

Abstract
This is the first report of the in vivo effectiveness of memantine as a neuroprotective agent against rotenone-induced retinal toxicity. We tested the hypothesis that uncompetitive NMDAR blockade with memantine prevents mitochondrial dysfunction-related neurodegeneration in vivo, using a mouse model of retinal ganglion cell layer (GCL) degeneration induced by rotenone, a mitochondrial complex I inhibitor. Rotenone induced an increase in cell death and oxidative stress in GCL compared to controls, and these changes were prevented by the co-administration of memantine. The neurotoxic effect of rotenone was also reflected as a decrease in total cell density in GCL and GCL + nerve fiber layer thickness. These changes were also prevented by co-administration of memantine in a dose-dependent manner. In addition, memantine induced an increase in long-term retinal energy metabolic capacity. The results suggest that NMDAR activation contributes to cell death induced by mitochondrial dysfunction and that uncompetitive NMDAR blockade may be used as a neuroprotective strategy against mitochondrial dysfunction in neurodegenerative diseases.
Section: Disease-related neuroscience
Key words: memantine, mitochondrial dysfunction, neurodegeneration, neuroprotection, rotenone, oxidative stress, energy metabolism
1. Introduction
Mitochondrial dysfunction and excitotoxicity are interrelated events that seem to have a critical role in neurodegeneration (Schinder et al., 1996, Sengpiel et al., 1998, Kanki et al., 2004). Inhibition of mitochondrial function has been shown to produce energy depletion, increase oxidative stress and induce apoptosis in several models (Sherer et al., 2003, Beretta et al., 2006, Zhang et al., 2006b). Energy depletion impairs membrane potential resetting and maintenance, which promotes constant neuronal excitation, massive influx of calcium mainly via NMDA receptor (NMDAR) activation, and the concomitant hyperactivation of proteases, lipases and endonucleases (Winkler, 1983, Henneberry et al., 1989, Chiu et al., 2005). Oxidative stress activates apoptosis signaling pathways and further impairs the function of components of both the mitochondrial and cell membranes (Cassina and Radi, 1996, Brookes et al., 1998, Rosenstock et al., 2004). Thus, mitochondrial dysfunction and excitotoxicity appear to be synergistic cellular events implicated in a vicious cycle enhancing occurrence of cell death and possibly having significant implications in the pathogenesis of neurodegenerative disorders.
Interruption of this vicious cycle may decrease activation of cell death signals, having an impact on the onset and progression of neurodegeneration. One of the candidate strategies to achieve this is by impairing NMDAR hyperactivation, a key event in excitotoxicity. Memantine is an amantadine derivative that exerts an uncompetitive antagonist effect on the NMDAR activation (Chen and Lipton, 1997). This means that its ion channel-blocking effect takes place only when the NMDAR has been previously activated by glutamate in a sustained manner. Thus, this antagonist might interfere with excitotoxic activity without significantly affecting normal function during synaptic transmission or long-term potentiation (Chen et al., 1998). This should represent an advantage when excitotoxic damage is to be prevented in a neuronal network subjected to the cellular stress introduced by mitochondrial dysfunction. In the present study, this hypothesis was tested using an in vivo toxicological model of retinal neurodegeneration induced by rotenone described previously (Zhang et al., 2002, Zhang et al., 2006a)‥ Rotenone is a specific inhibitor of mitochondrial complex I that may constitute a major environmental neurotoxin. It has been previously reported that a single intravitreal injection of rotenone causes retinal neurodegeneration by increasing reactive oxygen species production and by decreasing oxygen consumption (Zhang et al., 2006b). This model is suitable for efficiently testing the efficacy of candidate neuroprotective interventions, since it allows the assessment of in vivo biological effects in a relatively accessible part of the central nervous system, with the advantage of a within-subject control design. The present experiments evaluated the in vivo effects of memantine on cell death, morphological neurodegenerative changes, oxidative stress and energy metabolism induced by rotenone in the retina.
2. Results
2.1 Cell death and histopathology
Memantine prevented the increase in TUNEL+ cells in the ganglion cell layer induced by rotenone, as early as 1 hr post-treatment. Compared to vehicle treated controls (3 ± 0.3 TUNEL+ cells per section), rotenone induced an early significant increase in TUNEL+ cells in the retinal ganglion cell layer (GCL) (7.2 ± 2 TUNEL+ cells per section, p<0.01), a change that was detected 1 hr after injection. This effect was prevented by the co-administration of 70 µg/kg memantine (0.5 ± 0.3 TUNEL+ cells per section, F2,36=7.2, p<0.01) (Figure 1). There was no longer a difference in the mean number of TUNEL+ cells between control and rotenone-treated groups after 24 hr (F2,14=2.5, p=0.12). By 24 hr the TUNEL+ cells found at 1 hr post-rotenone may have disappeared due to the loss of GCL cells seen 24 hr after rotenone (Zhang et al., 2006a).
Figure 1.
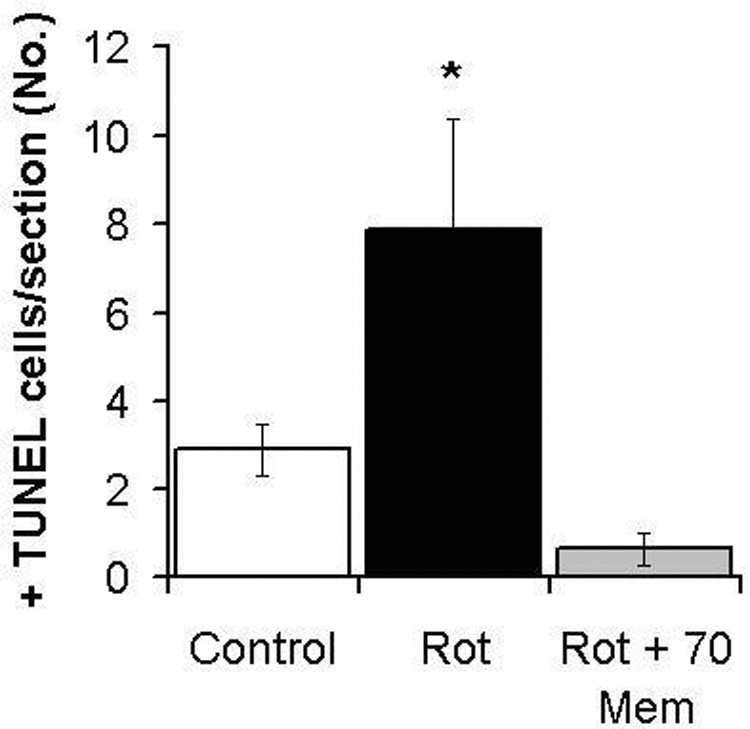
Effect of rotenone (Rot) alone or co-administered with memantine (Rot + Mem 70) on the number of TUNEL+ cells in the retinal ganglion cell layer. Memantine (70 µg/kg) prevented the increase in TUNEL+ cells in the ganglion cell layer induced by rotenone, as early as 1 hr post-treatment. The asterisk represents a significant group difference compared to control (vehicle-treated eyes). Dunnett’s test, p<0.01. A single asterisk indicates that only the rotenone alone group differed from control, while the group with rotenone plus memantine was not different from the vehicle-treated control.
Memantine prevented the decrease in ganglion cell layer cell density induced by rotenone at 24 hr post-treatment. Since only a small fraction of the GCL cell population was TUNEL+ by 1 hr post-injection, and based on previous morphometric analyses after rotenone intravitreal administration (Zhang et al., 2006a), a significant change in retinal morphology was not expected at 1 hr. Alteration of the retinal anatomy was evaluated 24 hr after injection, a time-point previously shown to be sufficient to detect alterations of retinal stereological parameters induced by rotenone (Zhang et al., 2002). The neurotoxic effect of rotenone was reflected as a 19.9% decrease in the mean total cell density in the GCL (26,792 ± 2,885 cells/mm3 vs. control 33,476 ± 3,694 cells/mm3, p<0.04). This change was prevented by the co-administration of memantine with cell densities of 40,427 ± 3,972, 42,680 ± 3,839 and 40,040 ± 6,181 cells/mm3 (respectively memantine 0.7, 7 and 70 µg/kg) (F4,65=2.6, p<0.05) (Figure 2).
Figure 2.
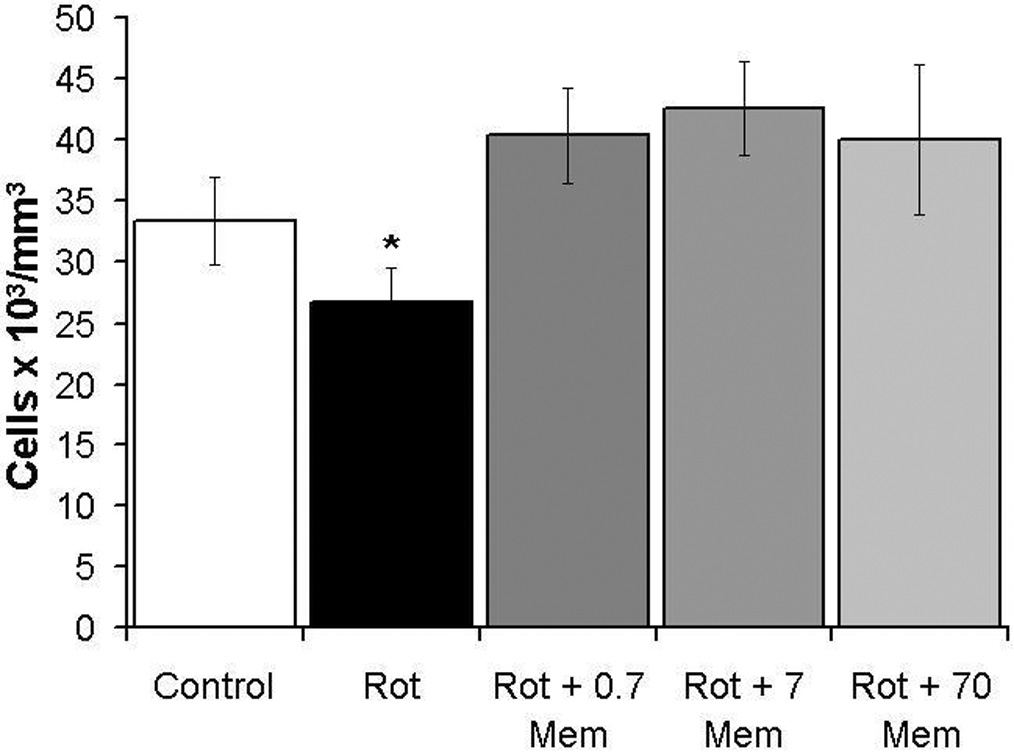
Memantine prevented the decrease in ganglion cell layer cell density induced by rotenone at 24 hr post-treatment. This neuroprotective effect on cell density was found for all memantine doses used (0.7, 7 and 70 µg/kg). The asterisk represents a significant group difference compared to control (vehicle-treated eyes). Dunnett’s test, p<0.05. A single asterisk indicates that only the rotenone alone group (Rot) differed from control, while the groups with rotenone plus memantine were not different from the vehicle-treated control.
Memantine visibly prevented the signs of retinal histopathology induced by rotenone at 24 hr post-treatment, as illustrated in optic pole samples of sagittal retinal sections stained for complex I activity (Figure 3). Rotenone induced a 32.6% decrease in the mean GCL + retinal nerve fiber layer (RNFL) thickness (16.3 ± 0.9 µm vs. control 24.2 ± 1.3 µm, p<0.001) (Figure 4). This reduction in GCL + RNFL thickness was prevented by memantine in a dose-dependent manner (16.9 ± 1, 18.9 ± 2, 21.8 ± 1 µm, for memantine 0.7, 7 and 70 µg/kg, respectively). Only high doses, but no a low dose of memantine were effective in preventing the reduction in GCL+RNFL thickness (F4,62=6.6, p<0.001). At 2 weeks, there was no group difference in the mean GCL+RNFL thickness (control 15.8 ± 1 µm, rotenone 11.8 ± 1 µm, 70 mg/kg memantine/rotenone 13.3 ± 4 µm, F2,16=0.61, p=0.5).
Figure 3.
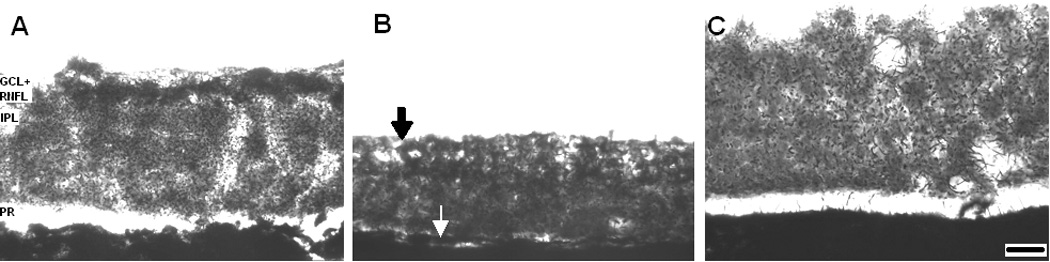
Memantine prevented the retinal histopathological changes induced by rotenone at 24 hr post-treatment. The figure shows optic pole samples of sagittal retinal sections stained with the complex I activity technique. Light Microscopy, 50X. Panel A. The ganglion cell layer and the retinal nerve fiber layer (GCL+RNFL) from control eyeballs present a strong staining compared to other layers such as the inner plexiform layer (IPL). Panel B. Rotenone reduces retinal thickness mainly at the expense of GCL+RNFL, which has a mild staining intensity and appears fenestrated (black arrow), but also by affecting other retinal regions such as the photoreceptor layer (PR) (white arrow). Panel C. Rotenone plus memantine-treated retina (70 µg/kg) showed no signs of histopathology. Scale bar=20 µm.
Figure 4.
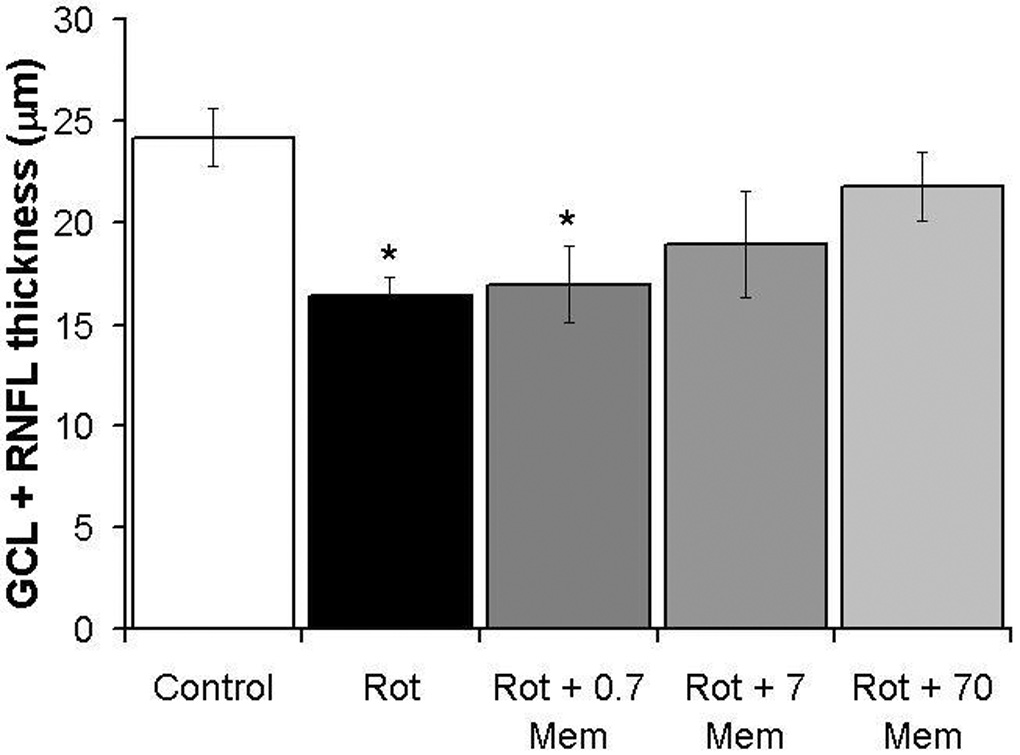
Dose-response neuroprotective effects of memantine on the thickness of ganglion cell layer and retinal nerve fiber layer treated with rotenone. High doses of memantine (7 and 70 µg/kg), but not a low dose (0.7 µg/kg) prevented the rotenone-induced ganglion cell layer and retinal nerve fiber layer (GCL+RNFL) neurodegeneration in the optic pole at 24 hr. Asterisks indicate a significant group difference compared to control (vehicle-treated eyes). Dunnett’s test, p<0.001. The asterisks indicate that both the rotenone alone group (Rot) and the group with the lowest memantine dose (Rot + 0.7 Mem) differed from control, while the groups with rotenone plus higher memantine doses were not different from the vehicle-treated control.
In addition, the rotenone-induced decrease in cell density was not the result of edema, since no significant changes at 24 hr were detected in terms of total retinal volume between groups (rotenone 0.58 mm3 vs. control 0.56 mm3, p=0.64) (Figure 5). This measurement reflects the volume of all layers in both the central and peripheral regions of the retina, as opposed to a localized sampling.
Figure 5.
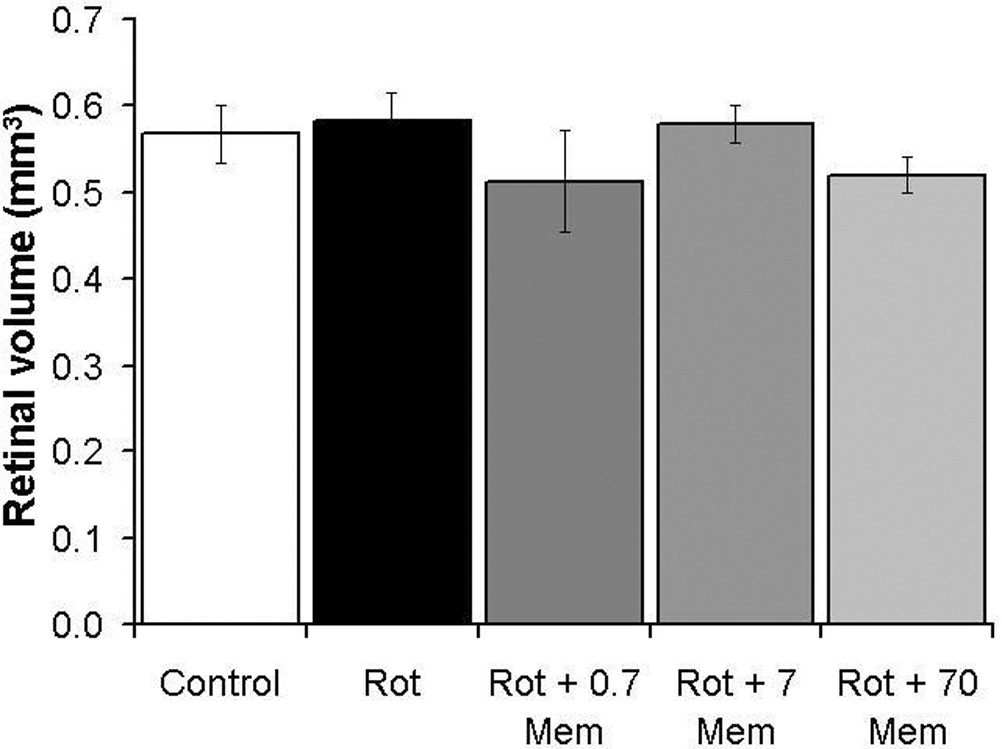
Total mean retinal volume at 24 hr post-treatment. No significant group differences were evident. Volume measurements include all retinal layers and all retinal regions (central and peripheral).
The use of unbiased stereological techniques maximized the accuracy and efficiency of the measurements. At 24 hr GCL + RNFL thickness was highly sensitive to rotenone’s neurotoxicity, as demonstrated by the large effect sizes (Table 1). The low coefficient of error and the low contribution of the stereological method to the observed variation between groups demonstrated that most of the variation observed is due to treatment effects (Table 1).
2.2 Retinal oxidative stress
The rotenone-induced histopathological changes described above were hypothesized to be mediated by an increase in oxidative stress exacerbated by NMDAR hyperactivation. We tested whether memantine would prevent the in situ increase in retinal oxidative stress induced by rotenone at 24 hr, by measuring dihydroethidium (DHE) fluorescence. Rotenone-treated retinas showed a 2-fold increase in the mean relative DHE fluorescence, compared to control (Figure 6). Only GCL cells appeared to show an increase in oxidative stress in response to rotenone, since DHE signal increased only in this layer. Memantine co-administration (70 µg/kg) was effective in reducing the increase in oxidative stress induced by rotenone, since memantine-treated retinas showed no difference in DHE fluorescence, compared to control (F2,14=3.6, p<0.05). This effect was also evident only in GCL, but not in other retinal layers (F2,14=0.5, p=0.61).
Figure 6.
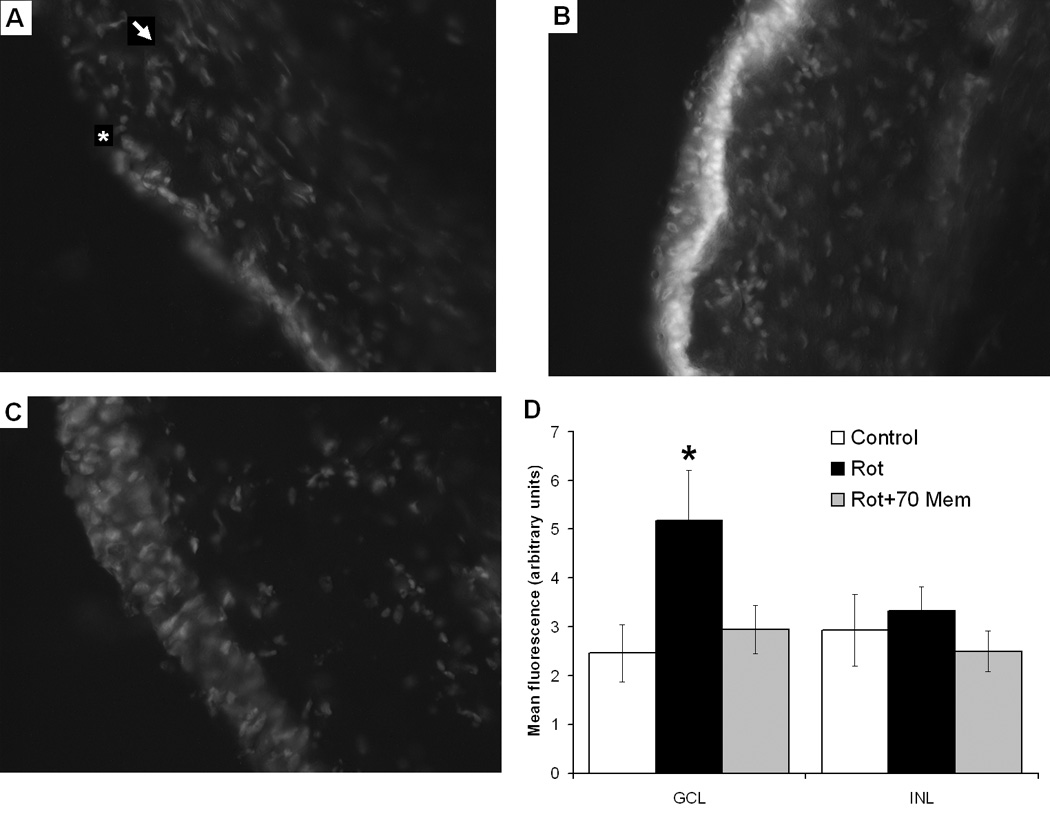
In situ detection of retinal oxidative stress by DHE fluorescence. Epifluorescence microscopy, 20X. Panel A: Vehicle-treated retinas show a weak signal in both the ganglion cell layer (GCL) (white asterisk) and outer retinal layers (white arrow). Panel B: Rotenone induced a marked increase in oxidative stress as seen by the increased fluorescence signal localized mainly in GCL (white layer). Panel C: The oxidative stress effect of rotenone alone was prevented by co-administration of 70 µg/kg memantine. Panel D: Summary group data of DHE fluorescence in GCL and inner nuclear layer (INL) relative to background for the control, rotenone (Rot) alone or co-administered with memantine (Rot + Mem 70). The black asterisk represents a significant group difference compared to control. Dunnett’s test, p<0.05.
2.3 Metabolic energy capacity
In addition to decreasing oxidative stress, NMDAR modulation by memantine was expected to allow the establishment of an adaptive response for neuronal survival. To test this hypothesis, we examined the retinal neuroplasticity in response to treatments, by means of the cytochrome oxidase activity histochemical staining procedure. Cytochrome oxidase is the terminal enzyme of the respiratory chain; its activity is the rate-limiting step for oxidative phosphorylation and it is tightly correlated with sustained neuronal metabolic demands, showing activity-dependent changes in function and expression (Hevner and Wong-Riley, 1993, Gonzalez-Lima and Cada, 1994). Thus, measurement of cytochrome oxidase activity was used to determine the effects of rotenone and memantine on retinal energy metabolism capacity. Memantine (70 µg/kg) plus rotenone-treated sections showed an increase in cytochrome oxidase activity relative to control at 2 weeks (248.2 ± 15 and 161.4 ± 21 µmol/min/mg, respectively, F2,14=3.4, p<0.05), whereas no change was detected at 24 hr (control, 132.7 ± 15 µmol/min/mg and memantine/rotenone, 142 ± 15 µmol/min/mg, F2,14=0.32, p=0.9) (Figure 7). Finally, the metabolic activity in both the control and rotenone groups at 24 hr was similar to their respective counterparts at 2 weeks. Nevertheless, the metabolic activity at 2 weeks in retinas co-treated with memantine and rotenone was 74.6 percent higher than the one at 24 hr receiving a similar co-treatment, a difference that was significant.
Figure 7.
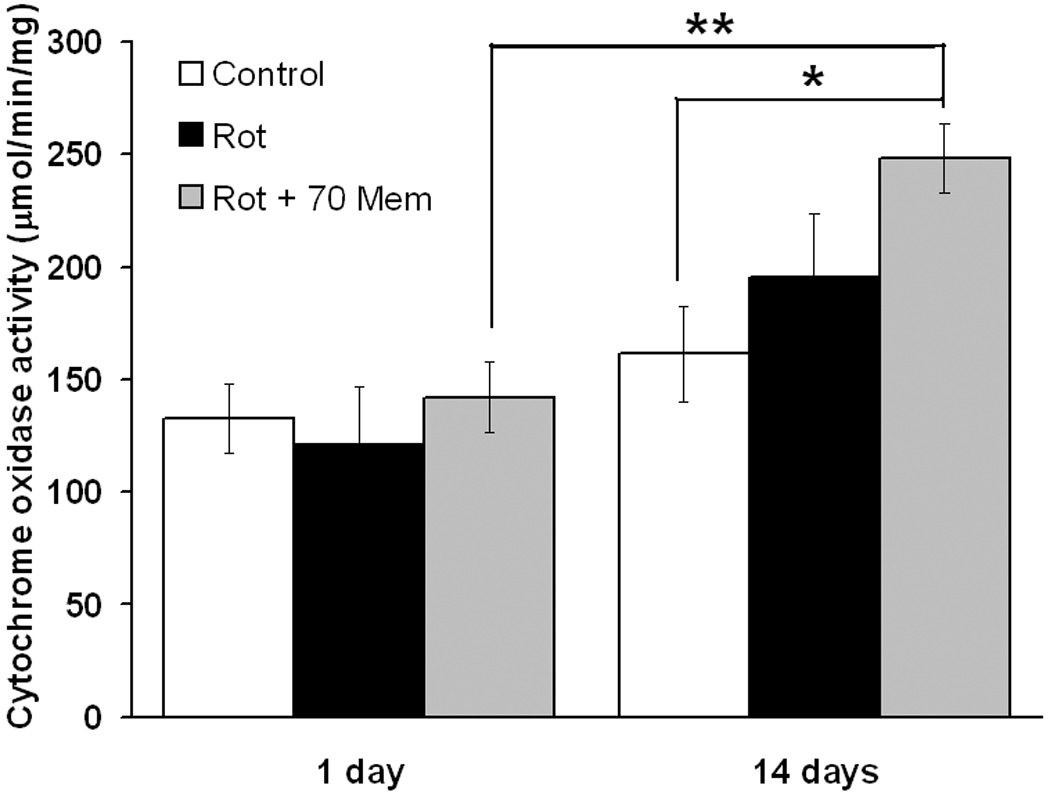
Retinal energy metabolic capacity evaluated by quantitative histochemistry of cytochrome oxidase activity. After 2 weeks, 70 mg/kg memantine/rotenone-treated retinas showed an increased metabolic capacity relative to control. The metabolic activity in the memantine/rotenone-treated group at 14 days was 76.4 percent higher than its counterpart at 1 day. No between group differences were evident at 24 hr. One asterisk indicates a significant group difference compared to control. Dunnett's test p<0.05. Two asterisks indicate a significant difference between 1 day and 14 days time points, ANOVA p<0.05.
3. Discussion
This study is the first to report the in vivo effectiveness of memantine as a neuroprotective agent against rotenone-induced retinal toxicity. Rotenone is a naturally occurring high-affinity inhibitor of mitochondrial complex I that impairs oxygen consumption (Zhang et al., 2006b) and oxidative phosphorylation (Ernster et al., 1963, Davey and Clark, 1996), and promotes oxidative stress (Koopman et al., 2005) and cell death (Sherer et al.,2003). Rotenone is a potential environmental neurotoxin. The United States is the greatest consumer of rotenone, with an annual usage estimated between 25,000 and 60,000 kg, most of which are used for pesticidal purposes, on both food and non-food crops, livestock and lakes (Ling, 2003).
In this study, low amounts of rotenone (6 µg per eyeball) rapidly triggered a 2.4-fold increase in the number of TUNEL positive cells after only one hour of exposure. The rapid toxicity of rotenone is a phenomenon that has been previously demonstrated in non-neural cells in in vitro systems where significant increases in apoptotic cells can be detected as early as 20 min after rotenone exposure (Isenberg and Klaunig, 2000). Our findings confirm that GCL cells are highly sensitive to the neurotoxic properties of rotenone in vivo. At 24 hr rotenone produced a ≈20% decrease in the GCL cell density and a ≈30% decrease in GCL + RNFL thickness. As previously shown, the reduction in thickness produced by rotenone at 24 hr is due to a ≈90% decrease in RNFL thickness and a 20% decrease in GCL thickness alone (Zhang et al., 2006a). The sensitivity of GCL cells was further supported by the fact that GCL showed a higher degree of oxidative stress compared to other retinal layers. Previous research has demonstrated that rotenone is particularly toxic to GCL cells in vitro, inducing cell death, impaired glutamate uptake, and increases in oxidative stress (Beretta et al., 2006). GCL cells seem to share a differential vulnerability to the oxidative damage induced by rotenone with cells in the striatum, midbrain and cortex, whereas other neuronal populations in the cerebellum or hippocampus appear to be more resistant to rotenone’s toxic effects (Sherer et al., 2003).
These data highlight that rotenone is not only relevant for the study of a causative role of environmental neurotoxins in human diseases, but also that this potent neurotoxic agent is ideal for promoting a context of retinal neurodegeneration in order to assess the effects of candidate neuroprotective interventions. The stereological estimates used for the morphometric analysis reported here displayed great accuracy and showed that the observed variability could be more attributable to biological factors and experimental conditions than the morphometric method itself. In addition, among the reported effect sizes for morphological estimates, the largest one was seen in layer thickness, which suggest not only that the status of nerve fibers is specifically vulnerable to complex I inhibition, but also that the complex I histochemical method exhibits exquisite sensitivity for detection of the layer thickness changes in the retina induced by the pharmacological treatments. Although drastic, the neurodegenerative changes in GCL+RNFL could only be detected with the application of this methodology, whereas the whole retinal volume analysis, which did not take into account the neurochemical properties of the tissue or a differential layer selection, did not reveal such a change.
Memantine is an FDA-approved drug that is clinically used to retard the cognitive decline in cases of moderate to severe Alzheimer’s disease. Memantine has unique pharmacodynamic features, with ion channel blocking effects that are limited to pathologically hyperactive NMDAR. Memantine reduces NMDA-activated currents, while having no effects on responses to GABA or AMPA (Parsons et al., 1993). Both its high selectivity and its antagonistic activity on active NMDAR contribute to increasing its therapeutic index, while maximizing its anti-excitotoxicity properties. Memantine is a versatile agent exhibiting neuroprotective effects in several models of neurodegeneration, including ischemia (Ehrlich et al., 1999), oxygen-glucose deprivation (Frankiewicz et al., 2000) and amyloid β-induced neurotoxicity (Miguel-Hidalgo et al., 2002, Yamada et al., 2005). Schulz et al (1996), reported that memantine prevented the volumetric changes in the mouse striatum following administration of malonate, a complex II inhibitor, demonstrating that memantine could be used to prevent the deleterious effects of mitochondrial dysfunction. In vitro studies have shown that memantine diminishes the toxicity induced by 3-nitropropionic acid, another complex II inhibitor, in cerebellar granular cell cultures (Volbracht et al., 2006), cholinergic neurons (Wenk et al., 1996), and hippocampal slices (Karanian et al., 2006). Despite the evidence supporting a relevant role of complex I inhibition in neurodegeneration, the effects of memantine against complex I inhibition have remained largely unexplored. The first demonstration of the neuroprotective properties of memantine against complex I inhibition using an in vitro system was recently reported by Volbracht and coworkers (2006). This group demonstrated that memantine preserved the neurite integrity and prevented cell death in cerebellar granular cell cultures exposed to the synthetic complex I inhibitor MPP+.
The present study provides evidence that memantine can prevent the in vivo morphological damage induced by complex I inhibition with the natural xenobiotic rotenone. The neuroprotective effects of memantine co-administered with rotenone were observed as early as 1 hr, manifested as no change in cell death in GCL as compare to the vehicle-treated control. At 24 hr, rotenone and memantine showed no effects on TUNEL+ cell numbers, which suggests that TUNEL+ cells found at 1 hr post-rotenone may have disappeared due to the loss of GCL cells seen 24 hr after rotenone (Zhang et al., 2006a).
Memantine may exert an early biological effect that may constitute a relevant mechanistic step in neuroprotection. Tremblay et al. (2000) demonstrated that pre-exposure of rat cortical neurons to memantine for 30 min is sufficient to protect against NMDA-mediated cell death, even when this lethal insult is applied 48 hr after the brief memantine exposure. Although animal studies have demonstrated that neuroprotection with memantine is achieved after relatively prolonged periods of steady plasma concentrations, it is possible that even transient exposures to the drug result in advantageous cellular effects. The observed effects of memantine on TUNEL+ cells found at 1 hr post-rotenone may be a consequence of the features of the model that was used. This model involves local intraocular administration of both rotenone and memantine. This approach minimizes issues of bioavailability associated with systemic drug administration and accelerates the onset of pharmacodynamic interactions of both compounds.
Memantine had remarkable neuroprotective effects against rotenone-induced morphometric tissue changes at 24 hr. It not only prevented the reduction in GCL cell density but also prevented the thinning of GCL + RNFL in a dose-dependent manner. This effect together with the high efficiency of the stereological estimates support that the complex I staining technique is highly sensitive for detecting the thickness changes of GCL + RNFL. Rotenone increased in situ retinal oxidative stress, which likely mediated the neurodegenerative morphological changes, and memantine prevented this increase in oxidative stress. Since memantine is known for its uncompetitive NMDAR antagonistic properties, the results suggest that excitotoxic damage mediates neurodegeneration induced by mitochondrial dysfunction. Energy depletion induced by rotenone is associated with mitochondrial and cell membrane depolarization (Rizzardini et al., 2006) and the later facilitates NDMAR activation, which may mediate excitotoxicity and further increases in oxidative stress (Sengpiel et al., 1998). NMDAR modulation with memantine may stop the excitotoxic response by decreasing the energy expenditure in otherwise energy-depleted cells, but most importantly may facilitate the expression of intracellular mechanisms to cope with oxidative stress, events that may promote neuronal survival.
The effects of memantine on long-term metabolic capacity were evaluated histochemically by analyzing changes in retinal cytochrome oxidase activity. At 2 weeks, memantine produced an increase in retinal metabolic capacity in the presence of rotenone, but no effect was observed at 24 hr. Changes in metabolic capacity are expected to parallel changes in neural energy demands (Gonzalez-Lima and Cada, 1994). The increase in metabolic capacity in memantine-treated retinas suggests that neurons were able to increase the long-term expression of functional proteins, which otherwise may be compromised in the presence of rotenone. From this perspective, neuroplasticity of energy-related proteins such as cytochrome oxidase would be essential to sustain further adaptive processes, showing the most robust plasticity effects (Hevner and Wong-Riley, 1993).
The results suggest that memantine may be used to prevent neurodegeneration promoted by mitochondrial dysfunction. Strategies against the immediate consequences of mitochondrial dysfunction (i.e. energy depletion and increased oxidative stress) include induction of antioxidant enzymes or administration of antioxidant xenobiotics. Although both of these strategies are theoretically compelling, the first one could lead to failure because it relies on gene expression and protein synthesis, cellular processes that might be compromised by energy depletion. The second strategy assumes that the xenobiotic antioxidant will have an optimal bioavailability in its adequate redox form through the blood-brain barrier and cell membranes to reach the effective intramitochondrial antioxidant concentrations, which may be problematic for some compounds. The results of this study suggest the existence of a critical interaction between the mitochondria and the cell membrane that can be manipulated through uncompetitive NMDAR blockade to prevent neurodegeneration induced by mitochondrial dysfunction. Hence, the results may be relevant for the management of some neurodegenerative illnesses, especially Leber’s optic neuropathy.
4. Experimental procedure
4. 1 Subjects and reagents
Adult male CBA/J mice (N=47, average weight 27 g) were obtained from Jackson Laboratory (Bar Harbor, ME). Animals were maintained in clear plastic cages with food and water ad libitum and subjected to standard light cycles (12 hr light/12 hr dark) in an animal facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Texas at Austin. Unless otherwise specified, all chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO).
4. 2 Intravitreal injections
Subjects were anesthetized by an intraperitoneal injection of a mixture of ketamine hydrochloride (45 mg/kg), xylazine hydrochloride (9 mg/kg), and acepromazine (1.5 mg/kg). Under a stereomicroscope, a small sclerotomy was performed in the superior temporal quadrant of the eye to give access to the vitreous body. The microinjection was done immediately underneath the corneoscleral junction with a 30-G dental injection needle (Monoject®, Sherwood Medical Company, Norfolk, NE). The needle was connected by polyethylene tubing (internal diameter=0.38 mm) to a 10 µl Hamilton microsyringe. The injections were delivered over 2 min using a microinjection pump (CMA microdialysis AB, North Chelmsford, MA) (0.5 µL, final volume) and the injection needle was left in place for an additional 40-sec period to allow for diffusion away from the needle tip and to avoid any efflux of fluid through the injection site. The edges of the injection point were gripped with forceps while the microsyringe was slowly withdrawn. One eye of each animal received an injection of 0.2 mg/kg rotenone in the vehicle dimethylsulfoxide (DMSO) plus either 0.7 µg/kg (n =8 eyes), 7 µg/kg (n=8 eyes), or 70 µg/kg memantine (n=24 eyes), while the other received either 0.2 mg/kg rotenone in DMSO (n=23 eyes) or DMSO only as control (n=25 eyes). DMSO was used as a rotenone solvent, because it has been previously reported that it does not induce changes in retinal morphology, compared to non-injected subjects (Zhang et al., 2002). Regarding rotenone, its threshold limit value time weighted average for aerial exposure in humans is 5 mg/m3, so the occupational exposure (operators dispersing the toxicant) is estimated to be below 0.7 mg/kg/day. This is a dose that compares to the one chosen for the experimental model presented here. Animals were decapitated 1 hr (n=10 mice), 24 hr (n=28 mice) or 2 weeks (n=9 mice) after the injections and eyeballs were rapidly removed and frozen in isopentane (−40°C). In order to control for cut angle differences, eyeballs were transferred to cubic aluminum foil containers (2 mm3), orienting their anterior-posterior axis parallel to surface. The containers were filled with Shandon M1 embedding matrix (Thermo Electron Corporation, Pitsburgh, PA) and stored at −40°C. Forty µm-thick sagittal eyeball sections parallel to the anterior-posterior axis were obtained in a 2800 Frigocut E Reichert-Jung cryostat (Leica Microsystems, Bannockburn, IL) at −18°C and were mounted on poly-L-lysine coated slides to create three adjacent series for histological analysis.
4. 3 TUNEL staining
Detection of cells undergoing cell death in the retinal ganglion cell layer (GCL) was determined using the TUNEL technique by means of a commercially available kit (FD NeuroApop, FD NeuroTechnologies Ellicott City, MD) (Gavrieli et al., 1992). Eyeball frozen sections were fixed in 4% paraformaldehyde, 0.5% glutaraldehyde, pH 7.4 for 45 min at room temperature and were subsequently treated according to the manufacturer’s directions. Sections were air dried for 5 min, counterstained in methyl green for 5 min (0.5% methyl green in 0.1 M sodium acetate buffer, pH 4.2), dehydrated in 95% and 100% ethanol, cleared in xylene and cover-slipped with Permount®.
4.4 Nissl staining
Nissl staining was used to obtain stereological estimates of GCL cell number and density. Frozen eyeball sections were delipidized in a series of 95% ethanol, 70% ethanol, distilled water, and 0.1 M sodium acetate buffer pH 4 (2 min each). Sections were stained in 0.1% cresyl violet in sodium acetate buffer pH 4 at 45°C for 7 min, differentiated in 70% and 95% ethanol and dehydrated in 95% and 100% ethanol. Slides were cleared in xylene and coverslipped with Permount®.
4.5 Complex I staining
Eyeball sections were stained for mitochondrial complex I activity following a well-established histochemical procedure based on the reduction of tetrazolium salts (Jung et al., 2002, Zhang et al., 2002). This staining technique allows the clear visualization of the retinal nerve fiber layer (RNFL) and was used to estimate retinal volume and the combined thickness of GCL and RNFL. Fresh-frozen sections were pre-fixed in a solution containing 0.5% glutaraldehyde and 10% sucrose PB for 7 min at room temperature, followed by 10% sucrose PB baths (3 × 5 min). Sections were then incubated at 37°C for 15–20 min with agitation in PB containing NADH (1 mg/ml), nitroblue tetrazolium (1.33 mg/ml), and sodium azide (0.065 mg/ml). After this, sections were rinsed, fixed in 10% paraformaldehyde for 30 min at room temperature, dehydrated in alcohol series, cleared in xylene and coverslipped with Permount®.
4.6 Stereological measurements
4.6.1 Retinal volume
Total retinal volume was estimated using the Cavalieri principle, which sampling methodology controls for biases introduced by object distortion and orientation (Henery and Mayhew, 1989). Sections stained for complex I activity were visualized using stage microprojection and a systematic array of test points was randomly superimposed on the image of every fourth section [area per point, a(p) = 0.0144 mm3, after adjustment for 25X magnification]. Points randomly lying on the structure of interest, which included all layers and regions (central and peripheral) of the retina, were counted (ΣP). Together with d, the average distance between designated sections (d = 240 µm), ΣP was used to estimate the total retinal volume, as Vret = ΣP × a(p) × d. Distance d was then calculated as the distance between every fourth section (20 µm × 4) by the number of designated sections (80 µm × 3). A total of three eyeball sections were used to calculate d. Although originally sections were obtained at 40 µm, tissue processing led to section shrinkage in the z plane so a microcator-based estimate of the real section thickness was obtained as the mean distance from the top to the bottom focal planes of one section per eyeball. The estimated average real thickness was 20 µm.
4.6.2 GCL cell density
Density of cells in the GCL (cells/volume) was estimated from cresyl violet stained sections using the optical disector methodology, in which cells (except those on the top of the sections) were identified at 50X through the thickness of a section and were counted when they first came into focus within an unbiased counting frame (Harding et al., 1994). Four samples per eyeball were obtained from a region adjacent to the optic pole at 200 µm intervals in the horizontal plane. This means that samples were restricted to the central retina and did not include the periphery. This sampling strategy was also used for thickness measurements. GCL cell density was calculated as Nvcells = ΣQ−/Σv(frame), where ΣQ− is the sum of cell counts per sample for one eye, and v(frame) is the area of the unbiased counting frame (0.0185 mm2, adjusted for 1500X magnification) multiplied by the average section thickness (d = 20 µm, as estimated for retinal volume measurements).
4.6.3 GCL + RNFL thickness
An estimate of ganglion cell layer + retinal nerve fiber layer (GCL+RNFL) thickness was obtained by means of a systematic analysis of complex I stained sections, which controlled for biases introduced by object shape (Gundersen et al., 1978, Mayhew, 1992). Additional control for object shape was obtained by using the same systematic sampling strategy used for GCL cell density. Eyeball sections selected for thickness estimates were those determined to have the largest diameter by 2X microscopic inspection, preferentially those in which the largest diameter of the lens, the pupil and the optic nerve were identified (Zhang et al., 2002). Within each section, and in order to maximize the efficiency of the estimate while avoiding the effect of retinal thickness by position in the medial-lateral axis of the eye, data were obtained from systematic samples 100 to 200 µm adjacent to the optic pole of the eyeball section. Microscopic retinal images at 20X were analyzed using a microscopic image-processing system including an Olympus BX40 microscope (Olympus America; Lake Success, NY), a CCD camera (Javelin Electronics; Torrance, CA), and JAVA imaging software (Jandel Scientific; Corte Madera, CA). An isotropic uniform random system of test lines was superimposed on captured images and distances between intercepts of each line with GCL and RNFL were calculated. An unbiased estimate of GCL + RNFL thickness was obtained as Tret=ΣLength of intercept/No. of intercepts.
4.7 Retinal oxidative stress
Dihydroethidium (DHE) fluorescence was used to determine the levels of oxidative stress in unfixed frozen retinal sections. DHE is a cell permeable oxidative fluorescent dye that is oxidized to ethidium bromide by superoxide. Ethidium bromide is then trapped into the nucleus and intercalates with DNA exhibiting red fluorescence (Dayal et al., 2002). On-the-slide sections were incubated in 10 µM DHE (Invitrogen, Carlsbad, CA) in PBS, pH 7.4 for 30 min at 37C in the dark. Fluid excess was removed and slides were coverslipped with ProGold antifading medium (Invitrogen, Carlasbad, CA). Images were captured with an epifluorescence microscope (BX61, Olympus, Center Valley, PA) at λ Ex: 490, Em: 520 nm, magnification: 20X, and with similar camera acquisition settings and exposure times for all sections. Digital images were analyzed for densitometry with Image J software (NIH). Data are expressed as the ratio of mean gray values in the region of interest/mean gray values of background.
4.8 Retinal energy metabolism
Retinal sections were stained with the quantitative cytochrome oxidase histochemistry technique as previously described (Gonzalez-Lima and Cada, 1994). Frozen sections were fixed in 0.5% glutaraldehyde/10% sucrose in PB, pH 7.6 at 4°C for 5 min, followed by three baths of 10% sucrose in PB (5 min each). The sections were then incubated for 10 min in a solution containing 50 mM Tris, 1.1 mM cobalt chloride, 10% sucrose and 0.5% DMSO. After a 5 min rinse in PB at room temperature, the sections were stained for 60 min at 37°C in a PB solution containing 1.3 mM 3,3’-diaminobenzidine tetrahydrochloride, 75 µg/ml cytochrome c, 20 µg/ml catalase, 5% sucrose and 0.25% DMSO. Sections were fixed in 4% formalin in PB at room temperature for 30 min, dehydrated in ethanol series, cleared in xylene (3 times, 5 min each) and coverslipped with Permount®. Cytochrome oxidase activity was quantified by optical densitometry. Stained retinal sections were placed on a DC-powered light box, digitized and analyzed using a CCD camera (Javelin Electronics; Torrance, CA), a Targa-M8 digitizer and JAVA imaging software (Jandel Scientific; Corte Madera, CA). Digitized images were corrected for slide and light box artifacts by means of background subtraction. Optical density was averaged across three retinal sections per subject. Cytochrome oxidase activity was reported as µmol/min/g of tissue.
4.9 Statistical analysis
Dunnett's tests were used to identify groups whose means were significantly different from the mean of the reference control group (DMSO vehicle-treated eyes). This test is specifically designed for situations where all groups are to be contrasted against one control group, and it was used after ANOVA has rejected the hypothesis of equality of the means of the distributions. Dunnett's tests the null hypothesis that no group has its mean significantly different from the mean of the control group. Effect sizes and Dunnett-corrected independent samples one-way ANOVAs were performed to test the significance of differences in total number of TUNEL+ cells, retinal volume, GCL cell density, GCL + RNFL thickness, DHE fluorescence and cytochrome oxidase activity, between rotenone, memantine and control groups. “n” was equal to total number of eyes per group. All tests used a two-tailed p<0.05 as criterion for significance. The precision of all estimates was described with the Matheron transitive coefficient of error method, as described previously (West and Gundersen, 1990). The coefficient of error (CE) was calculated with the formula:
where P is a single of the multiple measures of thickness, counted particles or volume estimates obtained from a given subject, A = Pi × Pi, B=Pi × Pi+1 and C = Pi × Pi+2. In addition, the variability of the estimate introduced by the stereological method itself was obtained with the formula:
where BV = Expected Coefficient of variation (ECV)/Observed coefficient of variation (OCV), OCV = Standard deviation/Mean and ECV = OCV−CE.
Acknowledgments
We thank two anonymous reviewers for constructive comments that helped improve the manuscript. We are also in debt with DeAnna Adkins for her technical advice and Dr. Theresa Jones for her supervision and use of her equipment. Supported in part by NIH Grant MH65728 to Dr. F. Gonzalez-Lima and CONACYT 187413 to Dr. Julio Rojas.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beretta S, Wood JP, Derham B, Sala G, Tremolizzo L, Ferrarese C, Osborne NN Relevance to Leber Hereditary Optic Neuropathy (LHON) Partial mitochondrial complex I inhibition induces oxidative damage and perturbs glutamate transport in primary retinal cultures. Neurobiol Dis. 2006;24:308–317. doi: 10.1016/j.nbd.2006.07.016. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Land JM, Clark JB, Heales SJ. Peroxynitrite and brain mitochondria: evidence for increased proton leak. J Neurochem. 1998;70:2195–2202. doi: 10.1046/j.1471-4159.1998.70052195.x. [DOI] [PubMed] [Google Scholar]
- Cassina A, Radi R. Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys. 1996;328:309–316. doi: 10.1006/abbi.1996.0178. [DOI] [PubMed] [Google Scholar]
- Chen HS, Lipton SA. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. J Physiol. 1997;499(Pt 1):27–46. doi: 10.1113/jphysiol.1997.sp021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HS, Wang YF, Rayudu PV, Edgecomb P, Neill JC, Segal MM, Lipton SA, Jensen FE. Neuroprotective concentrations of the N-methyl-D-aspartate open-channel blocker memantine are effective without cytoplasmic vacuolation following post-ischemic administration and do not block maze learning or long-term potentiation. Neuroscience. 1998;86:1121–1132. doi: 10.1016/s0306-4522(98)00163-8. [DOI] [PubMed] [Google Scholar]
- Chiu K, Lam TT, Ying Li WW, Caprioli J, Kwong Kwong JM. Calpain and N-methyl-D-aspartate (NMDA)-induced excitotoxicity in rat retinas. Brain Res. 2005;1046:207–215. doi: 10.1016/j.brainres.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Davey GP, Clark JB. Threshold effects and control of oxidative phosphorylation in nonsynaptic rat brain mitochondria. J Neurochem. 1996;66:1617–1624. doi: 10.1046/j.1471-4159.1996.66041617.x. [DOI] [PubMed] [Google Scholar]
- Dayal S, Brown KL, Weydert CJ, Oberley LW, Arning E, Bottiglieri T, Faraci FM, Lentz SR. Deficiency of glutathione peroxidase-1 sensitizes hyperhomocysteinemic mice to endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2002;22:1996–2002. doi: 10.1161/01.atv.0000041629.92741.dc. [DOI] [PubMed] [Google Scholar]
- Ehrlich M, Knolle E, Ciovica R, Bock P, Turkof E, Grabenwoger M, Cartes-Zumelzu F, Kocher A, Pockberger H, Fang WC, Wolner E, Havel M. Memantine for prevention of spinal cord injury in a rabbit model. J Thorac Cardiovasc Surg. 1999;117:285–291. doi: 10.1016/S0022-5223(99)70424-1. [DOI] [PubMed] [Google Scholar]
- Ernster L, Dallner G, Azzone GG. Differential effects of rotenone and amytal on mitochondrial electron and energy transfer. J Biol Chem. 1963;238:1124–1131. [Google Scholar]
- Frankiewicz T, Pilc A, Parsons CG. Differential effects of NMDA-receptor antagonists on long-term potentiation and hypoxic/hypoglycaemic excitotoxicity in hippocampal slices. Neuropharmacology. 2000;39:631–642. doi: 10.1016/s0028-3908(99)00168-9. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Lima F, Cada A. Cytochrome oxidase activity in the auditory system of the mouse: a qualitative and quantitative histochemical study. Neuroscience. 1994;63:559–578. doi: 10.1016/0306-4522(94)90550-9. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen TB, Osterby R. Distribution of membrane thickness determined by lineal analysis. J Microsc. 1978;113:27–43. doi: 10.1111/j.1365-2818.1978.tb00091.x. [DOI] [PubMed] [Google Scholar]
- Harding AJ, Halliday GM, Cullen K. Practical considerations for the use of the optical disector in estimating neuronal number. J Neurosci Methods. 1994;51:83–89. doi: 10.1016/0165-0270(94)90028-0. [DOI] [PubMed] [Google Scholar]
- Henery CC, Mayhew TM. The cerebrum and cerebellum of the fixed human brain: efficient and unbiased estimates of volumes and cortical surface areas. J Anat. 1989;167:167–180. [PMC free article] [PubMed] [Google Scholar]
- Henneberry RC, Novelli A, Cox JA, Lysko PG. Neurotoxicity at the n-methyl-D-aspartate receptor in energy-compromised neurons. An hypothesis for cell death in aging and disease. Ann N Y Acad Sci. 1989;568:225–233. doi: 10.1111/j.1749-6632.1989.tb12512.x. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Wong-Riley MT. Mitochondrial and nuclear gene expression for cytochrome oxidase subunits are disproportionately regulated by functional activity in neurons. J Neurosci. 1993;13:1805–1819. doi: 10.1523/JNEUROSCI.13-05-01805.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenberg JS, Klaunig JE. Role of the mitochondrial membrane permeability transition (MPT) in rotenone-induced apoptosis in liver cells. Toxicol Sci. 2000;53:340–351. doi: 10.1093/toxsci/53.2.340. [DOI] [PubMed] [Google Scholar]
- Jung C, Higgins CM, Xu Z. A quantitative histochemical assay for activities of mitochondrial electron transport chain complexes in mouse spinal cord sections. J Neurosci Methods. 2002;114:165–172. doi: 10.1016/s0165-0270(01)00524-6. [DOI] [PubMed] [Google Scholar]
- Kanki R, Nakamizo T, Yamashita H, Kihara T, Sawada H, Uemura K, Kawamata J, Shibasaki H, Akaike A, Shimohama S. Effects of mitochondrial dysfunction on glutamate receptor-mediated neurotoxicity in cultured rat spinal motor neurons. Brain Res. 2004;1015:73–81. doi: 10.1016/j.brainres.2004.04.044. [DOI] [PubMed] [Google Scholar]
- Karanian DA, Baude AS, Brown QB, Parsons CG, Bahr BA. 3-Nitropropionic acid toxicity in hippocampus: protection through N-methyl-D-aspartate receptor antagonism. Hippocampus. 2006;16:834–842. doi: 10.1002/hipo.20214. [DOI] [PubMed] [Google Scholar]
- Koopman WJ, Verkaart S, Visch HJ, van der Westhuizen FH, Murphy MP, van den Heuvel LW, Smeitink JA, Willems PH. Inhibition of complex I of the electron transport chain causes O2-. -mediated mitochondrial outgrowth. Am J Physiol Cell Physiol. 2005;288:C1440–C1450. doi: 10.1152/ajpcell.00607.2004. [DOI] [PubMed] [Google Scholar]
- Ling N. Rotenone-a review of its toxicity and use for fisheries management. Science for Conservation. 2003;211:5–40. [Google Scholar]
- Mayhew TM. A review of recent advances in stereology for quantifying neural structure. J Neurocytol. 1992;21:313–328. doi: 10.1007/BF01191700. [DOI] [PubMed] [Google Scholar]
- Miguel-Hidalgo JJ, Alvarez XA, Cacabelos R, Quack G. Neuroprotection by memantine against neurodegeneration induced by beta-amyloid(1–40) Brain Res. 2002;958:210–221. doi: 10.1016/s0006-8993(02)03731-9. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Gruner R, Rozental J, Millar J, Lodge D. Patch clamp studies on the kinetics and selectivity of n-methyl-D-aspartate recetor antagonism by memantine (1-amino-3,5,-dimethyladamantan) Neuropharmacology. 1993;32:1337–1350. doi: 10.1016/0028-3908(93)90029-3. [DOI] [PubMed] [Google Scholar]
- Rizzardini M, Lupi M, Mangolini A, Babetto E, Ubezio P, Cantoni L. Neurodegeneration induced by complex I inhibition in a cellular model of familial amyotrophic lateral sclerosis. Brain Res Bull. 2006;69:465–474. doi: 10.1016/j.brainresbull.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Rosenstock TR, Carvalho AC, Jurkiewicz A, Frussa-Filho R, Smaili SS. Mitochondrial calcium, oxidative stress and apoptosis in a neurodegenerative disease model induced by 3-nitropropionic acid. J Neurochem. 2004;88:1220–1228. doi: 10.1046/j.1471-4159.2003.02250.x. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Matthews RT, Henshaw DR, Beal MF. Neuroprotective strategies for treatment of lesions produced by mitochondrial toxins: implications for neurodegenerative diseases. Neuroscience. 1996;71:1043–1048. doi: 10.1016/0306-4522(95)00527-7. [DOI] [PubMed] [Google Scholar]
- Sengpiel B, Preis E, Krieglstein J, Prehn JH. NMDA-induced superoxide production and neurotoxicity in cultured rat hippocampal neurons: role of mitochondria. Eur J Neurosci. 1998;10:1903–1910. doi: 10.1046/j.1460-9568.1998.00202.x. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT. Mechanism of toxicity in rotenone models of Parkinson's disease. J Neurosci. 2003;23:10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay R, Chakravarthy B, Hewitt K, Tauskela J, Morley P, Atkinson T, Durkin JP. Transient NMDA receptor inactivation provides long-term protection to cultured cortical neurons from a variety of death signals. J Neurosci. 2000;20:7183–7192. doi: 10.1523/JNEUROSCI.20-19-07183.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volbracht C, van Beek J, Zhu C, Blomgren K, Leist M. Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. Eur J Neurosci. 2006;23:2611–2622. doi: 10.1111/j.1460-9568.2006.04787.x. [DOI] [PubMed] [Google Scholar]
- Wenk GL, Danysz W, Roice DD. The effects of mitochondrial failure upon cholinergic toxicity in the nucleus basalis. Neuroreport. 1996;7:1453–1456. doi: 10.1097/00001756-199606170-00001. [DOI] [PubMed] [Google Scholar]
- West MJ, Gundersen HJ. Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol. 1990;296:1–22. doi: 10.1002/cne.902960102. [DOI] [PubMed] [Google Scholar]
- Winkler BS. Relative inhibitory effects of ATP depletion, ouabain and calcium on retinal photoreceptors. Exp Eye Res. 1983;36:581–594. doi: 10.1016/0014-4835(83)90052-0. [DOI] [PubMed] [Google Scholar]
- Yamada K, Takayanagi M, Kamei H, Nagai T, Dohniwa M, Kobayashi K, Yoshida S, Ohhara T, Takuma K, Nabeshima T. Effects of memantine and donepezil on amyloid beta-induced memory impairment in a delayed-matching to position task in rats. Behav Brain Res. 2005;162:191–199. doi: 10.1016/j.bbr.2005.02.036. [DOI] [PubMed] [Google Scholar]
- Zhang X, Jones D, Gonzalez-Lima F. Mouse model of optic neuropathy caused by mitochondrial complex I dysfunction. Neurosci Lett. 2002;326:97–100. doi: 10.1016/s0304-3940(02)00327-0. [DOI] [PubMed] [Google Scholar]
- Zhang X, Jones D, Gonzalez-Lima F. Neurodegeneration produced by rotenone in the mouse retina: a potential model to investigate environmental pesticide contributions to neurodegenerative diseases. J Toxicol Environ Health A. 2006a;69:1681–1697. doi: 10.1080/15287390600630203. [DOI] [PubMed] [Google Scholar]
- Zhang X, Rojas JC, Gonzalez-Lima F. Methylene blue prevents neurodegeneration caused by rotenone in the retina. Neurotox Res. 2006b;9:47–57. doi: 10.1007/BF03033307. [DOI] [PubMed] [Google Scholar]