

Abstract
Accumulation of unfolded or malfolded proteins induces endoplasmic reticulum (ER) stress which elicits a complex network of interacting and parallel responses that dampen the stress. The ER stress response in the liver is controlled by intrinsic feedback effectors and is initially protective. However, delayed or insufficient responses or interplay with mito-chondrial dysfunction may turn physiological mechanisms into pathological consequences including apoptosis, fat accumulation and inflammation all of which have an important role in the pathogenesis of liver disorders such as genetic mutations, viral hepatitis, insulin resistance, ischemia/reperfusion injury, and alcoholic and non-alcoholic steatosis. In both alcohol and non-alcohol-induced ER stress, a common candidate is hyper-homocysteinemia. Betaine supplementation and/or expression of betaine-homocysteine methyltransferase (BHMT) promote removal of homocysteine and alleviate ER stress, fatty accumulation and apoptosis in cultured hepatocytes and mouse models. The rapidity and magnitude of homocysteine-induced activation of each of the main ER resident transmembrane sensors including inositol requiring enzyme 1 (IRE-lα), activating transcription factor 6 (ATF-6) and RNA-activated protein kinase (PKR)-like ER kinase (PERK) appear different in different experimental models. Dissection and differentiation of ER stress signaling may reveal clues on the specific importance of the ER stress response in contributing to liver injury and thus provide better strategies on prevention and treatment of liver disease.
Keywords: chaperone, fibrosis, liver cancer, mitochondrial stress, unfolded protein response
Introduction
The endoplasmic reticulum (ER) is an essential organelle which provides a specialized environment for the production and post-translational modifications of secretory and membrane proteins.1–3 The ER is also a site for storing Ca2+ which can be released in a controlled fashion to propagate cellular signal transduction. Nascent proteins are folded with the assistance of molecular chaperones and folding enzymes in the ER. Only properly folded proteins are allowed to reach their destiny. It has been estimated that as many as 30% of nascent proteins are unfolded or malfolded, retained in the ER, retrotranslocated to the cytoplasm by the machinery of ER-associated degradation (ERAD), and rapidly degraded by the ubiquin pathways.4 Accumulation of unfolded proteins occurs when there is a sudden increase in the translation of secretory proteins that exceeds the capacity of the folding apparatus and ERAD machinery or when there are perturbations in the ER environment, such as alterations in redox state, Ca2+ levels, or improper post-translational modifications. Unfolded proteins expose hydrophobic amino-acid residues and tend to form toxic protein aggregates disturbing homeostasis in the ER and creating a stress condition. To cope with ER stress, eukaryotic cells activate a series of signaling pathways referred to as unfolded protein response (UPR) which can be protective or detrimental. In the present review, I will summarize opposing effects of the UPR, briefly describe the role of ER stress in a number of experimental or naturally occurring models of liver disorders, and provide recent advances in dissecting ER stress signaling in alcohol or homocysteine-induced hepatic injury.
Protective role of the unfolded protein response
Accumulation of unfolded or malfolded proteins is initially sensed by three main resident transmembrane sensors in the ER: inositol requiring enzyme 1 (IRE-lα), activating transcription factor 6 (ATF-6), and RNA-activated protein kinase (PKR)-like ER kinase (PERK), which are normally held in an inactive and inhibited state by the binding of intraluminal ER chaperones, especially glucose regulated protein 78 (GRP78 or BiP).1–3 Upon UPR, GRP78 is displaced to deal with the exposed hydrophobic regions of the unfolded proteins, which facilitates folding through conformational change evoked by the hydrolysis of ATP by the ATPase domain. The displacement of GRP78 frees IRE-1α, PERK, and ATF-6, which are self-activated either by dimerization and auto-phosphorylation (IRE-lα and PERK) or transfer (ATF-6) to the Golgi apparatus for regulated intramembrane proteolysis (RIP). The activated IRE-lα functions as a nuclease which splices X box-binding protein 1 (XBP-1) mRNA; the resultant sXBP-l activates transcription through UPRE (UPR element) in promoters of genes which control ERAD. Another function of the nuclease activity of IRE-lα is to degrade mRNA of a variety of genes which encode for secretory and membrane proteins.5 After RIP, ATF-6 translocates to the nucleus, interacts with ER stress response element (ERSE) which upregulates chaperones/foldases such as GRP78, GRP94, IRE-1α, protein disulfide isomerase (PDI), ERp72, GRP58/ERp57, homocysteine-induced ER protein (HERP), calnexin, and calreticulin, all of which increase the capacity of folding in the ER.1–3 The activated PERK phosphorylates eukaryotic translation initiation factor 2, alpha subunit (eIF2α) to globally shut down protein translation halting protein loading while selectively increasing translation of certain mRNAs such as ATF-4 which upregulates chaperones. Activated PERK also phosphorylates NF-E2-related factor-2 (Nrf-2) which promotes antioxidant response and maintains redox homeostasis.6 In addition, recent studies have shown that the UPR induces autophagy possibly through an early c-jun-N-terminal kinase (JNK) activation or the target of rapamycin (TOR)-Atg (autophagy-related) signaling pathway which quickly removes superfluous or damaged organelles and portions of cytosol.7
Thus, by decreasing the load of nascent proteins, rapid unloading of unfolded proteins, increasing capacity of folding, or clearing out the damaged ER, the early UPR reverses ER stress resulting from accidental perturbations in the ER environment or from transient accumulation of large amounts of secretory proteins under physiological conditions in such cells as plasma cells, pancreatic β-cells, osteoblasts and hepatocytes (Fig. 1).2,3
Figure 1.
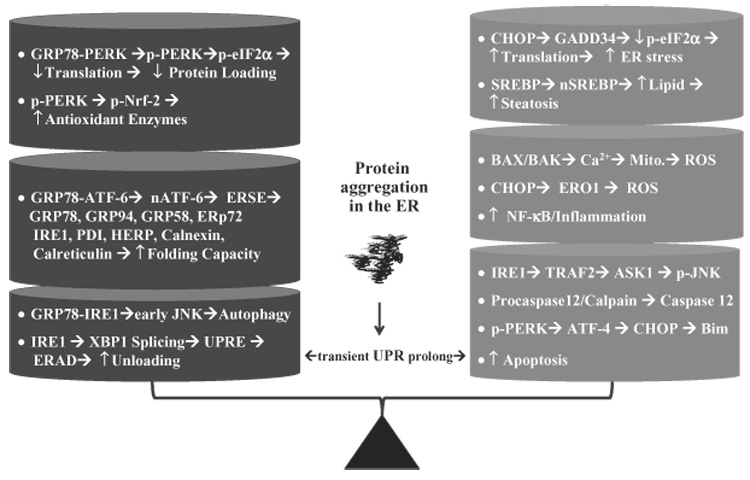
Opposing effects of the unfolded protein response (UPR): transient UPR leads to protection whereas prolonged UPR leads to injury. GRP78, glucose regulated protein 78; PERK, RNA-activated protein kinase (PKR)-like endoplasmic reticulum (ER) kinase; pPERK, phosphorylated PERK; eIF2α, eukaryotic translation initiation factor 2, alpha subunit; p-eIF2α, phosphorylated eIF2α; p-Nrf-2, phosphorylated nuclear factor (NF)-E2-related factor-2; ATF-6, activating transcription factor 6; nATF-6, activated ATF-6; ERSE, endoplasmic reticulum stress response element; IRE1, inositol requiring enzyme 1; PDI, protein disulfide isomerase; HERP, homocysteine-induced ER protein; JNK, c-jun-N-terminal kinase; p-JNK, phosphorylated JNK; XBP1, X box-binding protein 1; UPRE, unfolded protein response element; ERAD, ER-associated degradation; CHOP, C/EBP-homologous protein; SREBP, sterol regulatory element binding protein; nSREBP, nuclear SREBP; Mito, mitochondria; ROS, reactive oxygen species; ERO1, ER oxidase 1; TRAF2, TNF receptor- associated factor-2; ASK1, apoptosis signal regulated kinase 1; NF-κB, nuclear factor-κB.
Detrimental role of prolonged UPR
The early UPR dampens ER stress and ensures cell survival. However, prolonged or sustained UPR (alternatively called ER stress response) provokes a complex network of interacting and parallel responses leading to pathological consequences (Fig. 1).1–3 IRE-1α mediates recruitment of tumor necrosis factor (TNF) receptor-associated factor-2 (TRAF-2) which activates apoptosis signal regulated kinase 1 (ASK1) and/or JNK which promotes apoptosis. Bax/Bak, two proapoptotic members of the Bcl-2 protein family, translocate to the ER membrane causing Ca2+ release, leading to calpain activation or causing mitochondrial depolarization, promoting oxidative stress. The activated calpain may activate ER resident caspase-12 in rodents and caspase-4 in humans causing a cascade of activation of executioner caspases downstream of mitochondria which, along with the activated JNK, modulate apoptosis.
Sustained activation of PERK and subsequent eIF2α phosphorylation upregulates ATF-4 which increases expression of C/EBP-homologous protein (CHOP or GADD153).1,2,8 CHOP is a transcription factor and is of particular interest as its increased expression, along with GRP78, is a hallmark of the UPR and ER stress response. CHOP null cells are resistant to various stimuli of ER stress-induced apoptosis.8 How CHOP mediates apoptosis has so far been unclear. Increased expression of CHOP is associated with downregulation of anti-apoptotic Bcl-2, upregulation of ER oxidase 1 (ERO1) which promotes oxidative stress causing inflammatory response, and upregulation of GADD34 which dephosphorylates eIF2α causing a reversal of the translational attenuation by the early UPR and thus intensifying ER stress. Recently, Bim, a proapoptotic BH3-only member of the Bcl-2 family has been shown to be essential for ER stress-induced apoptosis in a diverse range of cell types both in culture and within the whole animal.9 In addition, parallel to ER stress, TNF and dsRNA (viruses) activate PKR, and amino acid deprivation activates general control of nitrogen protein kinase (GCN2). Both PKR and GCN2 phosphorylate elF-2α kinases which block translation, promoting apoptosis via theATF-4/CHOP pathway and sensitizing to TNF killing by inhibiting synthesis of nuclear factor (NF)-κB-dependent protective proteins.10
The prolonged UPR can result in increased production of triglyceride and cholesterol. Sterol regulatory element binding proteins (SREBP-lc and SREBP-2), which are ER-resident transcription factors, may play a key role in this consequence.2 SREBP are activated via RIP by site 1 protease (S1P) and site 2 protease (S2P). Activated SREBP translocate to the nucleus and promoter expression of genes which controls triglyceride and cholesterol synthesis. How ER stress promotes the activation of SREBP is not completely understood. As SREBP form a complex with SREBP cleavage activating protein (SCAP) which is retained in the ER by its interaction with Insig and Insig turns over fast,11,12 the ER stress-caused translational arrest may lead to a rapid decline in Insig protein, which would allow SREBP to migrate to the Golgi for activation. The ER stress-caused translational arrest may lead to a rapid decline in Insig protein, which would allow SREBP to migrate to the Golgi for activation. Another possibility is that the activation of JNK by ER stress response induces insulin resistance which increases transcriptional expression of SREBP. The effects of insulin on SREBP-1c transcription are opposed in the liver by glucagon.13 SREBP-1c mRNA expression is decreased in the livers of streptozotocin (STZ) diabetic rats and recovered by insulin treatment.14 The effects of insulin on SREBP-1c expression are mediated by a PI(3) kinase-dependent pathway. The downstream effectors remain unclear, with evidence suggesting that both PKB/ Akt and PKCλ may be involved.15,16
Role of ER stress in models of liver injury
The ER stress response has recently been recognized in a wide spectrum of experimental models of liver injury which is an emerging field of interest in the pathogenesis of nearly all types of human liver disease (Table 1).
Table 1.
Endoplasmic reticulum stress occurs in a variety of experimental models of hepatic injury.
| Etiology | ER components involved | Injury | References |
|---|---|---|---|
| Genetic deficiency | |||
| Fibrinogen | ERAD | Cirrhosis | 17 |
| Fumarylacetoacetate | GRP78, eIF2α, CHOP, caspase-12, | Liver failure, hepatocarcinogenesis | 18 |
| α1-antitrypsin | NF-κB overload response | Apoptosis, hepatocellular carcinoma | 19–21 |
| Hemochromatosis gene | GRP78, ATF-6, CHOP, EOR-NF-κB | Cirrhosis, liver cancer | 22 |
| Virus | |||
| HCV | GRP78 & 94, CHOP, Ca2+ depletion, NF-κB | ↑ALT, hepatitis, apoptosis, cancer | 2, 23–25 |
| HBV | GRP78, ATF-6, IRE-1-XBP1 | ↑ALT, hepatitis, cancer | 26, 27 |
| Drugs | |||
| Acetaminophen | ERp72, CHOP, ATF-6, caspase-12 | ↑ALT, apoptosis, liver failure | 28 |
| Methapyrilene | eIF2α, ATF-4, GRP78 | Hepatocarcinogenesis | 29, 30 |
| HIV protease inhibitor | CHOP, ATF-4, XBP-1, SREBP | ↑ALT, apoptosis, fat accumulation | 31 |
| Ischemia/reperfusion | GRP78, CHOP, GADD34, XBP1, PERK, eIF2α ATF-6, Bax inhibitor, caspase-12 | ↑ALT, apoptosis, inflammation, | 32–36 |
| Saturated fat, cholesterol | GRP78, CHOP, XBP-1, IRE-1, caspase-12 | Apoptosis, fatty liver, inflammation | 37–41 |
| Scurvy | GRP78, GRP94, PDI | Apoptosis | 42 |
| Reactive oxygen species (H2O2) | PERK, GRP78, XBP-1 | Keratin inclusions | 43 |
| Heavy metals (Cd, Ni, Co) | GRP78, CHOP, Alkaline phosphatase | ↑ALT | 44, 45 |
| Hyperhomocysteinemia | GRP78, CHOP, SREBP | ↑ALT, fatty liver, apoptosis | 46–50 |
| Lipopolysaccharide | IRE-1, ATF-6, eIF2α | Cirrhosis | 51 |
| Alcohol | GRP78, CHOP, SREBP, ATF-6 | ↑ALT, apoptosis, fatty liver, inflammation | 50, 52, 53 |
ALT, alanine aminotransferase, ATF-6, activating transcription factor 6; CHOP, C/EBP homologous protein; eIF2α, eukaryotic translation initiation factor 2, alpha subunit; EOR, endoplasmic reticulum (ER) overload response; ERAD, ER associated degradation; GRP78, glucose regulated protein 78; IRE-1, inositol requiring enzyme 1; NF- κB, nuclear factor- κB; PDI, protein disulfide isomerase; PERK, RNA-activated protein kinase (PKR)-like ER kinase; SREBP, sterol regulatory element binding protein; XBP1, X box-binding protein 1.
Genetic disorders
Accumulation of malfolding mutant proteins or metabolites from mutated enzymes often induces ER stress. Patients with variant fibrinogens develop accumulation of misfolded proteins in the ER and liver cirrhosis.17 Aberrant accumulation of fumarylacetoacetate due to fumarylacetoacetate hydrolase deficiency induced ER stress in cell culture.18 This deficiency leads to hereditary tyrosinemia type 1 disease and both ER stress and apoptosis were observed in mice in the absence of NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-1,3 cyclohexane dione) therapy. In α-antitrypsin deficiency (α1-AT) that can result in significant liver disease in 10% of all affected patients, non-polymer-forming mutants of α1-AT induced the UPR without detectable hepatic injury whereas the polymer-forming common mutant protein induced the UPR and elicited injurious components of the ER stress response such as EOR (NF-κB activation).19 Transgenic mice expressing the polymer-forming mutant protein had increased expression of CHOP and GRP78 and were more susceptible to liver fibrosis induced by cholestasis from bile duct ligation.20,21 Hereditary hemochromatosis (HH) is a genetic disease associated with iron overload which is caused by the retention of the mutant C282Y HFE protein in the ER. Expression of the mutant C282Y HFE protein in HEK293 cells triggered the UPR, EOR and apoptotic responses. Furthermore, the coexistence of the mutant proteins of both C282Y HFE and α1-AT acted as potential disease modifiers with respect to each other.22
Viral hepatitis and injury
Viral protein production during viral replication triggers the UPR which exerts either the protective role by limiting viral protein translation or the detrimental role by protecting against ER stress-induced cell death, allowing continued viral protein accumulation. 2 In addition, perhaps due to host–pathogen interactions, viruses have somehow evolved various mechanisms to cope with the UPR which also allow continued viral replication. Evidence from cell culture or transgenic models indicates that the hepatitis C virus (HCV) replicon or individual components induced expression of UPR genes as well as hepatic injury. For instance, HCV replicon induced sXBP-1 but inhibited induction of ERAD in Huh-7.23,24 HCV replicon decreased glycosylation preventing assembly of MHC-1, which promoted continued virus production, avoidance of immune clearance and ER stress-induced apoptosis leading to severe liver injury.25 HCV or hepatitis B virus (HBV) protein expression in cells induced an ER stress response resulting in Ca2+ release from the ER which activated cyclic AMP response element binding protein (CREB). The activated CREB upregulates protein phosphatase 2 Ac (PP2Ac) which impairs cell-cycle regulation and promotes apoptosis, leading to hepatocarcinogenesis. 26,27 In the infected human liver, the UPR/ER stress response could also lead to evasion of viral eradication and promotion of liver injury, as a rapidly progressive liver injury accompanying massive hepatic viral loading has been observed in post-orthotopic liver transplantation (OLT) or with concomitant HIV infection.
Drug-induced liver disease
Acetaminophen (AAP) is a widely used drug with analgesic and antipyretic effects. AAP overdose is a major and frequent cause of acute liver failure and the precise mechanism is not fully understood. Intraluminal redox imbalance of the ER and consequential activation of the ER stress signaling and proapoptotic events are believed to be involved in hepatocellular damage caused by AAP overdose. In fact, AAP overdose resulted in a decrease in microsomal total glutathione (GSH) and in the GSH/GSSG ratio.28 Redox state of thiols of ERp72 and PDI was shifted towards the oxidized form. ATF6, CHOP, and caspase-12 were also activated followed by an increased apoptosis of hepatocytes, all of which indicates a critical role of ER stress in AAP-induced liver injury.
Other drugs that induce typical ER stress are methapyrilene and HIV protease inhibitors (PI). Methapyrilene elicited hepatic damage that increased in severity with the number of doses. High-dose methapyrilene elicited thousands of gene changes including genes associated with ER stress.29,30 HIV PI including amprenavir, atazanavir, and ritonavir are applied in the treatment of HIV-infected patients. However, atazanavir and ritonavir cause serious lipid disturbances which are associated with increased levels of active SREBP, activation of the UPR signaling, induction of apoptosis, and formation of foam cells in macrophages.31 How protease inhibitors cause ER stress is unknown at present. Interestingly, amprenavir had no significant effect at the same concentrations. This may provide useful information to help predict clinical adverse effects in the development of new HIV PI and also provide a better understanding of the cellular mechanism of HIV PI-induced ER stress and dyslipidemia. Most likely, drug-induced ER stress contributes to the development of fatty liver and fatty liver, in turn, induces ER stress.
Ischemia-reperfusion injury
Hepatic ischemia-reperfusion injury (IRI) is a multistep process that involves oxidative stress, inflammation, and cell death. Dysfunction of mitochondria and the ER or pathophysiological interplay between the two organelles may be essential in IRI. Mitochondrial pathways in IRI involve reactive oxygen species (ROS), NF-κB activation, increased activity of major initiator (caspase-9) and effector (caspase-3) caspases. For example, cytochrome P450 2E1 (CYP2E1), known to be present in the ER and mitochondria,32 is downregulated during IR, contributing to oxidative stress. Mitochondrial dysfunction also leads to ATP depletion which causes Ca2+ release from the ER.33 Emerging evidence has shown that IRI is associated with features of ER stress, including increased GRP78, CHOP, sXBP-1 and PERK. Bax inhibitor-1 (BI-1) resides in the ER and suppresses ER stress-induced apoptosis. 34 BI-1 knock-out mice exhibited enhanced ER stress response and IRI.35 The small molecule chemical chaperone, sodium 4-phenylbutyrate protects against IR-induced ER stress-mediated apoptosis in the liver.36 Thus, combined protection of the ER and mitochondria may be a better therapeutic avenue to enhance liver graft viability and functional integrity.
Complications of liver steatosis and diabetes
The cause and effect between ER stress, liver steatosis and diabetes is complicated, but can be better understood as a dynamic triangle of ER stress-steatosis-diabetes. With regard to lipid and ER stress, high polyunsaturated or saturated fat in the diet may contribute to ER stress-related injury. It has been shown that high sucrose or saturated fat was associated with increased sXBP-1, GRP78 and CHOP as well as caspase-3 activities in rats.37 Increased saturated lipid content of ER directly compromises ER morphology and integrity.38 Treatment of mouse hepatocytes with palmitate induced sustained JNK activation and insulin resistance.39 However, ER stress-induced SREBP disrupt lipid homeostasis leading to liver steatosis.2 TNF-α involved in inflammation also induced steatosis by activating partially the Insulin-Insig-SREBP signaling pathway in the livers of mice.40 Furthermore, disruption of lipid homeostasis impairs immune response which could deteriorate steatosis. This is because the liver is enriched with natural killer T (NKT) cells that respond to lipid antigens which are presented by cluster of differentiation-1 (CD-1) molecules. The properly presented lipid antigens are required for optimal maturation and activation of the NKT cells. ER stress has been shown to decrease CD-1 and contribute to NKT cell dysregulation in ob/ob hepatocytes and livers.41 Thus, a vicious cycle between steatosis and ER stress facilitates the progression of steatosis to steatohepatitis.
The relationship between ER stress and diabetes is more substantial. ER stress induces tribbles 3 (TRB3) which inhibits Akt and induces JNK which inhibits insulin receptor substrate-1 (IRS-1). Inactivation of either Akt or IRS-1 induces insulin resistance in the liver.54 TRB3 expression is greatly increased through the cooperative effects of ATF-4 and CHOP in diabetic mice. Obesity (ob/ob or high fat diet)-caused ER stress in the liver is associated with hyperactivation of JNK and inactivation of IRS-1 in cell and mouse models. XBP-1 +/− mice fed high-fat diet exhibited insulin resistance and developed type 2 diabetes in the presence of increased ER stress response due to impaired UPR protection.55 Overexpression of oxygen-regulated protein 150 (ORP150), an ER chaperone, improved insulin resistance in db/db diabetic obese mice, whereas silencing ORP150 in normal mice decreased insulin sensitivity.56 Furthermore, treatment of ob/ob obese and diabetic mice with 4-phenylbutyrate or tauroursodeoxycholate (chemical chaperones) restored glucose tolerance, insulin sensitivity, and resolved fatty liver in parallel with marked inhibition of PERK, IRE-lα, and JNK activation.57 However, insulin resistance contributes to liver steatosis. Carbohydrate-responsive element-binding protein (ChREBP)-like SREBP-1c regulates lipogenic gene expression. ChREBP gene expression and ChREBP nuclear protein content are significantly increased in the livers of ob/ob mice with insulin resistance.58 Liver-specific inhibition of ChREBP markedly improved hepatic steatosis by specifically decreasing lipogenic rates.59
In brief, in the triangle of ER stress-steatosis-diabetes, ER stress mediates RIP of SREBP, apoptosis, and inflammatory and immune response leading to disruption of lipid homeostasis. Certain components of lipids can disrupt ER membrane integrity and impair UPR resulting in or exacerbating ER stress response. ER stress induces sustained JNK activation leading to insulin resistance which upregulates lipogenesis via ChREBP or SREBP, contributing to the development of liver steatosis (Fig. 2).
Figure 2.
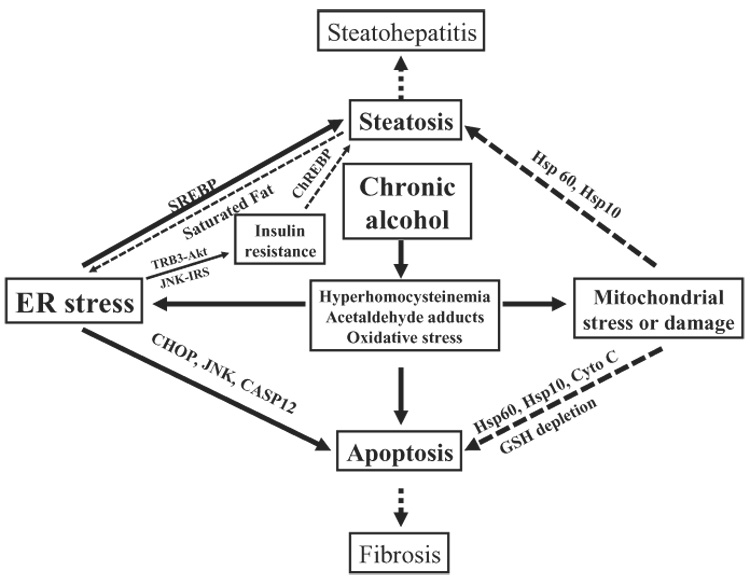
Proposed model elucidating the role of endoplasmic reticulum (ER) and/or mitochondrial stress in alcohol-induced liver injury. SREBPs, sterol regulatory element binding proteins; TRB3, tribbles 3; Akt, serinethreonine kinase (PKB); JNK, c-jun-N-terminal kinase; IRS, insulin-receptor substrate; ChREBP, carbohydrate-responsive element-binding protein; CHOP, C/EBP-homologous protein; CASP12, caspase-12; Hsp10, heat shock protein 10; Hsp60, heat shock protein 60; Cyto C, cytochrome c; GSH, glutathione.
Scurvy and damage by H2O2 and heavy metals
Insufficient ascorbate (vitamin C) intake causes scurvy in certain species deficient in gulonolactone oxidase that catalyzes the last step of ascorbate synthesis. Persistent ascorbate deficiency leads to ER stress, UPR, and apoptosis in the liver of guinea pigs, suggesting that insufficient protein processing participates in the pathology of scurvy.42 How ascorbate deficiency induces ER stress is not clear, but dysfunction of microsomal ascorbate oxidase due to ascorbate depletion can cause an enhanced oxidation of protein thiol groups which leads to malfolding of proteins. In addition, ascorbate can react with oxygen in a one-electron transfer and generates a ROS-altering redox state and affects proper protein folding in the ER. The link between ROS and ER stress has been further supported by recent evidence that generation of H2O2 from the glucose oxidase-treated HepG2 cells induced dysfunction of the ER and resulted in the formation of Mallory bodies.43
Heavy metals play essential roles in mediating enzymatic reactions as cofactors. However, heavy metal accumulation in organs especially in the liver and kidney via drinking water, foods and cigarette smoke causes injury. Recently, ER stress was shown to involve heavy metal-induced injury.44,45 Systemic exposure of mice to cadmium, nickel or cobalt caused rapid, transient and reversible induction of ER stress in vivo. Thus, therapeutic agents targeting the ER should be considered in dealing with heavy metal-induced liver injury.
Hyperhomocysteinemia and alcoholic liver injury
Homocysteine (Hcy) is an intermediate amino acid involved in methionine metabolism. Nutritional deficiency and mutations in some of the enzymes which remove Hcy lead to hyperhomocysteinemia (HHcy) which is often associated with fatty liver.2,46–50 Recent evidence in animal models has linked HHcy to ER stress that activates SREBP, contributing to fatty liver.46,47,50 There are a few potential mechanisms by which Hcy might induce ER stress. Nitrosylated Hcy can escape the proofing and editing by the methionine tRNA synthetase and can be wrongly incorporated into nascent proteins causing misfolding or unfolding. Under the function of methionine tRNA synthetase, Hcy can be converted to proactive thiolactone which either modifies lysine residues and other free amine groups on proteins or disrupts disulfide bond formation leading to malfolding in the ER. Increased Hcy thiolactone levels have been detected in humans with genetic disorders in Hcy metabolism and mice fed a high-methionine diet.60
Liver steatosis, steatohepatitis, fibrosis, and cirrhosis are characteristic of alcoholic liver disease (ALD). Factors contributing to ALD include, but are not limited to, alteration of redox state, oxidative stress, changes of cytokine milieu and signaling, impaired immune response, and polymorphisms in the genes encoding SOD2, CD14 endotoxin receptor, TNF-α, TGF-β, and angiotensinogen inhibition.61 The contribution of ER stress to ALD has been identified recently in animal models (Fig. 2). First, in intragastric alcohol-fed mice, severe steatosis, scattered apoptosis and necroinflammatory foci were observed.50 Microarray gene expression profiling of liver samples from the intragastric alcohol-fed versus pair-fed mice revealed initially altered expression of a set of genes relating to the UPR or ER stress response. Moderate upregulation of expression of SREBP-1c and SREBP-2 and their responsive genes was detected with reverse transcription–polymerase chain reaction (RT-PCR) and immunoblotting.50,62 SREBP-lc knock-out mice are protected against triglyceride accumulation but are not protected against increased cholesterol mediated by ethanol induction of SREBP-2. ER stress-caused block in the rapidly turning over Insig may contribute to the release and activation of SREBP in this model. CHOP knock-out mice fed ethanol exhibited no change in other markers of ER stress, or fatty liver. However, CHOP knock-out mice exhibited minimal apoptosis.63 This indicates that ER stress appears to be responsible for apoptosis through CHOP in this mouse model. Second, in another model of guinea-pigs fed alcohol, liver steatosis and apoptosis were shown to be accompanied by increased mRNA levels of CYP2E1, GRP78 and SREBP-1c, and protein levels of CYP2E1, GRP78, nSREBP, and activated caspase-12.52 In addition, the transcripts of lipogenic enzymes fatty acid synthase (FAS), acetyl-CoA carboxylase (ACC) and stearoyl-CoA desaturase (SCD) were elevated in the ethanol-fed micropigs. The evidence from micropigs fed alcohol supports a correlation of the ER stress response with the pathogenesis of ALD. Third, lipopolysaccharide (LPS) has been associated with ALD;61,64 recent evidence shows that the UPR is involved in LPS-induced injury. Rat cirrhotic livers exhibited partial UPR activation as indicated by eIF2α activation in the basal state and full UPR as indicated by activation of IRE1, ATF-6, and eIF2α after LPS challenge.51 LPS-induced accumulation of NF-κB-dependent anti-apoptotic proteins was not seen, suggesting that the UPR sensitized cirrhotic livers to LPS/TNF-α-mediated apoptosis. It is conceivable that the sensitization also occurs in alcohol-induced cirrhosis. Fourth, in a Lieber-DeCarli baboon model of ALD, a number of molecular pathways were identified to be altered using cDNAarray analysis.53 Although the ER stress mechanism was not recognized in this study, upregulation of calpain 2, calpain p94, and ERD21 and downregulation of eIF2α were among the genes listed in the supplemental data. In addition, the studies were extended to human ALD by comparison of gene expression profiles in non-diseased and cirrhotic liver tissue and upregulation of calpain and downregulation of calreticulin were also among genes revealed by the arrays. Therefore, the above several lines of evidence support that the ER stress response contributes to ALD.
What induces ER stress during ALD? HHcy, acetaldehyde adducts, and oxidative stress are important candidates (Fig. 2). Oxidative stress resulting mainly from alcohol-induced CYP2E1 and increased ratio of NADH/NAD+, in general, exerts many nonspecific effects, including the ER stress response. Acetaldehyde is able to induce ER stress in cultured HepG2 cells and activation of SREBP-1 in rat hepatoma cells.65,66 Hcy-induced ER stress in ALD is supported by specific in vivo evidence, which deserves further discussion. HHcy has been observed in alcoholic human subjects67 and 5–10% of the general population have HHcy mainly due to mutations in 5, 10-methylenetetrahydrofolate reductase (MTHFR) or folate deficiency.2 The intragastric alcohol feeding exhibited a striking five- to 10-fold increase in mouse plasma Hcy.50 The abrogation of ER stress in parallel with decreased alanine aminotransferase (ALT) and amelioration of liver steatosis and apoptosis were observed when the intragastric alcohol-fed mice were fed betaine which provides a methyl-donor to push the conversion of Hcy to methionine.50 The ER stress-induced SREBP-1c were reduced in guinea-pigs fed alcohol and folate-deficient diets supplemented with S-adenosylmethionine (SAM) which helps normalize Hcy metabolism.68 Activation of each of the main ER stress sensors including IRE-lα, ATF-6 and PERK was detected within 8 h after mice were fed homocysteine via gavages (15 mg/mouse). Acute feeding of alcohol via gavages (150 mg/mouse) did not induce elevation of Hcy or activation of the ER stress sensors similar to that by Hcy gavage (Ji C et al., unpubl. data, 2007). These results provide strong circumstantial evidence linking HHcy, ER stress and ALD.
Chronic alcohol-induced disturbance of methionine metabolism appears to contribute to HHcy. Hcy removal involves transsulfuration for GSH formation which is catalyzed by cystathionine β-synthase (CBS) and remethylation back to methionine which is catalyzed by methionine synthase (MS) and betaine-homocysteine methyltransferase (BHMT). Alterations of mRNA of MS, BHMT, and CBS were observed in the intragastric alcohol-fed mice.69 The mechanisms for the effects of ethanol are unknown but were independent of TNFα, as they were unaltered in TNF-R1 knock-out mice fed ethanol.69 BHMT remethylating approximately 50% of liver Hcy is of particular interest, as it is liver and kidney specific and vitamin independent.2 Severe reduction of BHMT mRNA was found in 90% of patients with HCV-induced cirrhosis and in approximately 50% of patients with chronic alcohol-induced cirrhosis. 70 The direct role of BHMT along with its methyl donorbetaine in liver steatosis and injury has been studied recently.71 BHMT overexpression in HepG2 cells inhibited Hcy-induced ER stress response, lipid accumulation, and cell death. Suppression of BHMT in primary mouse hepatocytes potentiated Hcy-induced but not tunicamycin-induced ER stress response and cell injury. Transgenic mice expressing human BHMT in organs peripheral to the liver are resistant toHHcy and fatty liver induced by chronic alcohol infusion or a high-methionine and low-folate diet (Ji C et al., unpubl. data, 2007). Furthermore, in the presence of betaine, expression of BHMT correlated with increased apolipoprotein B (apoB) expression and increased the SAM to S-adenosylhomocysteine (SAH) ratio. This suggests that beneficial effects of the BHMT/betaine system other than abrogating HHcy and ER stress may contribute to its protection against ALD.71
Does ER stress induce mitochondrial stress or damages contributing to ALD? Chronic alcohol-induced mitochondria stress is possible and is under our current investigation. Insufficient cellular ATP due to metabolizing alcohol and ATP consumption by the UPR in the ER can affect mitochondrial protein folding and degradation, which require ATP and molecular chaperones such as heat shock proteins (Hsp60/Hsp10) in mitochondria. In this respect, mitochondria stress and alterations of mitochondrial chaperones could have pathological consequences. For instance, Hsp60 expression has been linked to cytochrome c release, caspase-3 activation and apoptosis.72 It was also reported that Hsp10 exerted anti-inflammatory activity by inhibiting LPS-mediated Toll-like receptor signaling.73 Thus, changes in either Hsp60 or Hsp10 will exacerbate ALD as more Bax/Bak are released in response to ER stress and increased levels of gut-derived LPS are often detected after chronic alcohol feeding.61,64 With respect to the mitochondria damages, cholesterol accumulation in mitochondria mediated by the ER stress-induced SREBP-2 might be one of the causes. In HepG2 cells, acetaldehyde induced ER stress and accumulation of cholesterol in mitochondria.74 The cholesterol increased inner mitochondrial membrane viscosity and impaired mitochondrial GSH transport which led to mitochondrial GSH depletion and increased susceptibility to cell death in response to TNF and FasL signaling pathways.75 Therefore, the interplay between the ER and mitochondria can deteriorate ALD and both should be targeted for prevention and therapy.
Perspectives and future directions
Understanding the role of ER stress in the pathogenesis of various types of human liver disease is a rapidly emerging field of interest. Despite many advances made in recent years, important questions remain to be answered. How the upstream stress signal is recognized by the three sensors: IRE-1α, ATF-6 and PERK is still elusive and any structural basis for the recognition would provide clues for designing effective drugs. The three sensors can be simultaneously activated by severe ER stress under experimental conditions but may not be activated equally by lesser or gradual stress under myriad pathophysiological conditions. The rapidity and magnitude of the activation of the three sensors may determine the outcome of ER stress and its role in a particular illness. It is also conceivable that there are interactions between the ER stress signaling and other pathophysiological pathways. Besides, different types of liver cells (parenchymal versus non-parenchymal cells) may have different levels of sensitivity to ER stress and exhibit different levels of injury. Dissection and differentiation of the ER stress signaling may reveal clues on the specific importance of the ER stress response in contributing to liver injury.
With respect to the alcohol-induced HHcy, ER stress and liver injury, three questions are prominent. First, because of the wide range of elevated Hcy in alcoholics, low incidence of ALD among heavy drinkers, in addition to several lines of strong evidence to support association between HHcy and hepatic steatosis, should we assume that some of the common polymorphisms related to Hcy metabolism contribute to the susceptibility of a particular individual to ALD? The collection of large numbers of well-phenotyped cases and controls, which certainly require national and multinational collaborations, is needed to identify the possible genetic polymorphisms of Hcy metabolism involved in the pathogenesis of ALD. Second, although the ER stress response and the activation of SREBP-1c is often associated with alcoholic steatosis and lipotoxicity, it is not clear how ER stress induces the activation of SREBP. Structural analysis of other factors associated with SREBP on the ER membrane may be critical to answer this question. Third, does the ER stress response contribute to alcoholic fibrosis or even cirrhosis? If so what specific components of the ER stress signaling are involved besides the fact that ER stress-induced cell death could unleash fibrogenic factors in general?
Acknowledgments
This work was supported in part by NIH grants R01 AA014428, P50 AA11999, and P30 DK48522.
Footnotes
Conflict of interest: No conflict of interest has been declared by the author.
References
- 1.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 2.Ji C, Kaplowitz N. ER stress: can the liver cope? J. Hepatol. 2006;45:321–333. doi: 10.1016/j.jhep.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 2006;6:1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- 4.Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- 5.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 6.Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004;279(20):108–117. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- 7.Yorimitsu T, Klionsky DJ. Eating the endoplasmic reticulum: quality control by autophagy. Trends Cell Biol. 2007;17:279–285. doi: 10.1016/j.tcb.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 9.Puthalakath H, O’Reilly LA, Gunn P, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 10.Scheuner D, Patel R, Wang F, et al. Double-stranded RNA-dependent protein kinase phosphorylation of the oc-subunit of eukaryotic translation initiation factor 2 mediates apoptosis. J. Biol. Chem. 2006;281(21):458–468. doi: 10.1074/jbc.M603784200. [DOI] [PubMed] [Google Scholar]
- 11.Gong Y, Lee JN, Lee PC, Goldstein JL, Brown MS, Ye J. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. 2006;3:15–24. doi: 10.1016/j.cmet.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 12.Sun LP, Li L, Goldstein JL, Brown MS. Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J. Biol. Chem. 2005;280:26483–26490. doi: 10.1074/jbc.M504041200. [DOI] [PubMed] [Google Scholar]
- 13.Foretz M, Pacot C, Dugail I, et al. ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol. Cell Biol. 1999;19:3760–3768. doi: 10.1128/mcb.19.5.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimomura I, Bashmakov Y, Ikemoto S, Horton JD, Brown MS, Goldstein JL. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc. Natl Acad. Sci. USA. 1999;96(13):656–661. doi: 10.1073/pnas.96.24.13656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsumoto M, Ogawa W, Akimoto K, et al. PKClambda in liver mediates insulin-induced SREBP-1c expression and determines both hepatic lipid content and overall insulin sensitivity. J. Clin. Invest. 2003;112:935–944. doi: 10.1172/JCI18816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taniguchi CM, Kondo T, Sajan M, et al. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKClambda/zeta. Cell Metab. 2006;3:343–353. doi: 10.1016/j.cmet.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Kruse K, Dear A, Kaltenbrun E, et al. Mutant fibrinogen cleared from the endoplasmic reticulum via endoplasmic reticulum-associated protein degradation and autophagy: an explanation for liver disease. Am. J. Pathol. 2006;168:1299–1308. doi: 10.2353/ajpath.2006.051097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergeron A, Jorquera R, Orejuela D, Tanguay R. Involvement of endoplasmic reticulum stress in hereditary tyrosinemia type I. J. Biol. Chem. 2006;281:5329–5334. doi: 10.1074/jbc.M506804200. [DOI] [PubMed] [Google Scholar]
- 19.Hidvegi T, Schmidt B, Hale P, Perlmutter D. Accumulation of mutant al-antitrypsin Z in the endoplasmic reticulum activated caspases-4 and -12 NFkB, and BAP31 but not the unfolded protein response. J. Biol. Chem. 2005;280(39):002-15. doi: 10.1074/jbc.M508652200. [DOI] [PubMed] [Google Scholar]
- 20.Papp E, Szaraz P, Korcsmaros T, Csermely P. Changes of endoplasmic reticulum chaperone complexes, redox state, and impaired protein disulfide reductase activity in misfolding alpha1-antitrypsin transgenic mice. FASEB J. 2006;20:1018–1020. doi: 10.1096/fj.05-5065fje. [DOI] [PubMed] [Google Scholar]
- 21.Mencin A, Seki E, Osawa Y, et al. Alpha-1 antitrypsin Z protein (PiZ) increases hepatic fibrosis in a murine model of cholestasis (p NA) Hepatology. 2007;46:1443–1452. doi: 10.1002/hep.21832. [DOI] [PubMed] [Google Scholar]
- 22.Lawless MW, Mankan AK, White M, O’dwyer MJ, Norris S. Expression of hereditary hemochromatosis C282Y HFE protein in HEK293 cells activates specific endoplasmic reticulum stress responses. BMC Cell Biol. 2007;8:30. doi: 10.1186/1471-2121-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tardif KD, Mori K, Kaufman RJ, Siddiqui A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 2004;279:17158–17164. doi: 10.1074/jbc.M312144200. [DOI] [PubMed] [Google Scholar]
- 24.Zheng Y, Gao B, Ye L, et al. Hepatitis C virus non-structural protein NS4B can modulate an unfolded protein response. J. Microbiol. 2005;43:529–536. [PubMed] [Google Scholar]
- 25.Fournillier A, Wychowski C, Boucreux D, et al. Induction of hepatitis C virus E1 envelope protein-specific immune response can be enhanced by mutation of N-glycosylation sites. J. Virol. 2001;75(12):088–097. doi: 10.1128/JVI.75.24.12088-12097.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benali-Furet N, Chami M, Houel L, et al. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene. 2005;24:4921–4933. doi: 10.1038/sj.onc.1208673. [DOI] [PubMed] [Google Scholar]
- 27.Christen V, Treves S, Duong FH, Heim MH. Activation of endoplasmic reticulum stress response by hepatitis viruses up-regulates protein phosphatase 2A. Hepatology. 2007;46:558–565. doi: 10.1002/hep.21611. [DOI] [PubMed] [Google Scholar]
- 28.Nagy G, Kardon T, Wunderlich L, et al. Acetaminophen induces ER dependent signaling in mouse liver. Arch. Biochem. Biophys. 2007;459:273–279. doi: 10.1016/j.abb.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 29.Auman JT, Chou J, Gerrish K, et al. Identification of genes implicated in methapyrilene-induced hepatotoxicity by comparing differential gene expression in target and nontarget tissue. Environ. Health Perspect. 2007;115:572–578. doi: 10.1289/ehp.9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Craig A, Sidaway J, Holmes E, et al. Systems toxicology: integrated genomic, proteomic and metabonomic analysis of methapyrilene induced hepatotoxicity in the rat. J. Proteome Res. 2006;5:1586–1601. doi: 10.1021/pr0503376. [DOI] [PubMed] [Google Scholar]
- 31.Zhou H, Gurley E, Jarujaron S, et al. HIY protease inhibitors activate the unfolded protein response and disrupt lipid metabolism in primary hepatocytes. Am. J. Physiol. 2006;291:G1071–G1080. doi: 10.1152/ajpgi.00182.2006. [DOI] [PubMed] [Google Scholar]
- 32.Pahan K, Smith BT, Singh AK, Singh I. Cytochrome P-450 2E1 in rat liver peroxisomes: downregulation by ischemia/reperfusion-induced oxidative stress. Free Radic. Biol. Med. 1997;23:963–971. doi: 10.1016/s0891-5849(97)00017-8. [DOI] [PubMed] [Google Scholar]
- 33.Sakon M, Ariyoshi H, Umeshita K, Monden M. Ischemia-reperfusion of the liver with special reference to calcium-dependent mechanisms. Surg. Today. 2002;32:1–12. doi: 10.1007/s595-002-8105-8. [DOI] [PubMed] [Google Scholar]
- 34.Bailly-Maitre B, Fondevila C, Kaldas F, et al. Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic reticulum stress and ischemia-reperfusion injury. Proc. Natl Acad. Sci. USA. 2006;103:2809–2814. doi: 10.1073/pnas.0506854103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chae H, Kirn H, Xu C, et al. BI-1 regulates an apoptosis pathway linked to endoplasmic reticulum stress. Mol. Cell. 2004;15:355–366. doi: 10.1016/j.molcel.2004.06.038. [DOI] [PubMed] [Google Scholar]
- 36.Vilatoba M, Eckstein C, Bilbao G, et al. Sodium 4-phenybutyrate protects against liver ischemia reperfusion injury by inhibition of endoplasmic reticulum-stress mediated apoptosis. Surgery. 2005;138:342–351. doi: 10.1016/j.surg.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 37.Wang D, Wei Y, Pagliassotti M. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 2006;147:943–951. doi: 10.1210/en.2005-0570. [DOI] [PubMed] [Google Scholar]
- 38.Borradaile N, Han X, Harp J, et al. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 2006;47:2726–2737. doi: 10.1194/jlr.M600299-JLR200. [DOI] [PubMed] [Google Scholar]
- 39.Solinas G, Naugler W, Galimi F, Lee MS, Karin M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc. Natl Acad. Sci. USA. 2006;103(16):454–459. doi: 10.1073/pnas.0607626103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Endo M, Masaki T, Seike M, Yoshimatsu H. TNF-alpha induces hepatic steatosis in mice by enhancing gene expression of sterol regulatory element binding protein-1c (SREBP-1c) Exp. Biol. Med. (Maywood) 2007;232:614–621. [PubMed] [Google Scholar]
- 41.Yang L, Jhaveri R, Huang J, Qi Y, Diehl AM. Endoplasmic reticulum stress, hepatocyte CD1d and NKT cell abnormalities in murine fatty livers. Lab. Invest. 2007;87:927–937. doi: 10.1038/labinvest.3700603. [DOI] [PubMed] [Google Scholar]
- 42.Margittai E, Banhegyi G, Kiss A, et al. Scurvy leads to endoplasmic reticulum stress and apoptosis in the liver of Guinea pigs. J. Nutr. 2005;135:2530–2534. doi: 10.1093/jn/135.11.2530. [DOI] [PubMed] [Google Scholar]
- 43.Hanada S, Harada M, Kumemura H, et al. Oxidative stress induces the endoplasmic reticulum stress and facilitates inclusion formation in cultured cells. J. Hepatol. 2007;47:93–102. doi: 10.1016/j.jhep.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 44.Hiramatsu N, Kasai A, Du S, et al. Rapid, transient induction of ER stress in the liver and kidney after acute exposure to heavy metal: evidence from transgenic sensor mice. FEBS Lett. 2007;581:2055–2059. doi: 10.1016/j.febslet.2007.04.040. [DOI] [PubMed] [Google Scholar]
- 45.Dudley RE, Svoboda DJ, Klaassen CD. Time course of cadmium-induced ultrastructural changes in rat liver. Toxicol. Appl. Pharmacol. 1984;76:150–160. doi: 10.1016/0041-008x(84)90038-3. [DOI] [PubMed] [Google Scholar]
- 46.Werstuck G, Lentz S, Dayal S, et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Invest. 2001;107:1263–1273. doi: 10.1172/JCI11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamelet J, Demuth K, Paul J, Delabar J, Janel N. Hyperhomocysteinemia due to cystathionine beta synthase deficiency induces dysregulation of genes involved in hepatic lipid homeostasis in mice. J. Hepatol. 2007;46:151–159. doi: 10.1016/j.jhep.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe M, Osada J, Aratani Y, et al. Mice deficient in cystathionine beta synthase: animal models for mild and severe homocysteinemia. Proc. Natl Acad. Sci. USA. 1995;92:1585–1589. doi: 10.1073/pnas.92.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Z, Karaplis A, Ackerman S, et al. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum. Mol. Genet. 2001;10:433–443. doi: 10.1093/hmg/10.5.433. [DOI] [PubMed] [Google Scholar]
- 50.Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- 51.Tazi KA, Bieche I, Paradis V, et al. In vivo altered unfolded protein response and apoptosis in livers from lipopolysaccharide-challenged cirrhotic rats. J. Hepatol. 2007;46:1075–1088. doi: 10.1016/j.jhep.2007.01.034. [DOI] [PubMed] [Google Scholar]
- 52.Esfandiari F, Villanueva JA, Wong DH, French SW, Halsted CH. Chronic ethanol feeding and folate deficiency activate hepatic endoplasmic reticulum stress pathway in micropigs. Am. J. Physiol. Gastrointest. Liver Physiol. 2005;289:G54–G63. doi: 10.1152/ajpgi.00542.2004. [DOI] [PubMed] [Google Scholar]
- 53.Seth D, Leo MA, McGuinness PH, et al. Gene expression profiling of alcoholic liver disease in the baboon (Papio hamadryas) and human liver. Am. J. Pathol. 2003;163:2303–2317. doi: 10.1016/S0002-9440(10)63587-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–1577. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- 55.Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 56.Nakatani Y, Kaneto H, Kawamori D, et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J. Biol. Chem. 2005;280:847–851. doi: 10.1074/jbc.M411860200. [DOI] [PubMed] [Google Scholar]
- 57.Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dentin R, Benhamed F, Hainault I, et al. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes. 2006;55:2159–2170. doi: 10.2337/db06-0200. [DOI] [PubMed] [Google Scholar]
- 59.Postic C, Dentin R, Denechaud PD, Girard J. ChREBP, a Transcriptional Regulator of Glucose and Lipid Metabolism. Annu. Rev. Nutr. 2007;27:179–192. doi: 10.1146/annurev.nutr.27.061406.093618. [DOI] [PubMed] [Google Scholar]
- 60.Chwatko G, Boers GH, Strauss KA, Shih DM, Jakubowski H. Mutations in methylenetetrahydrofolate reductase or cystathionine beta-synthase gene, or a high-methionine diet, increase homocysteine thiolactone levels in humans and mice. FASEB J. 2007;21:1707–1713. doi: 10.1096/fj.06-7435com. [DOI] [PubMed] [Google Scholar]
- 61.Wilfred de Alwis NM, Day CP. Genetics of alcoholic liver disease and nonalcoholic fatty liver disease. Semin. Liver Dis. 2007;27:44–54. doi: 10.1055/s-2006-960170. [DOI] [PubMed] [Google Scholar]
- 62.Ji C, Chan C, Kaplowitz N. Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model. J. Hepatol. 2006;45:717–724. doi: 10.1016/j.jhep.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 63.Ji C, Mehrian-Shai R, Chan C, Hsu Y, Kaplowitz N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol. Clin. Exp. Res. 2005;29:1496–1503. doi: 10.1097/01.alc.0000174691.03751.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schenker S, Bay MK. Alcohol and endotoxin: another path to alcoholic liver injury? Alcohol. Clin. Exp. Res. 1995;19:1364–1366. doi: 10.1111/j.1530-0277.1995.tb01626.x. [DOI] [PubMed] [Google Scholar]
- 65.Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology. 2006;43:S63–S74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- 66.Lluis JM, Colell A, Garcia-Ruiz C, Kaplowitz N. Fernandez-Checa JC. Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology. 2003;124:708–724. doi: 10.1053/gast.2003.50089. [DOI] [PubMed] [Google Scholar]
- 67.Sakuta H, Suzuki T. Alcohol consumption and plasma homocysteine. Alcohol. 2005;37:73–77. doi: 10.1016/j.alcohol.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 68.Esfandiari F, You M, Villanueva JA, Wong DH, French SW, Halsted CH. S-adenosylmethionine attenuates hepatic lipid synthesis in micropigs fed ethanol with a folate-deficient diet. Alcohol. Clin. Exp. Res. 2007;31:1231–1239. doi: 10.1111/j.1530-0277.2007.00407.x. [DOI] [PubMed] [Google Scholar]
- 69.Ji C, Deng Q, Kaplowitz N. Role of TNF-α in ethanol-induced hyperhomocysteinemia and murine alcoholic liver injury. Hepatology. 2004;40:442–451. doi: 10.1002/hep.20309. [DOI] [PubMed] [Google Scholar]
- 70.Avila MA, Berasain C, Torres L, et al. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J. Hepatol. 2000;33:907–914. doi: 10.1016/s0168-8278(00)80122-1. [DOI] [PubMed] [Google Scholar]
- 71.Ji C, Shinohara M, Kuhlenkamp J, Chan C, Kaplowitz N. Mechanisms of protection by the betaine-homocysteine methyltransferase/betaine system in HepG2 cells and primary mouse hepatocytes (p NA) Hepatology. 2007;46:1586–1596. doi: 10.1002/hep.21854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arya R, Mallik M, Lakhotia SC. Heat shock genes-integrating cell survival and death. J. Biosci. 2007;32:595–610. doi: 10.1007/s12038-007-0059-3. [DOI] [PubMed] [Google Scholar]
- 73.Johnson BJ, Le TT, Dobbin CA, et al. Heat shock protein 10 inhibits lipopolysaccharide-induced inflammatory mediator production. J. Biol. Chem. 2005;280:4037–4047. doi: 10.1074/jbc.M411569200. [DOI] [PubMed] [Google Scholar]
- 74.Coll O, Colell A, Garcia-Ruiz C, et al. Sensitivity of the 2-oxoglutarate carrier to alcohol intake contributes to mitochondrial glutathione depletion. Hepatology. 2003;38:692–702. doi: 10.1053/jhep.2003.50351. [DOI] [PubMed] [Google Scholar]
- 75.Mari M, Caballero F, Collel A, et al. Mitochondrial free cholesterol loading sensitizes to TNF-and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]