

Abstract
Heterotrimeric G-proteins are integral to a conserved regulatory module that influences metazoan asymmetric cell division (ACD). In the Caenorhabditis elegans zygote, GOA-1 (Gαo) and GPA-16 (Gαi) are involved in generating forces that pull on astral microtubules and position the spindle asymmetrically. GPA-16 function has been analyzed in vivo owing notably to a temperature-sensitive allele gpa-16(it143), which, at the restrictive temperature, results in spindle orientation defects in early embryos. Here we identify the structural basis of gpa-16(it143), which encodes a point mutation (G202D) in the switch II region of GPA-16. Using Gαi1(G202D) as a model in biochemical analyses, we demonstrate that high temperature induces instability of the mutant Gα. At the permissive temperature, the mutant Gα was stable upon GTP binding, but switch II rearrangement was compromised, as were activation state-selective interactions with regulators involved in ACD, including GoLoco motifs, RGS proteins, and RIC-8. We solved the crystal structure of the mutant Gα bound to GDP, which indicates a unique switch II conformation as well as steric constraints that suggest activated GPA-16(it143) is destabilized relative to wild type. Spindle severing in gpa-16(it143) embryos revealed that pulling forces are symmetric and markedly diminished at the restrictive temperature. Interestingly, pulling forces are asymmetric and generally similar in magnitude to wild type at the permissive temperature despite defects in the structure of GPA-16(it143). These normal pulling forces in gpa-16(it143) embryos at the permissive temperature were attributable to GOA-1 function, underscoring a complex interplay of Gα subunit function in ACD.
G-protein-coupled receptors (GPCRs)5 mediate the actions of a variety of sensory and metabolic stimuli (1). Heterotrimeric G-proteins are molecular switches that transduce GPCR activation into intracellular changes and are composed of a Gα subunit and a Gβγ dimer (1, 2). GPCR-promoted activation of Gαβγ causes Gα to exchange GDP for GTP, which in turn causes Gα·GTP and Gβγ to dissociate. Gα·GTP and liberated Gβγ then regulate effector systems to alter cell physiology (1, 2). This canonical, GPCR-driven “G-protein cycle” is reset by the intrinsic GTP hydrolysis activity of Gα (1, 2).
A noncanonical, yet evolutionarily conserved, heterotrimeric G-protein regulatory module has been identified in Caenorhabditis elegans, Drosophila, and mammalian asymmetric cell division (ACD) that is important for generating diversity during development (1, 3–5). An initial step in ACD is generation of cell polarity. Cell fate determinants then segregate to different sides of the cell (3), and the mitotic spindle is positioned to permit proper distribution of determinants to daughter cells. Integral to accurate spindle positioning is a heterotrimeric G-protein module thought to be independent of GPCRs and involving GDP dissociation inhibitors (GoLoco motif proteins), guanine nucleotide exchange factors (i.e. RIC-8), and GTPase-accelerating proteins (RGS proteins) (1, 3, 4).
In the Caenorhabditis elegans zygote, G-protein involvement in ACD has been extensively studied using genetic and cell biological approaches (6). Two Gα subunits, GPA-16 and GOA-1, are important for generating pulling forces on astral microtubules critical for spindle positioning (7). These two Gα subunits are required for force generation in concert with the GoLoco motif proteins GPR-1/-2 and the coiled-coil protein LIN-5 (7). GPA-16 and GOA-1 exert partially redundant functions in ACD (8) but differ in some respects, e.g. RIC-8 is required for cortical localization of GPA-16 but not GOA-1 (9). The mechanisms by which GOA-1, GPA-16, and their binding partners mediate force generation during ACD are gradually being revealed (10–14) but remain incompletely understood.
Whereas the contribution of GOA-1 to ACD has been analyzed solely using null alleles and RNAi-mediated inactivation, the role of GPA-16 in ACD was discovered using both RNAi and a unique temperature-sensitive allele gpa-16(it143). At 25 °C, 70% of gpa-16(it143) worms die during embryogenesis, with a significant proportion of adult escapers showing reversal of left-right body axis asymmetry (15). At 16 °C, only 2% of gpa-16(it143) worms die during embryogenesis (15), suggesting that GPA-16(it143) somehow supports function at the permissive temperature; however, an analysis of ACD in one-cell stage embryos at this particular temperature has not been conducted previously. The gpa-16(it143) allele encodes a point mutation causing a glycine 202 to aspartate (G202D) change in switch II of GPA-16 (15). We have used enzymology, crystallography, genetics, and cell biology to delineate the molecular mechanism underlying the critical contribution of this residue to GPA-16 function during ACD. Purifying GPA-16 from baculovirus-infected insect cells yields only micrograms of purified protein (9), preventing detailed biochemical and, especially, x-ray diffraction crystallographic structural studies. We have thus also examined the G202D mutation in the context of the most rigorously characterized and experimentally tractable Gα subunit that is most closely related to GPA-16, namely mammalian Gαi1 (9).
EXPERIMENTAL PROCEDURES
Materials—Unless otherwise specified, all reagents were of the highest purity obtainable from Sigma or Fisher (Pittsburgh, PA). Site-directed mutagenesis was conducted using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA).
Protein Purification—Full-length His6-tagged human Gαi1 (wild type and G202D) was purified as described (16, 17). Full-length His6-tagged (wild type and G198D) chimeric Gαt/Gαi1 subunits (18) were purified using standard chromatography methods (16, 19). Care was taken to keep all Gα subunits GDP-bound and at 4 °C throughout their respective purification processes. A GST fusion of rat Ric-8A was purified as described (20). Recombinant Gβ1γ1 dimer incorporating the biotinylation sequence was produced and purified as described (21), with DNA coding for the biotin ligase substrate motif (GLNDIFEAQKIEWHE) inserted upstream of the Gγ1 coding sequence within the baculoviral shuttle vector pFASTBacHT (Invitrogen). Biotin protein ligase (BirA) was incubated with 50 μm Gβ1γ1 dimer for 24 h at 25 °C under the conditions described by the manufacturer (Avidity, Denver, CO). Resulting protein was buffer-exchanged into phosphate-buffered saline and stored at -80 °C. Full-length GST-tagged human PCP-2 was prepared as described (17). The RGS domain of rat RGS14 was prepared as a GST fusion protein as described (16). Wild type and scrambled RGS12 GoLoco motif synthetic peptides are described in Ref. 22. An N-terminal biotinylated peptide comprising amino acids 63–87 of bovine rod PDEγ is described in Ref. 23.
GTPγS Binding—[35S]GTPγS (PerkinElmer Life Sciences) binding was measured using a filter binding assay as described previously (10). Rate constants were obtained by subtracting nonspecific binding (counts/min obtained in the presence of 100 μm unlabeled GTPγS) and fitting binding curves to a single exponential function in GraphPad Prism version 4.0 (San Diego, CA).
C. elegans Strains and RNAi Conditions—The N2 strain was used as wild type and maintained according to established procedures (24). The gpa-16(it143) strain (15) was maintained at 16 °C and shifted for 20–24 h to 25 °C before analysis at the restrictive temperature. For inactivation of gpa-16 or goa-1 by RNAi, bacterial feeding strains (10) were used to feed L3/L4 worms for 36 h at 25 °C or for 48 h at 16 °C.
Coimmunoprecipitation and Western Blot Analysis—Generation of worm embryonic extracts and coimmunoprecipitation experiments were performed as described (10) with the following modifications. For each experiment, ∼1.5 mg of protein extract and 3 μg of GPA-16 antibodies (9) were utilized. As specified, GDP or GTPγS were included at final concentrations of 100 μm. For testing the interactions at 16 °C, lysates were incubated with antibodies and nucleotides at 16 °C for 40 min; 15 μl of protein G-Sepharose were added, and the incubation was continued at 4 °C overnight. Following immunoprecipitation, SDS-PAGE and Western blot analysis were performed according to standard procedures. RIC-8, GPR-1/2, and GPA-16 primary antibodies (9, 10) were diluted 1:1000 and horseradish peroxidase-conjugated goat anti-rabbit secondary antibodies (GE Healthcare) 1:2000; the signals were revealed by standard chemiluminescence (GE Healthcare).
Circular Dichroism—All CD experiments were performed in Buffer C (10 mm phosphate buffer (KH2PO4/K2HPO4), pH 7.5, 50 mm NaCl, 5 mm MgCl2). For CD experiments, Gα subunits were purified as described (16, 19), but the final purification step was by Sephacryl S200 gel filtration (GE Healthcare) using Buffer C. For CD measurements, 50 μm of Gα subunits were loaded with 100 μm GDP or GTPγS at 15 °C for the times determined to give ≈100% binding, based on rate constants (see Fig. 1B). Proteins were then diluted to 4.4 μm in Buffer C and kept at 4 °C. CD was measured using a PiStar-180 spectrophotometer (Applied Photophysics, Surrey, UK). CD was measured at 208 nm (slits 4.0 nm) for 30 s at each temperature. The temperature ramp was conducted using 1 °C steps with a tolerance of ±0.2 °C. Apparent melting temperatures were calculated as the minima of first derivatives with respect to the reciprocal of temperature (25). First differential minima were calculated using Rt-Plot (version 2.7, Horst Reichert, Eppstein, Germany) using the Akima interpolation with a weighting of 1 (26).
FIGURE 1.

Temperature-dependent nucleotide binding, protein stability, and protein interactions of wild type and G202D Gα subunits. A and B, time course of GTPγS binding by wild type and G202D Gαi1 measured at 30 °C (A) or 15 °C (B). 100 nm Gα was incubated with 1 μm [35S]GTPγS, and bound nucleotide was measured at indicated times. Data were fit to single exponential association curves (95% confidence intervals in brackets) as follows: 30 °C wild type, 0.017 (0.015–0.019) min-1; 30 °C G202D, data could not be fit; 15 °C wild type, 0.0025 (0.0021–0.0028) min-1; 15 °C G202D, 0.027 (0.021–0.032) min-1. C and D, CD in millidegrees (mdeg) of 4.4 μm wild type Gαi1 (C) or G202D Gαi1 (D) was measured at 208 nm in both GDP- and GTPγS-bound conformations. Thermal melting curves were generated by measuring CD values at 1 °C intervals. Data are graphed as mean ± S.E. The mean melting temperatures (S.E. in parenthesis; n = 3) for wild type Gαi1 (C) were GDP 50.2 °C (0.4) and GTPγS 77.2 °C (0.4) and for G202D Gαi1 (D) were GDP 49.5 °C (0.8) and GTPγS 50.1 °C (0.01). E, co-immunoprecipitation conducted at 16 °C using wild type or gpa-16 (it143) embryonic extracts and GPA-16 antibody, either without (w/o) exogenous nucleotides or in the presence of 100 μm GDP or GTPγS. Immunoprecipitated material was analyzed by Western blot using antibodies against RIC-8, GPR-1/-2, or GPA-16. Input corresponds to 1/70 of starting material.
Surface Plasmon Resonance—Surface plasmon resonance
experiments with GST fusion proteins and biotinylated proteins were performed
using a BIAcore 3000 (GE Healthcare) as described previously
(19,
27). Sensor surfaces for all
experiments were at 15 °C, and all proteins in the sample handler were
kept at 4 °C. The eluent buffer was 10 mm HEPES, pH 7.4, 150
mm NaCl, 5 mm MgCl2, and 0.005% (v/v) Nonidet
P-40. Nucleotide-specific conformations of Gα were obtained by
incubation with eluent buffer supplemented with 100 μm GDP (GDP)
or 100 μm GTPγS (GTPγS) or 100 μm GDP,
20 mm NaF, and 30 μm AlCl3
(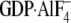 ). GTPγS
loading was conducted for 3 h at either 15 °C for
Gαi1(G202D) or 30 °C for Gαi1(wild
type). Loading levels for KD determination experiments
were as follows: biotin-Gβ1γ1 (1400 RU),
GST-Ric-8A (300 RU), GST-PCP-2 (400 RU), GST-RGS14 (400 RU), and
biotin-PDEγ (500 RU). Equilibrium binding KD
measurements were conducted at a flow rate of 20 μl/min using protocols
described previously (28).
Kinetic binding analyses were conducted as described previously
(29). Nonspecific binding was
determined using a biotinylated mNOTCH control peptide
(27) and GST alone
(19), respectively.
Nonspecific binding was subtracted from experimental data to give binding
curves using BIAevaluation software (version 3.0; Biacore).
). GTPγS
loading was conducted for 3 h at either 15 °C for
Gαi1(G202D) or 30 °C for Gαi1(wild
type). Loading levels for KD determination experiments
were as follows: biotin-Gβ1γ1 (1400 RU),
GST-Ric-8A (300 RU), GST-PCP-2 (400 RU), GST-RGS14 (400 RU), and
biotin-PDEγ (500 RU). Equilibrium binding KD
measurements were conducted at a flow rate of 20 μl/min using protocols
described previously (28).
Kinetic binding analyses were conducted as described previously
(29). Nonspecific binding was
determined using a biotinylated mNOTCH control peptide
(27) and GST alone
(19), respectively.
Nonspecific binding was subtracted from experimental data to give binding
curves using BIAevaluation software (version 3.0; Biacore).
Crystallization, Structure Determination, and Refinement—To aid crystallization of the labile G202D mutant of Gαi1, we were careful to ensure strict temperature control throughout protein induction (14 °C), purification (4 °C), and crystallization (18 °C). Moreover, crystallization trials were conducted immediately following concentration of purified protein, as freeze-thawed protein was incapable of reproducing crystal growth. Crystals of Gαi1(G202D) were obtained by vapor diffusion from hanging drops containing a 1:1 (v/v) ratio of protein solution (10–20 mg ml-1 in 50 mm HEPES buffer, pH 8.0, 1 mm EDTA, 100 μm GDP, and 5 mm dithiothreitol) to well solution (1.9 m ammonium sulfite and 100 mm sodium acetate, pH 6.0). Crystals (∼0.6 × 0.3 × 0.2 mm) were formed in 3–5 days in the space group I4 (a = b = 127.87 Å, c = 68.35 Å, α = β = γ = 90 °), with 1 molecule in the asymmetric unit. For data collection at 100 K, crystals were transferred to a solution containing well solution supplemented with 20% (w/v) glycerol for 90 s followed by immersion in liquid nitrogen. A native data set was collected on a single crystal using an R-Axis IV++ detector with Rigaku (The Woodlands, TX) rotating anode generator and osmic confocal “blue” optics at the University of North Carolina, Chapel Hill, x-ray facility. Diffraction data were scaled and indexed using HKL2000 (30). The structure of Gαi1·GDP (Protein Data Bank code 1AS3), excluding residues 177–184 and 195–220, GDP, waters, and other heterogeneous molecules, was used as a molecular replacement model for Gαi1(G202D) using Phaser in CCP4 (31). Model building was achieved using the programs O and Coot (32, 33). Model refinement was conducted using real space refinement protocols in Coot as well as a combination of rigid body, simulated annealing, energy minimization, and b-factor protocols in CNS (34). All structural images were made with PyMol (DeLano Scientific, San Carlos, CA).
Fluorescence Spectroscopy—Intrinsic tryptophan fluorescence was measured using an LS55 spectrometer (PerkinElmer Life Sciences). Excitation and emission wavelengths were 292 and 342 nm respectively, with slit widths of 2.5 nm. Fluorescence was measured in temperature-controlled cuvettes containing 1 ml of 10 mm Tris/HCl, pH 8.0, 1 mm EDTA, 5 mm MgCl2, 150 mm NaCl. Controls were performed to account for any nonspecific effects of GTPγS addition on fluorescence.
Microscopy and Spindle Severing—Preparation of embryos, time-lapse differential interference contrast microscopy, and spindle severing were performed as described (10, 35, 36). Experiments were conducted using a homemade device that consists of a thermostat and a cooling/heating element coupled to a fan blowing air at the appropriate temperature onto the objective and the microscope stage. The temperature of the embryo during the experiment was monitored using a thermometer inserted in the agarose pad. Measurements of peak velocities of spindle poles following spindle severing were performed essentially as described (36).
RESULTS AND DISCUSSION
G202D Mutation Gives Temperature-dependent Lability to the Activated Gα State—We purified wild type and G202D Gαi1 to homogeneity as assessed by SDS-PAGE (data not shown). Gαi1(G202D)·GDP migrated in size exclusion chromatography as a monomer and had a circular dichroism spectrum consistent with properly folded Gα (data not shown) (37). We first tested the ability of purified Gαi1 to bind nucleotide at 30 °C to approximate conditions found in vivo at the restrictive temperature. Wild type Gαi1 bound GTPγS in a saturable manner with an association rate comparable with published values (38) (Fig. 1A). By contrast, Gαi1(G202D) rapidly bound GTPγS but in a biphasic manner, peaking at 30 min then rapidly decaying over time. We also examined GTPγS binding at the permissive temperature (Fig. 1B). GTPγS binding by Gαi1(G202D) at 15 °C was rapid, monophasic, and stable for up to 15 h. These data suggest that Gαi1(G202D), and by extrapolation GPA-16(it143), undergoes nucleotide state-dependent inactivation at 30 °C but not 15 °C. The rate of GTPγS binding by Gαi1(G202D) at 15 °C was over an order of magnitude faster than the wild type protein (Fig. 1B), suggesting that the G202D mutation also enhances spontaneous GDP release.
High spontaneous GDP release engendered by the G202D mutation may contribute to Gα inactivation at the restrictive temperature by promoting the GTP-bound form of the protein. Alternatively, it is well described that nucleotide-free Gα subunits are inherently unstable (20, 39, 40); thus, increased residence of Gα(G202D) in the nucleotide-free state could also contribute to inactivation. This is suggested to be the mechanism of temperature sensitivity of the human Gαs(A366S) mutant, which bears an Ala-to-Ser point mutation in the GDP-binding pocket that increases spontaneous GDP release causing constitutive activity (40, 41). Uniquely, this mutation is permissive at the temperature of the human testis (32–33 °C) causing testotoxicosis because of the overproduction of testosterone. In other tissues, the protein is nonfunctional at 37 °C causing pseudohypoparathyroidism, typical of Gαs reduction of function (42).
Thermal melting of the Gαi family proteins under examination in this study gave a single cooperative transition from predominantly α-helical structure to random coil, as measured by circular dichroism. Normally, activated Gα is significantly more thermostable than inactive Gα; wild type Gαi1·GDP had a melting temperature of 50 °C, whereas wild type Gαi1·GTPγS had a melting temperature of 77 °C (Fig. 1C) consistent with structural data that the three switch regions of GDP-bound Gα are conformationally flexible, and GTPγS binding induces a distinct, stable switch region conformation (43). In contrast to wild type, GTPγS binding to Gαi1(G202D) did not induce a thermostable protein-nucleotide complex; both GDP- and GTPγS-bound Gαi1(G202D) had melting temperatures of 50 °C (Fig. 1D). These data suggest a mechanism for the temperature-sensitive loss-of-function in GPA-16(it143); at the permissive temperature, GPA-16(it143) is relatively stable in both the GDP- and GTP-bound forms, whereas at the restrictive temperature GPA-16(it143) is unstable in the GTP-bound state, becoming rapidly inactivated. Compatible with this view, the phenotype of gpa-16(it143) embryos at 25 °C is indistinguishable from that of embryos depleted of gpa-16 by RNAi (9, 10).
G202D Mutation Perturbs Nucleotide-dependent Interactions with
Gα Regulators—Gα switch regions are involved
in mediating interactions with various Gα regulators that are important
in ACD (7,
11,
43–48).
We examined the ability of Gαi1(G202D) to interact with
cognate regulatory proteins, performing these experiments at 15 °C to
ensure that stable Gαi1(G202D) was operative in all cases. As
assessed by surface plasmon resonance (SPR), wild type GDP-bound
Gαi1 exhibited robust interaction with immobilized,
biotinylated Gβ1γ1
(Fig. 2A;
Table 1), whereas GTPγS-
and 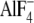 -bound wild type
Gαi1 had negligible binding, consistent with the known
nucleotide state-selective association of Gα with Gβγ
(49). In contrast,
Gαi1(G202D) exhibited strong interactions with
Gβ1γ1 irrespective of nucleotide state
(Fig. 2B;
Table 1). These results suggest
that the G202D mutation impairs the Gα switch region(s) from adopting
the activated conformation in response to the ligands GTPγS and
-bound wild type
Gαi1 had negligible binding, consistent with the known
nucleotide state-selective association of Gα with Gβγ
(49). In contrast,
Gαi1(G202D) exhibited strong interactions with
Gβ1γ1 irrespective of nucleotide state
(Fig. 2B;
Table 1). These results suggest
that the G202D mutation impairs the Gα switch region(s) from adopting
the activated conformation in response to the ligands GTPγS and
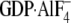 .
.
FIGURE 2.

Protein-protein interactions of wild type and Gly-to-Asp mutant Gα
subunits. Interactions between wild type (WT) or indicated
Gly-to-Asp mutated Gα subunits and
Gβ1γ1 (A and B), PCP-2
(C and D), Ric-8A (E and F), the RGS
domain of RGS14 (G and H), and PDEγ(aa 63–87)
(I and J) were measured using surface plasmon resonance.
Proteins were immobilized using biotin-streptavidin coupling (A, B,
I, and J) or anti-GST antibody capture (C–H).
Indicated concentrations of Gα subunits in the GDP (blue),
GTPγS(red), or
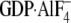 (green) loaded forms were injected over biosensor surfaces at a flow
rate of 20 μl/min as denoted by arrows. Binding curves were
generated after subtracting nonspecific binding to mNOTCH peptide (A, B,
I, and J) or GST (C–H) control surfaces.
(green) loaded forms were injected over biosensor surfaces at a flow
rate of 20 μl/min as denoted by arrows. Binding curves were
generated after subtracting nonspecific binding to mNOTCH peptide (A, B,
I, and J) or GST (C–H) control surfaces.
TABLE 1.
Dissociation constants for wild type and Gly-to-Asp-substituted Gα subunit interactions with binding partners
|
KD
(μm)a
|
Gαi1 wild type
|
Gαi1 G202D
|
||||
|---|---|---|---|---|---|---|
| GDP | GTPγS | 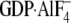 |
GDP | GTPγS | 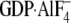 |
|
| Gβ1γ1 | 1.75b (1.7–1.8) | 38 (31–44) | NBc | 1.9 (1.3–2.6) | 2.8 (2.4–3.3) | 2.1 (1.3–2.9) |
| PCP-2 | 0.84 (0.68–1.0) | NB | 448 (130–760) | 7.5 (3.7–11.3) | 7.0 (4.6–9.3) | 7.7 (4.7–10.8) |
| Ric-8A | 0.39 (0.26–0.51) | 356 (41–670) | 107 (68–146) | 4.1 (1.2–7.0) | 5.5 (3.0–8.0) | 3.6 (1.8–5.3) |
| RGS14 (RGS) | NB | 310 (150–470) | 0.08 (0.05–0.1) | NB | NB | NB |
| Gαt/i1 wild type | Gαt/i1 G198D | |||||
| PDEγ (aa 63–87) | NB | NDd | 5.21 | NB | ND | NB |
Dissociation constants (KD in micromolar) were determined using surface plasmon resonance spectroscopy (see representative SPR sensorgrams in Fig. 2). For experiments with Gαi1, equilibrium resonance units of specific binding were graphed versus Gα concentration and fit to the equation Y = (Bmax·X)/(KD + X). Data are presented with 95% confidence intervals in parentheses. For Gαt/i1/PDEγ experiments, affinity was calculated by kinetic analysis (S.E. in parentheses): ka = 1.6 × 102 (0.9) m–1 s–1, kd = 8.33 × 10–4 (0.1) s–1
Note that the dissociation constant (KD) established for the binding of His6-Gαi1·GDP to immobilized Gβ1γ1 is higher than expected from comparable studies given our use of non-lipid-modified Gα and Gβγ subunits as well as a high concentration of free magnesium (Sarvazyan et al. 1998 JBC 273:7934; Higashijima et al. 1987 JBC 262:762)
NB indicates no binding was observed
ND indicates that affinity was not determined
Wild type Gαi1 also exhibited GDP-specific binding to the
GoLoco motif protein PCP-2 and the guanine nucleotide exchange factor Ric-8A
(Fig. 2, C and
E; Table
1), consistent with previous studies
(9,
17,
20). In contrast,
Gαi1(G202D) interacted in a nucleotide-independent manner
with PCP-2 and Ric-8A (Fig. 2, D
and F), and with markedly reduced affinities
versus wild type Gαi1·GDP
(KD for PCP-2 of 7–8 μm
versus 800 nm, KD for Ric-8A of
4–5 μm versus 400 nm;
Table 1). We and others have
previously shown RGS14 and Gαi subunits to be involved in
mammalian spindle formation and orientation (reviewed in Refs.
1,
5). Whereas wild type
Gαi1 exhibited a high affinity,
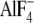 -dependent interaction with the
RGS14 RGS domain (Fig.
2G) as shown previously
(22), no binding was observed
between any form of Gαi1(G202D) and RGS14
(Fig. 2H). As RGS
domains and Gα effectors bind distinct
(44), but at times overlapping
(50), regions of switch II, we
examined if the G202D mutation also altered effector binding in the context of
a chimeric transducin/Gαi1 protein (Gαt/i1)
(18) known to exhibit
activation-dependent binding to a fragment of the γ subunit of cGMP
PDEγ (23). Wild type
Gαt/i1 bound, in an activation-dependent manner, to
immobilized PDEγ peptide as expected
(Fig. 2I;
Table 1); however, no binding
was seen between ground state (nor activated) Gαt/i1(G198D)
and PDEγ (Fig.
2J; Table
1), suggesting that the Gly-to-Asp substitution abrogated the
effector binding properties of Gαt/i1.
-dependent interaction with the
RGS14 RGS domain (Fig.
2G) as shown previously
(22), no binding was observed
between any form of Gαi1(G202D) and RGS14
(Fig. 2H). As RGS
domains and Gα effectors bind distinct
(44), but at times overlapping
(50), regions of switch II, we
examined if the G202D mutation also altered effector binding in the context of
a chimeric transducin/Gαi1 protein (Gαt/i1)
(18) known to exhibit
activation-dependent binding to a fragment of the γ subunit of cGMP
PDEγ (23). Wild type
Gαt/i1 bound, in an activation-dependent manner, to
immobilized PDEγ peptide as expected
(Fig. 2I;
Table 1); however, no binding
was seen between ground state (nor activated) Gαt/i1(G198D)
and PDEγ (Fig.
2J; Table
1), suggesting that the Gly-to-Asp substitution abrogated the
effector binding properties of Gαt/i1.
GPA-16(it143) Interacts with GPR-1/-2 and RIC-8 in a Nucleotide-independent Manner—The binding studies detailed above establish that the G202D mutation within switch II renders Gα unable to interact properly in vitro with many Gα regulators, including those that bind activated states (i.e. RGS domains, effectors). We confirmed that GPA-16(it143) exhibits this lack of proper nucleotide state-selective interactions by co-immunoprecipitation using C. elegans embryo extracts. Using this approach, we previously showed that wild type GPA-16 interacts robustly with the GoLoco motif proteins GPR-1/-2 in the presence of GDP but much less so in the presence of GTPγS, whereas wild type GPA-16 interacts with RIC-8 equally well in the presence of either nucleotide (9). Here we used embryonic extracts from worms grown at 16 °C and conducted co-immunoprecipitation at 16 °C to investigate the behavior of native complexes at the permissive temperature. The interaction between GPA-16(it143) and GPR-1/-2 was decreased compared with wild type GPA-16, whereas that between GPA-16(it143) and RIC-8 was not diminished (Fig. 1E). The interaction between GPA-16(it143) and GPR-1/-2 occurred equally well in the presence of GDP or GTPγS, in contrast to wild type GPA-16 (Fig. 1E). This finding is compatible with the in vitro results of Table 1 and supports the view that GPA-16(it143)·GDP and GPA-16(it143)·GTP can both associate with GPR-1/-2 at the permissive temperature in vivo. Overall, these findings suggest that the G202D substitution renders Gα proteins unable to interact properly in vivo with their regulatory proteins.
Structural Changes Caused by the G202D Mutation—We determined the crystal structure of Gαi1(G202D)·GDP to 2.2 Å (Fig. 3; supplemental Table 1). The overall structure of Gαi1(G202D) is largely unaltered from wild type Gαi1 (51) with an overall root mean square deviation of 0.38 Å (Fig. 3A). Surprisingly, switch I and III of GDP-bound Gαi1(G202D) adopt conformations similar to those of activated conformations of Gα (Fig. 3B). Arg178 and Thr181, important residues to GTP hydrolysis found in switch I (51), do not undergo significant alterations in side-chain conformation compared with activated Gα structures, despite a slight alteration in the switch I backbone conformation (Fig. 3B and data not shown). The conformation of switch II is unique and likely a direct result of the G202D mutation. Although residues Lys208–Trp211 of switch II are disordered, the β3/α2 loop (within which the G202D mutation resides) folds back toward the α2/α3 cleft and away from the nucleotide pocket (Fig. 3A). This altered conformation may underlie enhanced spontaneous GDP release seen with the G202D mutant (Fig. 1B), as the β3/α2 loop is thought to be an occlusive barrier to the release of GDP (2). Additionally, the orientation of the Asp202 side chain toward GDP (Fig. 3C) may introduce an electrostatic repulsion causing enhanced GDP release. Although the Asp202 side chain orients directly at the GDP β-phosphate, the distance separating its electronegative carboxylate from the β-phosphate (∼5 Å) is great enough to prevent an outright electrostatic or steric clash. However, GTP binding would present significant electrostatic and steric clashes with the Asp202 side chain (Fig. 3B). Such a clash may lead to protein instability, an effect likely exacerbated at elevated temperatures.
FIGURE 3.

Structural features of GDP-bound Gαi1(G202D) compared with GDP-bound and GTPγS-bound wild type Gαi1. A, superposition of GDP-bound Gαi1(G202D) (green) with wild type Gαi1·GDP/Gβ1γ2 heterotrimer (Gαi1 (yellow), Gβ1 (gray), Gγ2 (wheat); Protein Data Bank code 1GP2). Aside from the N-terminal helix, Gαi1(G202D) is largely unaltered compared with wild type. However, the β3/α2 loop containing the G202D mutation is displaced from the nucleotide-binding pocket relative to wild type, Gβγ-bound Gαi1. The partially ordered switch II region that proceeds from the β3/α2 loop does not assume the helical nature typical of Gβγ-bound and activated conformations of Gα. B, superposition of Gαi1(G202D)·GDP (green) and Gαi1·GTPγS (yellow; Protein Data Bank code 1AS0). Switch I and III regions are in a similar orientation; however, the orientations of switch II differ dramatically, most notably in the N-terminal portion (i.e. the β3/α2 loop). In wild type Gα, binding of GTPγS induces a rigid helical conformation in switch II (α2) that results in its movement toward the nucleotide-binding pocket. However, in the G202D mutant, switch II is deflected away from the nucleotide. Importantly, the Asp202 side chain demonstrates a significant steric and electrostatic clash with the γ-phosphate of the modeled GTPγS molecule. C, depiction of the GDP-binding pocket illustrating the orientation of the Asp202 side chain relative to GDP. Notably the acidic side chain of Asp202 is oriented directly toward the β-phosphate of GDP. Residues of switch I critical for GTP hydrolysis (Arg178 and Thr181) are shown along with Glu43 in the phosphate-binding loop region. The confidence of the structural model is highlighted by a 2Fo - Fc simulated annealing omit electron density map contoured at 1.0σ (gray mesh).
We also measured Gα conformational change upon activation by GTPγS. Gα subunits contain a tryptophan in switch II (Trp211 in Gαi1 and GPA-16) that shifts from being solvent-exposed when GDP is bound to a hydrophobic pocket when GTPγS is bound (reviewed in Ref. 52). Wild type Gαi1 gave a substantial increase in Trp fluorescence upon exposure to GTPγS (Fig. 4A); in contrast, Gαi1(G202D) gave a minimal increase in fluorescence upon incubation with GTPγS (Fig. 4A). This weak fluorescence enhancement was reproducible and specific (Fig. 4B), albeit severely diminished in magnitude (4% increase over GDP-bound basal fluorescence versus 57% increase of wild type).
FIGURE 4.

The G202D mutation compromises proper switch II rearrangement upon GTPγS binding. A, time course of Gαi1 intrinsic tryptophan fluorescence enhancement by GTPγS. The intrinsic fluorescence of buffer (No Protein), 100 nm wild type (WT) Gαi1, or 100 nm Gαi1(G202D) was measured at 30 °C. 100 μm GTPγS was added at 0 s as indicated by the arrow. RFU = relative fluorescence units. Data were fit to a single exponential association function to determine the activation rate constant (95% confidence intervals in parentheses): WT 0.0139 (0.0137–0.0142) min-1. B, time course of Gαi1(G202D) intrinsic tryptophan fluorescence enhancement by GTPγS. The intrinsic fluorescence of 100 nm Gαi1(G202D) was measured at 30 °C. 100 μm GTPγS was added at 0 s, as denoted. Data were normalized to percentage change in fluorescence. Data were fit to a single exponential association curve (95% confidence interval in parentheses): G202D+GTPγS 0.149 (0.13–0.16) min-1. Data from intrinsic tryptophan fluorescence experiments (as illustrated in A and B; n = 3 independent experiments) were also analyzed to determine mean percent fluorescence enhancement over GDP-bound basal state (with mean ± S.E. in parentheses as follows: wild type Gαi1, 57% (±4%); Gαi1(G202D), 4% (±1%).
Taken together, these findings indicate that Gαi1(G202D) alters the conformation of switch II in response to GTPγS binding, most likely by preventing complete rotation and translation toward the GTP-binding pocket (53, 54). Combined with the observation that switch I and III conformations in Gαi1(G202D)·GDP are similar to activated Gαi1, the G202D-induced changes in switch II conformation help resolve not only the reduced affinity of Gαi1(G202D) seen for certain binding partners (e.g. PCP-2, Ric-8A, and RGS14; Table 1) but also the loss of nucleotide selectivity for such interactions (Fig. 2).
Normal Pulling Forces Seen at the Permissive Temperature in gpa-16(it143) Embryos Are GOA-1-dependent—We also evaluated the consequences of the G202D mutation on GPA-16 function in vivo by analyzing pulling forces on astral microtubules in gpa-16(it143) embryos at 16 °C. In wild type embryos, the posterior aster undergoes characteristic oscillations transverse to the longitudinal axis, reflecting the extent of pulling forces acting on the spindle poles (Fig. 5A and supplemental movie 1) (7). Although oscillations are largely abolished in gpa-16(it143) embryos at 25 °C (Fig. 5C and supplemental movie 3) (9), they are indistinguishable from wild type oscillations in gpa-16(it143) embryos at 16 °C (Fig. 5D and supplemental movie 4), suggesting that pulling forces are intact at the permissive temperature. We conducted in vivo laser microbeam-mediated spindle severing to reveal the extent of net pulling forces acting on each spindle pole (36). Pulling forces in wild type embryos were reduced at 16 °C compared with 25 °C (Fig. 6 and supplemental Table 2), presumably a reflection of a global slowing at the lower temperature of biochemical reactions, such as microtubule dynamics known to be important for pulling force generation (14). However, net pulling forces were not decreased in gpa-16(it143) embryos at 16 °C compared with 25 °C and, more surprisingly, were even slightly increased on the anterior spindle pole in comparison with wild type at 16 °C (Fig. 6). Intact pulling forces in gpa-16(it143) embryos at 16 °C cannot be ascribed to GPA-16 being dispensable for pulling forces at this temperature, because gpa-16(RNAi) embryos at 16 °C did not exhibit oscillations (supplemental Movie 2, Fig. 5B, and supplemental movie 2) and had decreased pulling forces (Fig. 6).
FIGURE 5.

Spindle oscillations occur normally in gpa-16(it143) but not gpa-16(it143)/goa-1(RNAi) embryos at 16 °C. Images from time-lapse differential interference contrast microscopy of wild type (16 °C) (A), gpa-16(RNAi) (16 °C) (B), gpa-16(it143) (25 °C) (C), gpa-16(it143) (16 °C) (D), gpa-16(it143)/goa-1(RNAi) (16 °C) (E), and gpa-16(it143)/goa-1(RNAi) (25 °C) (F) embryos during anaphase (see corresponding supplemental movies 1–6). Black lines depict the distance between the two centrosomes of the spindles, and the dashed circles indicate the position of the posterior spindle pole. Elapsed time is indicated in minutes and seconds; embryos are about 50 μm long, and anterior is to the left, posterior to the right. Note that spindle oscillations occur in both wild type and gpa-16(it143) embryos at 16 °C, but not in embryos of the other genotypes (B, C, E, and F).
FIGURE 6.

Pulling forces on spindle poles at 16 °C are normal in gpa-16(it143) but not gpa-16(it143)/goa-1(RNAi) embryos. Average peak velocities of the anterior (A) and posterior (P) spindle poles (±S.E.) following spindle severing of C. elegans embryos of the indicated genotypes. Experiments were performed as indicated either at 16 °C (this study) or at 25 °C (as previously described in Refs. 7, 9, 10 or in this study for gpa-16(it143)/goa-1(RNAi) embryos). For values and statistical tests, see supplemental Table 2.
To test whether normal pulling forces at 16 °C in gpa-16(it143) embryos may be sustained by GOA-1 function, given that GPA-16 and GOA-1 are partially redundant for force generation (8), we inactivated goa-1 using RNAi in gpa-16(it143) embryos. Oscillations were absent and pulling forces substantially diminished in such embryos at 16 °C (Fig. 5, E and F, and Fig. 6; supplemental Table 2). Thus, the pulling forces observed in gpa-16(it143) embryos at the permissive temperature are entirely GOA-1-dependent. One likely possibility is that GPA-16(it143) at 16 °C has a dominant interfering function, trapping a negative regulator of force generation and thus allowing GOA-1 to generate more extensive pulling forces than normally found in the wild type embryo. As suggested by our biochemical and structural analyses, GPA-16(it143) may be permanently bound to Gβγ because of its inability to change switch II conformation upon GTP binding. Thus, at 16 °C, GPA-16(it143) could act to sequester Gβγ (Fig. 7). Compatible with this view, pulling forces on the anterior spindle pole in gpa-16(it143) embryos at 16 °C are even slightly higher than in wild type embryos, a phenotype reminiscent of depletion of the Gβ subunit GPB-1 (9, 10). The relationship between total levels of Gα and Gβγ is crucial for pulling forces, as depletion of GPB-1 alone or in combination with either GOA-1 or GPA-16 results in exaggerated pulling forces (9). Thus, at the permissive temperature, gpa-16(it143) may be thought of as a gain-of-function allele that increases the amount of GOA-1 freed from Gβγ, thus leading to higher pulling forces.
FIGURE 7.

A model for heterotrimeric G-protein function during C. elegans asymmetric cell division and functions of the GPA-16(it143) temperature-sensitive mutant at both the restrictive and permissive temperatures. A, this model, based on published data (7, 9, 10), assumes that Gα·GDP bound to GPR-1/2 is crucial for pulling force generation. In the wild type embryo, GOA-1·GDP and GPA-16·GDP interact with the GoLoco motif (“GL”) proteins GPR-1/2 at the cell cortex to mediate pulling forces. Gα·GDP/GPR-1/2 concentration is determined by an equilibrium between the levels of free Gα·GDP, the amount of free Gβγ, and the amount of Gα·GDP/Gβγ. For simplicity, we have omitted from this model other regulatory components that likely participate such as RIC-8 (10), RGS-7 (11), and LIN-5 (55). B, GPA-16(it143) is unstable at 25 °C and likely misfolds or has defects in tertiary structure. Loss of functional GPA-16(it143) prevents the formation of GPA-16·GDP/GPR-1/2, thereby decreasing pulling forces. Although not formally tested, it is possible that the loss of functional GPA-16(it143) protein may also lead to an increase in free Gβγ subunits, as illustrated here. This would increase the amount of GOA-1·GDP/Gβγ and consequently reduce the amount of GOA-1·GDP/GPR-1/2, thus decreasing pulling forces. C, GPA-16(it143) is stable at 16 °C and has lost the normal nucleotide-state dependence in its Gβγ and GoLoco motif interactions (denoted “GxP”; see Figs. 1E and 2 and Table 1). This leads to an increased amount of GPA-16/Gβγ, thereby reducing the amount of free Gβγ available for formation of GOA-1·GDP/Gβγ. The consequence of this is an increase in free GOA-1·GDP, a resultant increase in the amount of GOA-1·GDP/GPR-1/2, and thus increased pulling forces. (Although a strong nucleotide-independent interaction of GPA-16(it143) with Gβγ likely explains the observed phenotype at the permissive temperature, we cannot formally rule out that GPA-16(it143) alone (freed of Gβγ sequestration) may be competent for generating pulling forces. This notion is particularly interesting as GPA-16(it143) has appreciable, although nucleotide-state independent, binding to regulatory proteins such as RIC-8 and GPR-1/2. Thus, it is also possible that increased levels of GPA-16/GPR-1/2 at the permissive temperature may cause increased pulling forces.) D, loss of all GOA-1 protein by RNA interference prevents accumulation of GOA-1·GDP/GPR-1/2, thus decreasing pulling forces. Furthermore, increased amounts of free Gβγ sequesters the stable GPA-16(it143) mutant and prevents formation of GPA-16/GPR-1/2 complexes. Thus pulling forces are potentially reduced by two mechanisms.
Conclusions—Our structural, biochemical, and cell biological findings collectively suggest that, at the permissive temperature, GPA-16(it143) is stable but unable to interact properly with crucial regulators, leading to a dominant effect on the Gα-dependent force-generating pathway (Fig. 7). In contrast, at the restrictive temperature, our biochemical and functional analyses suggest that GTP binding destabilizes the protein, leading to a loss of activity. Moreover, enhanced spontaneous GDP release by the G202D mutation may contribute to inactivation at the restrictive temperature by promoting either the nucleotide-free form known to be highly unstable (39, 40) or the GTP-bound form, which we show here is also unstable at high temperature. Overall, at the restrictive temperature, gpa-16(it143) likely behaves as a null allele (Fig. 7B). Accordingly, gpa-16(it143) embryos at the restrictive temperature exhibit a clear reduction in pulling forces much like gpa-16(RNAi) embryos (9, 10). Future studies will help to further clarify the differential biochemistry and spatiotemporal dynamics of GPA-16 and GOA-1 in C. elegans embryos and thus better illuminate the conserved actions of Gα subunits in governing ACD across metazoan evolution.
Supplementary Material
Acknowledgments
We thank Ashutosh Tripathy for assistance with CD, Alex Singer and John Sondek for helpful discussions, and Keith Jones for a reprint.
The atomic coordinates and structure factors (code 2EBC) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grant GM074268 (to D. P. S.). This work was also supported by Swiss National Science Foundation Grant 3100A0-102087 (to P. G.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables 1 and 2 and Movies S1–S6.
Footnotes
The abbreviations used are: GPCR, G-protein coupled receptor; ACD, asymmetric cell division; GST, glutathione S-transferase; RU, resonance unit; GTPγS, guanosine 5′-3-O-(thio)triphosphate; RNAi, RNA interference; PDEγ, γ subunit of phosphodiesterase; cGMP, cyclic guanosine monophosphate.
References
- 1.Siderovski, D. P., and Willard, F. S. (2005) Int. J. Biol. Sci. 1 51-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnston, C. A., and Siderovski, D. P. (2007) Mol. Pharmacol. 72 219-230 [DOI] [PubMed] [Google Scholar]
- 3.Betschinger, J., and Knoblich, J. A. (2004) Curr. Biol. 14 R674-R685 [DOI] [PubMed] [Google Scholar]
- 4.Hampoelz, B., and Knoblich, J. A. (2004) Cell 119 453-456 [DOI] [PubMed] [Google Scholar]
- 5.Willard, F. S., Kimple, R. J., and Siderovski, D. P. (2004) Annu. Rev. Biochem. 73 925-951 [DOI] [PubMed] [Google Scholar]
- 6.Bellaiche, Y., and Gotta, M. (2005) Curr. Opin. Cell Biol. 17 658-663 [DOI] [PubMed] [Google Scholar]
- 7.Colombo, K., Grill, S. W., Kimple, R. J., Willard, F. S., Siderovski, D. P., and Gönczy, P. (2003) Science 300 1957-1961 [DOI] [PubMed] [Google Scholar]
- 8.Gotta, M., and Ahringer, J. (2001) Nat. Cell Biol. 3 297-300 [DOI] [PubMed] [Google Scholar]
- 9.Afshar, K., Willard, F. S., Colombo, K., Siderovski, D. P., and Gönczy, P. (2005) Development (Camb.) 132 4449-4459 [DOI] [PubMed] [Google Scholar]
- 10.Afshar, K., Willard, F. S., Colombo, K., Johnston, C. A., McCudden, C. R., Siderovski, D. P., and Gönczy, P. (2004) Cell 119 219-230 [DOI] [PubMed] [Google Scholar]
- 11.Hess, H. A., Roper, J. C., Grill, S. W., and Koelle, M. R. (2004) Cell 119 209-218 [DOI] [PubMed] [Google Scholar]
- 12.Tall, G. G., and Gilman, A. G. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 16584-16589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du, Q., and Macara, I. G. (2004) Cell 119 503-516 [DOI] [PubMed] [Google Scholar]
- 14.Nguyen-Ngoc, T., Afshar, K., and Gonczy, P. (2007) Nat. Cell Biol. 9 1294-1302 [DOI] [PubMed] [Google Scholar]
- 15.Bergmann, D. C., Lee, M., Robertson, B., Tsou, M. F., Rose, L. S., and Wood, W. B. (2003) Development (Camb.) 130 5731-5740 [DOI] [PubMed] [Google Scholar]
- 16.Willard, F. S., Kimple, A. J., Johnston, C. A., and Siderovski, D. P. (2005) Anal. Biochem. 340 341-351 [DOI] [PubMed] [Google Scholar]
- 17.Willard, F. S., McCudden, C. R., and Siderovski, D. P. (2006) Cell. Signal. 18 1226-1234 [DOI] [PubMed] [Google Scholar]
- 18.Pereira, R., and Cerione, R. A. (2005) J. Biol. Chem. 280 35696-35703 [DOI] [PubMed] [Google Scholar]
- 19.Willard, F. S., and Siderovski, D. P. (2004) Methods Enzymol. 389 320-338 [DOI] [PubMed] [Google Scholar]
- 20.Tall, G. G., Krumins, A. M., and Gilman, A. G. (2003) J. Biol. Chem. 278 8356-8362 [DOI] [PubMed] [Google Scholar]
- 21.Snyder, J. T., Singer, A. U., Wing, M. R., Harden, T. K., and Sondek, J. (2003) J. Biol. Chem. 278 21099-21104 [DOI] [PubMed] [Google Scholar]
- 22.Kimple, R. J., De Vries, L., Tronchere, H., Behe, C. I., Morris, R. A., Gist Farquhar, M., and Siderovski, D. P. (2001) J. Biol. Chem. 276 29275-29281 [DOI] [PubMed] [Google Scholar]
- 23.Johnston, C. A., Lobanova, E. S., Shavkunov, A. S., Low, J., Ramer, J. K., Blaesius, R., Fredericks, Z., Willard, F. S., Kuhlman, B., Arshavsky, V. Y., and Siderovski, D. P. (2006) Biochemistry 45 11390-11400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brenner, S. (1974) Genetics 77 71-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mergny, J. L., and Lacroix, L. (2003) Oligonucleotides 13 515-537 [DOI] [PubMed] [Google Scholar]
- 26.Akima, H. (1970) J. Assoc. Comp. Mach. 17 589-602 [Google Scholar]
- 27.Snow, B. E., Brothers, G. M., and Siderovski, D. P. (2002) Methods Enzymol. 344 740-761 [DOI] [PubMed] [Google Scholar]
- 28.McCudden, C. R., Willard, F. S., Kimple, R. J., Johnston, C. A., Hains, M. D., Jones, M. B., and Siderovski, D. P. (2005) Biochim. Biophys. Acta 1745 254-264 [DOI] [PubMed] [Google Scholar]
- 29.Willard, F. S., Low, A. B., McCudden, C. R., and Siderovski, D. P. (2007) Cell. Signal. 19 428-438 [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 31.Storoni, L. C., McCoy, A. J., and Read, R. J. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 432-438 [DOI] [PubMed] [Google Scholar]
- 32.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 33.Jones, T. A., Zou, J. Y., Cowan, S. W., and Kjeldgaard, M. (1991) Acta Crystallogr. Sect. A 47 110-119 [DOI] [PubMed] [Google Scholar]
- 34.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 35.Gönczy, P., Schnabel, H., Kaletta, T., Amores, A. D., Hyman, T., and Schnabel, R. (1999) J. Cell Biol. 144 927-946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grill, S. W., Gönczy, P., Stelzer, E. H., and Hyman, A. A. (2001) Nature 409 630-633 [DOI] [PubMed] [Google Scholar]
- 37.Streiff, J., Warner, D. O., Klimtchuk, E., Perkins, W. J., Jones, K., and Jones, K. A. (2004) Anesth. Analg. 98 660-667 [DOI] [PubMed] [Google Scholar]
- 38.Linder, M. E., Ewald, D. A., Miller, R. J., and Gilman, A. G. (1990) J. Biol. Chem. 265 8243-8251 [PubMed] [Google Scholar]
- 39.Chidiac, P., Markin, V. S., and Ross, E. M. (1999) Biochem. Pharmacol. 58 39-48 [DOI] [PubMed] [Google Scholar]
- 40.Posner, B. A., Mixon, M. B., Wall, M. A., Sprang, S. R., and Gilman, A. G. (1998) J. Biol. Chem. 273 21752-21758 [DOI] [PubMed] [Google Scholar]
- 41.Iiri, T., Herzmark, P., Nakamoto, J. M., van Dop, C., and Bourne, H. R. (1994) Nature 371 164-168 [DOI] [PubMed] [Google Scholar]
- 42.Spiegel, A. M., and Weinstein, L. S. (2004) Annu. Rev. Med. 55 27-39 [DOI] [PubMed] [Google Scholar]
- 43.Wall, M. A., Posner, B. A., and Sprang, S. R. (1998) Structure (Lond.) 6 1169-1183 [DOI] [PubMed] [Google Scholar]
- 44.Slep, K. C., Kercher, M. A., He, W., Cowan, C. W., Wensel, T. G., and Sigler, P. B. (2001) Nature 409 1071-1077 [DOI] [PubMed] [Google Scholar]
- 45.Kimple, R. J., Kimple, M. E., Betts, L., Sondek, J., and Siderovski, D. P. (2002) Nature 416 878-881 [DOI] [PubMed] [Google Scholar]
- 46.Tesmer, J. J., Sunahara, R. K., Gilman, A. G., and Sprang, S. R. (1997) Science 278 1907-1916 [DOI] [PubMed] [Google Scholar]
- 47.Tesmer, V. M., Kawano, T., Shankaranarayanan, A., Kozasa, T., and Tesmer, J. J. (2005) Science 310 1686-1690 [DOI] [PubMed] [Google Scholar]
- 48.Zwaal, R. R., Ahringer, J., van Luenen, H. G., Rushforth, A., Anderson, P., and Plasterk, R. H. (1996) Cell 86 619-629 [DOI] [PubMed] [Google Scholar]
- 49.Sarvazyan, N. A., Lim, W. K., and Neubig, R. R. (2002) Biochemistry 41 12858-12867 [DOI] [PubMed] [Google Scholar]
- 50.Hepler, J. R., Berman, D. M., Gilman, A. G., and Kozasa, T. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 428-432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Coleman, D. E., Berghuis, A. M., Lee, E., Linder, M. E., Gilman, A. G., and Sprang, S. R. (1994) Science 265 1405-1412 [DOI] [PubMed] [Google Scholar]
- 52.Kimple, R. J., Jones, M. B., Shutes, A., Yerxa, B. R., Siderovski, D. P., and Willard, F. S. (2003) Comb. Chem. High Throughput Screen 6 399-407 [DOI] [PubMed] [Google Scholar]
- 53.Mixon, M. B., Lee, E., Coleman, D. E., Berghuis, A. M., Gilman, A. G., and Sprang, S. R. (1995) Science 270 954-960 [DOI] [PubMed] [Google Scholar]
- 54.Lambright, D. G., Sondek, J., Bohm, A., Skiba, N. P., Hamm, H. E., and Sigler, P. B. (1996) Nature 379 311-319 [DOI] [PubMed] [Google Scholar]
- 55.Srinivasan, D. G., Fisk, R. M., Xu, H., and van den Heuvel, S. (2003) Genes Dev. 17 1225-1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.