

Abstract
Ionic sodium, obtained from dietary sources usually in the form of sodium chloride (NaCl, common table salt) is essential to physiological function, and in humans salt is generally regarded as highly palatable. This marriage of pleasant taste and physiological utility might appear fortunate – an appealing taste helps to ensure that such a vital substance is ingested. However, the powerful mechanisms governing sodium retention and sodium balance are unfortunately best adapted for an environment in which few humans still exist. Our physiological and behavioral means for maintaining body sodium and fluid homeostasis evolved in hot climates where sources of dietary sodium were scarce. For many reasons, contemporary diets are high in salt and daily sodium intakes are excessive. High sodium consumption can have pathological consequences. Although there are a number of obstacles to limiting salt ingestion, high sodium intake, like smoking, is a modifiable behavioral risk factor for many cardiovascular diseases. This review discusses the psychobiological mechanisms that promote and maintain excessive dietary sodium intake. Of particular importance are experience-dependent processes including the sensitization of the neural systems underlying sodium appetite and the effects of sodium balance on hedonic state and mood. Accumulating evidence suggests that plasticity within the central nervous system as a result of experience with high salt intake, sodium depletion, or a chronic unresolved sodium appetite fosters enduring changes in sodium related appetitive and consummatory behaviors.
Keywords: sodium intake, motivation, sensitization, hedonic behavior, hypertension
Introduction
In the course of evolution a successful transition from sea- to land-based dwelling rested on means for keeping bodily cells surrounded by a solution as salty as the seawater from which the first terrestrials emerged. Our more immediate hominid ancestors evolved in a hot, dry environment and subsisted primarily on herbivorous diets that were bereft of ionic sodium [1]. Such circumstances provoked a strong need for sodium conservation and consequently favored the adaptation of complex physiological and behavioral strategies for ensuring an ocean-like environment for our cells. Extracellular fluids needed to be kept sodium-rich, intracellular fluids relatively free of sodium, and cellular and vascular volumes maintained. Sodium and water needed to be retained and ingested when physiologically significant amounts of these substances were lost.
In most mammals studied, experience with a depletion of body sodium leads not only to rapid physiological (i.e., autonomic and endocrine) adjustments for defending homeostasis but also to profound behavioral changes. The most frequently studied behavioral changes are associated with increased intake of sodium that follows loss of this cation [2-4]. Sodium (or salt) appetite refers to the state and behaviors associated with seeking and ingesting salty substances, usually taken as hypertonic NaCl solutions in laboratory experiments. It is striking that under conditions of sodium deficiency animals will accept concentrated hypertonic saline solutions that are normally avoided when sodium replete. This indicates that a change occurs in the nervous system to transform a substance perceived as aversive into one considered pleasurable or rewarding.
Most animals fed laboratory chow and most humans eating contemporary Western diets are likely to be in a state of chronic sodium excess. Hence, a moderate loss of sodium need not present a significant homeostatic challenge per se. It has been suggested that body and/or plasma sodium levels are regulated around a set point [5,6]. Because plasma sodium levels tend to be maintained at a concentration of approximately 145 mmol/L and because a number of negative feedback mechanisms are initiated following sodium depletion, the argument for a set point seems reasonable. However, many, but not all, sodium deficient animals often consume far more sodium than needed to restore homeostasis [7]. Thus it can be argued that in many cases such a set point has little influence on an animal’s overt behavior. Mammalian behavior in the case of sodium homeostasis is largely out of step with actual need in spite of evidence that excessive sodium intake over the long-term has pathological consequences (e.g., hypertension, heart failure) [8-12].
In cases of experimentally-induced sodium depletion, animals generally “overshoot” and ingest significantly more sodium than needed to replace what was lost (Figure 1). Starr and Rowland [13] have demonstrated that while volume depleted rats overconsume salt when given access to saline from a drinking spout these same rats do not overingest saline when given access in a progressive ratio task. However, rats depleted of sodium multiple times show a higher breakpoint on a progressive ratio reinforcement schedule [14] suggesting that this effect is context specific. Particularly documented in certain strains of rats, so-called need-free ad libitum intakes are also increased in the days and weeks following a depletion [15 but also see 16]. Clinical and experimental observations in humans, along with recent animal studies suggest that a persistent and unresolved sodium appetite can induce behavioral characteristics that are qualitatively similar to psychological depression as well as induce plasticity in brain regions implicated in motivation, reward, and drug sensitization and withdrawal [17-26]. Such observations raise important questions about the psychobiological nature of sodium appetite. Do animals increase need-free intakes to defend against potential future depletions, given that the animal has experienced depletion in the present environment? Does the perceived reward value of sodium change as a function of experience with deficiency or depletion? Are there mood-related consequences of sodium loss in an animal that expects a sodium-rich diet?
Figure 1.
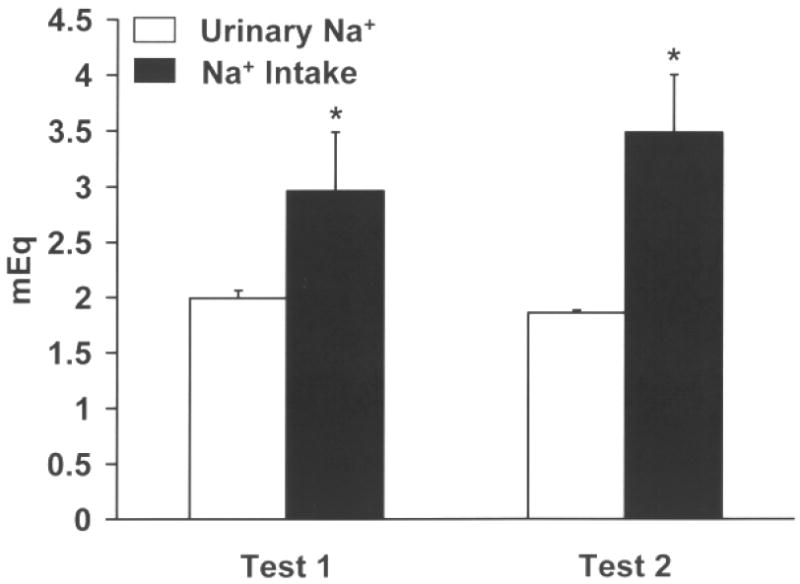
Mean (+) SEM urine (Na+) output (white bars) and saline (Na+) intake (black bars) values expressed as mEq of sodium after two acute furosemide depletions. Rats were sodium depleted two times, each depletion spaced one week apart. Twenty four hour urine samples were collected beginning immediately following the diuretic/natriuretic treatment. Rats with water available continuously were given access to 0.3 M saline for 2 h the next day and intake was recorded. Intakes of saline were compared against urine output. During tests 1 and 2, rats consumed more than Na+ was necessary to restore the sodium deficit caused by furosemide treatment. * p<0.05
The primary purpose of this review is to discuss mechanisms that support and maintain excessive dietary sodium intake. We will discuss the nature of neural plasticity that results from experience with sodium depletion and also recent evidence from animal studies and human clinical observations indicating that sodium deficiency can negatively impact hedonic (i.e., sensitivity to or interaction with rewarding stimuli) and affective states. Escape from such negative states of mood may, along with neural plasticity, play a role in promoting homeostatically inappropriate excesses in sodium consumption.
The physiological and behavioral costs of high and low sodium intake
Sodium is essential to mammalian physiology
In 1940, a case study reported by Wilkins and Richter [27] described a child with adrenal insufficiency and abnormally low synthesis of the sodium-retaining adrenal mineralocorticoid (MC) aldosterone (ALDO) that displayed an remarkable sodium appetite. The boy began licking the salt from crackers at age 1, and one of his first words was salt. Later he began eating salt directly from the shaker. His parents were often awakened in the middle of the night to the sounds of their son struggling to climb a chair or stool to reach a cabinet to find salt. Lacking the capacity to synthesize MCs, which made it impossible for his kidneys to retain sodium, the boy turned to his only available means of survival -- the intake of massive amounts of salt. Tragically, the boy died following admission to the hospital, as the hospital diet was low in salt and his crafty methods of obtaining sodium at home were no longer available. At that time the young boy’s condition could not be properly diagnosed, although his behavior under the circumstances was clearly appropriate.
A lack of sodium intake or the inability to retain what is ingested is incompatible with survival. Sodium is critical for determining membrane potentials in excitable cells and participates in various metabolic reactions in the body [10]. An adequate intake of sodium is required for optimal growth. Rats maintained on low sodium diets exhibit decreased bone and muscle weights, and required a daily intake of 300 μEq Na+ for normal growth of fat, bone, and muscle tissues [28]. In a study conducted by Bursey and Watson [29] sodium restriction during gestation in rats increased the number of stillborn pups, led to smaller brain size and amount of protein per unit of wet brain tissue, and decreased total brain RNA. Severe sodium restriction may negatively affect glucose metabolism and disturb normal blood viscosity [30]. Distribution of intracellular and extracellular fluid volumes are dictated by sodium, and either a deficit or excess of sodium will alter overall fluid balance and distribution. Under normal circumstances deviations from optimal body fluid homeostasis are corrected primarily by the kidneys, and proper renal handling of sodium is necessary for normal cardiovascular function [10]. Given that survival and normal mammalian development are dependent on adequate sodium intake and retention, the question is – how much sodium is sufficient?
Daily sodium requirement
The minimum sodium requirement for humans is arguable, but it is clear that the average daily intake in developed countries far exceeds what is needed for survival. The worldwide average salt intake per individual is approximately 10 g/day, which is greater than the FDA recommended intake by about 4 g, and may exceed what is actually necessary by more than 8 g [10,31].
Beginning around 2000 BC the use of salt as a preservative, particularly for meats, was likely among the most important factors along the path to excessive sodium intake [1,31]. Since that time it appears that salt intake has risen steadily, reaching an average of 18 g/day in 19th century Europe. Due to salted fish being a dietary staple, it is believed that intakes as high as 100 g/day were reached in 16th century Sweden [31]! The advent of mechanical refrigeration in the 19th century eased reliance on salt as a preservative, which probably played a role in reducing the average daily salt intake to its current levels [1,31].
There are cultural differences in the amount of sodium that is added to foods and the salt content considered palatable in different societies. Although for five million years our ancestors survived on a diet to which no salt was added, examples of present day communities that exhibit very low salt intakes are rare and almost always occur in isolated primitive groups [7,32-35]. For example, the New Guinea Highlanders average daily salt intake is around 0.5 g/day, and there is no evidence they are unhealthy. In fact they show far less cardiovascular disease than populations that consume the worldwide norm [31,35]. Individuals in these groups often show an initial aversion to salt when it is first introduced as an additive, but after repeated experiences with salty foods become, as some authors claim, addicted in a fashion similar to what is commonly seen as a result of consumption of coffee or nicotine [31,32]. A similar effect has been reported in chimpanzees (Pan troglodytes). In the wild, chimpanzees exist on a diet mainly of fruits and vegetables (i.e., a low salt/high potassium diet). A study by Denton and coworkers [36] employed a laboratory diet composed of fruit, vegetables, and biscuits which were high in sodium that yielded a daily salt intakes (6-12 g/day) similar to the human diet. An attempt to change the chimpanzees’ diet by reducing the sodium content of the biscuits was not well received. The chimpanzees refused to eat the low sodium alternative and rapidly lost weight.
Cordain and colleagues state, “the evolutionary collision of our ancient genome with the nutritional qualities of recently introduced foods may underlie many of the chronic diseases of Western civilization” [37]. Paleolithic era diets were vastly different (and likely much healthier) in comparison to contemporary fare in terms of fat content, high glycemic load carbohydrates, sodium-to-potassium ratio, macro- and micro-nutrient content, and fiber content. In the United States and most Western countries diet related diseases are the largest cause of morbidity and mortality, yet these rarely occur in primitive groups [37]. In the context of our evolutionary history, the addition of large quantities of salt to the diet is a recent phenomenon (approximately the last 5000 years). It appears that evolution has rendered us best adapted to handle far less sodium than we now consume, perhaps less than one-tenth as was the case with our hunter-gatherer ancestors [7,31,33,34,38]. A strictly vegetarian diet contains about 750 mg of salt per day [34]. The estimated Paleolithic diet of approximately 1000-1500 mg of salt per day, which was clearly sufficient for survival, falls short of the FDA recommended amount by approximately 300% [1,31,34].
Pathophysiology of excess sodium intake
In his book Salt: A World History, Mark Kurlansky [39] states that throughout history salt has proven to be a substance so valuable it served as currency, it has influenced the establishment of trade routes and cities, provoked and financed wars, secured empires and inspired revolutions. In the middle ages, salt was used as a treatment for ailments as diverse as toothaches, upset stomachs, and “heaviness of mind” [39]. The Chinese emperor Huang Ti, approximately 4000 years ago, is credited with being the first to recognize a relationship between high dietary intake of this once treasured commodity and negative health consequences, characterized as a hardness of the pulse [1].
In the middle of the last century, Dahl recognized that primitive societies subsisted on low daily salt intakes exhibited little if any prevalence of hypertension [33]. Also, Kempner discovered that a rice and fruit diet, basically devoid of salt (and rather unpalatable), dramatically reduced blood pressure in hypertensives [40]. Presently, the evidence relating high sodium diets to hypertension is compelling. The results of INTERSALT, a worldwide epidemiological study of the relationship between sodium intake/excretion and hypertension, highlight the relationship between sodium intake and hypertension at the population level [12,35]. Among the 52 populations studied, the Yananomo and Xingu Indians of Brazil, and rural populations in Kenya and Papua New Guinea had the lowest blood pressures (average systolic pressure 103 mmHg vs. 120 mmHg for the other 52 groups), and by far the lowest average salt consumption (1-3 g/day). The remaining populations averaged 9 g/day. It was concluded that 100 mmol per day lower sodium intake was correlated with a 9 mmHg lower rise in systolic pressure between the ages 25 to 55. Populations boasting the lowest salt intake had virtually no incidence of high blood pressure and no age-related increase in blood pressure. Other large scale studies have agreed with the basic conclusions of the INTERSALT study -- that high dietary salt intake promotes hypertension, and is a risk factor that can be reduced by modification of behavior [8,9,41,42]. A controlled study assessing the effects of the popular Dietary Approaches to Stop Hypertension (DASH) diet in hypertensive patients found that a low sodium diet (1500 mg/day) led to a blood pressure reduction of around 7 mmHg in only 30 days [42]. It has been estimated that reducing sodium intake by half will save 150,000 lives per year and $1.5 trillion over 20 years in the United States [43]. Along with hypertension and an accelerated risk of mortality in congestive heart failure, the disorders of atherosclerosis, stroke, renal disease, and osteoporosis are also associated with excessive sodium intake [31,44,45].
In many animal models of hypertension, dietary salt is either necessary, or drastically exacerbates, the rise in arterial blood pressure. The deoxycorticosterone acetate (DOCA)-salt model of experimental hypertension, for instance, relies on chronic treatments with the MC agonist DOCA and provision of a source of additional salt, typically isotonic or slightly hypertonic saline solutions [46-48] (see Figure 2). Importantly, DOCA treatment alone coupled with a standard laboratory rodent diet has little effect on blood pressure or requires a much longer period of treatment with MC [47]. Dahl salt-sensitive rats, a genetic line developed from the Sprague-Dawley strain by selecting for NaCl-induced elevated blood pressure, respond to normal salt diets by becoming hypertensive [49]. About half of human hypertensives are salt-sensitive, meaning that they respond to high dietary salt intake with large increases in blood pressure. In the absence of any other manipulation, it has been shown that high sodium intake alone can promote hypertension in rats, chimpanzees, pigs, and African Green Monkeys [36,50-54]. Both human and animal studies have provided evidence that there may be a strong genetic component underlying the susceptibility to salt-sensitive hypertension [52,55-57].
Figure 2.

(A) Mean (+ SEM) deoxycorticosterone acetate (DOCA)-induced (10 mg/kg, sc) 0.3 M saline intake plotted as the average daily intake for weeks 1, 2, 3, and 4. DOCA treatment typically induces an increase in saline intake within 2-3 days. DOCA-induced saline intakes were elevated relative to vehicle (VEH)-injected rats at all four time points. (B) Effects of 4 weeks of DOCA treatment (10 mg/kg, sc) on mean arterial pressure (MAP) in rats that have 0.3 M saline continuously available (DOCA-WS), never available during treatment (DOCA-NS), and in vehicle treated rats (VEH). DOCA-WS rats displayed a significantly elevated MAP relative to DOCA-NS and VEH-treated rats. *p<0.05 vs. VEH. †p< .05 vs. DOCA-NS group.
A voluntary reduction of daily sodium intake requires a considerable effort. Most processed foods are rich in sodium, and there is little incentive for the food industry to minimize the sodium content of processed food as salt facilitates manufacturing procedures and aids in preservation. Around 77% of our salt intake comes from processed and restaurant foods [43]. For example, many of the Swanson Hungry-Man frozen dinners contain in excess of 3000 mg of sodium, which is more than the recommended daily intake for an individual. A tablespoon of La Choy soy sauce is stocked with 1260 mg sodium; Maruchan Instant Lunch ramen noodles with vegetables 1410 mg sodium; a McDonalds Big Mac 1010 mg; a salt bagel from Dunkin Donuts 4520 mg. There are countless examples of how difficult it is to avoid high sodium intake when consuming a contemporary diet that relies heavily upon processed foods (Table 1).
Table 1.
Examples of sodium content from popular food items.
| Product | Serving Size | Sodium content (mg) | % Daily Value |
|---|---|---|---|
| Aunt Jemima Original Complete Pancake and Waffle Mix | 1/3 cup | 470 | 20 |
| Banquet Crock Pot Classics Homestyle Pork | 2/3 cup | 600 | 25 |
| Campbell’s Condensed Chicken Noodle Soup | 1/2 cup | 890 | 37 |
| Chef Boyardee Spaghetti and Meatballs | 7.5 oz | 820 | 34 |
| Chex Mix Chocolate Turtle Mix | 2/3 cup | 230 | 10 |
| Chunky Ragu Garden Combination | 1/2 cup | 520 | 22 |
| Healthy Choice Tomato Basil and Mozzarella Panini | 1 sandwich | 600 | 25 |
| Hostess Cup Cakes | 2 cakes | 580 | 24 |
| Hostess Fruit Pie – Apple | 1 pie | 440 | 18 |
| Hot Pocket Philly Steak and Cheese | 1 piece | 740 | 31 |
| Jack Link’s Original Beef Jerky | 1 oz | 590 | 25 |
| Kraft Macaroni and Cheese | 70 g | 580 | 24 |
| Kraft Ranch Dressing | 2 tablespoons | 310 | 13 |
| Mr. & Mrs. T Rich & Spicy Bloody Mary | 8 oz | 970 | 40 |
| Oberto Classics Bite Size Teriyaki Snacks | 5 pieces | 810 | 34 |
| Rold Gold Tiny Twists Pretzels | 18 pretzels | 420 | 18 |
| Sara Lee Virginia Brand Baked Ham | 4 slices | 550 | 23 |
| Stouffer’s Chicken a la King | 1 package | 800 | 33 |
| Tina’s Quality Baked Goods Gourmet Cinnamon Roll | 155 g | 315 | 13 |
| Weight Watcher Smart Ones Fire Grilled Chicken and Vegetable | 1 package | 730 | 30 |
Experimental Sodium Appetite
Induction and mechanisms of sodium appetite
Sodium appetite is often induced experimentally by depleting extracellular fluid volume, which results in the activation of sympathetic and endocrine responses that cooperatively function to defend against further losses in volume and instigate thirst- and sodium appetite-related behaviors to replace the lost volume [58]. Sodium depletion can be achieved through the administration of diuretic agents (e.g., furosemide), intraperitoneal dialysis, or injections of inert colloids (e.g., polyethylene glycol) into the peritoneal cavity or subcutaneously which provoke sodium appetite by directly depleting extracellular volume or functionally by sequestering extracellular fluid in localized edematous depots [17,59,60]. A disruption in body fluid homeostasis leads to activation of the renin-angiotensin-aldosterone system (RAAS) which is also important in cardiovascular regulation [61,62]. Sodium depletion provides a stimulus for the activation of renin which ultimately leads to the generation of angiotensin II (ANG II). ANG II then serves as a signal for ALDO to be released from the adrenal cortex.
Curt Richter [63] first discovered the importance of MCs in the maintenance of body fluid homeostasis. Surgical removal of the adrenal glands in rats led to copious drinking of NaCl solutions. Denying rats access to salt resulted in loss of appetite, ensuing loss of weight and death. Richter appropriately concluded that some adrenal factor absent in the adrenalectomized (ADX) rats was necessary for maintaining sodium balance. A few years later, Rice and Richter [64] found that injections of DOCA, an adrenal corticoid that acts as a MC agonist by being metabolized to form ALDO, paradoxically increases salt intake in intact rats. Intact animals given DOCA or ALDO ingest saline despite the fact that they retain sodium. In ADX rats low (i.e., replacement) doses of MCs decrease sodium appetite, but progressively increasing the dose to pharmacological levels produces a marked salt intake [65].
The discovery of the role of ANG II in sodium appetite occurred through a more circuitous route. Tigerstedt and Bergman [66] found that crude kidney extracts produced a pressor response in anesthetized rabbits. The renal factor was appropriately named renin, and years later it was discovered that the enzymatic activity of renin ultimately leads to the generation of an effector peptide, ANG II (see [67] for complete review). ANG II is an octapeptide that acts as a direct, potent vasoconstrictor and also within the central nervous system to promote both water [68,69] and sodium intake [70,71]. Bolus injections or continuous central infusion(s) of ANG II stimulate large intakes of 1.8% or 3% saline [70-72] which can be attenuated by central infusions of losartan, a potent angiotensin type 1 receptor (AT1) antagonist [73].
Epstein and colleagues [61,74] investigated the effects of combining the natriorexigenic actions of both ALDO and ANG II. The results of these studies led to the synergy hypothesis positing that ANG II and ALDO enhance sodium appetite induced by either agent alone through actions in the central nervous system [61,74]. Blockade of ALDO, with the MC receptor antagonist RU-28318, or reducing ANG II availability with a angiotensin converting enzyme inhibitor attenuates sodium intake induced by sodium depletion [61,62]. Concurrent blockade of MC receptors and ANG II synthesis completely blocks depletion-induced sodium appetite demonstrating that the combination of ALDO and ANG II is necessary to express a full blown appetitive response [61].
It has been suggested that the synergy hypothesis is incomplete in the sense that cardiopulmonary and arterial baroreceptor input plays an important role in the induction of hypovolemia/hypotension-related ingestive behaviors [3,58,75-77]. Losses of extracellular fluid volume are accompanied by a decrease in venous pressure and if the loss is severe enough also arterial pressure. Hypovolemia and/or hypotension trigger the activation of ANG II and vasopressin (i.e., antidiuretic hormone), which are potent vasoconstrictors that partially mitigate the negative consequences associated with a loss in fluid volume. Neural input provided by baroreceptors located on the great veins, atria, carotid sinus, and aortic arch activate sympathetic and endocrine responses to changes in blood volume. Producing hypovolemia and hypotension through administration of furosemide in conjunction with a hypotensive drug (e.g., angiotensin converting enzyme inhibitor or a direct acting vasodilator) results in high circulating levels of ANG II and ALDO as well as unloading of both cardiopulmonary and arterial baroreceptors and a rapidly developing sodium appetite [78,79]. Systemic administration of phenylephrine, which blocks the decrease in arterial blood pressure seen after furosemide/captopril treatment, significantly and selectively reduces saline intake [79]. Thus synergy results not only from the interaction between ALDO and ANG II but from the contribution of these hormonal stimuli along with afferent input from high and low pressure baroreceptors.
Key neural substrates for MC-mediated sodium appetite appear to be located in the subfornical organ (SFO), amygdala, brainstem, and the periventricular tissue surrounding the anteroventral third ventricle (i.e., AV3V) region which encompasses the organum vasculosum of the lamina terminalis (OVLT), median preoptic nucleus (MnPO), the anteroventral periventricular nuclei, and the periventricular hypothalamic nuclei [80-84]. ANG II binds to circumventricular organs (CVOs) which are effectively situated on the blood side of the blood-brain barrier. The SFO, OVLT, and the area postrema (AP) are CVOs that are thought to be critical for detecting disruption of sodium homeostasis and for initiating behavioral responses to sodium depletion [58,85]. These brain regions are particularly important for ANG II-induced sodium appetite and thirst-related behaviors, and they are rich in AT1 [86]. (For review of the neural circuitry mediating sodium appetite see [58,87].) Treatment with the diuretic furosemide and low doses of captopril increases circulating ANG II and ALDO, raises afferent activity from the systemic baroreceptors, and elevates Fos-immunoreactivity (Fos-ir) in the SFO, OVLT, and MnPO as well as the supraoptic and paraventricular nuclei (PVN) [88]. Furosemide alone increases the expression of angiotensin Type 1A receptor (AT1A) mRNA in the SFO, and in the MnPO and PVN of the hypothalamus suggesting that these nuclei exhibit plasticity in response to perturbations in body fluid balance [89]. Furosemide treatment also increases AT1A binding sites in these regions [89].
Taste and sodium appetite
The sense of taste provides the oral gateway to the body with a means of rapidly identifying essential nutrients and detrimental commodities. The four classic taste qualities of sweet, sour, bitter and salty are subserved by receptors and transduction mechanisms that are specific for sugars, acids, alkaloids, and sodium ions, respectively. Sweet objects (ripe fruits and vegetables) are generally salutary whereas sour and bitter tastes are often associated with toxic agents (bacteria laden, spoiled food and plant alkaloid toxins). The utility of a sensory system able to rapidly discriminate what is likely to be beneficial from ingesta that are likely to induce sickness is obvious. The fact that one of the four major taste submodalities is devoted to sensing salty substances may be a reflection of the evolutionary importance of being able to identify sources of sodium in environments where it was once generally scarce.
The receptors for salt taste are located primarily on the anterior portion of the tongue and palatal receptors which are innervated by the chorda tympani (CT) and greater superficial petrosal (GSP) branches of the facial nerve, respectively. The transduction of sodium involves increasing intracellular Na+ by entry of the cation through epithelial type sodium channels (ENac) to depolarize the receptor and a subsequent increase in intracellular Na+ and Ca2+ [90]. Inhibiting the depolarization of sodium taste receptors by blocking sodium channels with amiloride produces a significant impairment of sodium ingestion in depleted rats [91-93] and also reduces responsivity to NaCl in sodium replete rats in the CT and GSP nerves [94]. This effect is compounded when combined with a CT transection or a CT plus GSP transection [92,95].
The gustatory neural pathways are anatomically very similar for rodents and primates with one exception. In both species the CT projects taste information first to the rostral portion of the nucleus of the solitary tract (NTS) [96]. In rodents efferents from the NTS are then sent to the medial parabrachial nucleus. However, in primates gustatory information bypasses the parabrachial nucleus and is sent instead to the central tegmental tract [97,98]. Gustatory relays from the parabrachial nucleus and central tegmental tract in rodents and primates, respectively, are sent to the parvocellular ventral posteromedial thalamic nucleus (VPMpc), central nucleus of the amygdala (CeA), bed nucleus of the stria terminalis (BNST), and the lateral hypothalamus. From the VPMpc there are projections to insular gustatory cortex and related cortical areas [99,100]. The neural circuitry subserving taste is not only important for identifying sodium but is also critical in the ingestion of salt in sodium hungry rats. Transection of the CT nerve leads to a significant decrease in salt appetite in sodium depleted rats [101-103]. Electrolytic lesions of gustatory portions of the NTS and parabrachial nucleus abolish need-induced sodium ingestion [104-106]. Severing connections from the parabrachial nucleus and NTS through decerebration significantly attenuates DOCA and furosemide-induced sodium appetite [107] while CeA, lateral BNST and lateral hypothalamic (LH) lesions attenuate MC-induced sodium appetite [84,108]. It is important to note that the BNST, CeA, and LH are implicated in the mediation of drive, reward, mood, and addiction [109-112]. The fact that sodium taste as well as the aforementioned processes converge at these limbic forebrain sites suggests that taste and motivational/affective processes overlap and influence behavioral states related to sodium homeostasis.
Salt becomes more palatable as a consequence of sodium loss, and thus in times of severe sodium deficiency, taste plays a fundamental role in identifying sources of the cation to resolve the ionic deficit. Sodium-deficient humans typically report having a peculiar sensation in the mouth that is more commonly associated with thirst than with sodium appetite [25]. Patients with adrenal insufficiency losing large amounts of urinary sodium also do not report a specific craving for salt but instead report a longing for salty foods [27,113]. Humans do, however, demonstrate changes in preference for sodium under sodium deficiency or restriction conditions. Sodium depletion induced by the loop diuretics, furosemide or ethacrynic acid, increases preference for salty foods and also induces acute cravings for salty foods (e.g., chili dogs with cheese) [114]. Long-term sodium restriction in hypertensive patients reduces the sensory threshold for sodium detection and also increases the pleasantness ratings of salty foods [115]. The enhanced pleasure of salt in states of sodium depletion may hinder compliance with low sodium diets that are often prescribed for these patients. An extracellular volume deficit produced by intermittent light exercise increased palatability ratings for 1.0 M and 0.3 M saline solutions and coincided with an increase in plasma ALDO [116]. Sodium depletion induced by rigorous exercise has also been reported to condition a flavor preference for a novel drink when paired with a salt supplement [117]. Moreover, pleasantness responses are correlated with the amount of sodium loss such that a significant sodium depletion increases positive preference-aversion ratings for drinks paired with a salt supplement [117].
Physiological sodium deficits profoundly change behavioral responses to salt in rats such that any deficiency produces a subsequent change in the acceptability of more concentrated saline solutions. Sodium replete rats find isotonic concentrations of saline more palatable than hypertonic solutions. However, the intake of hypertonic saline increases once rats are depleted of sodium and typically aversive hypertonic saline becomes more palatable when a need for sodium arises. The palatability of solutions can be assessed in rats using the occurrence of species-typical behaviors in what are referred to as taste reactivity tests. Rats are implanted with intraoral cannulas which allows for the infusion of various tastants directly into the mouth. Berridge, Flynn, Schulkin, and Grill [118] have demonstrated that sodium depletion significantly enhances positive appetitive responses (e.g., lateral tongue protrusions, paw licking) to 0.5 M saline [118,119]. These oral/facial behaviors are similar to the responses seen in rats ingesting palatable concentrations of sucrose, while sodium replete rats display predominantly aversive responses (e.g., gapes, flails) to 0.5 M saline (Figure 3). The capacity of the status of the internal environment to influence the pleasant or unpleasantness of sensory stimuli has been described for various sensory modalities (e.g., taste; temperature sensation) and homeostatic end points (e.g., energy balance; body temperature) and has been defined by Cabanac as alliesthesia [120].
Figure 3.
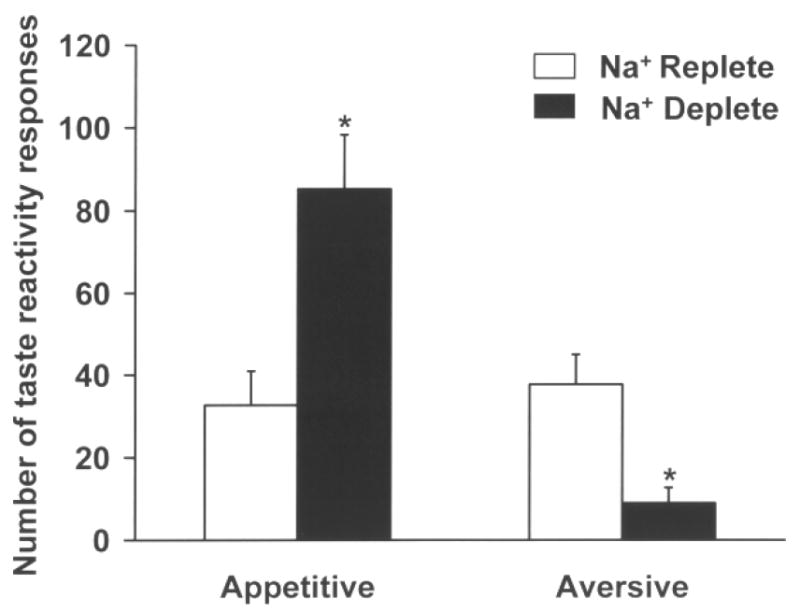
Mean (+) SEM appetitive and aversive responses during a 5 min taste reactivity test. Rats implanted with intraoral cannulas predominantly displayed aversive behaviors when sodium replete and infused intraorally with 0.3 M saline. Sodium depletion however increased appetitive responses (e.g., paw licking, lateral and rhythmic tongue protrusions) and decreased aversive responses (e.g., gapes, flails, face wipes, head shakes) to intraoral 0.3 M saline infusion. * p<0.05.
Sodium deficiency not only affects behavioral responses to sodium but also the electrophysiological profiles of neurons involved in encoding taste as well as reward. Sodium deprivation for 2 days increases lick rate responses in brief access tests [121], while prolonged sodium deprivation for 10 to 13 days decreases taste responsiveness in NTS neurons [122,123] to various concentrations of saline. Importantly, it was discovered that while responses to sodium decreased by 80% among NTS salt-sensitive neurons, responses by sugar-sensitive neurons increased by almost 10 times compared with normal controls. Forty-six percent of sugar-sensitive neurons responded to sodium compared to 7% of salt-sensitive neurons. These results suggest a shift in the way sodium taste is encoded under conditions of deficiency, shifting from salt-sensitive neurons to include sugar-sensitive neurons. The sudden responsiveness of NTS sugar-sensitive neurons to salt taste may partially underlie the behavioral shift in the acceptability of aversive concentrations of saline in sodium depleted/deprived rats. A similar shift is seen when a rapid sodium appetite is induced through DOCA administered systemically in conjunction with central renin injections [124] thereby demonstrating that sodium appetite regardless of its method of induction produces significant changes in responsiveness to sodium in NTS neurons. Sodium deprivation of pregnant mothers early in development (from embryonic day 3 to 12) results in plasticity of the CT leading to a dramatic restructuring of terminal field volumes in the gustatory brain stem in the offspring of sodium deprived rats [125].
The ventral pallidum (VeP; i.e., the medial globus pallidus) is another nucleus whose neuronal firing rates are affected by sodium appetite. The VeP is associated with the mesolimbic reward system and according to Berridge, mediates the liking or palatable response to sensory stimuli such as sucrose [126]. The increase in VeP neural activity seen in sodium-depleted rats given a 1.5 M saline solution is comparable to the neural activity produced in response to sucrose solutions. This effect is not seen in replete rats [127]. These data support the notion that sodium appetite produces neural activity related to the enhancement of the palatability of NaCl so that in the state of sodium appetite, neural responses seem to mirror those responses associated with palatable substances like sucrose.
Sodium Appetite Sensitization
Plasticity and sodium appetite
Sensitization refers to an increase in the strength (i.e., frequency, amplitude, duration) of a response to a stimulus induced by past experience with the same or related stimuli. This type of response modification reflects alterations in nervous system function. Repeated bouts of sodium deficit sensitize salt appetite such that an increase in sodium ingestion is seen over the course of the initial depletions [15,17,26,128,129] (Figure 4). An enhanced appetitive response for saline occurs over 2 to 4 sodium depletions at which point intakes plateau and remain stable over the course of additional instances of induced deficits [15,130]. Sodium appetite is an excellent model to study the sensitization of a complex behavior and the neural mechanisms underlying it. Moreover, understanding sodium appetite sensitization may be informative for identifying mechanisms underlying excess sodium consumption in humans.
Figure 4.

Total acute 0.3 M saline intake (± SEM) in rats sodium depleted 2 times compared to vehicle treated rats. Rats were sodium or sham depleted 3 times, each depletion spaced one week apart and given access to 0.3 M saline 24 h after the first 2 depletions. Total intakes were recorded over a 2 h period. Acute saline intake was significantly greater during the second sodium depletion as compared with the first in 3F rats (3F=3 furosemide treatments; 3V = 3 vehicle treatments). Rats were sacrificed for immunohistochemical processing of brain tissue after the third sodium depletion and were not given access to saline.*p < 0.05 as compared to first sodium appetite test.
Falk and Herman [59] were the first to report sodium appetite sensitization, which they induced via intraperitoneal dialysis (IPD).A 5% glucose solution injected into the peritoneal cavity sequesters isotonic extracellular fluid which can be withdrawn after several hours to produce an absolute sodium deficiency and robust sodium appetite. Rats given another identical IPD-induced intake test display enhanced 3% saline ingestion on the second depletion as compared to the first. Falk and Herman inferred from these data that a central mechanism sensitized by sodium depletion directly contributes to this overt change in intake. Moreover, Falk [131] discovered in a separate group of rats that repleting sodium loss through a stomach load of 3% saline rather than by conventional oral ingestion produced the same behavioral plasticity -- in other words, enhanced sodium intake following a second IPD challenge. The experience of tasting or orally ingesting saline, when sodium deficient, is not required for sensitization.
Sakai et al. [129] demonstrated that rats depleted multiple times with furosemide also express a greater avidity for 3% saline as evidenced by an ever decreasing latency to consume saline as well as an enhanced ingestive response. Sakai and co-workers also assessed various physiological consequences of multiple sodium depletions including urine output, urinary and plasma sodium, and hematocrit. They found that acute diuretic-induced urine loss was not differentially affected (i.e., enhanced) by multiple sodium depletions. Plasma sodium concentration and hematocrit also did not change over repeated furosemide treatments. Therefore it appears that multiple sodium depletions differentially affect sodium intake, although this appears to be context and strain dependent [128], while loss remains consistent. Sakai and associates also found that rats do not require sodium ingestion to induce salt appetite sensitization. Moreover, a history of sodium depletions produces an increase in daily 3% saline intake that is independent of a deficit state. This phenomenon is referred to as need free sodium intake [129]. Pretreatment with systemic ALDO and central ANG II sensitizes rats to sodium in a manner similar to that seen in rats with a history of sodium depletions [129]. Systemic blockade of ALDO and ANG II with a MC receptor antagonist, RU 28318, and an angiotensin converting enzyme inhibitor, captopril, respectively, prevents the development of sodium appetite sensitization [129]. These data suggest that the mechanisms associated with the elicitation of sodium appetite may also play a prominent role in facilitating sodium appetite sensitization. The excessive sodium consumption is not necessarily reflective of restoring sodium imbalance but may be indicative of either a change in the palatability of sodium as a consequence of repeated sodium depletions, an increase in the craving for sodium, or simply a dysregulation of those physiological mechanisms associated with stimulating sodium appetite.
Prenatal development is affected by the physiological and psychological well being of pregnant mothers. Severe disruptions in the intrauterine environment caused by stress, malnutrition, or dysregulated endocrine systems alter the development of the progeny. Particularly interesting in the context of this review are studies that have examined the effects of high and low sodium diets on the development of sodium appetite in the resulting offspring. In general, pregnancy induces increased preference for salt in women [132] and in rats [133]. Infants whose mothers experience moderate to severe morning sickness during the first 14 weeks of pregnancy demonstrate a preference for saline as compared to infants whose mothers did not [134]. Subjecting pregnant rats to sodium depletion by subcutaneous injection of an inert large molecular weight colloid, polyethylene glycol, results in an increase in 3% NaCl intake in their offspring under ad libitum conditions (need-free intake) and after acute depletions [135]. Moreover, the offspring of pregnant rats depleted repeatedly with a natriuretic/diuretic showed both elevated need-free intake and enhanced ingestion of saline in response to acute sodium depletion [136]. Partial aortic ligation produces an avid sodium appetite in pregnant dams which subsequently also enhances need-free and depletion-induced sodium appetite in the offspring [137].
Interestingly, studies have shown that ingestion of a high salt diet during pregnancy has similar effects on the offspring to those observed following sodium deficiency in the mother. In other words there is an increase in need-free hypertonic saline intakes in the offspring. Pregnant dams fed a high salt diet during gestation produce offspring that express an elevated preference for hypertonic saline solutions compared to rats whose mothers ate a moderate sodium diet [138]. These data have implications for cardiovascular disease as high sodium diets during gestation significantly increase mean arterial pressure in the offspring [139].
Neural substrates and the mediation of the rewarding aspects of salty substances
The nucleus accumbens (NAc) and dopaminergic signaling are implicated in mediating reward-associated behaviors such as feeding and drug use [140-142]. In general, the NAc is activated during the ingestion of palatable foods [109,143] and pleasurable activities such as wheel running [144]. In addition, the NAc is implicated in drug abuse. Extracellular levels of dopamine are increased following the administration of virtually all addictive drugs [145], and excitotoxic lesions of the NAc disrupt drug seeking behavior and the psychostimulant effects of cocaine [146]. In microdialysis studies of the NAc, Hoebel and colleagues [147] found that sodium depleted rats also show increased levels of extracellular dopamine following saline intake. These findings are consistent with the observation that dopamine transporter (DAT) activity in the NAc of furosemide treated rats is decreased [22,26,148] and that ALDO treatment in in vitro reparations decreases DAT activity in the NAc [148]. Administration of a dopamine D2 receptor antagonist blocks increased sham drinking (i.e., where ingested fluid is drained from the stomach through a fistula to eliminate the post-ingestive effects of saline intake) of hypertonic saline in sodium-depleted rats [149]. This suggests that dopaminergic systems are involved in the perceived increase in reward value attributed to sodium when animals are sodium-depleted. Neural activity as represented by Fos-ir in the NAc is also changed as a consequence of furosemide treatment but only in sodium depleted rats sham drinking hypertonic saline [23]. Decreases in DAT activity suggest an increase in extracellular dopamine in the NAc, which in turn should increase binding to dopamine receptors in the NAc. These results suggest that a strong motivational state such as sodium appetite produces neurochemical changes in the NAc which is likely to be an important structure within the neural network underlying the motivation to obtain sodium and/or in determining its reward value.
Multiple sodium depletions lead to morphological changes in medium spiny neurons in the shell of the NAc [26]. Rats with a history of sodium depletions exhibit significantly more dendritic branches and spines than naïve control rats, an effect also evident in rats sensitized to cocaine and amphetamine [150,151]. It has also been shown that the effects of repeated furosemide treatments will cross-sensitize with the psychomotor effects of amphetamine providing further evidence of shared neural mechanisms activated by sodium appetite and administered amphetamine. Interestingly, we have recently found that DOCA treatment with no access to saline increases voluntary running wheel activity (unpublished results), an effect possibly analogous to the well-documented increases in this locomotor response following food restriction [152].
A recent experiment in our laboratory has elaborated on the potential neural mechanisms involved in facilitating sodium appetite sensitization [17]. We have found that experience with multiple sodium depletions differentially changes neural activity as a function of experiencing repeated deficits. Rats were depleted three times and were given a sodium appetite test after the first two depletions. The animals were not given access to sodium during the third depletion and were sacrificed to examine Fos-ir. Fos protein is the product of an immediate early gene, c-fos, that reflects a generalized increase in neuronal activity. An increase in Fos-ir was demonstrated in the SFO of rats depleted of sodium multiple times as compared to sham depleted rats and those depleted of sodium just once (Figures 5&6). Increased neural activity was correlated with the animals’ behavior, as incremental increases in 0.3 M saline intake were observed with each successive sodium depletion. The distribution of Fos-positive neurons was localized to the core portion of the SFO, an area of the SFO that expresses high levels of AT1A mRNA as assessed by in situ hybridization [153]. AT1A mRNA and Fos protein expression have been shown to co-localize in the core region of the SFO in hypovolemic rats [154], thereby suggesting that the increase in Fos-ir seen in rats with a history of sodium depletions may be associated with changes in AT1 mRNA expression in the SFO.
Figure 5.

Photomicrographs of nucleus accumbens shell (first row), and subfornical organ (second row) depicting Fos-immunoreactivity (Fos-ir). Na+ depleted rats with a prior history of three Na+ depletions had increased levels of Fos-ir in the shell of the nucleus accumbens and in the subfornical organ compared to rats depleted once and vehicle-treated controls. A&E from rat treated with furosemide 3 times (3F); B&F from rat with two vehicle treatments + 1 furosemide treatment (2V1F); C&G from rat treated with vehicle 3 times (3V); D&H schematic drawings of areas counted [205], hatched bars define boundaries. First row: AcbC = nucleus accumbens, core; AcbSh = nucleus accumbens, shell; ICj = islands of Calleja; LAcbSh = nucleus accumbens, shell, lateral division; VP = ventral pallidum (bregma 1.20 mm). Second row: cc = corpus callosum; D3V = dorsal 3rd ventricle; Fi = fimbria of the hippocampus; LV = lateral ventricle; SFO = subfornical organ (bregma -1.40 mm). (Reprinted from [17] with permission.)
Figure 6.

(A) Mean (± SEM) Fos-immunoreactivity (Fos-ir) counts/mm2 for furosemide- and vehicle-treated rats with no access to saline and water. BLA=basolateral amygdala; CeA=central amygdala; mPFC=medial prefrontal cortex; NAc=nucleus accumbens Rats with multiple sodium depletions had significantly more Fos-ir in the BLA and NAc compared to rats depleted once and sham depleted rats. (B); PVN=paraventricular nucleus; SFO=subfornical organ; SON=supraoptic nucleus. 3V=3 vehicle treatments; 2V1F=2 vehicle treatments + 1 furosemide treatment; 3F=3 furosemide treatments. Rats depleted multiple times with furosemide had significantly more Fos-ir in the SFO than rats depleted once or sham depleted rats.*Significantly different as compared to 3V and as †compared to 2V1F. (Reprinted from [17] with permission.)
Rats with a history of sodium depletions also had increased Fos-ir expression in the NAc and basolateral amygdala (BLA) as compared to rats depleted only once and compared to sodium replete rats (Figures 5&6). This is the first experiment implicating the BLA in sodium appetite sensitization and also the first experiment demonstrating that the BLA and NAc may interact as part of a neural network mediating sodium appetite sensitization. It can be hypothesized that the activity in the SFO, BLA, and NAc reflects neural plasticity associated with sensitization of sodium intake following multiple depletions and also the increase in need free intake that persists long after restoration of sodium balance. When considered in conjunction with the aforementioned findings regarding DAT/Fos-ir, these data suggest that the need for sodium induces neural plasticity at central sites associated not only with body fluid balance, but also with motivation, reward, and mood.
Hedonic and mood-related effects of sodium deficiency
Changes in mood are among the initial signs of a subadequate diet. With vitamin deficiency, psychological symptoms such as depression and hysteria may appear [155]. Deficiency of vitamin B12 and folate are associated with psychiatric symptoms, including dementia and depression [156]. Iron deficiency, which is not uncommon in third world countries, has occasionally been associated with symptoms of depression, and if anemia is present, poor mood, lethargy, and difficulties sustaining attention [157]. In a double-blind study, vitamin supplementation for one year was shown to improve mood in 129 healthy adults [158]. Appetites for specific ions (e.g., Na+, K+, Ca2+, PO4-) have scarcely been addressed in relation to affective state. A relationship between magnesium deficiency and signs of depressed mood have been reported in humans and in mice [159,160]. Individuals that work day-to-day in extremely hot environments losing copious amounts of sodium through sweating commonly complain of fatigue, headache, difficulty concentrating, and sleep disturbances-- symptoms which are often associated with psychological depression [24].
The classic study by McCance in 1936 was among the first experimental investigations of the effects of sodium deficiency in humans [25]. Using sodium-free diets and sweating, it took about 7 days to make subjects sodium deficient. The experiments were carried out over 11 days, therefore all of the effects reported were present within the 4 days of sodium deficiency. The participants reported that they experienced extreme, unquenchable thirst. One participant reported that he experienced a longing for salt and often went to sleep thinking about it. McCance himself, however, reported feeling, “no specific craving for salt” [25]. With regard to mood-related symptoms, subjects reported a loss of appetite, anhedonia, difficulty concentrating, excessive fatigue, and a general sense of exhaustion. A failure to detect any changes in blood pressure or pulse rate, and the absence of cramps suggests that the treatments did not severely disturb physiological functioning. Therefore, it is possible that changes in mood and appetite are among the first noticeable manifestations accompanying sodium deficiency.
Feeling exhausted is the primary symptom of chronic fatigue syndrome (CFS), a disease with a poorly understood etiology. CFS is characterized by excessive fatigue lasting at least 6 months, lightheadedness, and difficulty concentrating [161]. A study by Bou-Holaigah and colleagues [161] reported on a group of CFS patients that presented the above symptoms in conjunction with orthostatic hypotension and presyncopal signs during an upright tilt test. The treatment regimen was primarily aimed at ameliorating the symptoms associated with the presyncopal state and hypotension due to insufficient sympathetic activation by encouraging patients not to restrict sodium intake and by administering, in most cases, the drug fludrocortisone which has sodium retaining properties. Following the debriefing of the subjects at the conclusion of the study, it was learned that 61% of the CFS patients had voluntarily imposed low sodium diets upon themselves (presumably before the onset of CFS symptoms) and had tried to avoid salt and salty food in an effort, they thought, to be healthy. Sixteen of the 21 patients reported a favorable response to therapy by manifesting reduced CFS symptoms and improved clinical signs indicating reduced orthostatic hypotension. Especially noteworthy was the observation that the patients also improved scores on a test of general well-being that likely reflected improved mood. It can only be speculated, but an increase in sodium ingestion and retention may have contributed to the mood improvements. In light of the McCance study, this interpretation seems viable. Fludrocortisone has been shown to be an efficacious treatment for CFS in other studies [162].
McEwen and coworkers [20-22] reported a series of studies that suggest that mesolimbic dopaminergic and opioidergic systems may be involved in sodium appetite. These researchers found that both diuretic-induced sodium depletion and DOCA administration for 11 days led to distinct neurochemical and neuropeptidergic profiles in the ventral striatum that were dependent upon availability of saline for ingestion [20-22]. With no access to saline, rats that were sodium depleted and rats that were DOCA-treated showed a decrease in enkephalin mRNA and an increase in dopamine transporter activity throughout the ventral striatum, and particularly in the shell region of the NAc. The authors concluded that dopaminergic and opioidergic neurotransmission within ventral striatal brain regions associated with reward may be important for salt craving. The authors also hypothesized that animals with a prolonged sodium appetite or animals treated with DOCA without access to saline may be anhedonic (i.e., display decreased responsiveness to previously rewarding stimuli) due to low levels of dopamine in the NAc. Anhedonia is a core symptom of major depressive disorder in humans [163].
Recent experiments from our laboratory have tested the hypothesis that a persistent, unattenuated sodium appetite can produce anhedonia. In the first experiment we found that peripheral DOCA treatment (10 mg/kg) for 6-11 days with no access to saline produced a decrease in sensitivity to two rewards – lateral hypothalamic self-stimulation (LHSS), and the consumption of a palatable 2% sucrose solution [18] (Figure 7). Importantly, DOCA treatment alone had little effect on hedonic behavior as the presence of 0.3 M saline during DOCA treatment prevented the hedonic deficits (Figure 7). There was no evidence of a motor impairment in the animals as they achieved maximal response rates for LHSS during DOCA-treatment that were not statistically different from their baseline responding. We have also found that a similar treatment protocol (daily DOCA treatment, 10 mg/kg, for 4 weeks) actually increases voluntary running wheel activity (unpublished observations). A second experiment found that furosemide-induced sodium depletion also produced a decrease in sensitivity to LHSS reward when the animals were denied saline for 48 hours and that the presence of saline or subsequent repletion prevented or reversed, respectively, the rightward shifts in the midpoint (ECu50) of LHSS current-response functions [19] (Figure 8). Sodium depleted rats that were denied access to saline also exhibited decreased heart rate variability compared to rats provided saline. Decreased heart rate variability is often observed in depressed patients and in rats with experimentally-induced anhedonia (i.e., chronic mild stress-induced depression) [164-167]. Electrical stimulation of the medial forebrain bundle and lateral hypothalamus, as well as sucrose ingestion, has been shown to elicit increases in dopamine release in the NAc [147,168]. Changes in dopaminergic neurotransmission within the NAc as a result of chronic DOCA treatment or sodium depletion without the ability to quench the appetite via saline ingestion could potentially diminish responding for stimuli that utilize the NAc as a neural substrate for processing reward related information. We have recently reported a decrease in Fos-ir in the NAc in rats made anhedonic via the chronic mild stress paradigm compared to control rats following ingestion of a fixed volume of a palatable sucrose solution suggesting that neural activity in the NAc may indeed reflect or underlie hedonic deficits in rats [169].
Figure 7.

(A). Mean lateral hypothalamic self stimulation (LHSS) current-response curves for deoxycorticosterone acetate (DOCA) treated rats during baseline (solid line), no saline (dotted line), and with saline (dashed line) phases showing a parallel rightward shift during the no saline period relative to the baseline and with saline phases. Animals were treated with DOCA for 6 days with 0.3 M saline continuously available and then for 6 days with no saline available, and these treatment orders were counterbalanced. Data are displayed as sigmoid curves fit to mean values. Closed and open circles indicate the mean of the effective current 50 (ECu50; i.e., the calculated current level required to produce responding at 50% of maximum rate) for each animal with corresponding vertical and horizontal SEM bars. (B) Mean (± SEM) 1-hr intakes of 2% sucrose solution by DOCA- and VEH-treated rats. “Baseline” sucrose intakes were taken as the average of five one-hour tests conducted on alternate days; “test” intakes were taken as the average of five tests conducted on alternate days beginning on day 3 of DOCA or VEH treatment. DOCA-treated rats’ intakes were significantly reduced relative to their respective baselines. *p<0.05. (Reprinted from [18] with permission.)
Figure 8.

Mean (± SEM) effective current 50 (ECu50) values for sodium depleted rats, relative to this group’s respective baseline ECu50 value, 48 hours following sodium depletion and 96 hours following sodium recovery. The sodium depletion value was significantly different than the baseline value. *p <0.05 baseline value vs. sodium depletion. The sodium recovery value was not significantly different from either the baseline value or the sodium depletion value. (Reprinted from [19] with permission.)
Although the results of the Morris et al. study [18] discussed above suggest that it was the prolonged sodium appetite, rather than MC treatment, that led to hedonic deficits, it has been suggested that increased ALDO may either be a state marker for depression or may be involved in its etiology [170-172]. Clinically depressed patients have been shown to have increased levels of ALDO with no evidence of increased plasma renin [170,171]. A recent study found that the DOCA-salt hypertension paradigm produces apoptosis in limbic brain regions, particularly the hippocampus, a brain region often implicated in depression in humans [173-175]. There are reports of patients with Conn’s disease, a disorder characterized by high levels of ALDO, displaying symptoms characteristic of clinical depression [176], and ALDO has also been proposed as a potential mediator of the affective disturbances that frequently accompany cardiovascular disease [165]. Rats with congestive heart failure (CHF), induced experimentally via coronary artery ligation, display an activated RAAS, which provokes a sodium appetite and ultimately extracellular volume expansion that further compromises cardiac function [177,178]. Rats with experimentally-induced CHF show a decline in intake of 1% sucrose solutions, an increase in immobility time in the forced swim test, and reduced social exploration, characteristics indicative of “depressive” behavior [179,180]. Grippo and colleagues [181] reported that CHF rats are anhedonic as evidenced by rightward shifts in LHSS current-response functions, and an increased ECu50 in CHF rats relative to their own baselines. The animals in that study were denied saline solution access, however it has been shown that CHF rats exhibit a persistent sodium appetite [178]. It is possible that the disturbances in RAAS activity, and/or the chronically increased sodium appetite in CHF relates to the disturbances in affective state, however to our knowledge no experiments directly addressing these issues have been conducted.
A small body of literature regarding hypertensive patients with major depression suggests that the angiotensin converting enzyme inhibitor captopril may have mood enhancing properties [182-185]. ANG II has multiple pressor actions, and blockade of its formation can reduce blood pressure. Case studies have shown that elevations and depressions in mood, respectively, accompany the administration and withdrawal of captopril, and this effect appears to be dose-dependent [184]. Croog and coworkers [185] used measures of general well-being in 626 men with mild to moderate hypertension to conclude that captopril improved affective state and had fewer side effects than other pharmacological treatments. Other treatments, such as methyldopa, although efficacious in reducing blood pressure, did not lead to mood improvements. ANG II has been implicated in the stress response and in excitation of the hypothalamic-pituitary adrenal (HPA) axis [186]. Chronic hyperactivity of the HPA system has frequently been found in depressed patients [187-189]. It is possible that the positive effects of captopril reported in these studies may be related to diminished HPA activity [186,190]. ANG II is also a primary stimulus for ALDO release from the adrenal cortex, and therefore ANG II could promote hedonic disturbances indirectly by stimulating increases in circulating ALDO. It is also possible that ANG II itself may have negative effects on mood. While we are not aware of any studies directly investigating the effects of ANG II on hedonic state, it has been shown that treatment with the AT1 blocker losartan decreases immobility time in the Porsolt forced swim test [191], an effect observed with most clinically effective antidepressant drugs [192].
Concluding Remarks
One goal of the present review is to highlight potential mechanisms underlying excessive sodium intake following sodium depletion or restriction. Although a number of researchers have argued that we as a society are addicted to sodium [31,193-195], much of the data in support of this assertion is based on cross-cultural studies, anecdotes, and self-reports that have provided very little insight into the neural underpinnings of this abuse of sodium. Emerging evidence from animal studies suggests that sodium is similar to other natural reinforcers (e.g., sex, voluntary exercise, fats, carbohydrates, chocolate) in its possessing addictive qualities. There appears to be a significant overlap in the neural substrates involved in drug addiction and sodium appetite sensitization, and cross-sensitization has been demonstrated between sodium depletion and psychostimulant treatment [26,196]. Rats depleted of sodium multiple times show a neurochemical profile similar to that seen following sensitization to drugs of abuse [17,26] and demonstrate a craving for sodium as exhibited by an increased breakpoint in a progressive fixed ratio operant task [14]. These findings suggest that changes in sodium status can alter the chemistry and anatomy of putative reward pathways in the brain, the same pathways impacted by drugs of abuse and potentially involved in maintaining addiction.
Our laboratory has recently discovered that two models of sodium appetite induction in rats, DOCA-induced and diuretic-induced sodium appetite, produce decreases in reward sensitivity in animals that are denied saline access [18,19] (Figures 7 & 8). Koob and colleagues have reported analogous findings following withdrawal from cocaine, morphine, and nicotine in rats [197-199]. Resolution of a chronic sodium appetite through the intake of salty substances may ameliorate the negative effects of a sodium deficit on hedonic and affective states. We suggest that hedonic and/or affective consequences of major fluctuations in sodium balance along with neural plasticity that follows disturbed sodium homeostasis may play a role in promoting excessive sodium intake. It seems likely that the brain regions underlying the sensitization of sodium appetite are also involved in mediating the hedonic changes (e.g., amygdala; NAc) although more research is needed to test these hypotheses.
It is well known that for many people salt enhances taste. Although how such a strong preference for sodium develops is still under scrutiny, it is possible that our preference for sodium is a learned behavior. For example, newborn infants generally display an aversion or indifference to moderate levels of salt, and it is not until age 2-3 that they prefer salty tastes [31,200,201]. Similar effects are observed in low salt intake groups following initial experience with high salt diets suggesting that at any point during the lifespan the introduction of a high salt diet induces an initial aversion and thereafter requires a period of habituation [31,32]. Our preference for sodium is often so compelling that patients with hypertension or CHF have difficulty adhering to a low sodium diet due to issues with palatability (e.g., food becomes bland and unappetizing) despite the recognized health benefits of doing so [10,193,202,203].
Walter Cannon, in his famous book The Wisdom of the Body, highlights a number of examples of the elegance of the controlling processes involved in the homeostatic regulation of physiological variables [204]. However given that dietary sodium intake is widely agreed to be excessive (perhaps extreme), it begs the question whether or not the body is exhibiting wisdom or foolishness with regard to sodium intake. The discovery and mechanistic analysis of the neural substrates governing the enduring behavior changes associated with sodium appetite are likely to increase our understanding of topics as diverse as homeostatic regulation, addiction, affective disorders, sensitization, and learning and memory.
Acknowledgments
Research from the authors’ laboratory reported in this manuscript and support for preparation of this review was provided by grants from the National Heart, Lung, and Blood Institute HL14388 and HL57472, National Institute of Diabetes and Digestive and Kidney Diseases DK66086 (AKJ), and American Heart Association 0615313Z (MJM).
The authors thank Dr. Connie L. Grobe for her valuable comments on an earlier draft of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cirillo M, Capasso G, Di Leo VA, De Santo NG. A history of salt. Am J Nephrol. 1994;14:426–431. doi: 10.1159/000168759. [DOI] [PubMed] [Google Scholar]
- 2.Daniels D, Yee DK, Fluharty SJ. Angiotensin II receptor signalling. Exp Physiol. 2007;92:523–527. doi: 10.1113/expphysiol.2006.036897. [DOI] [PubMed] [Google Scholar]
- 3.Johnson AK. The sensory psychobiology of thirst and salt appetite. Med Sci Sports Exerc. 2007;39:1388–1400. doi: 10.1249/mss.0b013e3180686de8. [DOI] [PubMed] [Google Scholar]
- 4.Fitzsimons JT. Angiotensin, thirst, and sodium appetite. Physiol Rev. 1998;78:583–686. doi: 10.1152/physrev.1998.78.3.583. [DOI] [PubMed] [Google Scholar]
- 5.Hollenberg NK. Surfeit, deficit, and the set point for sodium homeostasis. Kidney Int. 1982;21:883–884. doi: 10.1038/ki.1982.114. [DOI] [PubMed] [Google Scholar]
- 6.Strauss MB, Lamdin E, Smith WP, Bleifer DJ. Surfeit and deficit of sodium; a kinetic concept of sodium excretion. AMA Arch Intern Med. 1958;102:527–536. doi: 10.1001/archinte.1958.00260210013003. [DOI] [PubMed] [Google Scholar]
- 7.Denton D. The Hunger for Salt: An Anthropological, Physiological, and Medical Analysis. New York: Springer Verlag; 1982. [Google Scholar]
- 8.Hollenberg NK. The influence of dietary sodium on blood pressure. J Am Coll Nutr. 2006;25:240S–246S. doi: 10.1080/07315724.2006.10719573. [DOI] [PubMed] [Google Scholar]
- 9.Law M. Salt, blood pressure and cardiovascular diseases. J Cardiovasc Risk. 2000;7:5–8. doi: 10.1177/204748730000700102. [DOI] [PubMed] [Google Scholar]
- 10.Michell AR. The Clinical Biology of Sodium. New York: Elsevier; 1995. [Google Scholar]
- 11.Sanders PW. Salt-sensitive hypertension: lessons from animal models. Am J Kidney Dis. 1996;28:775–782. doi: 10.1016/s0272-6386(96)90265-6. [DOI] [PubMed] [Google Scholar]
- 12.Stamler J. The INTERSALT Study: background, methods, findings, and implications. Am J Clin Nutr. 1997;65:626S–642S. doi: 10.1093/ajcn/65.2.626S. [DOI] [PubMed] [Google Scholar]
- 13.Starr LJ, Rowland NE. Characteristics of salt appetite in chronically sodium-depleted rats using a progressive ratio schedule of procurement. Physiol Behav. 2006;88:433–442. doi: 10.1016/j.physbeh.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 14.Clark JJ, Bernstein IL. Sensitization of salt appetite is associated with increased “wanting” but not “liking” of a salt reward in the sodium-deplete rat. Behav Neurosci. 2006;120:206–210. doi: 10.1037/0735-7044.120.1.206. [DOI] [PubMed] [Google Scholar]
- 15.Sakai RR, Frankmann SP, Fine WB, Epstein AN. Prior episodes of sodium depletion increase the need-free sodium intake of the rat. Behav Neurosci. 1989;103:186–192. doi: 10.1037//0735-7044.103.1.186. [DOI] [PubMed] [Google Scholar]
- 16.Dietz DM, Curtis KS, Contreras RJ. Taste, salience, and increased NaCl ingestion after repeated sodium depletions. Chem Senses. 2006;31:33–41. doi: 10.1093/chemse/bjj003. [DOI] [PubMed] [Google Scholar]
- 17.Na ES, Morris MJ, Johnson RF, Beltz TG, Johnson AK. The neural substrates of enhanced salt appetite after repeated sodium depletions. Brain Res. 2007;1171:104–110. doi: 10.1016/j.brainres.2007.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morris MJ, Na ES, Grippo AJ, Johnson AK. The effects of deoxycorticosterone-induced sodium appetite on hedonic behaviors in the rat. Behav Neurosci. 2006;120:571–579. doi: 10.1037/0735-7044.120.3.571. [DOI] [PubMed] [Google Scholar]
- 19.Grippo AJ, Moffitt JA, Beltz TG, Johnson AK. Reduced hedonic behavior and altered cardiovascular function induced by mild sodium depletion in rats. Behav Neurosci. 2006;120:1133–1143. doi: 10.1037/0735-7044.120.5.1133. [DOI] [PubMed] [Google Scholar]
- 20.Lucas LR, Pompei P, McEwen BS. Correlates of deoxycorticosterone-induced salt appetite behavior and basal ganglia neurochemistry. Ann N Y Acad Sci. 1999;897:423–428. doi: 10.1111/j.1749-6632.1999.tb07912.x. [DOI] [PubMed] [Google Scholar]
- 21.Lucas LR, Pompei P, McEwen BS. Salt appetite in salt-replete rats: involvement of mesolimbic structures in deoxycorticosterone-induced salt craving behavior. Neuroendocrinology. 2000;71:386–395. doi: 10.1159/000054559. [DOI] [PubMed] [Google Scholar]
- 22.Lucas LR, Grillo CA, McEwen BS. Involvement of mesolimbic structures in short-term sodium depletion: in situ hybridization and ligand-binding analyses. Neuroendocrinology. 2003;77:406–415. doi: 10.1159/000071312. [DOI] [PubMed] [Google Scholar]
- 23.Voorhies AC, Bernstein IL. Induction and expression of salt appetite: effects on Fos expression in nucleus accumbens. Behav Brain Res. 2006;172:90–96. doi: 10.1016/j.bbr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 24.McEwen OR. Salt loss as a common cause of ill-health in hot climates. Lancet. 1935;225:1015. [Google Scholar]
- 25.McCance RA. Experimental sodium chloride deficiency in man. Proc R Soc Lond B Biol Sci. 1936;119:245–268. [Google Scholar]
- 26.Roitman MF, Na E, Anderson G, Jones TA, Bernstein IL. Induction of a salt appetite alters dendritic morphology in nucleus accumbens and sensitizes rats to amphetamine. J Neurosci. 2002;22:RC225. doi: 10.1523/JNEUROSCI.22-11-j0001.2002. 221-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilkins L, Richter CP. A great craving for salt by a child with corticoadrenal insufficiency. JAMA. 1940;114:866–868. [Google Scholar]
- 28.Fine BP, Ty A, Lestrange N, Levine OR. Sodium deprivation growth failure in the rat: alterations in tissue composition and fluid spaces. J Nutr. 1987;117:1623–1628. doi: 10.1093/jn/117.9.1623. [DOI] [PubMed] [Google Scholar]
- 29.Bursey RG, Watson ML. The effect of sodium restriction during gestation of offspring brain development in rats. Am J Clin Nutr. 1983;37:43–51. doi: 10.1093/ajcn/37.1.43. [DOI] [PubMed] [Google Scholar]
- 30.Lind L, Lithell H, Gustafsson IB, Pollare T, Ljunghall S. Metabolic cardiovascular risk factors and sodium sensitivity in hypertensive subjects. Am J Hypertens. 1992;5:502–505. doi: 10.1093/ajh/5.8.502. [DOI] [PubMed] [Google Scholar]
- 31.MacGregor GA, De Wardener HE. Salt, Diet, and Health. Cambridge: Cambridge University Press; 1998. [Google Scholar]
- 32.Kaunitz H. Causes and consequences of salt consumption. Nature. 1956;178:1141–1144. doi: 10.1038/1781141a0. [DOI] [PubMed] [Google Scholar]
- 33.Dahl LK. Salt intake and salt need. N Engl J Med. 1958;258:1205–1208. doi: 10.1056/NEJM195806122582406. [DOI] [PubMed] [Google Scholar]
- 34.Lev-Ran A, Porta M. Salt and hypertension: a phylogenetic perspective. Diabetes Metab Res Rev. 2005;21:118–131. doi: 10.1002/dmrr.539. [DOI] [PubMed] [Google Scholar]
- 35.Stamler J, Rose G, Stamler R, Elliott P, Dyer A, Marmot M. INTERSALT study findings. Public health and medical care implications. Hypertension. 1989;14:570–577. doi: 10.1161/01.hyp.14.5.570. [DOI] [PubMed] [Google Scholar]
- 36.Denton D, Weisinger R, Mundy NI, Wickings EJ, Dixson A, Moisson P, Pingard AM, Shade R, Carey D, Ardaillou R, Paillard F, Chapman J, Thillet J, Michel JB. The effect of increased salt intake on blood pressure of chimpanzees. Nat Med. 1995;1:1009–1016. doi: 10.1038/nm1095-1009. [DOI] [PubMed] [Google Scholar]
- 37.Cordain L, Eaton SB, Sebastian A, Mann N, Lindeberg S, Watkins BA, O’Keefe JH, Brand-Miller J. Origins and evolution of the Western diet: health implications for the 21st century. Am J Clin Nutr. 2005;81:341–354. doi: 10.1093/ajcn.81.2.341. [DOI] [PubMed] [Google Scholar]
- 38.Beevers DG. The epidemiology of salt and hypertension. Clin Auton Res. 2002;12:353–357. doi: 10.1007/s10286-002-0061-7. [DOI] [PubMed] [Google Scholar]
- 39.Kurlansky M. Salt: A world history. New York: Walker Publishing Company, Inc.; 2002. [Google Scholar]
- 40.Kempner W. Treatment of kidney disease and hypertensive vascular disease with rice diet I. N C Med J. 1944;5:125–133. [PubMed] [Google Scholar]
- 41.Fodor JG, Whitmore B, Leenen F, Larochelle P. Lifestyle modifications to prevent and control hypertension. 5. Recommendations on dietary salt. Canadian Hypertension Society, Canadian Coalition for High Blood Pressure Prevention and Control, Laboratory Centre for Disease Control at Health Canada, Heart and Stroke Foundation of Canada. CMAJ. 1999;160:S29–34. [PMC free article] [PubMed] [Google Scholar]
- 42.Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, Obarzanek E, Conlin PR, Miller ER, Simons-Morton DG, Karanja N, Lin PH. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N Engl J Med. 2001;344:3–10. doi: 10.1056/NEJM200101043440101. [DOI] [PubMed] [Google Scholar]
- 43.Jacobson MF. Salt: The forgotten killer. Washington, D.C: Center for Science in the Public Interest; 2005. [Google Scholar]
- 44.Devine A, Criddle RA, Dick IM, Kerr DA, Prince RL. A longitudinal study of the effect of sodium and calcium intakes on regional bone density in postmenopausal women. Am J Clin Nutr. 1995;62:740–745. doi: 10.1093/ajcn/62.4.740. [DOI] [PubMed] [Google Scholar]
- 45.Woolfson RG, de Wardener HE. Primary renal abnormalities in hereditary hypertension. Kidney Int. 1996;50:717–731. doi: 10.1038/ki.1996.370. [DOI] [PubMed] [Google Scholar]
- 46.Gomez-Sanchez EP, Zhou M, Gomez-Sanchez CE. Mineralocorticoids, salt and high blood pressure. Steroids. 1996;61:184–188. doi: 10.1016/0039-128x(96)00010-4. [DOI] [PubMed] [Google Scholar]
- 47.Selye H. The Stress of Life. New York: McGraw-Hill; 1976. [Google Scholar]
- 48.Funder JW. Aldosterone, salt and cardiac fibrosis. Clin Exp Hypertens. 1997;19:885–899. doi: 10.3109/10641969709083193. [DOI] [PubMed] [Google Scholar]
- 49.Knudsen KD, Dahl LK, Thompson K, Iwai J, Heine M, Leitl G. Effects of chronic excess salt ingestion. Inheritance of hypertension in the rat. J Exp Med. 1970;132:976–1000. doi: 10.1084/jem.132.5.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cherchovich GM, Capek K, Jefremova Z, Pohlova I, Jelinek J. High salt intake and blood pressure in lower primates (Papio hamadryas) J Appl Physiol. 1976;40:601–604. doi: 10.1152/jappl.1976.40.4.601. [DOI] [PubMed] [Google Scholar]
- 51.Corbett WT, Kuller LH, Blaine EH, Damico FJ. Utilization of swine to study the risk factor of an elevated salt diet on blood pressure. Am J Clin Nutr. 1979;32:2068–2075. doi: 10.1093/ajcn/32.10.2068. [DOI] [PubMed] [Google Scholar]
- 52.Strazzullo P, Galletti F. Genetics of salt-sensitive hypertension. Curr Hypertens Rep. 2007;9:25–32. doi: 10.1007/s11906-007-0006-6. [DOI] [PubMed] [Google Scholar]
- 53.Srinivasan SR, Dalferes ER, Wolf RH, Radhakrishnamurthy B, Foster TA, Berenson GS. Variability in blood pressure response to dietary sodium intake among African green monkeys (Cercopithecus aethiops) Am J Clin Nutr. 1984;39:792–796. doi: 10.1093/ajcn/39.5.792. [DOI] [PubMed] [Google Scholar]
- 54.Elliott P, Walker LL, Little MP, Blair-West JR, Shade RE, Lee DR, Rouquet P, Leroy E, Jeunemaitre X, Ardaillou R, Paillard F, Meneton P, Denton DA. Change in salt intake affects blood pressure of chimpanzees: implications for human populations. Circulation. 2007;116:1563–1568. [Google Scholar]
- 55.Grim CE, Cowley AW, Jr, Hamet P, Gaudet D, Kaldunski ML, Kotchen JM, Krishnaswami S, Pausova Z, Roman R, Tremblay J, Kotchen TA. Hyperaldosteronism and hypertension: ethnic differences. Hypertension. 2005;45:766–772. doi: 10.1161/01.HYP.0000154364.00763.d5. [DOI] [PubMed] [Google Scholar]
- 56.de Wardener HE, MacGregor GA. Harmful effects of dietary salt in addition to hypertension. J Hum Hypertens. 2002;16:213–223. doi: 10.1038/sj.jhh.1001374. [DOI] [PubMed] [Google Scholar]
- 57.Fisher ND, Hurwitz S, Jeunemaitre X, Hopkins PN, Hollenberg NK, Williams GH. Familial aggregation of low-renin hypertension. Hypertension. 2002;39:914–918. doi: 10.1161/01.hyp.0000013784.18175.51. [DOI] [PubMed] [Google Scholar]
- 58.Johnson AK, Thunhorst RL. The neuroendocrinology of thirst and salt appetite: visceral sensory signals and mechanisms of central integration. Front Neuroendocrinol. 1997;18:292–353. doi: 10.1006/frne.1997.0153. [DOI] [PubMed] [Google Scholar]
- 59.Falk JL, Herman TS. Specific appetite for NaCl without postingestional repletion. J Comp Physiol Psychol. 1961;54:405–408. doi: 10.1037/h0046547. [DOI] [PubMed] [Google Scholar]
- 60.Smith CA, Curtis KS, Smith JC, Stricker EM. Presystemic influences on thirst, salt appetite, and vasopressin secretion in the hypovolemic rat. Am J Physiol Regul Integr Comp Physiol. 2007;292:R2089–2099. doi: 10.1152/ajpregu.00595.2006. [DOI] [PubMed] [Google Scholar]
- 61.Sakai RR, Nicolaidis S, Epstein AN. Salt appetite is suppressed by interference with angiotensin II and aldosterone. Am J Physiol. 1986;251:R762–768. doi: 10.1152/ajpregu.1986.251.4.R762. [DOI] [PubMed] [Google Scholar]
- 62.Weiss ML, Moe KE, Epstein AN. Interference with central actions of angiotensin II suppresses sodium appetite. Am J Physiol. 1986;250:R250–259. doi: 10.1152/ajpregu.1986.250.2.R250. [DOI] [PubMed] [Google Scholar]
- 63.Richter CP. Increased salt appetite in adrenalectomized rats. Am J Physiol. 1936;115:155–161. [Google Scholar]
- 64.Rice KK, Richter CP. Increased sodium chloride and water intake of normal rats treated with desoxycorticosterone acetate. Endocrinology. 1943;33:106–115. [Google Scholar]
- 65.Fregly MJ, Waters W. Effect of mineralocorticoids on spontaneous sodium chloride appetite of adrenalectomized rats. Physiol Behav. 1966;1:65–74. [Google Scholar]
- 66.Tigerstedt R, Bergman P. Niere und Kreislauf. Scand Arch Physiol. 1898;8:223–271. [Google Scholar]
- 67.Page IH, Bumpus FM. Angiotensin. Physiol Rev. 1961;41:331–390. doi: 10.1152/physrev.1961.41.2.331. [DOI] [PubMed] [Google Scholar]
- 68.Epstein AN, Fitzsimons JT, Simons BJ. Drinking caused by the intracranial injection of angiotensin into the rat. J Physiol. 1969;200:98P–100P. [PubMed] [Google Scholar]
- 69.Epstein AN, Fitzsimons JT, Rolls BJ. Drinking induced by injection of angiotensin into the brain of the rat. J Physiol. 1970;210:457–474. doi: 10.1113/jphysiol.1970.sp009220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Avrith DB, Fitzsimons JT. Increased sodium appetite in the rat induced by intracranial administration of components of the renin-angiotensin system. J Physiol. 1980;301:349–364. doi: 10.1113/jphysiol.1980.sp013210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buggy J, Fisher AE. Evidence for a dual central role for angiotensin in water and sodium intake. Nature. 1974;250:733–735. doi: 10.1038/250733a0. [DOI] [PubMed] [Google Scholar]
- 72.Bryant RW, Epstein AN, Fitzsimons JT, Fluharty SJ. Arousal of a specific and persistent sodium appetite in the rat with continuous intracerebroventricular infusion of angiotensin II. J Physiol. 1980;301:365–382. doi: 10.1113/jphysiol.1980.sp013211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beresford MJ, Fitzsimons JT. Intracerebroventricular angiotensin II-induced thirst and sodium appetite in rat are blocked by the AT1 receptor antagonist, Losartan (DuP 753), but not by the AT2 antagonist, CGP 42112B. Exp Physiol. 1992;77:761–764. doi: 10.1113/expphysiol.1992.sp003643. [DOI] [PubMed] [Google Scholar]
- 74.Zhang DM, Stellar E, Epstein AN. Together intracranial angiotensin and systemic mineralocorticoid produce avidity for salt in the rat. Physiol Behav. 1984;32:677–681. doi: 10.1016/0031-9384(84)90325-1. [DOI] [PubMed] [Google Scholar]
- 75.Toth E, Stelfox J, Kaufman S. Cardiac control of salt appetite. Am J Physiol. 1987;252:R925–929. doi: 10.1152/ajpregu.1987.252.5.R925. [DOI] [PubMed] [Google Scholar]
- 76.Johnson AK, Thunhorst RL. Sensory mechanisms in the behavioral control of body fluid balance: thirst and salt appetite. Prog Psychobiol Physiol Psychol. 1995;16:145–176. [PubMed] [Google Scholar]
- 77.Johnson AK, Thunhorst RL. The neuroendocrinology, neurochemistry and molecular biology of thirst and salt appetite. In: Lajtha A, Blaustein JD, editors. Handbook of Neurochemistry and Molecular Neurobiology: Behavioral Neurochemistry, Neuroendocrinology and Molecular Neurobiology. 3. New York: Springer; 2007. pp. 641–687. [Google Scholar]
- 78.Fitts DA, Masson DB. Forebrain sites of action for drinking and salt appetite to angiotensin or captopril. Behav Neurosci. 1989;103:865–872. doi: 10.1037/h0092457. [DOI] [PubMed] [Google Scholar]
- 79.Thunhorst RL, Johnson AK. Renin-angiotensin, arterial blood pressure, and salt appetite in rats. Am J Physiol. 1994;266:R458–465. doi: 10.1152/ajpregu.1994.266.2.R458. [DOI] [PubMed] [Google Scholar]
- 80.Antunes-Rodrigues J, Gentil CG, Negro-Vilar A, Covian MR. Role of adrenals in the changes of sodium chloride intake following lesions in the central nervous system. Physiol Behav. 1970;5:89–93. doi: 10.1016/0031-9384(70)90018-1. [DOI] [PubMed] [Google Scholar]
- 81.Rolls BJ, Rolls ET. Effects of lesions in the basolateral amygdala on fluid intake in the rat. J Comp Physiol Psychol. 1973;83:240–247. doi: 10.1037/h0034413. [DOI] [PubMed] [Google Scholar]
- 82.Carithers J, Bealer SL, Brody MJ, Johnson AK. Fine structural evidence of degeneration in supraoptic nucleus and subfornical organ of rats with lesions in the anteroventral third ventricle. Brain Res. 1980;201:1–12. doi: 10.1016/0006-8993(80)90770-2. [DOI] [PubMed] [Google Scholar]
- 83.Geerling JC, Kawata M, Loewy AD. Aldosterone-sensitive neurons in the rat central nervous system. J Comp Neurol. 2006;494:515–527. doi: 10.1002/cne.20808. [DOI] [PubMed] [Google Scholar]
- 84.Zardetto-Smith AM, Beltz TG, Johnson AK. Role of the central nucleus of the amygdala and bed nucleus of the stria terminalis in experimentally-induced salt appetite. Brain Res. 1994;645:123–134. doi: 10.1016/0006-8993(94)91645-4. [DOI] [PubMed] [Google Scholar]
- 85.Johnson AK, Gross PM. Sensory circumventricular organs and brain homeostatic pathways. FASEB J. 1993;7:678–686. doi: 10.1096/fasebj.7.8.8500693. [DOI] [PubMed] [Google Scholar]
- 86.Allen AM, Zhuo J, Mendelsohn FA. Localization and function of angiotensin AT1 receptors. Am J Hypertens. 2000;13:31S–38S. doi: 10.1016/s0895-7061(99)00249-6. [DOI] [PubMed] [Google Scholar]
- 87.Johnson AK, de Olmos J, Pastuskovas CV, Zardetto-Smith AM, Vivas L. The extended amygdala and salt appetite. Ann N Y Acad Sci. 1999;877:258–280. doi: 10.1111/j.1749-6632.1999.tb09272.x. [DOI] [PubMed] [Google Scholar]
- 88.Thunhorst RL, Xu Z, Cicha MZ, Zardetto-Smith AM, Johnson AK. Fos expression in rat brain during depletion-induced thirst and salt appetite. Am J Physiol. 1998;274:R1807–1814. doi: 10.1152/ajpregu.1998.274.6.r1807. [DOI] [PubMed] [Google Scholar]
- 89.Charron G, Laforest S, Gagnon C, Drolet G, Mouginot D. Acute sodium deficit triggers plasticity of the brain angiotensin type 1 receptors. FASEB J. 2002;16:610–612. doi: 10.1096/fj.01-0531fje. [DOI] [PubMed] [Google Scholar]
- 90.Sugita M. Taste perception and coding in the periphery. Cell Mol Life Sci. 2006;63:2000–2015. doi: 10.1007/s00018-006-6100-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roitman MF, Bernstein IL. Amiloride-sensitive sodium signals and salt appetite: multiple gustatory pathways. Am J Physiol. 1999;276:R1732–1738. doi: 10.1152/ajpregu.1999.276.6.R1732. [DOI] [PubMed] [Google Scholar]
- 92.Brot MD, Watson CH, Bernstein IL. Amiloride-sensitive signals and NaCl preference and appetite: a lick-rate analysis. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1403–1411. doi: 10.1152/ajpregu.2000.279.4.R1403. [DOI] [PubMed] [Google Scholar]
- 93.McCutcheon NB. Sodium deficient rats are unmotivated by sodium chloride solutions mixed with the sodium channel blocker amiloride. Behav Neurosci. 1991;105:764–766. doi: 10.1037//0735-7044.105.5.764. [DOI] [PubMed] [Google Scholar]
- 94.Sollars SI, Hill DL. Taste responses in the greater superficial petrosal nerve: substantial sodium salt and amiloride sensitivities demonstrated in two rat strains. Behav Neurosci. 1998;112:991–1000. doi: 10.1037//0735-7044.112.4.991. [DOI] [PubMed] [Google Scholar]
- 95.Blonde GD, Garcea M, Spector AC. The relative effects of transection of the gustatory branches of the seventh and ninth cranial nerves on NaCl taste detection in rats. Behav Neurosci. 2006;120:580–589. doi: 10.1037/0735-7044.120.3.580. [DOI] [PubMed] [Google Scholar]
- 96.Halpern BP, Nelson LM. Bulbar gustatory responses to anterior and to posterior tongue stimulation in the rat. Am J Physiol. 1965;209:105–110. doi: 10.1152/ajplegacy.1965.209.1.105. [DOI] [PubMed] [Google Scholar]
- 97.Spector AC, Travers SP. The representation of taste quality in the mammalian nervous system. Behav Cogn Neurosci Rev. 2005;4:143–191. doi: 10.1177/1534582305280031. [DOI] [PubMed] [Google Scholar]
- 98.Simon SA, de Araujo IE, Gutierrez R, Nicolelis MA. The neural mechanisms of gustation: a distributed processing code. Nat Rev Neurosci. 2006;7:890–901. doi: 10.1038/nrn2006. [DOI] [PubMed] [Google Scholar]
- 99.Daniels D, Fluharty SJ. Salt appetite: a neurohormonal viewpoint. Physiol Behav. 2004;81:319–337. doi: 10.1016/j.physbeh.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 100.Pastuskovas CV, Cassell MD, Johnson AK, Thunhorst RL. Increased cellular activity in rat insular cortex after water and salt ingestion induced by fluid depletion. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1119–R1125. doi: 10.1152/ajpregu.00189.2002. [DOI] [PubMed] [Google Scholar]
- 101.Breslin PA, Spector AC, Grill HJ. Chorda tympani section decreases the cation specificity of depletion-induced sodium appetite in rats. Am J Physiol. 1993;264:R319–323. doi: 10.1152/ajpregu.1993.264.2.R319. [DOI] [PubMed] [Google Scholar]
- 102.Breslin PA, Spector AC, Grill HJ. Sodium specificity of salt appetite in Fischer-344 and Wistar rats is impaired by chorda tympani nerve transection. Am J Physiol. 1995;269:R350–356. doi: 10.1152/ajpregu.1995.269.2.R350. [DOI] [PubMed] [Google Scholar]
- 103.Spector AC, Grill HJ. Salt taste discrimination after bilateral section of the chorda tympani or glossopharyngeal nerves. Am J Physiol. 1992;263:R169–176. doi: 10.1152/ajpregu.1992.263.1.R169. [DOI] [PubMed] [Google Scholar]
- 104.Flynn FW, Grill HJ, Schulkin J, Norgren R. Central gustatory lesions: II. Effects on sodium appetite, taste aversion learning, and feeding behaviors. Behav Neurosci. 1991;105:944–954. doi: 10.1037//0735-7044.105.6.944. [DOI] [PubMed] [Google Scholar]
- 105.Scalera G, Spector AC, Norgren R. Excitotoxic lesions of the parabrachial nuclei prevent conditioned taste aversions and sodium appetite in rats. Behav Neurosci. 1995;109:997–1008. [PubMed] [Google Scholar]
- 106.Spector AC, Scalera G, Grill HJ, Norgren R. Gustatory detection thresholds after parabrachial nuclei lesions in rats. Behav Neurosci. 1995;109:939–954. [PubMed] [Google Scholar]
- 107.Grill HJ, Schulkin J, Flynn FW. Sodium homeostasis in chronic decerebrate rats. Behav Neurosci. 1986;100:536–543. doi: 10.1037//0735-7044.100.4.536. [DOI] [PubMed] [Google Scholar]
- 108.Schulkin J, Marini J, Epstein AN. A role for the medial region of the amygdala in mineralocorticoid-induced salt hunger. Behav Neurosci. 1989;103:179–185. doi: 10.1037/0735-7044.103.1.178. [DOI] [PubMed] [Google Scholar]
- 109.Kelley AE, Baldo BA, Pratt WE, Will MJ. Corticostriatal-hypothalamic circuitry and food motivation: integration of energy, action and reward. Physiol Behav. 2005;86:773–795. doi: 10.1016/j.physbeh.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 110.Everitt BJ, Parkinson JA, Olmstead MC, Arroyo M, Robledo P, Robbins TW. Associative processes in addiction and reward. The role of amygdala-ventral striatal subsystems. Ann N Y Acad Sci. 1999;877:412–438. doi: 10.1111/j.1749-6632.1999.tb09280.x. [DOI] [PubMed] [Google Scholar]
- 111.Everitt BJ, Cardinal RN, Parkinson JA, Robbins TW. Appetitive behavior: impact of amygdala-dependent mechanisms of emotional learning. Ann N Y Acad Sci. 2003;985:233–250. [PubMed] [Google Scholar]
- 112.Robbins TW, Everitt BJ. Limbic-striatal memory systems and drug addiction. Neurobiol Learn Mem. 2002;78:625–636. doi: 10.1006/nlme.2002.4103. [DOI] [PubMed] [Google Scholar]
- 113.Thorn GW, Forsham PH, Frawley TF, Wilson DL, Renold AE, Fredrickson DS, Jenkins D. Advances in the diagnosis and treatment of adrenal insufficiency. Am J Med. 1951;10:595–611. doi: 10.1016/0002-9343(51)90330-0. [DOI] [PubMed] [Google Scholar]
- 114.Beauchamp GK, Bertino M, Burke D, Engelman K. Experimental sodium depletion and salt taste in normal human volunteers. Am J Clin Nutr. 1990;51:881–889. doi: 10.1093/ajcn/51.5.881. [DOI] [PubMed] [Google Scholar]
- 115.Bertino M, Beauchamp GK, Engelman K. Long-term reduction in dietary sodium alters the taste of salt. Am J Clin Nutr. 1982;36:1134–1144. doi: 10.1093/ajcn/36.6.1134. [DOI] [PubMed] [Google Scholar]
- 116.Takamata A, Mack GW, Gillen CM, Nadel ER. Sodium appetite, thirst, and body fluid regulation in humans during rehydration without sodium replacement. Am J Physiol. 1994;266:R1493–1502. doi: 10.1152/ajpregu.1994.266.5.R1493. [DOI] [PubMed] [Google Scholar]
- 117.Wald N, Leshem M. Salt conditions a flavor preference or aversion after exercise depending on NaCl dose and sweat loss. Appetite. 2003;40:277–284. doi: 10.1016/s0195-6663(03)00013-8. [DOI] [PubMed] [Google Scholar]
- 118.Berridge KC, Flynn FW, Schulkin J, Grill HJ. Sodium depletion enhances salt palatability in rats. Behav Neurosci. 1984;98:652–660. doi: 10.1037//0735-7044.98.4.652. [DOI] [PubMed] [Google Scholar]
- 119.Na E, Morris MJ, Johnson AK. Naloxone treatment increases aversive responses to hypertonic saline in sodium deplete rats. Atlanta, GA: Society for Neuroscience; 2006. [Google Scholar]
- 120.Cabanac M. Physiological role of pleasure. Science. 1971;173:1103–1107. doi: 10.1126/science.173.4002.1103. [DOI] [PubMed] [Google Scholar]
- 121.Curtis KS, Krause EG, Contreras RJ. Altered NaCl taste responses precede increased NaCl ingestion during Na(+) deprivation. Physiol Behav. 2001;72:743–749. doi: 10.1016/s0031-9384(01)00422-x. [DOI] [PubMed] [Google Scholar]
- 122.Jacobs KM, Mark GP, Scott TR. Taste responses in the nucleus tractus solitarius of sodium-deprived rats. J Physiol. 1988;406:393–410. doi: 10.1113/jphysiol.1988.sp017387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tamura R, Norgren R. Repeated sodium depletion affects gustatory neural responses in the nucleus of the solitary tract of rats. Am J Physiol. 1997;273:R1381–1391. doi: 10.1152/ajpregu.1997.273.4.R1381. [DOI] [PubMed] [Google Scholar]
- 124.McCaughey SA, Scott TR. Rapid induction of sodium appetite modifies taste-evoked activity in the rat nucleus of the solitary tract. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1121–1131. doi: 10.1152/ajpregu.2000.279.3.R1121. [DOI] [PubMed] [Google Scholar]
- 125.Mangold JE, Hill DL. Extensive reorganization of primary afferent projections into the gustatory brainstem induced by feeding a sodium-restricted diet during development: less is more. J Neurosci. 2007;27:4650–4662. doi: 10.1523/JNEUROSCI.4518-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Berridge KC. Food reward: brain substrates of wanting and liking. Neurosci Biobehav Rev. 1996;20:1–25. doi: 10.1016/0149-7634(95)00033-b. [DOI] [PubMed] [Google Scholar]
- 127.Tindell AJ, Smith KS, Pecina S, Berridge KC, Aldridge JW. Ventral pallidum firing codes hedonic reward: when a bad taste turns good. J Neurophysiol. 2006;96:2399–2409. doi: 10.1152/jn.00576.2006. [DOI] [PubMed] [Google Scholar]
- 128.Leshem M, Kavushansky A, Devys JM, Thornton S. Enhancement revisited: the effects of multiple depletions on sodium intake in rats vary with strain, substrain, and gender. Physiol Behav. 2004;82:571–580. doi: 10.1016/j.physbeh.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 129.Sakai RR, Fine WB, Epstein AN, Frankmann SP. Salt appetite is enhanced by one prior episode of sodium depletion in the rat. Behav Neurosci. 1987;101:724–731. doi: 10.1037//0735-7044.101.5.724. [DOI] [PubMed] [Google Scholar]
- 130.Hubbell CL, McCutcheon NB. Opioidergic manipulations affect intake of 3% NaCl in sodium-deficient rats. Pharmacol Biochem Behav. 1993;46:473–476. doi: 10.1016/0091-3057(93)90382-4. [DOI] [PubMed] [Google Scholar]
- 131.Falk JL. Serial sodium depletion and NaCl solution intake. Physiol Behav. 1966;1:75–77. [Google Scholar]
- 132.Brown JE, Toma RB. Taste changes during pregnancy. Am J Clin Nutr. 1986;43:414–418. doi: 10.1093/ajcn/43.3.414. [DOI] [PubMed] [Google Scholar]
- 133.Barelare B, Richter CP. Increased sodium chloride appetite in pregnant rats. Am J Physiol. 1937;121:185–188. [Google Scholar]
- 134.Crystal SR, Bernstein IL. Infant salt preference and mother’s morning sickness. Appetite. 1998;30:297–307. doi: 10.1006/appe.1997.0144. [DOI] [PubMed] [Google Scholar]
- 135.Nicolaidis S, Galaverna O, Metzler CH. Extracellular dehydration during pregnancy increases salt appetite of offspring. Am J Physiol. 1990;258:R281–283. doi: 10.1152/ajpregu.1990.258.1.R281. [DOI] [PubMed] [Google Scholar]
- 136.Galaverna O, Nicolaidis S, Yao SZ, Sakai RR, Epstein AN. Endocrine consequences of prenatal sodium depletion prepare rats for high need-free NaCl intake in adulthood. Am J Physiol. 1995;269:R578–583. doi: 10.1152/ajpregu.1995.269.3.R578. [DOI] [PubMed] [Google Scholar]
- 137.Arguelles J, Brime JI, Lopez-Sela P, Perillan C, Vijande M. Adult offspring long-term effects of high salt and water intake during pregnancy. Horm Behav. 2000;37:156–162. doi: 10.1006/hbeh.1999.1567. [DOI] [PubMed] [Google Scholar]
- 138.Contreras RJ, Kosten T. Prenatal and early postnatal sodium chloride intake modifies the solution preferences of adult rats. J Nutr. 1983;113:1051–1062. doi: 10.1093/jn/113.5.1051. [DOI] [PubMed] [Google Scholar]
- 139.Contreras RJ. Differences in perinatal NaCl exposure alters blood pressure levels of adult rats. Am J Physiol. 1989;256:R70–77. doi: 10.1152/ajpregu.1989.256.1.R70. [DOI] [PubMed] [Google Scholar]
- 140.Kelley AE. Functional specificity of ventral striatal compartments in appetitive behaviors. Ann N Y Acad Sci. 1999;877:71–90. doi: 10.1111/j.1749-6632.1999.tb09262.x. [DOI] [PubMed] [Google Scholar]
- 141.Wise RA. Brain reward circuitry: insights from unsensed incentives. Neuron. 2002;36:229–240. doi: 10.1016/s0896-6273(02)00965-0. [DOI] [PubMed] [Google Scholar]
- 142.Robinson TE, Berridge KC. Incentive-sensitization and addiction. Addiction. 2001;96:103–114. doi: 10.1046/j.1360-0443.2001.9611038.x. [DOI] [PubMed] [Google Scholar]
- 143.Kelley AE, Bakshi VP, Haber SN, Steininger TL, Will MJ, Zhang M. Opioid modulation of taste hedonics within the ventral striatum. Physiol Behav. 2002;76:365–377. doi: 10.1016/s0031-9384(02)00751-5. [DOI] [PubMed] [Google Scholar]
- 144.Werme M, Messer C, Olson L, Gilden L, Thoren P, Nestler EJ, Brene S. Delta FosB regulates wheel running. J Neurosci. 2002;22:8133–8138. doi: 10.1523/JNEUROSCI.22-18-08133.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- 146.Ito R, Robbins TW, Everitt BJ. Differential control over cocaine-seeking behavior by nucleus accumbens core and shell. Nat Neurosci. 2004;7:389–397. doi: 10.1038/nn1217. [DOI] [PubMed] [Google Scholar]
- 147.Hoebel BG, Hernandez L, Schwartz DH, Mark GP, Hunter GA. Microdialysis studies of brain norepinephrine, serotonin, and dopamine release during ingestive behavior. Theoretical and clinical implications. Ann N Y Acad Sci. 1989;575:171–193. doi: 10.1111/j.1749-6632.1989.tb53242.x. [DOI] [PubMed] [Google Scholar]
- 148.Roitman MF, Patterson TA, Sakai RR, Bernstein IL, Figlewicz DP. Sodium depletion and aldosterone decrease dopamine transporter activity in nucleus accumbens but not striatum. Am J Physiol Regul Integr Comp Physiol. 1999;276:R1339–R1345. doi: 10.1152/ajpregu.1999.276.5.R1339. [DOI] [PubMed] [Google Scholar]
- 149.Roitman MF, Schafe GE, Thiele TE, Bernstein IL. Dopamine and sodium appetite: antagonists suppress sham drinking of NaCl solutions in the rat. Behav Neurosci. 1997;111:606–611. doi: 10.1037//0735-7044.111.3.606. [DOI] [PubMed] [Google Scholar]
- 150.Robinson TE, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology. 2004;47(Suppl 1):33–46. doi: 10.1016/j.neuropharm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 151.Robinson TE, Kolb B. Persistent structural modifications in nucleus accumbens and prefrontal cortex neurons produced by previous experience with amphetamine. J Neurosci. 1997;17:8491–8497. doi: 10.1523/JNEUROSCI.17-21-08491.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Epling WF, Pierce WD. Activity-based anorexia: A biobehavioral perspective. Int J Eat Disord. 1988;7:475–485. [Google Scholar]
- 153.Lenkei Z, Corvol P, Llorens-Cortes C. The angiotensin receptor subtype AT1A predominates in rat forebrain areas involved in blood pressure, body fluid homeostasis and neuroendocrine control. Brain Res Mol Brain Res. 1995;30:53–60. doi: 10.1016/0169-328x(94)00272-g. [DOI] [PubMed] [Google Scholar]
- 154.Grob M, Trottier JF, Mouginot D. Heterogeneous co-localization of AT1A receptor and Fos protein in forebrain neuronal populations responding to acute hydromineral deficit. Brain Res. 2004;996:81–88. doi: 10.1016/j.brainres.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 155.Mattson MP. Diet-Brain Connections. Boston: Kluwer Academic; 2002. [Google Scholar]
- 156.Hector M, Burton JR. What are the psychiatric manifestations of vitamin B12 deficiency? J Am Geriatr Soc. 1988;36:1105–1112. doi: 10.1111/j.1532-5415.1988.tb04397.x. [DOI] [PubMed] [Google Scholar]
- 157.Fordy J, Benton D. Does low iron status influence psychological functioning? J Hum Nutr Diet. 1994;7:127–133. [Google Scholar]
- 158.Benton D, Haller J, Fordy J. Vitamin supplementation for 1 year improves mood. Neuropsychobiology. 1995;32:98–105. doi: 10.1159/000119220. [DOI] [PubMed] [Google Scholar]
- 159.Murck H. Magnesium and affective disorders. Nutr Neurosci. 2002;5:375–389. doi: 10.1080/1028415021000039194. [DOI] [PubMed] [Google Scholar]
- 160.Singewald N, Sinner C, Hetzenauer A, Sartori SB, Murck H. Magnesium-deficient diet alters depression- and anxiety-related behavior in mice--influence of desipramine and Hypericum perforatum extract. Neuropharmacology. 2004;47:1189–1197. doi: 10.1016/j.neuropharm.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 161.Bou-Holaigah I, Rowe PC, Kan J, Calkins H. The relationship between neurally mediated hypotension and the chronic fatigue syndrome. JAMA. 1995;274:961–967. [PubMed] [Google Scholar]
- 162.Peterson PK, Pheley A, Schroeppel J, Schenck C, Marshall P, Kind A, Haugland JM, Lambrecht LJ, Swan S, Goldsmith S. A preliminary placebo-controlled crossover trial of fludrocortisone for chronic fatigue syndrome. Arch Intern Med. 1998;158:908–914. doi: 10.1001/archinte.158.8.908. [DOI] [PubMed] [Google Scholar]
- 163.American-Psychiatric-Association. Diagnostic and Statistical Manual of Mental Disorders. 4. Washington, D.C: American Psychiatric Association; 2000. Text Revision. [Google Scholar]
- 164.Udupa K, Sathyaprabha TN, Thirthalli J, Kishore KR, Lavekar GS, Raju TR, Gangadhar BN. Alteration of cardiac autonomic functions in patients with major depression: a study using heart rate variability measures. J Affect Disord. 2007;100:137–141. doi: 10.1016/j.jad.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 165.Grippo AJ, Johnson AK. Biological mechanisms in the relationship between depression and heart disease. Neurosci Biobehav Rev. 2002;26:941–962. doi: 10.1016/s0149-7634(03)00003-4. [DOI] [PubMed] [Google Scholar]
- 166.Grippo AJ, Moffitt JA, Johnson AK. Cardiovascular alterations and autonomic imbalance in an experimental model of depression. Am J Physiol Regul Integr Comp Physiol. 2002;282:R1333–1341. doi: 10.1152/ajpregu.00614.2001. [DOI] [PubMed] [Google Scholar]
- 167.Grippo AJ, Beltz TG, Johnson AK. Behavioral and cardiovascular changes in the chronic mild stress model of depression. Physiol Behav. 2003;78:703–710. doi: 10.1016/s0031-9384(03)00050-7. [DOI] [PubMed] [Google Scholar]
- 168.You ZB, Chen YQ, Wise RA. Dopamine and glutamate release in the nucleus accumbens and ventral tegmental area of rat following lateral hypothalamic self-stimulation. Neuroscience. 2001;107:629–639. doi: 10.1016/s0306-4522(01)00379-7. [DOI] [PubMed] [Google Scholar]
- 169.Grippo AJ, Na ES, Johnson RF, Beltz TG, Johnson AK. Sucrose ingestion elicits reduced Fos expression in the nucleus accumbens of anhedonic rats. Brain Res. 2004;1019:259–264. doi: 10.1016/j.brainres.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 170.Emanuele E, Geroldi D, Minoretti P, Coen E, Politi P. Increased plasma aldosterone in patients with clinical depression. Arch Med Res. 2005;36:544–548. doi: 10.1016/j.arcmed.2005.03.046. [DOI] [PubMed] [Google Scholar]
- 171.Murck H, Held K, Ziegenbein M, Kunzel H, Koch K, Steiger A. The renin-angiotensin-aldosterone system in patients with depression compared to controls--a sleep endocrine study. BMC Psychiatry. 2003;3:15. doi: 10.1186/1471-244X-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Johnson AK, Grippo AJ. Sadness and broken hearts: neurohumoral mechanisms and co-morbidity of ischemic heart disease and psychological depression. J Physiol Pharmacol. 2006;57(Suppl 11):5–29. [PubMed] [Google Scholar]
- 173.Pietranera L, Saravia F, Gonzalez Deniselle MC, Roig P, Lima A, De Nicola AF. Abnormalities of the hippocampus are similar in deoxycorticosterone acetate-salt hypertensive rats and spontaneously hypertensive rats. J Neuroendocrinol. 2006;18:466–474. doi: 10.1111/j.1365-2826.2006.01436.x. [DOI] [PubMed] [Google Scholar]
- 174.Sapolsky RM. Depression, antidepressants, and the shrinking hippocampus. Proc Natl Acad Sci U S A. 2001;98:12320–12322. doi: 10.1073/pnas.231475998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci U S A. 1996;93:3908–3913. doi: 10.1073/pnas.93.9.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Khurshid KA, Weaver ME. Conn’s syndrome presenting as depression. Am J Psychiatry. 2005;162:1226. doi: 10.1176/appi.ajp.162.6.1226. [DOI] [PubMed] [Google Scholar]
- 177.Katz AM. Heart failure: Pathophysiology, molecular biology, and clinical management. Philadelphia: Lippincott, Williams, & Wilkins; 2000. [Google Scholar]
- 178.Francis J, Weiss RM, Wei SG, Johnson AK, Beltz TG, Zimmerman K, Felder RB. Central mineralocorticoid receptor blockade improves volume regulation and reduces sympathetic drive in heart failure. Am J Physiol Heart Circ Physiol. 2001;281:H2241–2251. doi: 10.1152/ajpheart.2001.281.5.H2241. [DOI] [PubMed] [Google Scholar]
- 179.Wann BP, Bah TM, Boucher M, Courtemanche J, Le Marec N, Rousseau G, Godbout R. Vulnerability for apoptosis in the limbic system after myocardial infarction in rats: a possible model for human postinfarct major depression. J Psychiatry Neurosci. 2007;32:11–16. [PMC free article] [PubMed] [Google Scholar]
- 180.Schoemaker RG, Smits JF. Behavioral changes following chronic myocardial infarction in rats. Physiol Behav. 1994;56:585–589. doi: 10.1016/0031-9384(94)90305-0. [DOI] [PubMed] [Google Scholar]
- 181.Grippo AJ, Francis J, Weiss RM, Felder RB, Johnson AK. Cytokine mediation of experimental heart failure-induced anhedonia. Am J Physiol Regul Integr Comp Physiol. 2003;284:R666–673. doi: 10.1152/ajpregu.00430.2002. [DOI] [PubMed] [Google Scholar]
- 182.Germain L, Chouinard G. Treatment of recurrent unipolar major depression with captopril. Biol Psychiatry. 1988;23:637–641. doi: 10.1016/0006-3223(88)90010-8. [DOI] [PubMed] [Google Scholar]
- 183.Germain L, Chouinard G. Captopril treatment of major depression with serial measurements of blood cortisol concentrations. Biol Psychiatry. 1989;25:489–493. doi: 10.1016/0006-3223(89)90203-5. [DOI] [PubMed] [Google Scholar]
- 184.Zubenko GS, Nixon RA. Mood-elevating effect of captopril in depressed patients. Am J Psychiatry. 1984;141:110–111. doi: 10.1176/ajp.141.1.110. [DOI] [PubMed] [Google Scholar]
- 185.Croog SH, Levine S, Testa MA, Brown B, Bulpitt CJ, Jenkins CD, Klerman GL, Williams GH. The effects of antihypertensive therapy on the quality of life. N Engl J Med. 1986;314:1657–1664. doi: 10.1056/NEJM198606263142602. [DOI] [PubMed] [Google Scholar]
- 186.Armando I, Seltzer A, Bregonzio C, Saavedra JM. Stress and angiotensin II: novel therapeutic opportunities. Curr Drug Targets CNS Neurol Disord. 2003;2:413–419. doi: 10.2174/1568007033482661. [DOI] [PubMed] [Google Scholar]
- 187.Arborelius L, Owens MJ, Plotsky PM, Nemeroff CB. The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol. 1999;160:1–12. doi: 10.1677/joe.0.1600001. [DOI] [PubMed] [Google Scholar]
- 188.De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19:269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- 189.Mason BL, Pariante CM. The effects of antidepressants on the hypothalamic-pituitary-adrenal axis. Drug News Perspect. 2006;19:603–608. doi: 10.1358/dnp.2006.19.10.1068007. [DOI] [PubMed] [Google Scholar]
- 190.Rivier C, Vale W. Effect of angiotensin II on ACTH release in vivo: role of corticotropin-releasing factor. Regul Pept. 1983;7:253–258. doi: 10.1016/0167-0115(83)90018-6. [DOI] [PubMed] [Google Scholar]
- 191.Gard PR, Mandy A, Sutcliffe MA. Evidence of a possible role of altered angiotensin function in the treatment, but not etiology, of depression. Biol Psychiatry. 1999;45:1030–1034. doi: 10.1016/s0006-3223(98)00101-2. [DOI] [PubMed] [Google Scholar]
- 192.Porsolt RD, Le Pichon M, Jalfre M. Depression: a new animal model sensitive to antidepressant treatments. Nature. 1977;266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- 193.Tekol Y. Salt addiction: A different kind of drug addiction. Med Hypotheses. 2006;67:1233–1234. doi: 10.1016/j.mehy.2006.04.041. [DOI] [PubMed] [Google Scholar]
- 194.Michell AR. Sums and assumptions about salt. Perspect Biol Med. 1984;27:221–233. [Google Scholar]
- 195.Bernstein IL. Interaction between natural motivational systems and those which respond to drugs of abuse. Appetite. 2003;41:333–334. doi: 10.1016/j.appet.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 196.Clark JJ, Bernstein IL. Reciprocal cross-sensitization between amphetamine and salt appetite. Pharmacol Biochem Behav. 2004;78:691–698. doi: 10.1016/j.pbb.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 197.Markou A, Koob GF. Bromocriptine reverses the elevation in intracranial self-stimulation thresholds observed in a rat model of cocaine withdrawal. Neuropsychopharmacology. 1992;7:213–224. [PubMed] [Google Scholar]
- 198.Kenny PJ, Markou A. Conditioned nicotine withdrawal profoundly decreases the activity of brain reward systems. J Neurosci. 2005;25:6208–6212. doi: 10.1523/JNEUROSCI.4785-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Schulteis G, Markou A, Gold LH, Stinus L, Koob GF. Relative sensitivity to naloxone of multiple indices of opiate withdrawal: a quantitative dose-response analysis. J Pharmacol Exp Ther. 1994;271:1391–1398. [PubMed] [Google Scholar]
- 200.Beauchamp GK, Cowart BJ, Mennella JA, Marsh RR. Infant salt taste: developmental, methodological, and contextual factors. Dev Psychobiol. 1994;27:353–365. doi: 10.1002/dev.420270604. [DOI] [PubMed] [Google Scholar]
- 201.Beauchamp GK, Bertino M, Engelman K. Failure to compensate decreased dietary sodium with increased table salt usage. JAMA. 1987;258:3275–3278. [PubMed] [Google Scholar]
- 202.Bentley B, De Jong MJ, Moser DK, Peden AR. Factors related to nonadherence to low sodium diet recommendations in heart failure patients. Eur J Cardiovasc Nurs. 2005;4:331–336. doi: 10.1016/j.ejcnurse.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 203.Mattes RD, Westby E, De Cabo R, Falkner B. Dietary compliance among salt-sensitive and salt-insensitive normotensive adults. Am J Med Sci. 1999;317:287–294. doi: 10.1097/00000441-199905000-00004. [DOI] [PubMed] [Google Scholar]
- 204.Cannon W. Wisdom of the Body. New York: W.W Norton, Inc; 1932. [Google Scholar]
- 205.Paxinos G, Watson C. Rat Brain in Stereotaxic Coordinates. New York: Elsevier; 1998. [DOI] [PubMed] [Google Scholar]