

Abstract
The mechanisms that permit adult tissues to regenerate when injured are not well understood. Initiation of liver regeneration requires the injury-related cytokines, tumor necrosis factor (TNF) α and interleukin (IL) 6, and involves the activation of cytokine-regulated transcription factors such as NF-κβ and STAT3. During regeneration, TNFα and IL-6 promote hepatocyte viability, as well as proliferation, because interventions that inhibit either cytokine not only block hepatocyte DNA synthesis, but also increase liver cell death. These observations suggest that the cytokines induce hepatoprotective factors in the regenerating liver. Given evidence that nitric oxide can prevent TNF-mediated activation of the pro-apoptotic protease caspase 3 and protect hepatocytes from cytokine-mediated death, cytokine-inducible nitric oxide synthase (iNOS) may be an important hepatoprotective factor in the regenerating liver. In support of this hypothesis we report that the hepatocyte proliferative response to partial liver resection is severely inhibited in transgenic mice with targeted disruption of the iNOS gene. Instead, partial hepatectomy is followed by increased caspase 3 activity, hepatocyte death, and liver failure, despite preserved induction of TNFα, IL-6, NF-κβ, and STAT3. These results suggest that during successful tissue regeneration, injury-related cytokines induce factors, such as iNOS and its product, NO, that protect surviving cells from cytokine-mediated death.
In adult vertebrates, the capacity for regeneration is limited to a few tissues, such as the liver. Furthermore, in mammals, even tissues that can regenerate sometimes do not regenerate when injured. Achieving a wider range and greater degree of regeneration in injured tissues will require a deeper understanding of the cellular and molecular mechanisms that permit cells to survive and proliferate in a potentially lethal microenvironment (1). Interestingly, the pluripotent, injury-related cytokine, tumor necrosis factor (TNF) α, can induce either hepatocyte death or proliferation, and, thus, is thought to influence both liver injury and liver regeneration (2). Identification of the signals that modulate the cellular response to TNFα may permit the development of treatments that create a regenerative environment in injured tissues.
When large pieces of liver are amputated by two-thirds (partial) hepatectomy (PH), increased local expression of TNFα triggers the production of other cytokines, including interleukin (IL) 6, and is required to initiate subsequent hepatocyte proliferation (3, 4). Post-PH liver regeneration and IL-6 induction are markedly inhibited in mice with targeted disruption of the TNF receptor type 1 (TNFR-1) gene. In addition, such mice exhibit excessive lipid accumulation in hepatocytes and increased mortality after PH (4). IL-6-null mice produce TNFα and express TNFRs (5) but respond to PH like TNFR-1-null mice (6). Interestingly, injection of recombinant TNFα (7), but not IL-6 (4), provokes hepatocyte proliferation in normal healthy mice. However, pretreatment with IL-6 normalizes post-PH liver regeneration and prevents mortality in both TNFR-1-null (4) and IL-6-null mice (6). Taken together, these results suggest that IL-6 may help to induce factors that permit hepatocytes to proliferate, rather than die, after exposure to TNFα.
Injury-related cytokines regulate the transcription of many hepatocyte genes, including inducible nitric oxide synthase (iNOS), the enzyme that catalyzes the formation of NO from arginine (8–13). In vivo footprint analysis of the murine iNOS gene indicates that neither TNFα nor IL-6 alone are sufficient to activate iNOS transcription but that when combined these two cytokines up-regulate iNOS transcription as much as lipopolysaccharide (LPS), a potent iNOS inducer (12, 14). There is growing evidence that NO may prevent TNF-mediated hepatocyte injury in LPS-treated animals and in cultured hepatocytes (15–17). Thus, it is possible that IL-6 may protect hepatocytes in the regenerating liver from TNFα, at least in part, by promoting the induction of iNOS. Others have shown that iNOS mRNAs accumulate in the liver during the prereplicative period after PH (18–21). During this time period, iNOS induction appears to occur preferentially in hepatocytes because NO production was found almost exclusively in these cells, and not in liver nonparenchymal cells that were isolated from rat livers from 1–16 hr after PH (18, 19). There is a striking temporal correlation between peak NO production, iNOS mRNA, and protein accumulation (18–21) and the induction of IL-6, all of which occur in the liver around 3–8 hr after PH. However, the functional implications of iNOS induction in hepatocytes entering the cell cycle is not well understood.
We hypothesize that the induction of iNOS in regenerating liver cells protects them from lethal effects of endotoxemia and increased TNFα, which also occur in the prereplicative period after PH (22, 23). Thus, PH may provoke liver injury, rather than regeneration, when induction of iNOS is prevented. The present study tests this hypothesis by comparing several markers of hepatocyte proliferation and death in transgenic mice with targeted disruption of the iNOS gene and wild-type controls. Regenerative induction of TNFα, IL-6, NF-κβ, and STAT3 also have been evaluated to determine whether these critical components of the cytokine signaling cascades are preserved in the NO-deficient animals.
METHODS
PH Experiments in Mice.
The mice used were MF-1, C129 hybrids, males between 8 and 10 weeks old at the start of the experiments (courtesy of F. Y. Liew, University of Glasgow and C. Lowenstein, Johns Hopkins University). iNOS-null mice and MF-1, C129 hybrid controls of the same age and sex, were subjected to PH according to the method of Higgins and Anderson (24). All experiments were done in accordance with National Institutes of Health and Johns Hopkins University guidelines for the humane use of laboratory animals.
Evaluation of Liver Regeneration.
BrdUrd labeling. To evaluate hepatocyte DNA synthesis, BrdUrd (10 μg/g body weight) was injected i.p. 2 hr before sacrifice. The remnant liver was harvested at 0, 24, 36, 48, 72, and 96 hr after PH, fixed, sectioned and stained with antibodies to BrdUrd. Positive, dark-stained hepatocyte nuclei were counted in 10 different 400× fields/tissue section. Tissue sections from at least 4–6 different iNOS-deficient and a similar number of wild-type control mice were evaluated at each time point after PH.
Hepatocyte mitotic activity.
To evaluate hepatocyte replication, mitotic figures in hepatocytes were counted in 10 different 400× fields from 4–6 mice/group at each time point.
Liver mass regenerated.
At the time of PH, the resected liver was weighed, and this information was used to calculate the initial liver weight for each mouse based on the assumption that the resected liver = 0.70 × (total liver). When mice were sacrificed after PH, the remnant livers were weighed. For each mouse, the weight of the remnant liver was divided by that animal’s initial liver weight to derive the percentage of liver weight that had been reconstituted after PH.
Evaluation of Hepatocellular Damage.
Serum markers of hepatocellular damage. Serum alanine aminotransferase activity and bilirubin concentration were determined by the clinical chemistry laboratory of the Johns Hopkins Hospital in samples from 4–6 iNOS-null mice and their wild-type littermates at several different time points after PH.
Histological evaluation of liver injury.
For morphological assessment, hematoxylin/eosin-stained liver sections were inspected for evidence of liver injury. The number of necro-inflammatory foci were counted in at least five different 400× fields/tissue section. Sections from four different mice/group were evaluated at each time point. TUNEL staining (terminal deoxynucleotidyltransferase-mediated-UTP nick end labeling) in formalin-fixed liver tissue sections was performed to assess the presence of hepatocyte apoptosis as described by Gavreili and associates (25). In brief, the sections were dewaxed in xylene, rehydrated through graded series of alcohols, and washed in double distilled water. After optimal digestion with 20 mg/ml of proteinase K, the sections were incubated with the nick end-labeling solution using digoxigenin-11-dUTP (Boehringer Mannheim) at 37°C for 60 min in a humid chamber. Specific antiserum to digoxigenin conjugated with fluorescein (Boehringer Mannheim) was used to visualize apoptotic nuclei. Negative controls included serial sections incubated without enzyme or substrate. Positive controls were performed in commercially available sections (Promega) of regressing mouse mammary gland. The number of fluorescein-labeled apoptotic nuclei were counted in 10 high-power fields by two independent observers in slides obtained from three animals per time point (24, 36, and 48 hr after PH) in each group.
Activation of the pro-apoptotic protease, caspase 3.
In vivo cleavage of U1–70kD (70-kDa fragment of the U-1 ribonucleosomal protein), a specific substrate for caspase 3, was used as a marker of caspase activity. Liver samples were homogenized in lysis buffer containing 10 mM Hepes/KOH, pH 7.4, 2 mM EDTA, 5 mM DTT, 1% Nonidet P-40, and the protease inhibitors phenylmethylsulfonyl fluoride, antipain, leupeptin, and pepstatin A. Protein concentrations were determined by Bio-Rad assay (Amersham) using BSA as standard (26). Western blot analysis was done to investigate the in vivo cleavage of U1–70kD. One hundred micrograms of protein homogenate was loaded per lane. Samples were electrophoresed on 10% SDS-polyacrylamide gels containing 0.087% bisacrylamide. Cleavage of intact U1–70kD was assayed by immunoblotting with monospecific human sera recognizing U1–70kD and its signature 40-kDa apoptotic fragment (27).
Evaluation of Cytokines.
Cytokine expression. Serum concentrations of TNFα and IL-6 were measured by commercially available ELISAs (BioSource International, Camarillo, CA). Samples from 4–6 different mice/group were evaluated at each time point. Each assay was performed in triplicate with murine recombinant cytokine as standard. In addition, separate iNOS-deficient and wild-type mice were injected i.p. with 3 ml of thioglycollate, and 4 days later peritoneal macrophages were harvested. Cells were cultured overnight in DMEM with 10% fetal bovine serum and then Escherichia coli-derived lipopolysaccharide 0111:B4 (Sigma) (1 mg/ml) was added to half of the cultures. After 90 min, the medium was removed and the cells were harvested. Total RNA was isolated from the cell pellets as described (26), and aliquots (20 mg/lane) were separated by electrophoresis on 1% agarose gels under denaturing conditions. After transfer to nylon membranes by capillary blotting, blots were hybridized overnight with cDNA probes to either murine TNFα (obtained from Bruce Beutler, Univ. of Texas Southwestern, Dallas; ref. 28) or murine IL-6 (obtained from Genaro Ciliberto, Istituto di Ricerche di Biologia Molecolare, Pompezia, Rome) (29). Blots were washed under stringent conditions and exposed to Kodak film with intensifying screens. Results were obtained by laser scanning densitometry (Molecular Dynamics) and normalized to the 18S band for each individual lane.
Cytokine-regulated transcription factors.
Liver nuclear proteins were isolated at various time points after PH according to the methods of Lavery and Schibler (30) as described (26). Electrophoretic mobility-shift assays were performed to evaluate the DNA binding activities of NF-κβ and STAT3 (31). The 32P-labeled, double-stranded oligonucleotide probe used in the NF-κβ assays was the NF-κβ consensus sequence in the Ig enhancer (26). STAT3 assays used a 32P-labeled, double-strand, 20-bp oligonucleotide that included the cis-inducible element binding sites. One milliliter of commercially available (Santa Cruz Biotechnology) antisera to NF-κβ p50 or p65 or to total STAT3 or phosphorylated STAT3 was added to some reaction mixtures to evaluate the specificity of DNA complex formation by supershift analysis. Eight milligrams of nuclear protein was loaded per lane. STAT3 protein levels were analyzed by Western blot, as described (26, 31). In brief, proteins were separated on 12% SDS/PAGE and transferred onto Immobilon-P membranes (Millipore). The blots were probed with commercially available specific antisera (Santa Cruz Biotechnology) for either total or phosphorylated STAT3, and proteins were visualized by the ECL detection system (Amersham). Blots obtained from 3–4 unique experiments were evaluated by scanning laser densitometry (Molecular Dynamics).
RESULTS
Eight- to 10-week-old, male iNOS-null mice (32) and wild-type (MF1, C129 hybrid) mice of the same sex and age were subjected to PH (24). Mice of similar genetic background were used to minimize potential inter-strain differences in the response to liver injury or PH. Liver weight, histology, and serum aminotransferase levels were normal in iNOS-null mice before PH. Mortality during the initial 96 hr after PH was only slightly higher in the iNOS-null mice (13 ± 2%) than in controls (4 ± 2%). However, liver regeneration was significantly inhibited in all surviving iNOS-null mice, as evidenced by decreased hepatocyte incorporation of BrdUrd (Fig. 1 A and B), mitoses (Fig. 1C), and restitution of liver mass (Fig. 1D). In addition, compared with control mice that developed minor (grade 1) periportal lipid accumulation after PH, iNOS-null mice developed severe (grade 3–4) panlobular microvesicular steatosis in hepatocytes within 24–48 h after PH (Fig. 2). This steatosis was accompanied by histologic evidence of liver injury variously manifested as necrotic foci, polymorphonuclear cell infiltration, and hepatocyte apoptosis on hematoxylin/eosin-stained liver sections (Fig. 3 A–C). The presence of increased apoptosis was confirmed by TUNEL staining, which clearly illustrated nuclear fragmentation in fluorescein isothiocyanate-labeled hepatocytes and nonparenchymal cells (Fig. 4A). A graphical representation of the increased numbers of apoptotic liver cells on TUNEL-stained sections is shown in Fig. 4B, demonstrating a significant increase in apoptosis at 24 hr in iNOS-null mice when compared with controls.
Figure 1.
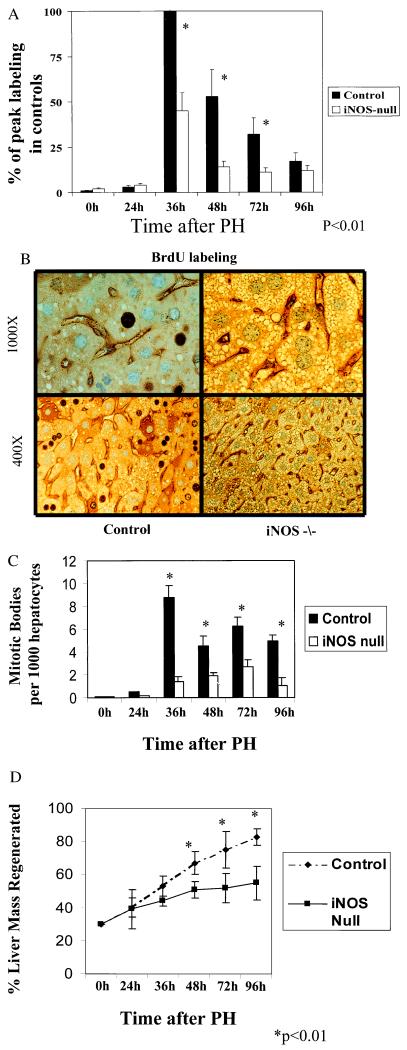
Liver regeneration is inhibited in iNOS-null mice. (A) The average number of BrdUrd-labeled hepatocytes was determined for each mouse by counting the number of positive hepatocytes in 10 different fields. Samples from 4–6 different mice/group were evaluated at each time point. Results shown are the mean ± SE of the averages from 4–6 mice/group per time point and are expressed as a percentage of the peak BrdUrd labeling in control mice. (B) Representative photomicrographs are shown from control and iNOS-deficient mice at two different magnifications [×400 (Lower) and ×1,000 (Upper)]. The dark-stained nuclei indicate BrdUrd-labeled hepatocytes. (C) Hepatocyte mitoses are expressed as the number of mitotic bodies/100 hepatocytes, and results are shown as the mean ± SE for each group. (D) Liver weights were used as another measure of liver regeneration. Results shown are the mean ± SE of data from 4–6 different mice/group per time point.
Figure 2.
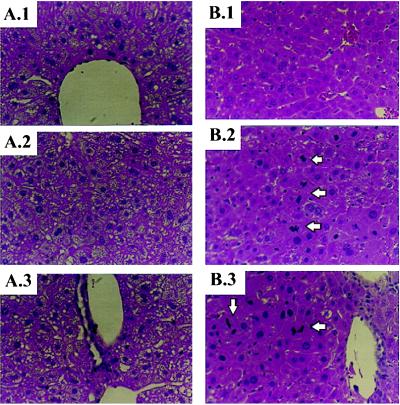
iNOS-deficient mice develop excessive lipid accumulation in hepatocytes after PH. Representative photomicrographs of hematoxylin/eosin-stained sections of livers from iNOS-deficient mice (A) and control mice (B) 48 hr after PH. (A.1 and B.1) Zone 3 (pericentral region) of the hepatic acinus. (A.2 and B.2) Mid-acinar region (zone 2). (A.3 and B.3) Zone 1 (periportal region) of the hepatic acinus. Note that mitotic figures (arrows) are easily identified in the periportal and mid-acinar regions of the livers of control mice but are not apparent when similar fields are inspected in iNOS-deficient livers. (Magnification = ×400.)
Figure 3.
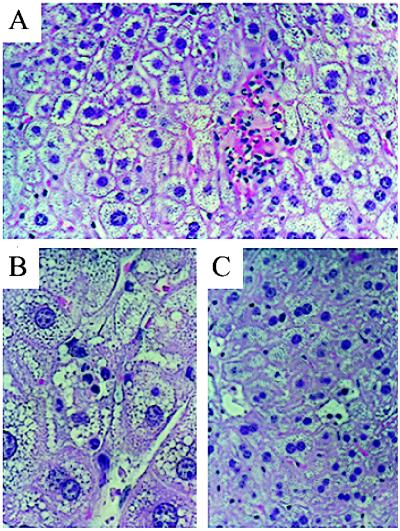
PH leads to foci of liver injury. Liver injury in iNOS-deficient mice may be manifest on hematoxylin/eosin-stained sections as (A) focal necrotic areas with hemorrhage and polymorphonuclear cell infiltration, (B) diffuse steatosis with inflammatory cell infiltration, or (C) distinct morphologic features of hepatocyte apoptosis with chromatin margination and/or condensation. (Magnification: A and C, ×400; B, ×1,000).
Figure 4.
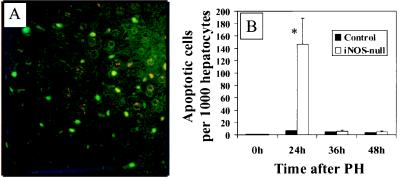
PH leads to hepatocyte apoptosis in iNOS-deficient mice. (A) Representative ×400 photomicrograph from TUNEL-stained liver sections from an iNOS-deficient mouse 24 hr after PH. Apoptotic cells are indicated by bright green fluorescence (FITC+). (B) Increased apoptotic activity in hepatocytes after PH in iNOS-deficient mice. A graphical representation of apoptotic activity in liver tissue sections obtained at 24, 36, and 48 hr from control and iNOS-deficient mice is shown. The percentage of hepatocytes with positive staining by TUNEL and classic features of chromatin margination and/or condensation and nuclear fragmentation are depicted on the x axis.
The extent and predominant type of liver cell death varied dramatically with time. The number of apoptotic liver cells in iNOS-null mice was only greater than controls at 24 hr post-PH. TUNEL stains at that time point demonstrated increased apoptosis in both hepatocytes and liver nonparenchymal cells. By 36 hr after PH, apoptosis was rare in both iNOS-null and control mice. However, small foci of necrotic-appearing hepatocytes were evident in iNOS-null animals, but not in controls from 36 to 48 hr post-PH. Areas of necrosis often were associated with focal hemorrhage. Liver infiltration with inflammatory cells was sparse and tended to colocalize with the small isolated foci of necrotic hepatocytes. Consistent with this histologic evidence of liver injury, iNOS-null mice exhibited elevated alanine aminotransferase activity in serum and features of liver failure, including hyperbilirubinemia (Table 1), which was severe enough to produce overt jaundice in the surviving mice (data not shown).
Table 1.
Alanine aminotransferase and bilirubin levels in sera from iNOS-deficient and control mice
| Time after PH | Serum bilirubin
|
Serum ALT
|
||
|---|---|---|---|---|
| Control | iNOS-null | Control | iNOS-null | |
| 0 h | 0.2 ± 0.05 | 0.25 ± 0.04 | 33 ± 8 | 30 ± 5 |
| 24 h | 1.2 ± 0.3 | 4.4 ± 1.1* | 987 ± 67 | 714 ± 105 |
| 36 h | 1.8 ± 0.6 | 4.9 ± 1.2* | 854 ± 221 | 1,752 ± 352* |
| 48 h | 0.9 ± 0.4 | 6.4 ± 2.1* | 360 ± 76 | 589 ± 82* |
| 72 h | 0.6 ± 0.3 | 18.5 ± 4.9* | 121 ± 42 | 477 ± 79* |
| 96 h | 0.5 ± 0.1 | 17.2 ± 6.2* | 54 ± 21 | 402 ± 63* |
Sera were obtained from iNOS-deficient and control mice at several different time points after PH (n = 4 or more mice/group per time point). ∗, P < 0.01 iNOS-deficient group vs. control group at the same time point. ALT, alanine aminotransferase.
Although the appearance of hepatic steatosis and decreased hepatocyte proliferation in the iNOS-null mice resembled similar features that were reported in TNFR-1-null (4) and IL-6-null (6) mice after PH, regenerative induction of TNFα and IL-6 in iNOS mice was not impaired. In fact, serum concentrations of these cytokines were 2- to 5-fold greater than those of control mice at every post-PH time point evaluated. Consistent with this finding, cultured peritoneal macrophages from healthy iNOS-null mice expressed more TNFα and IL-6 mRNAs than controls after lipopolysaccharide stimulation (Fig. 5<zct;F5). Thus, there is no evidence that iNOS-null mice were deficient in either of the cytokines that are required for liver regeneration. Furthermore, post-PH induction of the cytokine-regulated transcription factors (NF-κβ and STAT3) occurred normally in the livers of the iNOS-null animals. As shown in Fig. 6, the DNA binding activity of STAT3 (Fig. 6A) and intrahepatic induction of STAT3 protein (Fig. 6B) after PH in iNOS-null mice was comparable to control animals at all time points evaluated. The DNA binding activity of NF-κβ was also similar in iNOS-null mice and controls at all time points evaluated (data not shown). These results indicate that the immediate downstream signaling pathways of TNFα and IL-6 were unaffected by iNOS deficiency and responded normally to induction by PH.
Figure 5.

Macrophage production of TNFα and IL-6 is not decreased in iNOS-null mice. Autoradiographs of representative Northern blots for TNFα (Top) and IL-6 (Middle) are shown. (Bottom) The 18S RNA band on the same ethidium-bromide-stained blot.
Figure 6.

Induction of STAT3 in iNOS-null mice at 3 hr after PH occurs normally, when compared with controls. (A) STAT3 DNA binding activity was determined by electrophoretic mobility-shift assay (EMSA) and supershift assays at various time points after PH. Maximal STAT3 binding activity occurred at 3 hr post-PH. This representative EMSA depicts STAT3 binding activity at baseline (time 0) and 3 hr after PH in controls and iNOS-null animals. Lanes 1 a-c contain time 0 extracts from control animals with added preimmune sera (a), total STAT3 antibody (b), or phosphorylated STAT3 antibody (c). Lanes 2 a-c contain extracts from controls at 3 hr post-PH in a similar order; lanes 3 a-c and 4 a-c contain extracts from iNOS-null animals at time 0 and 3 hr post-PH, respectively. (B) Total and phosphorylated STAT3 protein levels were determined by Western blot assay in control and iNOS-null mice at baseline (time 0), 30 min, and 3 hr after PH. Lanes 1 and 2 represent two individual animals used for the experiment at each time point.
Subsequent efforts were directed toward identifying potential mechanisms that may contribute to post-PH liver injury in the iNOS-null mice. Because several groups have reported that NO inhibits TNF-dependent activation of caspase 3 (15–17, 33, 34), and this protease is known to be a proximal mediator of TNF-initiated cell death (17, 35), caspase 3 activity was compared in iNOS-null mice and controls at several time points after PH. Fig. 7 demonstrates that after PH, the caspase 3 substrate, Ul-70kD, is cleaved to generate the signature apoptotic 40-kDa fragment in the livers of iNOS-null mice. Such proteolysis is not observed in control livers after PH. Because both TNFα (22, 23) and NO production (18–21) normally increase in the regenerating liver after PH, the latter finding suggests that one function of iNOS-generated NO is to prevent TNFα from activating caspase 3 in regenerating hepatocytes. Although not proven, this interpretation is supported by the finding that caspase 3 activity is increased after PH in iNOS-deficient mice.
Figure 7.

PH induces proteolytic cleavage of a constitutively expressed 70-kDa hepatic protein in iNOS-null mice. To investigate hepatocyte apoptotic activity, in vivo cleavage of U1–70kD was assayed by immunoblotting with monospecific human sera recognizing U1–70kD. A signature 40-kDa peptide indicates apoptotic cleavage of the intact protein. Results shown are representative of those found in four different mice/group at 24 or 36 hr after PH. In this blot, each lane contains a sample from a different mouse at 36 hr after PH.
DISCUSSION
This comparison of the regenerative response to liver resection in transgenic iNOS-deficient mice and control mice with a similar genetic background provides evidence that regeneration requires the induction of factors that protect proliferating cells from death. This finding is likely to have general relevance and may help to explain why some tissues can regenerate themselves after injury, while most others cannot replace dead cells and heal by scarring. The present results are particularly informative when interpreted in the context of abundant recent evidence that injury-related cytokines are required for hepatocytes to re-enter the cell cycle after the liver has been damaged. Studies in mice treated with neutralizing anti-TNF antibodies before PH and in hepatectomized mice with targeted disruption of the TNFR-1 gene indicate that TNFα plays a crucial role in activating growth-regulatory kinase cascades and several growth-related immediate early genes in the early prereplicative period after PH (3, 4). Apparently both TNFR-1 and TNFR-2 are involved in this response and work by Fausto’s group (4) suggests that, although TNFR-1 activation is essential for hepatocyte proliferation, there is some redundancy in the signaling initiated by TNFR-2 in this setting (53). For example, both receptors are capable of mediating the activation of NF-κβ (36).
In addition to initiating growth-related signaling in hepatocytes, TNFα plays a major role in the induction of IL-6 that occurs after PH (3, 4). However, studies by Cressman and colleagues (6) prove that TNFα is not sufficient for liver regeneration because regeneration is inhibited in transgenic mice that lack IL-6. Interestingly, PH leads to increased hepatic steatosis and high rates of mortality in both TNFR-1-null and IL-6 null mice (4, 6). Neither of the previous studies of those transgenic animals reported whether or not PH increased hepatocyte apoptosis. However, both document that supplemental IL-6 restored the hepatocyte proliferative response and completely prevented PH-related mortality.
More recently, Brenner and colleagues (37) reported that enforced overexpression of I-κβ inhibited NF-κβ activation and resulted in massive hepatocyte apoptosis after PH (37). Because regenerative activation of NF-κβ is attenuated in TNFR-1-null mice, the former may have contributed to increased liver injury and post-PH mortality in TNFR-1-null mice. However, it is difficult to explain why supplemental IL-6 protected those animals, because IL-6 is not known to activate NF-κβ. Similarly, IL-6-null mice would not be expected to have decreased induction of NF-κβ after PH because TNFα is predominantly responsible for inducing NF-κβ in the regenerating liver (4) and IL-6 null mice express TNF and TNFRs. Yet, similar to TNFR-1-null mice, IL-6-null mice develop liver injury and death after PH (6). These paradoxical observations may be reconciled by evidence generated during the present work, which demonstrates that the cytokine-inducible gene, iNOS, is necessary to achieve the normal regenerative response to PH.
In vivo footprint analysis of the iNOS gene indicates that neither TNFα nor IL-6 is capable of inducing iNOS transcription as a single agent. However, a combination of these two cytokines up-regulates iNOS transcription as much as lipopolysaccharide (14). These observations are consistent with evidence that both NF-κβ (a TNF-activated factor) and IRF-1 (an IL-6 inducible, STAT3-dependent factor) are required to induce iNOS gene transcription (11–14, 38). iNOS expression and activity normally increase in the liver during the prereplicative period after PH (18–21). However, increases in iNOS mRNAs do not occur after PH until around the time that the IL-6-inducible transcription factors STAT3 and IRF-1 are known to increase (20, 21). Previous studies using l-NAME, a nonspecific NOS inhibitor, indicated that NO is involved in the regenerative response because inhibition of NOS resulted in decreased hepatocyte proliferation after PH (39). The present study suggests why NO is important for liver regeneration by demonstrating that hepatocytes undergo necrosis and apoptosis instead of proliferating when the induction of iNOS is prevented in the damaged liver. These negative outcomes occur in iNOS-deficient animals despite an abundance of the two cytokines (TNF and IL-6) that are necessary for the initiation of the proliferative response to liver resection and despite preservation of the cytokine-initiated signals that lead to NF-κβ and STAT3 activation during early liver regeneration.
NO is an extremely reactive molecule and is known to have pleiotropic effects (9, 32, 40, 41). Thus, it is conceivable that it has many actions during liver regeneration. Recently described growth-regulatory actions of NO may be particularly relevant in this setting. For example, NO has been shown to activate extracellular signal-regulated kinases (Erks 1 and 2) in vascular endothelial cells (42). It is also capable of inhibiting the activation of NF-κβ in neuronal cells (43). Because our iNOS-deficient mice developed evidence of massive liver injury after PH, we focused on specific well-documented mechanisms that are thought to explain how NO can prevent cell death.
In some cell types, including hepatocytes, NO is known to protect against apoptotic death induced by TNF (17, 34, 44). During apoptosis, pro-caspase 3 is cleaved to a biologically active protease in which Cys-163, a highly conserved amino acid among IL-1 β-converting enzyme-like and the caspase subfamily of cysteine proteases, is required for full proteolytic function (45). NO is known to nitrosylate Cys-163 and, thus, can inhibit the activity of caspase 3 by a direct posttranslational mechanism (34). In many cells, NO also promotes the accumulation of cGMP, and treatment with cGMP has been shown to inhibit TNF-dependent induction of caspase 3 activity, whereas treatment with a cGMP-dependent kinase inhibitor abolishes this effect of NO. Thus, NO also may prevent caspase 3 activation via a cGMP-dependent mechanism (17). Given growing evidence that caspase 3 plays a critical role in TNF-mediated hepatocyte death (46, 47), caspase 3 seems to be a logical target for NO in the regenerating liver, where hepatocytes are exposed to increased levels of TNFα (22, 23, 48). Consistent with this possibility, we found that caspase 3 activity is increased in transgenic mice with targeted disruption of the iNOS gene compared with control mice, which presumably exhibit induction of iNOS during their normal regenerative response. U1–7OkD is a substrate for caspase 3 (27), and therefore an excellent marker for the activity of this enzyme. However, this assay does not permit elucidation of the precise mechanism by which iNOS deficiency results in increased caspase 3 activity after PH. Either decreased nitrosylation of Cys-163 or inhibition of cGMP-regulated suppression of caspase 3 activity could account for this result. Furthermore, because we did not specifically compare all of the conceivable responses that may have been influenced by NO deficiency in iNOS-null and control mice, it is impossible to exclude contributions from other potential intracellular effects (e.g., inhibited extracellular signal-regulated kinase activation) or extracellular effects (e.g., changes in liver blood flow) of NO deficiency. Indeed, because hepatocyte necrosis and steatosis also were increased in iNOS-null mice suggests that NO has many effects in the regenerating liver, because steatosis and the two types of cell death each may be mediated by a different mechanism. However, there is no doubt that the prevention of iNOS induction has a profound effect on the liver’s ability to regenerate after it has been damaged. In the absence of iNOS, PH provokes predominantly hepatocyte death instead of the typical increase in hepatocyte proliferative activity. Nonetheless, many iNOS mice survived this insult and exhibited some evidence of liver regeneration, although it was delayed and attenuated. The latter suggests that other protective mechanisms can compensate, at least partially, for the NO deficiency in this setting. Other candidate cytokine-inducible protective factors include bcl-x and Hsp-70, which are induced around the same time that iNOS mRNAs and activity increase after PH (49, 50).
The ability of cytokine-inducible molecules, like NO, to “gate” the actions of cytokines that accumulate during tissue damage may have broad implications. In organisms that are as divergent as urodeles and humans, tissue regeneration depends on the local extracellular environment after amputation or wounding and the ability of proliferation-competent cells to re-enter the cell cycle from the differentiated state. However, regardless of the species or organ, successful regeneration of damaged tissues also requires a temporary suspension of normal, adult, homeostatic mechanisms that restrict neoplasia by coupling cell cycle re-entry to cell death (51, 52). Evidence that iNOS activity is an important prerequisite for net liver growth after partial hepatic resection suggests that NO may help to convert an injury environment into one that favors regeneration by aborting the activation of molecules that promote cell death.
ABBREVIATIONS
- iNOS
inducible NO synthase
- PH
partial hepatectomy
- TNF
tumor necrosis factor
- TNFR
TNF receptor
- IL
interleukin
- TUNEL
terminal deoxynucleotidyltransferase-mediated-UTP nick end labeling
- U1–70kD = 70-kDa fragment of the U-1 ribonucleosomal protein.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Stocum D L. Science. 1997;276:15. doi: 10.1126/science.276.5309.15. [DOI] [PubMed] [Google Scholar]
- 2.Tracey K J, Cerami A. Annu Rev Cell Biol. 1993;9:317–343. doi: 10.1146/annurev.cb.09.110193.001533. [DOI] [PubMed] [Google Scholar]
- 3.Akerman P, Cote P, Yang S Q, McClain C, Nelson S, Bagby G J, Diehl A M. Am J Physiol. 1992;263:G579–G585. doi: 10.1152/ajpgi.1992.263.4.G579. [DOI] [PubMed] [Google Scholar]
- 4.Yamada Y, Kirillova I, Peschon J J, Fausto N. Proc Natl Acad Sci USA. 1997;94:1441–1446. doi: 10.1073/pnas.94.4.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, Faggioni R, Luini W, van Hinsbergh V, Sozzani S, et al. Immunity. 1997;6:315–325. doi: 10.1016/s1074-7613(00)80334-9. [DOI] [PubMed] [Google Scholar]
- 6.Cressman D E, Greenbaum L E, DeAngelis R A, Ciliberto G, Furth E E, Poli V, Taub R. Science. 1996;274:1369–1383. doi: 10.1126/science.274.5291.1379. [DOI] [PubMed] [Google Scholar]
- 7.Mealy K, Wilmore D W. Br J Surg. 1991;78:331–333. doi: 10.1002/bjs.1800780320. [DOI] [PubMed] [Google Scholar]
- 8.Aono K, Isobe K, Kiuchi K, Fan Z H, Ito M, Takeuchi A, Miyachi M, Nakashima I, Nimura Y. J Cell Biochem. 1997;65:349–358. [PubMed] [Google Scholar]
- 9.Hierholzer C, Harbrecht B, Menezes J M, Kane J, MacMicking J, Nathan C F, Peitzman A B, Billiar T R, Tweardy D J. J Exp Med. 1998;187:917–928. doi: 10.1084/jem.187.6.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geller D A, Lowenstein C J, Shapiro R A, Nussler A K, Di Silvio M, Wang S C, Nakayama D K, Simmons R L, Snyder S H, Billiar T R. Proc Natl Acad Sci USA. 1993;90:3491–3495. doi: 10.1073/pnas.90.8.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldring C E, Narayanan R, Lagadec P, Jeannin J F. Biochem Biophys Res Commun. 1995;209:73–79. doi: 10.1006/bbrc.1995.1472. [DOI] [PubMed] [Google Scholar]
- 12.Goldring C E, Reveneau S, Algarte M, Jeannin J F. Nucleic Acids Res. 1996;24:1682–1687. doi: 10.1093/nar/24.9.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinugawa K, Takahashi T, Kohmoto O, Yao A, Aoyagi T, Momomura S, Hirata Y, Serizawa T. Circ Res. 1994;75:285–295. doi: 10.1161/01.res.75.2.285. [DOI] [PubMed] [Google Scholar]
- 14.Spink J, Evans T. J Biol Chem. 1997;272:24417–24425. doi: 10.1074/jbc.272.39.24417. [DOI] [PubMed] [Google Scholar]
- 15.Bohlinger I, Leist M, Barsig J, Uhlig S, Tiegs G, Wendel A. Toxicol Lett. 1995;82–83:227–231. doi: 10.1016/0378-4274(95)03480-3. [DOI] [PubMed] [Google Scholar]
- 16.Bohlinger I, Leist M, Barsig J, Uhlig S, Tiegs G, Wendel A. Hepatology. 1995;22:1829–1837. [PubMed] [Google Scholar]
- 17.Kim Y M, Talanian R V, Billiar T R. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 18.Obolenskaya M, Vanin A F, Mordvintcev P I, Mulsch A, Decker K. Biochem Biophys Res Commun. 1994;202:571–576. doi: 10.1006/bbrc.1994.1966. [DOI] [PubMed] [Google Scholar]
- 19.Obolenskaya M, Schulze-Specking A, Plaumann B, Frenzer K, Freudenberg N, Decker K. Biochem Biophys Res Commun. 1994;204:1305–1311. doi: 10.1006/bbrc.1994.2605. [DOI] [PubMed] [Google Scholar]
- 20.Diaz-Guerra M J, Velasco M, Martin-Sanz P, Bosca L. Biochem J. 1997;326:791–797. doi: 10.1042/bj3260791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diaz-Guerra M J M, Velasco M, Martin-Sanz P, Bosca L. J Biol Chem. 1996;271:30114–30120. doi: 10.1074/jbc.271.47.30114. [DOI] [PubMed] [Google Scholar]
- 22.Rai R M, Yang S Q, McClain C, Karp C L, Klein A S, Diehl A M. Am J Physiol Gastrointestinal Liver Physiol. 1996;33:G909–G918. doi: 10.1152/ajpgi.1996.270.6.G909. [DOI] [PubMed] [Google Scholar]
- 23.Rai R M, Loffreda S, Karp C L, Yang S Q, Lin H Z, Diehl A M. Hepatology. 1997;25:889–895. doi: 10.1002/hep.510250417. [DOI] [PubMed] [Google Scholar]
- 24.Higgins G M, Anderson R M. Arch Pathol. 1931;12:186–202. [Google Scholar]
- 25.Gavreili Y, Sherman Y, Ben-Sasson S. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin M, Yang S Q, Lin H Z, Lane M D, Chatterjee S, Diehl A M. J Biol Chem. 1996;271:17974–17978. doi: 10.1074/jbc.271.30.17974. [DOI] [PubMed] [Google Scholar]
- 27.Casciola-Rosen L, Nicholson D W, Chong T, Rowan K R, Thornberry N A, Miller D K, Rosen A. J Exp Med. 1996;183:1957–1964. doi: 10.1084/jem.183.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caput D, Beutler B, Hartog K, Thayer R, Brown-Shimer S, Cerami A. Proc Natl Acad Sci USA. 1986;83:1670–1674. doi: 10.1073/pnas.83.6.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poli V, Balena R, Fattori E, Markatos A, Yamamoto M, Tanaka H, Ciliberto G, Rodan G A, Costantini F. EMBO J. 1994;13:1189–1196. doi: 10.1002/j.1460-2075.1994.tb06368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lavery D J, Schibler U. Genes Dev. 1993;7:1871–1884. doi: 10.1101/gad.7.10.1871. [DOI] [PubMed] [Google Scholar]
- 31.Zeldin G, Yang S Q, Yin M, Lin H Z, Rai R, Diehl A M. Alcohol Clin Exp Res. 1996;20:1639–1645. doi: 10.1111/j.1530-0277.1996.tb01710.x. [DOI] [PubMed] [Google Scholar]
- 32.Wei X Q, Charles I G, Smith A, Ure J, Feng G J, Huang F P, Xu D, Muller W, Moncada S, Liew F Y. Nature (London) 1995;375:408–411. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]
- 33.Mohr S, Zech B, Lapetina E G, Brune B. Biochem Biophys Res Commun. 1997;238:387–391. doi: 10.1006/bbrc.1997.7304. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Billiar T R, Talanian R V, Kim Y M. Biochem Biophys Res Commun. 1997;240:419–424. doi: 10.1006/bbrc.1997.7672. [DOI] [PubMed] [Google Scholar]
- 35.Inayat-Hussain S H, Couet C, Cohen G M, Cain K. Hepatology. 1997;25:1516–1526. doi: 10.1002/hep.510250634. [DOI] [PubMed] [Google Scholar]
- 36.Mukherjee R, Singh S, Chaturvedi M M, Aggarwal B B. J Interferon Cytokine Res. 1998;18:117–123. doi: 10.1089/jir.1998.18.117. [DOI] [PubMed] [Google Scholar]
- 37.Iimuro Y, Nishiura T, Hellerbrand C, Behrns K E, Schoonhoven R, Grisham J W, Brenner D A. J Clin Invest. 1998;101:802–811. doi: 10.1172/JCI483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hecker M, Preiss C, Schini-Kerth V B. Br J Pharmacol. 1997;120:1067–1074. doi: 10.1038/sj.bjp.0701026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hortelano S, Dewez B, Genaro A M, Diaz-Guerra M J, Bosca L. Hepatology. 1995;21:776–786. [PubMed] [Google Scholar]
- 40.Parratt J R. J Physiol Pharmacol. 1997;48:493–506. [PubMed] [Google Scholar]
- 41.Rockey D C, Chung J J. Am J Physiol. 1997;273:G124–G130. doi: 10.1152/ajpgi.1997.273.1.G124. [DOI] [PubMed] [Google Scholar]
- 42.Parenti A, Morbidelli L, Cui X L, Douglas J G, Hood J D, Granger H J, Ledda F, Ziche M. J Biol Chem. 1998;273:4220–4226. doi: 10.1074/jbc.273.7.4220. [DOI] [PubMed] [Google Scholar]
- 43.Togashi H, Sasaki M, Frohman E, Taira E, Ratan R R, Dawson T M, Dawson V L. Proc Natl Acad Sci USA. 1997;94:2676–2680. doi: 10.1073/pnas.94.6.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor B S, Kim Y M, Wang Q, Shapiro R A, Billiar T R, Geller D A. Arch Surg. 1997;132:1177–1183. doi: 10.1001/archsurg.1997.01430350027005. [DOI] [PubMed] [Google Scholar]
- 45.Dimmeler S, Haendeler J, Nehls M, Zeiher A M. J Exp Med. 1997;185:601–607. doi: 10.1084/jem.185.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patel T, Gores G J. Hepatology. 1995;21:1725–1741. doi: 10.1002/hep.1840210635. [DOI] [PubMed] [Google Scholar]
- 47.Jones B A, Gores G J. Am J Physiol. 1997;273:G1174–G1188. doi: 10.1152/ajpgi.1997.273.6.G1174. [DOI] [PubMed] [Google Scholar]
- 48.Loffreda S, Rai R, Yang S Q, Lin H Z, Diehl A M. Gastroenterology. 1997;112:2089–2098. doi: 10.1053/gast.1997.v112.pm9178702. [DOI] [PubMed] [Google Scholar]
- 49.Dimmeler S, Zeiher A M. Nitric Oxide. 1997;1:275–281. doi: 10.1006/niox.1997.0133. [DOI] [PubMed] [Google Scholar]
- 50.Wong H R, Ryan M, Wispe J R. Biochem Biophys Res Commun. 1997;231:257–263. doi: 10.1006/bbrc.1997.6076. [DOI] [PubMed] [Google Scholar]
- 51.Michalopoulos G K, DeFrances M C. Science. 1997;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 52.Brockes J P. Science. 1997;276:81–87. doi: 10.1126/science.276.5309.81. [DOI] [PubMed] [Google Scholar]
- 53.Yamada Y, Webber E M, Kirillova I, Peschon J J, Fausto N. Hepatology. 1998;28:959–970. doi: 10.1002/hep.510280410. [DOI] [PubMed] [Google Scholar]