

Abstract
Using the intra tumoral aromatase xenograft model, we have observed that despite long lasting growth inhibition tumors eventually begin to grow during continued letrozole treatment. In cells isolated from these Long Term Letrozole Treated tumors (LTLT-Ca), ERα levels were decreased whereas signaling proteins in the MAPK cascade were upregulated along with Her-2. In the current study we evaluated the effect of discontinuing the letrozole treatment on the growth of letrozole resistant cells and tumors. The cells formed tumors equally well in the absence or presence of letrozole and had similar growth rates. After treatment was discontinued for six weeks, letrozole was administered again. Marked tumor regression was observed with this second course of letrozole treatment. Similarly in MCF-7Ca xenografts, a six-week break in letrozole treatment prolonged the responsiveness of the tumors to letrozole. To understand the mechanisms of this effect, LTLT-Ca cells were cultured in the absence of letrozole for 16 weeks. The resulting cell line (RLT-Ca) exhibited properties similar to MCF-7Ca cells. The cell growth was inhibited by letrozole and stimulated by estradiol. The expression of p-MAPK was reduced and ERα and aromatase increased compared to levels in LTLT-Ca cells and were similar to the levels in MCF-7Ca cells. These results indicate that discontinuing treatment can reverse letrozole resistance. This could be a beneficial strategy to prolong responsiveness to AIs for breast cancer patients.
Keywords: aromatase inhibitors, estrogen, letrozole, ERα, Her-2
Introduction
About 75% of the breast cancer patients have hormone dependent breast cancer and are treated with endocrine therapy. Aromatase inhibitors are proving to be more effective than the antiestrogen tamoxifen. Nevertheless, as with all forms of cancer therapy, not all patients respond and some relapse due to development of resistance to aromatase inhibitor treatment. To study the hormone responsive breast cancer, we created a model system in which tumors of human ER positive breast cancer cells stably transfected with the aromatase gene (MCF-7Ca) are grown in immune suppressed ovariectomized mice (1, 2). This model system has provided us with results (3) that have predicted the outcomes of several clinical trials (4-7). We have previously reported that during treatment with letrozole, the MCF-7Ca xenografts increased expression of Her-2 and proteins in the downstream MAPK pathway. In order to investigate the mechanisms in the loss of sensitivity of the tumors to AIs, we developed a cell line isolated from tumors of MCF-7Ca cells treated long term with letrozole (8, 9). These cells also exhibited lower expression of ERα and increase in Her-2 as well as apparent “estradiol independent” growth (8). In our previous studies, we found that inhibition of these growth factor pathways leads to down-regulation of Her-2/MAPK activation and restoration of ERα mediated signaling suggesting that signaling pathways such as Her-2/MAPK are the key regulators of growth of letrozole refractory cells and tumors. Furthermore, trastuzumab, an inhibitor of Her-2 pathway reverses the resistance of LTLT-Ca cells to letrozole (10). This suggests cross-talk between the Her-2/MAPK and the ERα pathway is controlling the growth of cancer cells.
In this study, we evaluated the effect of discontinuing letrozole treatment on these signaling pathways and whether hormone sensitivity could be restored in LTLT-Ca cells and tumors. Treatment with letrozole shifted the balance towards the growth factor pathway whereas when the treatment is stopped, compensatory signaling via the Her-2/MAPK pathway declined, ERα levels were restored and the cells regain their original sensitivity to letrozole.
Materials and Methods
Materials
Dulbecco's Modified Eagle Medium (DMEM), Modified Improved Minimum Essential medium (IMEM), penicillin/streptomycin solution (10,000IU each), 0.25% trypsin–1 mM EDTA solution, Dulbecco's phosphate-buffered saline (DPBS), and geneticin (G418) were obtained from Invitrogen (Carlsbad, CA). Androstenedione (Δ4A) was obtained from Sigma Chemical Company (St. Louis, MO).
Cell culture
MCF-7Ca cells were routinely cultured in DMEM supplemented with 5% FBS, 1% Penicillin/Streptomycin, 700μg/mL G418. LTLT-Ca cells were isolated from the tumors of mice treated with letrozole for 56 weeks as previously described (8). In brief, small pieces of tumors were disrupted by repeatedly drawing into a pipette. The tissue was incubated at 37°C, stirring overnight with collagenase type 1-A, hyalyronidase type 1-S, amphotericin B and polymixin B sulfate. The cells were filtered and plated into Petri dishes. After a week, phenol red–free trypsin was added to remove fibroblast and washed with DPBS. Epithelial cells were cultured in medium with 750 μg/mL G418 and 1 μM of letrozole. Over the next 2 to 3 weeks, serum levels in the medium were reduced to 5% charcoal stripped serum and penicillin/streptomycin level to 1%. The cells were then propagated in phenol red–free IMEM supplemented with 5% CSS, 1% penicillin/streptomycin, 750 μg/mL G418 and 1 μM of letrozoleRLT-Ca cells were derived by culturing LTLT-Ca cells supplementing with Δ4A and in absence of letrozole for at least 4 months.
Cell proliferation assays were performed using the MTT assay as described earlier (8, 11). The results were expressed as a percentage of the absorbance of the controls. IC50 values for inhibitors were calculated from the non-linear regression line of the plot of cell viability (percentage of control) versus inhibitor concentration.
Tumor Growth in Ovariectomized Female Athymic Nude Mice
All animal studies were performed according to the guidelines and approval of the Animal Care Committee of the University Of Maryland School Of Medicine. Female ovariectomized athymic nude mice 4−6 weeks of age were obtained from the National Cancer Institute (Frederick, MD). The mice were housed in a pathogen-free environment under controlled conditions of light and humidity and received food and water ad libitum.
The tumor xenografts of MCF-7Ca cells were grown in the mice as previously described (3, 9). Mice were assigned to groups for treatment so that there was no statistically significant difference in tumor volume among the groups at the beginning of treatment (p = 0.97). Tumors were measured weekly with calipers and volumes were calculated using the formula (4/3) π r12 r2 (r1 ≤ r2). The doses of letrozole and Δ4A used are as previously determined and reported. Mice in the Δ4A group (n= 6) were treated for 7 weeks after which they were sacrificed due to large tumor volumes. The mice in letrozole group (n = 30) were continued on treatment. When the tumor volumes of these mice reached double their starting size, the mice were regrouped into 2 groups. One group (n = 10) stayed on continued letrozole treatment and the other group received only Δ4A (n = 20). This group was assigned as “Off Letrozole”. Out of these 20 mice, 10 mice were switched back to letrozole after 6-weeks of discontinuation of treatment. This group was assigned “Back On”.
LTLT-Ca xenografts were grown similarly as the MCF-7Ca xenografts (8). However, after inoculating LTLT-Ca cells into the mice, they were not supplemented with any steroidal agent. After 4 months off treatment, the mice were grouped so that there was no statistically significant difference in the tumor volumes (p=0.56).
Western blotting
The protein extracts from tumor tissues were prepared by homogenizing the tissue in ice-cold DPBS containing protease inhibitors and lysates from cells were prepared as described previously and separated by SDS-PAGE. (11).
3H2O release assay for aromatase activity measurement
The radiometric 3H2O release assay was performed as described previously (12). The activity of the enzyme is corrected for protein concentration in the tumor homogenates and cells.
Statistics: The tumor volumes were analyzed with S-PLUS (7.0, Insightful Corp.) to estimate and compare an exponential parameter (βi) controlling the growth rate for each treatment groups as described previously (13). All p values less than 0.05 were considered statistically significant.
Results
AEs, AIs produce inhibitory effects and E2 induces mitogenic effects on RLT-Ca cells
LTLT-Ca cells were cultured without letrozole for 4 months. These cells were designated as RLT-Ca cells. As shown in Figure 1A, the IC50 of letrozole was 44.3pM in the parental MCF-7Ca cells and 1.85nM in RLT-Ca cells, whereas LTLT-Ca cells were resistant to growth inhibitory effects of letrozole. Similar effects were observed with other AIs, anastrozole, exemestane and AE fulvestrant and tamoxifen. The IC50 values of these agents in RLT-Ca cells are comparable to the parental MCF-7Ca cells (Table 1). However, tamoxifen was found to be the least effective compound of all studied (IC50 > 19.8 μM). Our findings indicate that when letrozole resistant cells (LTLT-Ca) are no longer exposed to letrozole for several weeks (RLT-Ca), they regain their original sensitivity to AIs and AEs whereas LTLT-Ca cells exhibit complete cross-resistance to all the endocrine agents (data not shown). The reversal of response to letrozole was accompanied by increased aromatase activity in RLT-Ca cells (Figure 1B) compared to LTLT-Ca cells (p<0.01) and which was higher than in MCF-7Ca cells (p<0.05).
Figure 1. A: Effect of letrozole on proliferation of MCF-7Ca, LTLT-Ca and RLT-Ca cells in vitro.

Viability of cells was measured by MTT assay after 6 day treatment with letrozole as described in Materials and Methods. The treatment with letrozole is significantly more effective in MCF-7Ca and RLT-Ca cells compared to LTLT-Ca cells (p = 0.0003).
Figure 1B: Aromatase activity of MCF-7Ca, LTLT-Ca and RLT-Ca cells: Aromatase activity was measured by 3H2O release assay as described in Materials and Methods. RLT-Ca cells exhibit significantly higher aromatase activity compared to MCF-7Ca (*a, p<0.05) and LTLT-Ca cells (*b, p<0.001).
Table 1. IC50 values of fulvestrant, tamoxifen anastrozole, letrozole and exemestane in MCF-MCF-7Ca, LTLT-Ca and RLT-Ca cells.
Cell viability of cells was measured by MTT assay after 6 day treatment with described in Materials and Methods. IC50 values were calculated from the non-linear regression of the plot of cell viability (% of control) versus log inhibitor concentration.
| IC50 (nM) | MCF-7Ca | LTLT-Ca | RLT-Ca |
|---|---|---|---|
| Letrozole | 0.0443 | > 1μM | 1.85 |
| Anastrozole | 28 | > 1μM | 58 |
| Exemestane | 59 | > 1μM | 71 |
| Tamoxifen | 128 | > 1μM | 19.8 μM |
| Fulvestrant | 5.4 | > 1μM | 28 |
In addition, RLT-Ca cells respond to mitogenic effects of Δ4A and E2. Figure 2A shows that Δ4A stimulated the growth of RLT-Ca cells at concentrations of 10−11 and 10−9M (p<0.01) a response similar to that of MCF-7Ca cells. Interestingly, the growth of LTLT-Ca cells was decreased at all Δ4A concentrations (Figure 2A). This effect may be due to specific androgenic effect of Δ4A as it was not converted to estrogen owing to low level of aromatase activity in LTLT-Ca cells (Figure 1B) (14). E2 was ineffective in stimulating the growth of LTLT-Ca cells, as shown in Figure. 2B. However, the growth of RLT-Ca cells was significantly stimulated by E2 at concentrations 10−12 - 10−10M (p<0.01). The biphasic dose response of Δ4A and E2 observed in RLT-Ca cells is similar to the response of MCF-7Ca cells. RLT-Ca cells show changes in the activation and expression of several signal transduction proteins. Thus, compared to LTLT-Ca cells, Her-2, p-MAPK levels were decreased and ERα increased to levels approaching those of MCF-7Ca cells. (Figure 2C).
Figure 2. A: Effect of Δ4A on proliferation of LTLT-Ca and RLT-Ca cells.

Viability of cells was measured by MTT assay after 6 day treatment as described in Materials and Methods. Compared to LTLT-Ca cells, RLT-Ca cells exhibit a significantly marked stimulation of proliferation in response to Δ4A at concentrations of 10−11M and 10−10M (* p <0.001). LTLT-Ca cells are markedly inhibited by Δ4A at all concentrations (‡ p<0.01), but only at concentration of 10−6M Δ4A in RLT-Ca cells (†p<0.05) and 10−5M and 10−4M (* p <0.001)
Figure 2B: Effect of estradiol on proliferation of LTLT-Ca and RLT-Ca cells: Viability of cells was measured by MTT assay after 6 day treatment as described in Materials and Methods. Compared to LTLT-Ca cells, RLT-Ca cells exhibit a significantly marked stimulation of proliferation in response to E2 at concentrations of 10−12M through 10−10M (* p <0.001). E2 markedly inhibited growth of LTLT-Ca cells at concentrations 10−6M through 104M (‡ p<0.01), but only at 10−4M E2 in RLT-Ca cells (p<0.01).
Figure 2C: Protein expression profile of RLT-Ca cells compared to MCF-7Ca and LTLT-Ca cells: Expression of proteins was examined using western imunoblotting as described in Materials and Methods. Blot shows phospho-MAPK at 42−44 kDa, Her-2 at 185 kDa, ERα at 66 kDa and β-actin at 45 kDa. The blots were stripped and re-probed for β-actin to verify equal loading. The blots show a single representative of three independent experiments.
Reversal of letrozole resistance in LTLT-Ca xenografts
As shown in Figure 3A, when LTLT-Ca xenografts were grown in nude mice in absence of letrozole for a period of 4 months, they responded to a second course of letrozole treatment for up to 12 weeks. The growth rate (βi) of tumors treated with letrozole (6.5±7.1) was significantly (p = 0.04) lower compared to control tumors (48.8±16.8). The mean tumor volume of letrozole treated mice at week 24 was found to be 196±169.4 mm3 whereas without letrozole treatment the mice had mean tumor volume of 1261.9±409.3 mm3 (p=0.03). As shown in Figure 3B the tumor weights of these two groups were also significantly different. Furthermore, the low uterine weight in the letrozole treated group suggests that letrozole was effective in inhibiting synthesis of estrogen production (Figure. 3C).
Figure 3. A: Effect of letrozole treatment at on the growth of LTLT-Ca xenografts after 4 months off letrozole.


LTLT-Ca xenografts were grown in female OVX nude mice as described in Materials and Methods. The mice were kept off letrozole treatment for a period of 4 months. After which they were grouped into two groups, shown as on and off letrozole. The mice in the control and letrozole treated group exhibited significantly different growth rate. The difference in the exponential parameter governing growth was 42.3, p = 0.04.
Figure 3B: Effect of letrozole on the tumor weight of the LTLT-Ca xenografts: The mean tumor weight of control mice was 607.5 ± 225.5 mg which was significantly different from those of the letrozole treated mice (112.5 ± 84.77 mg); p = 0.024. The graph shows mean ± SEM.
Figure 3C: Effect of letrozole on the uterine weight of the LTLT-Ca xenografts: The mean uterine weight of control mice was 83 ± 21.45 mg which was significantly different from those of the letrozole treated mice (39.67 ± 9.14 mg); p<0.01. The graph shows mean ± SEM.
Figure 3D: Discontinuous treatment prolongs responsiveness of MCF-7Ca xenografts to letrozole: The tumors of MCF-7Ca cells were grown as described in materials and methods. The letrozole and off groups were sacrificed on week 34. The graph shows mean tumor volume ± SEM versus time (weeks).
Reversal of letrozole resistance in MCF-7Ca xenografts
Consistent with our earlier report, letrozole treatment in MCF-7Ca xenografts caused marked tumor regression, which was maintained for a prolonged period. The mice treated with letrozole versus Δ4A (control) had significantly lower growth rates (p<0.0001) and tumor volumes (p<0.0001). After 22 weeks letrozole was discontinued in one group of mice (the “off” group). Tumors of these mice had a similar growth rate to that of the group continued on letrozole over weeks 22−34. The difference in the exponential parameter governing growth rate (βi) was 0.002 ± 0.12, p = 0.99. This suggests that when resistance to letrozole develops, discontinuing treatment does not affect the growth rate of tumors significantly. When the mice in the “off” group were put “back on” letrozole the tumor growth was significantly reduced. The difference in the exponential parameter governing growth rate was 0.436 ± 0.12 (p = 0.0005). Also, the “back on” group had a significantly lower growth rate compared to the “letrozole” group. The difference in the exponential parameter governing growth rate was 0.434 ± 0.13 (p = 0.0009). (Figure 3D).
On week 34, the mice were sacrificed due to large tumor volumes of “letrozole” and “off” groups. Tumors and uteri were excised, cleaned, weighed, and stored at −80°C. The tumor weights of the mice in control, letrozole and “off” group were not statistically different (p = 0.46) (Figure. 4A).
Figure 4. A: Effect of letrozole on/off treatment the tumor weight of the MCF-7Ca xenografts.

The mean tumor weight of control mice was 1.38 ± 0.39 g, letrozole treated mice was 0.92 ± 0.36 g and off letrozole mice was 0.74 ± 0.34g. The graph shows mean ± SEM. The mean tumor weights were not significantly across the groups (p = 0.46).
Figure 4B: Effect of letrozole on/off treatment the uterus weight of the MCF-7Ca xenografts: The mean uterus weight of letrozole treated mice was 10.71 ± 1.658 mg which was significantly different from those of the control mice (50.67 ± 12.28 mg); p = 0.0006 (*a) and “off letrozole” mice (43.63 ± 13.44 mg); p = 0.02 (*b). The mean uterine weight of control and off letrozole mice was not significantly different (p = 1). The graph shows mean ± SEM.
Pearson correlation coefficient for uterine weight and tumors in letrozole group was −0.19 and 0.14 (left and right tumor respectively); p= 0.68 and 0.76- no correlation.
Pearson correlation coefficient for uterine weight and tumors in control group was 0.96 and 0.96 (left and right tumor respectively); p = 0.003 and 0.002 – very strong positive correlation is present.
Pearson correlation coefficient for uterine weight and tumors in off group was 0.95 and 0.84 (left and right tumor respectively); p= 0.0002 and 0.009– very strong positive correlation is present.
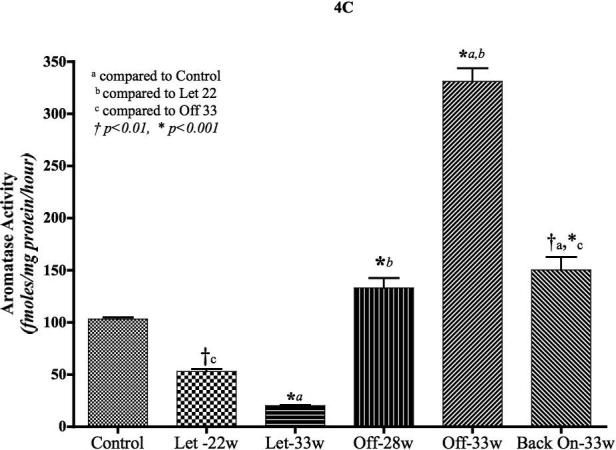
Figure 4C: Aromatase activity in the tumors of MCF-7Ca cells treated with letrozole: Aromatase activity was measured using 3H2O release assay as described in materials and methods. Control tumors were collected at week 7; letrozole treated tumors were collected at week 22 (when the group was split into on and off letrozole) and week 33 (when experiment was terminated); off letrozole tumors at week 28 (when group was split into off and back on letrozole) and week 33 (when experiment was terminated); back on letrozole at week 33. The graph represents mean ± SEM. One Way ANOVA with post-hoc Tukey multiple comparison test was performed to examine statistical significance.
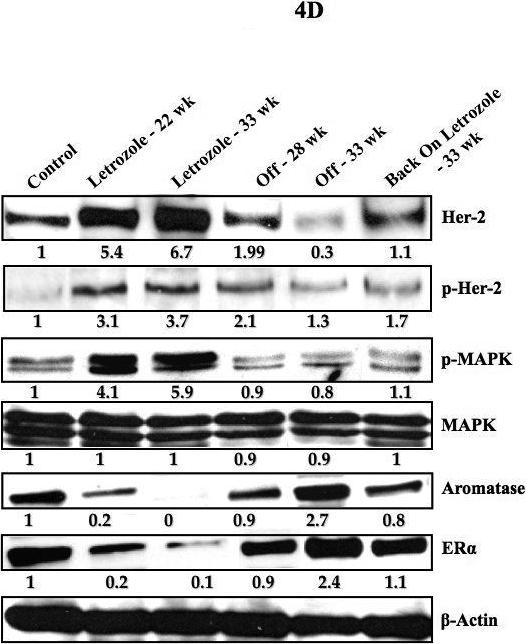
Figure 4D: Protein expression profile of the tumors of MCF-7Ca cells treated with letrozole: Expression of proteins was examined using western imunoblotting as described in Materials and Methods. Lane 1 Δ4A treated control, lane 2 letrozole treated tumor at week 22, lane 3 letrozole treated tumors at week 33, lane 4 tumor from “off” group at week 28, lane 5 “off” tumors at week 33 and lane 6 tumor from “back on” group at week 33. Blot shows Her-2 and p-Her-2 at 185 kDa, phospho-MAPK and MAPK at 42−44 kDa, aromatase at 55kDa, ERα at 66 kDa and β-actin at 45 kDa. The blots were stripped and re-probed for β-actin to verify equal loading. The blots show a single representative of three independent experiments.
However, the uterine weights exhibit significant differences (Figure. 4B). As the mouse uterus is very sensitive to estrogens, the measurement of uterine weight is a useful bioassay that indicates the level of circulating estrogens in the mouse. The mice in the “letrozole” group had significantly lower uterine weights compared to the control group (p = 0.0006) and “off” (p = 0.02) group indicating that aromatase was effectively inhibited by letrozole in this group. However, the mice in control and “off” group had similar uterine weights (p = 1). Furthermore, the uterine weights of mice in the control and “off” group correlated well with their tumor weights. In contrast, the uterine weights of mice in the letrozole group were markedly reduced and did not correlate with the tumor weights. This suggests that estrogen production in control and “off” groups was sufficient to stimulate tumor growth. Although estrogen synthesis was suppressed as evidenced by low uterine weight in the letrozole group, the growth of tumors in letrozole group was independent of estrogen.
Protein expression and activity after letrozole withdrawal in MCF-7Ca xenografts
MCF-7Ca xenografts were examined for changes in the aromatase activity after treatment with letrozole. Tumors used for this assay were collected at different time points during the course of treatment (Figure 4C). When treated with letrozole, aromatase activity of the tumors was significantly lower compared to control (p<0.01, week 22) and (p<0.001, week 33). However, after discontinuing the treatment (“Off” – 22 weeks), aromatase activity increased and was similar to the control level (p<0.001 compared to letrozole at week 22 and 33). The aromatase activity however had increased further by week 33, and it was significantly higher than the control level (p<0.001). When the mice were put “back on” letrozole, the aromatase activity ceased to increase and remained at the same level as the “Off” group at week 28 but it was still higher than control (p<0.01).
When protein expression was examined (Figure 4D), tumors treated with letrozole (22 weeks) exhibited higher expression of Her-2, p-Her-2 and p-MAPK than controls. The levels increased further by 33 weeks. In contrast, the tumors from the “off” group showed decreased levels of Her-2, p-Her-2 and p-MAPK compared to letrozole treated tumors and were similar to control tumors. On the other hand, ERα and aromatase expression decreased with letrozole treatment and was restored in the “off” groups (week 28 and 33). These results suggest that tumors may adapt using the Her-2/MAPK signaling pathway during letrozole treatment but revert to the ERα pathway when letrozole is withdrawn.
Discussions
Despite significant advances in the treatment of breast cancer since the development of AIs, (15-17) some patients eventually become resistance to treatment. Thus, it is important to understand the mechanisms of the resistance and determine how resistance to treatment can be overcome. We have developed a xenograft model in order to investigate how cells adapt and survive the effects of treatment. Although letrozole caused marked inhibition of tumor growth initially, tumors eventually acquired the ability to grow in the presence of letrozole and were refractory to estrogen stimulation and to second-line therapy with antiestrogens (3, 8, 9).
Previous studies of cells isolated from the long term letrozole treated tumors (LTLT-Ca) revealed that overexpression of Her-2 and activation of the MAPK signaling pathway accompanied the resistance to AEs, AIs, Δ4A and E2 (8, 18). In these cells, ER levels were reduced and cell growth was independent of estrogen indicating, cross-talk between ERα and HER-2 (10). These results suggest that acquisition of resistance and progression to hormone independence represents a shift in the balance that leans towards growth factor signaling in the presence of letrozole and back to hormonal signaling upon withdrawal of the aromatase inhibitor. These results led to the hypothesis that Her-2/MAPK pathway provides a compensatory signaling mechanism for the cells when estrogen levels are extremely low (10).
Analysis of the protein expression and aromatase activity in RLT-Ca cells revealed that Her-2 and the downstream signaling proteins as well as ERα had returned to the levels in the original phenotype, MCF-7Ca cells. Thus, RLT-Ca cells respond to E2, Δ4A, AIs and fulvestrant. The EC50 values for these RLT-Ca cells were similar to those previously reported for MCF-7Ca cells (19). The response to tamoxifen however was not completely restored in RLT-Ca cells.
The in vivo findings were consistent with the in vitro results. When mice were inoculated with MCF-7Ca cells and treated with letrozole, tumor growth was suppressed for several weeks but eventually tumors began to grow despite continued treatment. By 22 weeks the tumor volume reached twice the initial volume. The mice were then divided into two groups. One group continued on letrozole whereas treatment was then stopped in the other group for 6-weeks. At week-28, half of the animals in the latter group began treatment with letrozole again while the others continued on Δ4A until week-33. When letrozole treatment was discontinued, aromatase activity returned to MCF-7Ca control levels by 28 weeks. However at 33 weeks aromatase activity was more than 3-fold higher than control. Although the mechanism of this increase is unclear, previous studies have reported increased aromatase activity after letrozole withdrawal that has been explained by stabilization and reduced degradation of the aromatase protein. Interestingly, growth of tumors in “off” group was not significantly different from those of the animals continuing on letrozole. Although, the increase in aromatase activity may subsequently lead to increase in tumor growth in the “Off” group, further increase in Her-2/MAPK signaling could drive the growth of tumors on continuous letrozole treatment.
In conclusion, our results suggest that resistance to letrozole can be reversed by discontinuing treatment for 6-weeks. These findings suggest that alternating “on-off” letrozole treatment may be an effective way to delay resistance and prolong sensitivity to AIs. This strategy could have advantages for the patients relapsing from aromatase inhibitor treatment.
Acknowledgements
MCF-7 human breast cancer cells stably transfected with the human aromatase gene (MCF-7Ca) were provided by Dr. S. Chen (City of Hope, Duarte, CA). Letrozole (Femara®, CGS 20267) was provided by Dr. D. Evans (Novartis Pharma, Basel, Switzerland).
Note: Supported by grant CA-62483 to Dr. Brodie from the National Cancer Institute, National Institute of Health. Presented in part at 89th Annual Endocrine Society Meeting, Toronto, Ontario, Canada, June 2007.
Abbreviations used
- ERα
Estrogen Receptor α
- Δ4A
Androstenedione
- E2
Estradiol
- Her-2
Human Epidermal Growth factor Receptor- 2
- MAPK
Mitogen Activated Protein KInase
- AIs
Aromatase Inhibitors
- AEs
Antiestrogens
References
- 1.Yue W, Brodie A. MCF-7 human breast carcinomas in nude mice as a model for evaluating aromatase inhibitors. J Steroid Biochem Mol Biol. 1993;44:671–3. doi: 10.1016/0960-0760(93)90278-5. [DOI] [PubMed] [Google Scholar]
- 2.Yue W, Zhou D, Chen S, Brodie A. A new nude mouse model for postmenopausal breast cancer using MCF-7 cells transfected with the human aromatase gene. Cancer Res. 1994;54:5092–5. [PubMed] [Google Scholar]
- 3.Long BJ, Jelovac D, Handratta V, et al. Therapeutic strategies using the aromatase inhibitor letrozole and tamoxifen in a breast cancer model. Journal of the National Cancer Institute. 2004;96:456–65. doi: 10.1093/jnci/djh076. [DOI] [PubMed] [Google Scholar]
- 4.Goss PE. Preventing relapse beyond 5 years: the MA.17 extended adjuvant trial. Seminars in oncology. 2006;33:S8–12. doi: 10.1053/j.seminoncol.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 5.Goss PE, Ingle JN, Martino S, et al. Efficacy of Letrozole Extended Adjuvant Therapy According to Estrogen Receptor and Progesterone Receptor Status of the Primary Tumor: National Cancer Institute of Canada Clinical Trials Group MA.17. J Clin Oncol. 2007 doi: 10.1200/JCO.2006.09.4482. [DOI] [PubMed] [Google Scholar]
- 6.Baum M. The ATAC (Arimidex, Tamoxifen, Alone or in Combination) adjuvant breast cancer trial in postmenopausal patients: factors influencing the success of patient recruitment. Eur J Cancer. 2002;38:1984–6. doi: 10.1016/s0959-8049(02)00154-5. [DOI] [PubMed] [Google Scholar]
- 7.Baum M, Budzar AU, Cuzick J, et al. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. Lancet. 2002;359:2131–9. doi: 10.1016/s0140-6736(02)09088-8. [DOI] [PubMed] [Google Scholar]
- 8.Jelovac D, Sabnis G, Long BJ, et al. Activation of mitogen-activated protein kinase in xenografts and cells during prolonged treatment with aromatase inhibitor letrozole. Cancer Res. 2005;65:5380–9. doi: 10.1158/0008-5472.CAN-04-4502. [DOI] [PubMed] [Google Scholar]
- 9.Long BJ, Jelovac D, Thiantanawat A, Brodie AM. The effect of second-line antiestrogen therapy on breast tumor growth after first-line treatment with the aromatase inhibitor letrozole: long-term studies using the intratumoral aromatase postmenopausal breast cancer model. Clin Cancer Res. 2002;8:2378–88. [PubMed] [Google Scholar]
- 10.Sabnis GJ, Schayowitz A, Goloubeva O, Brodie AMH. Trastuzumab increases sensitivity of hormone dependent and hormone refractory breast cancer cells to endocrine agents. AACR Meeting Abstracts. 2007 Abstract 991. [Google Scholar]
- 11.Sabnis GJ, Jelovac D, Long B, Brodie A. The role of growth factor receptor pathways in human breast cancer cells adapted to long-term estrogen deprivation. Cancer Res. 2005;65:3903–10. doi: 10.1158/0008-5472.CAN-04-4092. [DOI] [PubMed] [Google Scholar]
- 12.Long BJ, Tilghman SL, Yue W, et al. The steroidal antiestrogen ICI 182,780 is an inhibitor of cellular aromatase activity. J Steroid Biochem Mol Biol. 1998;67:293–304. doi: 10.1016/s0960-0760(98)00122-8. [DOI] [PubMed] [Google Scholar]
- 13.Sabnis G, Goloubeva O, Jelovac D, Schayowitz A, Brodie A. Inhibition of the Phosphatidylinositol 3-Kinase/Akt Pathway Improves Response of Long-term Estrogen-Deprived Breast Cancer Xenografts to Antiestrogens. Clin Cancer Res. 2007;13:2751–7. doi: 10.1158/1078-0432.CCR-06-2466. [DOI] [PubMed] [Google Scholar]
- 14.Macedo LF, Guo Z, Tilghman SL, et al. Role of androgens on MCF-7 breast cancer cell growth and on the inhibitory effect of letrozole. Cancer Res. 2006;66:7775–82. doi: 10.1158/0008-5472.CAN-05-3984. [DOI] [PubMed] [Google Scholar]
- 15.Boccardo F, Rubagotti A. Switching to aromatase inhibitors in early breast cancer. Lancet. 2007;369:533–5. doi: 10.1016/S0140-6736(07)60201-3. [DOI] [PubMed] [Google Scholar]
- 16.Howell A. Adjuvant aromatase inhibitors for breast cancer. Lancet. 2005;366:431–3. doi: 10.1016/S0140-6736(05)67036-5. [DOI] [PubMed] [Google Scholar]
- 17.Swain SM. Aromatase inhibitors--a triumph of translational oncology. N Engl J Med. 2005;353:2807–9. doi: 10.1056/NEJMe058273. [DOI] [PubMed] [Google Scholar]
- 18.Jelovac D, Sabnis G, Long BJ, Macedo L, Brodie A. Strategies to oppose loss of sensitivity to hormone therapy in breast cancer cells. AACR Meeting Abstracts. 2005;2005:1032–b. [Google Scholar]
- 19.Thiantanawat A, Long BJ, Brodie AM. Signaling pathways of apoptosis activated by aromatase inhibitors and antiestrogens. Cancer Res. 2003;63:8037–50. [PubMed] [Google Scholar]