-163 (48). This apparent discrepancy might result from the different methodologies and reaction conditions used to measure DNA binding in these studies. Our data suggest that the three positively charged arginine residues on the flexible loops positioned adjacent to the active sites bind DNA first, most likely to the negatively charged phosphodiester backbone, but interactions with the bases cannot be ruled out from the current data. The flexible nature of the binding loops could provide a mechanism whereby TREX2 scans the DNA phosphodiester backbone facilitated by the Arg-163, Arg-165, and Arg-167 contacts to identify polynucleotide discontinuities and to locate available 3′ termini. The DNA binding loop amino acid sequences of TREX2 are highly conserved from various species and unique when compared with the DNA binding loop sequences of TREX1 (20, 21). This unique TREX2 DNA binding loop region appears to be ideally positioned on the exterior surface of each protomer to facilitate the initial DNA binding event leading to a “scanning-like” process by which TREX2 identifies available 3′ termini. Such a binding mechanism would be analogous to that of other DNA processing enzymes that first contact DNA nonspecifically and are then directed to more sequence- or structure-specific enzyme-DNA interactions by a diffusion mechanism as has been described for many enzymes that utilize polymers such as DNA as substrates (17, 49–52). TREX2 differs from many DNA-binding enzymes in that there is no apparent DNA-binding groove or closed structure in the protein to contribute to DNA binding. Thus, TREX2 likely represents the class of proteins that initially contact DNA nonspecifically, and upon encountering the appropriate structure (i.e. an available 3′ terminus for TREX2), the disordered loop region undergoes a folding transition to facilitate the DNA structure-specific binding event and subsequent catalysis (53). This partial separation of the DNA binding and exonuclease activities within TREX2 was also suggested in a previous study (48).
The equilibrium binding study presented here provides new insights into the precise binding mechanism of TREX2 to ssDNA polynucleotides, and a model is proposed to describe this binding process (Fig. 8). There are two regions on each of the TREX2 protomers that contribute to DNA binding in the dimer, the arginine-containing α6-α7 loops and the four active site DEDD carboxylate residues positioned to coordinate two Mg2+ ions in the active sites for catalysis. A major challenge in these binding studies was to identify appropriate conditions to measure TREX2 DNA binding contributions by these two regions while preventing catalytic degradation of the DNA. To perform DNA binding studies under conditions that we previously showed optimal for catalysis, two important considerations were made. First, our TREX2 DNA binding analysis was performed at low monovalent salt concentration to optimize the TREX2-DNA interaction that is mostly governed by the arginines on the TREX2 loop regions. DNA-binding proteins in general are known to be affected by monovalent salt concentrations, and the driving forces behind these effects are complex (54). Our previous data show that TREX2 exonuclease activity is sensitive to monovalent salts above 50 mm (7). Second, these DNA binding studies were performed in the presence of the divalent ion Ca2+, which provides the appropriate active site coordination chemistry without activating the TREX2 exonuclease (Fig. 3C). This was demonstrated in binding studies using a TREX2 variant in which the active site His-188 was mutated to Ala to prevent generation of the nucleophile but to allow the Mg2+-mediated DNA binding within the active site. Our studies show that maximal TREX2 DNA binding efficiency is achieved at monovalent salt concentrations below 50 mm (data not shown) and in the presence of a divalent ion such as Mg2+ or Ca2+ (Fig. 3) consistent with our previous TREX2 exonuclease activity analysis (7). Thus, our systematic approach identified appropriate conditions to maximize TREX2 DNA binding. Additional analyses are required to identify the nature of the TREX2 salt sensitivity with respect to activity and DNA binding.
FIGURE 8.

Model of TREX2 binding to ssDNA. The data presented in this study indicate that TREX2 binds first nonspecifically to the DNA through contacts between the α6-α7 loop arginine residues and the DNA phosphodiester backbone (A). This initial binding would allow TREX2 to “scan” the DNA backbone to identify 3′ termini. Upon identification of an available 3′ terminus the DNA is positioned into the active site for potential 3′-exonuclease action (B). Increased amounts of TREX2 result in increased TREX2 binding via the arginine loop residues until saturation of the 30-mer DNA with one TREX2 bound to approximately 10 nucleotides of ssDNA (C). The second protomer in the TREX2WT dimer is accessible for binding to a second ssDNA 30-mer in contrast to the TREX2WT/R163A,R165A,R167A heterodimer, which accounts for the approximately half-maximal observed anisotropy Aobs measured using the TREX2WT/R163A,R165A,R167A heterodimer relative to the TREX2WT dimer (see Fig. 4).
A model is proposed for TREX2 DNA binding of an ssDNA polynucleotide (Fig. 8). Our finding that multiple TREX2 dimers bind to an ssDNA polymer provides a framework to understand the potential mechanism of how TREX2 identifies 3′ termini in DNA. TREX2 likely first contacts the DNA phosphodiester backbone using the α6-α7 loop arginines and slides freely along the DNA sampling for available 3′ termini (Fig. 8). Titrations of increased concentrations of TREX2 using the 30-mer ssDNA oligonucleotide reveal that up to three TREX2 dimers bind to the polynucleotide allowing one to calculate a 10-nucleotide DNA binding site-size for TREX2. The method employed here to derive a quantitative measure of the TREX2-binding site size has been used previously to examine binding to a polynucleotide lattice (47) and has provided considerable insight into the mechanisms of protein-nucleic acid interactions (42, 43). The sigmoidal shape of the TREX2 DNA binding curves indicates cooperativity in the binding process. A comparative analysis of TREX2WT with the TREX2WT/R163A,R165A,R167A heterodimer shows that the sigmoidal binding isotherm is retained in the heterodimer despite the nearly complete loss of DNA binding capacity in one of the TREX2 protomers (Fig. 4B). This observation indicates that the cooperative binding in TREX2 is not likely a result of allosteric effects transmitted across the dimer interface. Instead, these data are most indicative of a cooperative TREX2 binding effect between TREX2 dimers bound along the ssDNA. Furthermore, the observed anisotropy of TREX2WT is approximately double that of the TREX2WT/R163A,R165A,R167A heterodimer consistent with the potential to bind two ssDNA 30-mer polymers in the TREX2WT and only one in this heterodimer. The TREX2 dimer at the 3′ end of the ssDNA 30-mer is positioned to load approximately four nucleotides of DNA into the active site DNA binding pocket. This DNA binding mechanism is supported by the equilibrium binding and single turnover studies presented here, and suggest some processivity in TREX2 DNA binding that might be limited to steps before catalysis. Our data do not distinguish between a two-step binding mechanism in which rapid equilibrium binding occurs first by the Arg163–167 followed by a rate-limiting conformational change to position the 3′ end into the active site or a single binding step.
A strategy to generate heterodimeric TREX2 variants has revealed coordinated exonuclease action within the dimer. In an attempt to test the requirement of the dimeric structure on TREX2 activity, several mutagenesis approaches were tried without success to generate a monomeric TREX2 enzyme. A major contribution to the stability of the TREX2 dimer structure results from the centrally located extended β-sheet that connects the individual β-sheets of each protomer at the dimer interface. Amino acid substitutions in TREX2 designed to replace salt bridges between the protomers with repulsive centers resulted in inactivation of the TREX2 enzyme but failed to disrupt the dimeric structure (data not shown). To demonstrate coordinated catalysis within the TREX2 dimer, we measured exonuclease activity in one TREX2 protomer that was in complex with a variant TREX2 protomer. The kinetic stability of the TREX2 heterodimer was considered, because potential subunit exchange could influence interpretation of our heterodimeric TREX2 enzyme studies. Although we cannot formally rule out some level of subunit exchange leading to redistribution of the TREX2 protomers after purification, no changes in TREX2 heterodimer activities that might accompany such an exchange have been detected in the heterodimer preparations indicating kinetic stability in the TREX2 dimer under the conditions used in these studies. A 7-fold decrease in exonuclease activity was detected in the TREX2 dimer when one protomer was catalytically defective but fully capable of binding DNA. In contrast, if the defective TREX2 protomer lacks DNA binding activity, there is no measurable effect on the opposing wild-type TREX2 protomer. These data suggest a coordinated catalytic mechanism communicated across the dimer interface from one active site to the other that is dependent upon DNA binding and catalysis in the opposing protomer. When one protomer binds DNA appropriately but fails to catalyze nucleotide excision, the required structural signal is not communicated to the opposing protomer to affect excision activity. The coordination of the TREX2 active sites that are ∼35 Å apart is likely communicated through the salt bridge pairs of Glu-191:Lys-59 positioned between the active sites at the dimer interface of TREX2. The position of the nucleophile-generating His-188 is on a mobile loop at the end of a helix that is likely affected by the Glu-191 participation in the salt bridge across the dimer interface. Our TREX2 structural studies show that the Nζ of Lys-59 is shifted by ∼5.1 Å dependent upon the presence of DNA in the active site supporting this mechanism of communication in the TREX2 dimer.4 It is likely that this coordinated excision mechanism is fine-tuned for optimal activity on the true biological substrate of TREX2, which remains to be identified. Our studies indicate that biological substrates of the TREX2 dimer might include duplex DNA containing nicked strands where TREX2 might act to melt the duplex and generate the single-stranded DNA for entry into the active site.
Supplementary Material
Acknowledgments
We thank Dr. Wlodzimierz Bujalowski (University of Texas Medical Branch) for helpful suggestions during the course of this study.
This work was supported, in whole or in part, by National Institutes of Health Grant GM069962 (to F. W. P.). This work was also supported by American Cancer Society Grant RSG-04-187-01-GMC (to T. H.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3 and additional references.
Footnotes
F. W. Perrino and S. Harvey, unpublished data.
The abbreviations used are: FAM, 5′-fluorescein; BD, binding density; WT, wild type; MUT, mutant; ssDNA, single-stranded DNA.
U. de Silva, F. W. Perrino, and T. Hollis, submitted for publication.
References
- 1.Shevelev, I., and Hubscher, U. (2002) Nat. Rev. 3 364-375 [DOI] [PubMed] [Google Scholar]
- 2.Marti, T. M., and Fleck, O. (2004) Cell. Mol. Life Sci. 61 336-354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindahl, T., Gally, J. A., and Edelman, G. M. (1969) J. Biol. Chem. 244 5014-5019 [PubMed] [Google Scholar]
- 4.Perrino, F. W., Miller, H., and Ealey, K. A. (1994) J. Biol. Chem. 269 16357-16363 [PubMed] [Google Scholar]
- 5.Mazur, D. J., and Perrino, F. W. (1999) J. Biol. Chem. 274 19655-19660 [DOI] [PubMed] [Google Scholar]
- 6.Hoss, M., Robins, P., Naven, T. J., Pappin, D. J., Sgouros, J., and Lindahl, T. (1999) EMBO J. 18 3868-3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mazur, D. J., and Perrino, F. W. (2001) J. Biol. Chem. 276 17022-17029 [DOI] [PubMed] [Google Scholar]
- 8.Freemont, P. S., Friedman, J. M., Beese, L. S., Sanderson, M. R., and Steitz, T. A. (1988) Proc. Natl. Acad. Sci. U. S. A. 85 8924-8928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beese, L. S., and Steitz, T. A. (1991) EMBO J. 10 25-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barnes, M. H., Spacciapoli, P., Li, D. H., and Brown, N. C. (1995) Gene (Amst.) 165 45-50 [DOI] [PubMed] [Google Scholar]
- 11.Strauss, B. S., Sagher, D., and Acharya, S. (1997) Nucleic Acids Res. 25 806-813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taft-Benz, S. A., and Schaaper, R. M. (1998) Nucleic Acids Res. 26 4005-4011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zuo, Y. H., and Deutscher, M. P. (2001) Nucleic Acids Res. 29 1017-1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inamdar, K. V., Yu, Y., and Povirk, L. F. (2002) Radiat. Res. 157 306-311 [DOI] [PubMed] [Google Scholar]
- 15.Harrigan, J. A., Fan, J., Momand, J., Perrino, F. W., Bohr, V. A., and Wilson, D. M. (2007) Mech. Ageing Dev. 128 259-266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller, H., and Perrino, F. W. (1996) Biochemistry 35 12919-12925 [DOI] [PubMed] [Google Scholar]
- 17.Breyer, W. A., and Matthews, B. W. (2000) Nat. Struct. Biol. 12 1125-1128 [DOI] [PubMed] [Google Scholar]
- 18.DeRose, E. F., Li, D. W., Darden, T., Harvey, S., Perrino, F. W., Schaaper, R. M., and London, R. E. (2002) Biochemistry 41 94-110 [DOI] [PubMed] [Google Scholar]
- 19.Hamdan, S., Carr, P. D., Brown, S. E., Ollis, D. L., and Dixon, N. E. (2002) Structure (Lond.) 10 535-546 [DOI] [PubMed] [Google Scholar]
- 20.Perrino, F. W., Harvey, S., McMillin, S., and Hollis, T. (2005) J. Biol. Chem. 280 15212-15218 [DOI] [PubMed] [Google Scholar]
- 21.deSilva, U., Choudhury, S., Bailey, S. L., Harvey, S., Perrino, F. W., and Hollis, T. (2007) J. Biol. Chem. 282 10537-10543 [DOI] [PubMed] [Google Scholar]
- 22.Brucet, M., Querol-Audi, J., Serra, M., Ramirez-Espain, X., Bertlik, K., Ruiz, L., Lloberas, J., Macias, M. J., Fita, I., and Celada, A. (2007) J. Biol. Chem. 282 14547-14557 [DOI] [PubMed] [Google Scholar]
- 23.Mazur, D. J., and Perrino, F. W. (2001) J. Biol. Chem. 276 14718-14727 [DOI] [PubMed] [Google Scholar]
- 24.Crow, Y. J., Hayward, B. E., Parmar, R., Robins, P., Leitch, A., Ali, M., Black, D. N., van Bokhoven, H., Brunner, H. G., Hamel, B. C., Corry, P. C., Cowan, F. M., Frints, S. G., Klepper, J., Livingston, J. H., Lynch, S. A., Massey, R. F., Meritet, J. F., Michaud, J. L., Ponsot, G., Voit, T., Lebon, P., Bonthron, D. T., Jackson, A. P., Barnes, D. E., and Lindahl, T. (2006) Nat. Genet. 38 917-920 [DOI] [PubMed] [Google Scholar]
- 25.Morita, M., Stamp, G., Robins, P., Dulic, A., Rosewell, I., Hrivnak, G., Daly, G., Lindahl, T., and Barnes, D. E. (2004) Mol. Cell. Biol. 24 6719-6727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang, Y. G., Lindahl, T., and Barnes, D. E. (2007) Cell 131 873-886 [DOI] [PubMed] [Google Scholar]
- 27.Chowdhury, D., Beresford, P. J., Zhu, P. C., Zhang, D., Sung, J. S., Demple, B., Perrino, F. W., and Lieberman, J. (2006) Mol. Cell 23 133-142 [DOI] [PubMed] [Google Scholar]
- 28.Rice, G., Newman, W. G., Dean, J., Patrick, T., Parmar, R., Flintoff, K., Robins, P., Harvey, S., Hollis, T., O'Hara, A., Herrick, A. L., Bowden, A. P., Perrino, F. W., Lindahl, T., Barnes, D. E., and Crow, Y. J. (2007) Am. J. Hum. Genet. 80 811-815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee-Kirsch, M. A., Chowdhury, D., Harvey, S., Gong, M., Senenko, L., Engel, K., Pfeiffer, C., Hollis, T., Gahr, M., Perrino, F. W., Lieberman, J., and Hubner, N. (2007) J. Mol. Med. 85 531-537 [DOI] [PubMed] [Google Scholar]
- 30.Lee-Kirsch, M. A., Gong, M., Chowdhury, D., Senenko, L., Engel, K., Lee, Y. A., deSilva, U., Bailey, S. L., Witte, T., Vyse, T. J., Kere, J., Pfeiffer, C., Harvey, S., Wong, A., Koskenmies, S., Hummel, O., Rohde, K., Schmidt, R. E., Dominiczak, A. F., Gahr, M., Hollis, T., Perrino, F. W., Lieberman, J., and Hubner, N. (2007) Nat. Genet. 39 1065-1067 [DOI] [PubMed] [Google Scholar]
- 31.Richards, A., van den Maagdenberg, A., Jen, J. C., Kavanagh, D., Bertram, P., Spitzer, D., Liszewski, M. K., Barilla-LaBarca, M. L., Terwindt, G. M., Kasai, Y., McLellan, M., Grand, M. G., Vanmolkot, K. R. J., de Vries, B., Wan, J., Kane, M. J., Mamsa, H., Schafer, R., Stam, A. H., Haan, J., Paulus, T., Storimans, C. W., van Schooneveld, M. J., Oosterhuis, J. A., Gschwendter, A., Dichgans, M., Kotschet, K. E., Hodgkinson, S., Hardy, T. A., Delatycki, M. B., Hajj-Ali, R. A., Kothari, P. H., Nelson, S. F., Frants, R. R., Baloh, R. W., Ferrari, M. D., and Atkinson, J. P. (2007) Nat. Genet. 39 1068-1070 [DOI] [PubMed] [Google Scholar]
- 32.Rice, G., Patrick, T., Parmar, R., Taylor, C. F., Aeby, A., Aicardi, J., Artuch, R., Montalto, S. A., Bacino, C. A., Barroso, B., Baxter, P., Benko, W. S., Bergmann, C., Bertini, E., Biancheri, R., Blair, E. M., Blau, N., Bonthron, D. T., Briggs, T., Brueton, L. A., Brunner, H. G., Burke, C. J., Carr, I. M., Carvalho, D. R., Chandler, K. E., Christen, H. J., Corry, P. C., Cowan, F. M., Cox, H., D'Arrigo, S., Dean, J., De Laet, C., De Praeter, C., Dery, C., Ferrie, C. D., Flintoff, K., Frints, S. G. M., Garcia-Cazorla, A., Gener, B., Goizet, C., Goutieres, F., Green, A. J., Guet, A., Hamel, B. C. J., Hayward, B. E., Heiberg, A., Hennekam, R. C., Husson, M., Jackson, A. P., Jayatunga, R., Jiang, Y. H., Kant, S. G., Kao, A., King, M. D., Kingston, H. M., Klepper, J., van der Knaap, M. S., Kornberg, A. J., Kotzot, D., Kratzer, W., Lacombe, D., Lagae, L., Landrieu, P. G., Lanzi, G., Leitch, A., Lim, M. J., Livingston, J. H., Lourenco, C. M., Lyall, E. G. H., Lynch, S. A., Lyons, M. J., Marom, D., McClure, J. P., McWilliam, R., Melancon, S. B., Mewasingh, L. D., Moutard, M. L., Nischal, K. K., Ostergaard, J. R., Prendiville, J., Rasmussen, M., Rogers, R. C., Roland, D., Rosser, E. M., Rostasy, K., Roubertie, A., Sanchis, A., Schiffmann, R., Scholl-Burgi, S., Seal, S., Shalev, S. A., Corcoles, C. S., Sinha, G. P., Soler, D., Spiegel, R., Stephenson, J. B. P., Tacke, U., Tan, T. Y., Till, M., Tolmie, J. L., Tomlin, P., Vagnarelli, F., Valente, E. M., Van Coster, R. N. A., van der Aa, N., Vanderver, A., Vles, J. S. H., Voit, T., Wassmer, E., Weschke, B., Whiteford, M. L., Willemsen, M. A. A., Zankl, A., Zuberi, S. M., Orcesi, S., Fazzi, E., Lebon, P., and Crow, Y. J. (2007) Am. J. Hum. Genet. 81 713-72517846997 [Google Scholar]
- 33.Goldsby, R. E., Lawrence, N. A., Hays, L. E., Olmsted, E. A., Chen, X., Singh, M., and Preston, B. D. (2001) Nat. Med. 7 638-639 [DOI] [PubMed] [Google Scholar]
- 34.Goldsby, R. E., Hays, L. E., Chen, X., Olmsted, E. A., Slayton, W. B., Spangrude, G. J., and Preston, B. D. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 15560-15565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McElhinny, S. A. N., Pavlov, Y. I., and Kunkel, T. A. (2006) Cell Cycle 5 958-962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pavlov, Y. I., Frahm, C., McElhinny, S. A. N., Niimi, A., Suzuki, M., and Kunkel, T. A. (2006) Curr. Biol. 16 202-207 [DOI] [PubMed] [Google Scholar]
- 37.Albertson, T. M., and Preston, B. D. (2006) Curr. Biol. 16 R209-R211 [DOI] [PubMed] [Google Scholar]
- 38.Chen, M. J., Dumitrache, L. C., Wangsa, D., Ma, S. M., Padilla-Nash, H., Ried, T., and Hasty, P. (2007) Cancer Res. 67 9077-9083 [DOI] [PubMed] [Google Scholar]
- 39.Perrino, F. W., Krol, A., Harvey, S., Zheng, S. L., Horita, D. A., Hollis, T., Meyers, D. A., Isaacs, W. B., and Xu, J. F. (2004) Adv. Enzyme Regul. 44 37-49 [DOI] [PubMed] [Google Scholar]
- 40.Higuchi, R. (1990) in PCR Protocols: A Guide to Methods and Applications (Innis, M. A., Gelfand, D. H., Shinsky, J. J., and White, T. J., eds) pp. 177-183, Academic Press, San Diego
- 41.Jezewska, M. J., and Bujalowski, W. (1996) Biochemistry 35 2117-2128 [DOI] [PubMed] [Google Scholar]
- 42.Bujalowski, W. (2006) Chem. Rev. 106 556-606 [DOI] [PubMed] [Google Scholar]
- 43.Lohman, T. M., and Bujalowski, W. (1991) Methods Enzymol. 208 258-290 [DOI] [PubMed] [Google Scholar]
- 44.Jezewska, M. J., Galletto, R., and Bujalowski, W. (2003) Biochemistry 42 5955-5970 [DOI] [PubMed] [Google Scholar]
- 45.Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D., and Bairoch, A. (2005) in The Proteomics Protocols Handbook (Walker, J. M., ed) pp. 571-607, Humana Press Inc., Totowa, NJ
- 46.Hill, T. L. (1985) Cooperativity Theory in Biochemistry, Springer-Verlag Inc., New York
- 47.McGhee, J. D., and Hippel, P. H. V. (1974) J Mol. Biol. 86 469-489 [DOI] [PubMed] [Google Scholar]
- 48.Chen, M. J., Ma, S. M., Dumitrache, L. C., and Hasty, P. (2007) Nucleic Acids Res. 35 2682-2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jezewska, M. J., Rajendran, S., and Bujalowski, W. (1998) J. Mol. Biol. 284 1113-1131 [DOI] [PubMed] [Google Scholar]
- 50.Beard, W. A., and Wilson, S. H. (2000) Mutat. Res. 460 231-244 [DOI] [PubMed] [Google Scholar]
- 51.Halford, S. E., and Marko, J. F. (2004) Nucleic Acids Res. 32 3040-3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Townson, S. A., Samuelson, J. C., Bao, Y. M., Xu, S. Y., and Aggarwal, A. K. (2007) Structure (Lond.) 15 449-459 [DOI] [PubMed] [Google Scholar]
- 53.Spolar, R. S., and Record, M. T. (1994) Science 263 777-784 [DOI] [PubMed] [Google Scholar]
- 54.Saecker, R. M., and Record, M. T. (2002) Curr. Opin. Struct. Biol. 12 311-319 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.