

Abstract
Eukaryotic ribonuclease (RNase) P and RNase MRP are evolutionary related RNA-based enzymes involved in metabolism of various RNA molecules, including tRNA and rRNA. In contrast to the closely related eubacterial RNase P, which is comprised of an RNA component and a single small protein, these enzymes contain multiple protein components. Here we report the results of footprinting studies performed on purified Saccharomyces cerevisiae RNase MRP and RNase P holoenzymes. The results identify regions of the RNA components affected by the protein moiety, suggest a role of the proteins in stabilization of the RNA fold, and point to substantial similarities between the two evolutionary related RNA-based enzymes.
Keywords: ribonuclease MRP, ribonuclease P, RNA–protein interactions, footprinting
INTRODUCTION
Ribonuclease P (RNase P) is a ubiquitous RNA-based enzyme (ribozyme) found in all three kingdoms of life (Guerrier-Takada et al. 1983; Altman and Kirsebom 1999). RNase MRP is a universal eukaryotic site-specific endoribonuclease (Chang and Clayton 1987; Karwan et al. 1991). RNase MRP is closely related to RNase P, but it has evolved to have a different specificity. RNase P is responsible for the 5′-end maturation of tRNA and is involved in processing of other RNAs, including rRNA (Kazantsev and Pace 2006; Kirsebom 2007); RNase P was also shown to have a role in transcription (Reiner et al. 2006). The vast majority of RNase MRP is located in the nucleolus (Kiss and Filipowicz 1992). Nucleolar RNase MRP is involved in the maturation of rRNA (Schmitt and Clayton 1993; Henry et al. 1994; Lygerou et al. 1996). In Saccharomyces cerevisiae, RNase MRP was also shown to be involved in mRNA decay, cleaving the 5′-untranslated region of CLB2 mRNA (which encodes Cyclin B2), thus triggering degradation of this mRNA and aiding cell cycle progression (Gill et al. 2004). In humans, defects in the RNA component of RNase MRP were shown to be the cause of a severe autosomal multisystemic disorder, cartilage hair hypoplasia (CHH) (Ridanpaa et al. 2001).
Eubacterial RNase P consists of a large (typically 200–450 nucleotides [nt]) RNA molecule and a small (12–14 kDa) basic protein (Stark et al. 1978; Kazantsev and Pace 2006). The RNA component of eubacterial RNase P is catalytically active in vitro under conditions of high ionic strength or in the presence of polyamines; the protein component is required for the activity of eubacterial RNase P under conditions of low ionic strength and is essential in vivo (Guerrier-Takada et al. 1983; Guerrier-Takada and Altman 1984, Reich et al. 1988). Archaeal RNase P consists of an RNA component and four to five proteins. While the RNA components of some archaeal RNases P can be catalytically active without any proteins, others require proteins to exhibit noticeable activity (Haas et al. 1996; Pannucci et al. 1999); binding of the individual protein components enhances the catalytic properties of archaeal RNase P RNA (Tsai et al. 2006). The RNA component of eukaryotic RNase P resembles RNA from the eubacterial RNase P with several key elements (presumably involved in substrate recognition and catalysis) demonstrating a high degree of phylogenetic conservation. However, the eukaryotic RNase P is much more complex than its eubacterial and archaeal counterparts and contains a large number of protein components. S. cerevisiae RNase P has nine protein components: Pop1, Pop3, Pop4, Pop5, Pop6, Pop7, Pop8, Rpp1, and Rpr2; all these proteins are essential for the activity of the enzyme in vivo (Chamberlain et al. 1998). The RNA component of eukaryotic RNase P has been shown to be capable of low-level specific cleavage of a tRNA substrate in vitro without any proteins (Kikovska et al. 2007), demonstrating its similarity to the bacterial enzymes.
The putative catalytic domain of the eukaryotic RNase P RNA has a striking similarity to a domain of the RNase MRP RNA (the putative catalytic domain of RNase MRP) in both secondary structure (Forster and Altman 1990; Li et al. 2002; Piccinelli et al. 2005) and nucleotide sequence (Zhu et al. 2006; Gopalan 2007). Moreover, essential structural elements of RNases MRP and P (specifically the P3 helices) were shown to be interchangeable (Lindahl et al. 2000). The similarity of the putative catalytic domains in RNase MRP and P strongly suggests that, similar to RNase P, it is the RNA moiety of RNase MRP that is responsible for catalysis, although no direct proof has been reported (Gopalan 2007). The putative substrate recognition (specificity) domain of eukaryotic RNase P has a high degree of similarity with the specificity domain of the eubacterial RNase P (Loria and Pan 1996), but it has no resemblance to the corresponding region of RNase MRP. The difference between the putative substrate recognition (specificity) domains in RNases MRP and P is consistent with the divergent specificity of the two enzymes. The S. cerevisiae RNase MRP has 10 essential protein components, eight of which (Pop1, Pop3, Pop4, Pop5, Pop6, Pop7, Pop8, and Rpp1) are also found in RNase P (Chamberlain et al. 1998). One of the two unique protein components of RNase MRP, Snm1 (Schmitt and Clayton 1994), is a homolog of the RNase P Rpr2 subunit. The second unique protein, Rmp1 (Salinas et al. 2005), shows no homology with RNase P proteins. Similar to RNase P, the protein components of RNase MRP are required for the activity of the enzyme in vivo and are essential for viability in yeast (Schmitt and Clayton 1994; Chamberlain et al. 1998; Salinas et al. 2005).
The reasons for the increased protein dependence and complexity of RNase MRP and eukaryotic RNase P are not clear. In the case of the well-studied eubacterial RNase P, the single protein component plays a many faceted role: It is suggested to influence the function of eubacterial RNase P by enhancing substrate binding through the reduction of electrostatic repulsion, by stabilizing the active conformation, by promoting transition from the intermediate to the native fold, by helping catalysis by discrimination between substrate and product, by altering the substrate specificity and enhancing catalytic efficiency, and by enhancing metal ion activity in the active site (for review, see Smith et al. 2007). The roles of the proteins in RNase MRP and eukaryotic RNase P have been much less studied. The protein components of eukaryotic RNase P were suggested to contribute to the architecture of the catalytic site or participate in interactions with the substrate (True and Celander 1998). Several human proteins (corresponding to homologs of Pop1, Pop3, Pop4, and Pop8 in yeast) were shown to accumulate in the nucleolus, suggesting that they might be involved in localization of RNase MRP and P (Jarrous et al. 1999; van Eenennaam et al. 2001). Several overexpressed human RNase P proteins were shown to selectively bind to precursor tRNA, suggesting that they might be involved in interaction with the substrate in the holoenzyme (Jarrous 2002). Filter binding assays (Aspinall et al. 2007) suggested that individually expressed yeast proteins Pop4, Pop6, Rmp1, and Snm1 might interact with the RNase MRP pre-rRNA substrate.
To help clarify the roles of the protein components in RNase MRP and eukaryotic RNase P, we purified the two holoenzymes from S. cerevisiae, subjected them to footprinting analysis to identify the regions of the RNA moieties protected by the proteins, and compared the results obtained for the two evolutionarily related RNA-based enzymes.
RESULTS AND DISCUSSION
Active RNase MRP and RNase P were purified from yeast using tandem affinity purification (Riqaut et al. 1999) with a tag attached to the C terminus of the Pop4 protein (Salinas et al. 2005). Tandem affinity purification (TAP-tag) was previously used to purify RNase P and RNase MRP as well as other ribonucleoprotein complexes (Chamberlain et al. 1998; Puig et al. 2001; Salinas et al. 2005). Since Pop4 is a protein component of both RNase MRP and RNase P, the tag allowed for the simultaneous purification of both enzymes. Separation of RNases MRP and P is not required for footprinting studies involving the use of primer extension with a reverse transcriptase since the RNA components of RNases MRP and P are sufficiently different. In addition, despite their overall similarity, RNases MRP and P are well known not to be directly interacting with each other and can be biochemically separated (Srisawat and Engelke 2001; Salinas et al. 2005). The purified product contained three different RNA molecules corresponding to the RNA components of RNase MRP, RNase P, and pre-RNase P (Srisawat et al. 2002), which were present in a molar ratio of about 0.9:1.0:0.2, respectively (data not shown). The RNase MRP to RNase P ratio is reproducible and may reflect the relative abundance of the enzymes in yeast.
To identify regions of RNase MRP and RNase P RNAs affected by protein binding, we performed chemical and enzymatic probing of the RNA components in the holoenzymes and compared the sensitivity to the probing agents with that for the RNA-only controls. As the RNA-only controls we used the RNA components of RNases MRP and P, which were purified from yeast, as parts of the holoenzymes, but later deproteinated with proteinase K (Tranguch et al. 1994) (Materials and Methods). The footprinting profiles obtained for the deproteinated RNase MRP RNA were generally consistent with the results reported for in vitro transcribed RNA, except for the region corresponding to the proposed “P7” domain (Walker and Avis 2004). As the chemical probing agents we used (a) hydroxyl ions produced by Fenton reaction (Fe-EDTA; Powers and Noller 1995), which attack the exposed sugar residues (likely at the C4′ position), thus revealing the exposure of the RNA backbone to the solvent; (b) dimethyl sulfate (DMS; Ehresmann et al. 1987), which modifies the exposed N1 in adenines and N3 in cytosines; and (c) 1-cyclohexyl-3-(2-morpholinoethyl) carbodiimide metho-p-toluene sulfonate (CMCT; Ehresmann et al. 1987), which preferentially modifies the exposed N3 in uridines and, to a lesser extent, N1 in guanines. Kethoxal, which reacts with exposed N1, N2 of guanines (Ehresmann et al. 1987), was also used in some experiments. As enzymatic probes we used RNase A (cleaving RNA at accessible single-stranded pyrimidines) and RNase V1 (cleaving exposed double-stranded or stacked RNA). The enzymatic probes are much more bulky than the chemical probes and therefore require a higher degree of exposure of RNA to the solvent for the cleavage. Typical results of footprinting experiments are shown in Figure 1. The results of the footprinting studies of RNase MRP and RNase P are summarized in Figure 2. The results of the footprinting studies for the purified RNase P are generally consistent with the previously reported results obtained using the crude yeast extract (Tranguch et al. 1994). The major differences were observed in the following regions of RNase P RNA: eP19, CR-IV, eP15, P7, P10/11, CR-II, CR-III, P12, and P3. These differences can be attributed to the purity of the samples (crude yeast extract versus purified holoenzyme) as well as to the addition of Fe-EDTA probing in our study.
FIGURE 1.

Examples of the footprinting assays. (A) Fe-EDTA footprinting of RNase MRP. (Lane 1) deproteinated RNase MRP RNA control (no Fe-EDTA); (lane 2) holoenzyme control (no Fe-EDTA); (lane 3) deproteinated RNase MRP RNA (Fe-EDTA); (lane 4) holoenzyme (Fe-EDTA); (lanes 5,6) sequence marker (sequenced RNase MRP RNA). Numbers on the right show nucleotide numbering. Primer RTP4 was used in the primer extension reactions. (B) DMS footprinting of RNase MRP. (Lane 1) deproteinated RNase MRP RNA control (no DMS); (lane 2) holoenzyme control (no DMS); (lanes 3–5) deproteinated RNase MRP RNA (various concentrations of DMS); (lanes 6–8) RNase MRP holoenzyme (various concentrations of DMS); (lanes 9,10) sequence marker (sequenced plasmid DNA). Numbers on the right show nucleotide numbering. Primer RTP25 was used in the primer extension reactions.
FIGURE 2.

Regions of RNase MRP (A) and RNase P (B) RNAs affected by protein binding in the holoenzyme. Red markings signify higher level of protection in the holoenzyme versus deproteinated RNA; green markings indicate no change in sensitivity upon deproteination; blue markings indicate lower sensitivity upon deproteination. Highlighting of nucleotides describes the results of the Fe-EDTA footprinting assays (indicative of the exposure of the RNA backbone). Diamonds describe the results of chemical probing (indicative of the exposure of the bases; D- DMS modifications; C- CMCT modifications; K- kethoxal modifications). Triangles describe the results of enzymatic probing (A- RNase A; V- RNase V1). Empty red diamonds or triangles correspond to weaker (1.5× to 4.5×) protection; filled red diamonds or triangles correspond to stronger (5× and up) protection. The diagrams and nomenclature of the structural elements are generally based on the work of Frank et al. (2000), Li et al. (2002), and Walker and Avis (2004) with minor corrections (Materials and Methods).
Comparison of the regions affected by protein binding in RNase MRP (Fig. 2A) and RNase P (Fig. 2B) demonstrates a striking similarity between parts of the two enzymes. The putative catalytic domains of RNase MRP and eukaryotic RNase P (Fig. 2, domains 1) share the same general architecture as the catalytic domain (Loria and Pan 1996) in eubacterial RNase P. Since RNases MRP and P also share most of their protein components, it is reasonable to expect that the shared protein components interact predominantly with this domain and in a conserved manner between the two RNAs. Our results suggest that this is the case, indeed. Most of the core nucleotides in domain 1 are protected from the probing agents by protein components in both holoenzymes. However, analysis of the regions that are not affected by the proteins or become more sensitive (exposed to solvent) in the holoenzyme reveals a high degree of similarity between RNase MRP and RNase P.
The P3 helix is a key conserved element of RNase MRP and RNase P in all eukaryotes (Ziehler et al. 2001), but its role is not clear. The pattern of the protein-induced protection of the P3 helix in RNase MRP and RNase P holoenzymes is very similar (Fig. 2): The left helical stems (nucleotides 45–64 in RNase MRP and 47–71 in RNase P) are mostly unaffected by proteins while the right helical stems are completely protected in the presence of proteins. The internal P3 loops of RNase MRP and RNase P demonstrate nearly identical protected and sensitive regions. This overall conservation is consistent with the previously reported observation that RNase MRP and RNase P P3 helices can be interchanged between the yeast enzymes (Lindahl et al. 2000).
In S. cerevisiae RNases MRP and P RNAs, the P3 helix was shown to directly interact with the protein components Pop6 and Pop7 (Perederina et al. 2007); similar results were obtained for the human RNase MRP (Welting et al. 2007). Pop6 and Pop7 were shown to form a heterodimer that binds P3, protecting a segment of the lower strand of the internal loop of P3 and part of the adjacent helical stem (nucleotides 30–38, RNase MRP numbering) (Perederina et al. 2007). The results obtained for the holoenzymes (Fig. 2) suggest that the RNA–protein interactions involving P3 are not limited to the interactions with the Pop6/Pop7 heterodimer, and that other protein component(s) must interact with this extended helix. About half of the nucleotides of the upper strand of the P3 internal loop as well as several nucleotides of the lower strand are not protected by Pop6/Pop7 binding (Perederina et al. 2007), but are protected in the holoenzyme. It is possible that protection of some of these nucleotides is caused by conformational changes in the P3 helix of the holoenzyme that differ when compared to the conformation of RNA in the reconstituted Pop6/Pop7-RNA complex (Perederina et al. 2007); however, given the magnitude of the additional protection, involvement of additional protein(s) seems likely. It was previously suggested that in S. cerevisiae RNases MRP and P, the P3 helix was involved in specific interactions with the protein component Pop1 (Ziehler et al. 2001). In addition, a mutant in the RNase MRP RNA that deletes nucleotides 45–64 of the distal P3 stem–loop was suppressed by overexpression of Pop1 (M.E. Schmitt, unpubl. data). It is highly likely that the deletion of nucleotides 45–64 disrupts the fold of the P3 loop. Therefore, it is not surprising that this deletion may affect protein binding even though the deleted region itself is not protected. The regions of the additional protection could indicate the sites of the Pop1–RNA interactions.
Schizosaccharomyces pombe RNase P RNA was shown to be able to bind the substrate tRNA in the absence of proteins, and cross-linking analysis of RNase P RNA–tRNA conjugates was reported (Marquez et al. 2006). Three out of seven nucleotides that were shown to cross-link to the 5′ or 3′ ends of mature tRNA belonged to the P3 internal loop. Cross-linking studies indicate that a nucleotide of the upper strand of the P3 internal loop (C54 in S. pombe RNase P, corresponding to approximately G66 in S. cerevisiae RNase MRP or G73 in RNase P) is in close proximity to both 5′ and 3′ ends of the bound tRNA (Marquez et al. 2006). Our footprinting assays (Fig. 2) indicate that this region of P3 is not protected by proteins in the holoenzyme, suggesting that this part of the P3 internal loop is available for direct RNA–RNA interactions with the substrate. The two additional P3 nucleotides that cross-link to the substrate (U33, C34 in S. pombe RNase P) (Marquez et al. 2006) are located in the lower strand of the P3 internal loop. Their location corresponds to the edge of the region protected by a Pop6/Pop7 heterodimer (nucleotide C38 in S. cerevisiae RNases MRP) (Perederina et al. 2007). The immediate proximity of the cross-linking site to the Pop6/Pop7 binding site suggests a possible involvement of the Pop6/Pop7 heterodimer in interactions with the substrate in RNase P and, given the similarity between the two enzymes, likely in RNase MRP as well. Nucleotide C38 is protected in the presence of the proteins, but additional studies are needed to determine whether this protection is due to a change of the P3 conformation or due to direct protein binding. In any case, Pop6/Pop7 heterodimer appears to be positioned near the cleavage site. Since Pop6/Pop7 and Pop1 are shared between RNases MRP and P, their role may be one of general substrate binding as opposed to providing substrate specificity. The suggested role of the Pop6/Pop7 is consistent with the results of filter binding assays (Aspinall et al. 2007) that indicate possible Pop6–substrate interactions.
It is interesting to note that the stems eP19 show a similar degree of protection in RNase P and RNase MRP holoenzymes: The protected region extends about 5–6 bp from the start of the stem, further supporting the idea of conservation between the catalytic domains of the two enzymes. The observed protection of a part of the eP19 terminal loop in RNase MRP may be due to a protein-mediated tertiary interaction in the holoenzyme, which is disrupted when the proteins are removed.
The secondary structure of the core of domain 1 (stems P1, P2, and P4, CR-IV region, helical part of P3, and non-helical junctions) is highly conserved in RNases P from all kingdoms of life (Kazantsev and Pace 2006) as well as in RNase MRP (Li et al. 2002) and is believed to contain the catalytic core of the enzymes. In eubacterial RNase P this region was suggested to be involved in direct interactions with the protein component (Buck et al. 2005); the results obtained for the human RNase MRP suggest that this region is involved in interaction with the protein component Pop4 (Welting et al. 2004). The footprinting analysis in yeast shows that this core is heavily protected in the presence of protein components in both RNase P and RNase MRP (Fig. 2). It is not clear whether this protection is due to direct interactions of RNA with the protein components or protein-mediated structural rearrangements (tighter packing) of the RNA. The available crystal structures of the eubacterial RNases P RNA (Kazantsev et al. 2005; Torres-Larios et al. 2005) indicate that RNA packing in this area is sufficiently tight to account for protection of a substantial part of the RNA. Given the high degree of conservation in this region, it is likely that the RNA packing in the eukaryotic enzymes would be similarly tight. Hence, the observed protection in this region in the holoenzymes versus deproteinated RNA may be at least partly caused by protein-mediated stabilization of the RNA fold.
The secondary structures of domain 2 in RNases MRP and P are very diverse, likely reflecting the different substrate specificities of the two related enzymes. Since S. cerevisiae RNase P and RNase MRP have only one and two unique protein components, respectively (Rpr2 in RNase P, Snm1 and Rmp1 in RNase MRP), it is reasonable to suggest that the nonconserved domains 2 are mostly responsible for binding these unique proteins, while the conserved domains 1 contain the primary binding sites for the remaining eight (shared) proteins. Our observation that domain 2 appears to be much less affected by proteins than domain 1 in both RNase MRP and RNase P (Fig. 2) is consistent with this idea.
In RNase P, domain 2 has two major areas that are protected in the presence of proteins: conserved regions CR-II/CR-III and stems eP8/eP9. The internal loop CR-II/CR-III contains several nucleotides that are highly conserved in all RNases P from all kingdoms of life (Kazantsev and Pace 2006). In eubacterial RNase P the conserved parts of each strand of the CR-II/CR-III region (corresponding to nucleotides A207–A211 and U243–A247 in S. cerevisiae) were shown (Krasilnikov et al. 2003, 2004) to fold into loops closely resembling the T-loop of tRNA (T-loop motif) (Krasilnikov and Mondragon 2004). The T-loop motifs from the two strands fold into a complicated three-dimensional structure forming the substrate recognition interface. This interface appears to be universally conserved in all RNases P (Krasilnikov et al. 2004; Torres-Larios et al. 2006). Because the CR-II/CR-III region by itself is flexible (Krasilnikov et al. 2003), its position relative to the rest of the RNA molecule needs to be stabilized through tertiary interactions (Krasilnikov et al. 2004; Torres-Larios et al. 2006). The RNA elements responsible for this stabilization in eubacterial RNase P are missing in eukaryotes, suggesting that the corresponding tertiary interaction is mediated by a protein (or proteins). Drawing parallels between the eubacterial and eukaryotic enzymes, one can suggest that the heavily protected by proteins stems eP8 and eP9 may be involved in such interactions in S. cerevisiae RNase P, playing a role similar to the role of the P10.1 stem/tetraloop receptor in the Bacillus subtilis RNase P (Krasilnikov et al. 2003). A large portion of the CR-II/CR-III is protected in the presence of proteins. Analysis of the available crystallographic data (Materials and Methods) obtained for the corresponding part of eubacterial RNases P (Krasilnikov et al. 2003, 2004) shows that the nucleotides forming the two T-loop motifs (corresponding to A207–A211 and U243–A247 in S. cerevisiae) should be protected (except G245) from the probes used in the footprinting analysis due to RNA packing alone. This suggests that the observed protection of the corresponding region in the presence of proteins in yeast (Fig. 2B) can be attributed to stabilization of the RNA fold by protein(s), but not necessarily to an extensive interaction of this region with the protein moiety. It is interesting to note that the nucleotide G245 is the only nucleotide in this area that has its base exposed to the solvent regardless of the presence of proteins. G245 in S. cerevisiae corresponds to G220 in B. subtilis, A172 in Thermus thermophilus, or G147 in Thermotoga maritima. This is a phylogenetically conserved purine nucleotide that is located at the top of the T-loop motif in eubacterial RNase P (thus it is exposed to the solvent) (Krasilnikov et al. 2003, 2004; Torres-Larios et al. 2005). This nucleotide is predicted to be involved in a key stacking interaction with the TψC loop of the tRNA substrate (Torres-Larios et al. 2005). The phylogenetic conservation of this region in RNase P suggests that a similar substrate recognition interface is formed in all RNases P, including yeast. The exposure of the G245 in yeast RNase P supports this idea.
Genetic studies of yeast RNase MRP (Schmitt and Clayton 1994) indicated that stem ymP5 of domain 2 might be involved in the binding of the protein component Snm1. Mutation of G122 resulted in a conditional phenotype, likely due to a failure to bind the Snm1 protein well (Cai and Schmitt 2001). This mutation could be suppressed by overexpression of Snm1 (Schmitt and Clayton 1994). The results of the footprinting studies show that the corresponding region is indeed protected in the presence of proteins and point to the base of the stem ymP5 and the regions in the immediate vicinity as the potential Snm1 binding site. The protected region also includes the C149–A150 bulge, which could be involved in the recognition of the stem by the protein. Mutation of this bulge resulted in a conditional phenotype (Li et al. 2004). Protection of nucleotides 224–233 and the base of the stem eP15 may be attributed to the binding of Rmp1, the second unique protein component of RNase MRP (K. Salinas and M.E. Schmitt, unpubl.), but further studies are needed to support this hypothesis.
Comparison of the results of the footprinting analysis obtained for RNase MRP with available mutational studies (Shadel et al. 2000; Li et al. 2004) shows a very strong correlation. A gradual shortening of individual RNA stems is tolerated without apparent phenotypic changes as long as the deletions do not involve the regions of the stems that we identified as protected by proteins in the holoenzyme (Fig. 2A). Further shortening of the stems results in conditional phenotypes or lethality. This suggests that the changes observed in the mutational studies are caused to a large extent by disruption of RNA–protein interactions in RNase MRP. At the same time it should be noted that deletion of the terminal loops of the stems eP19, ymP5, and ymP7 did not result in apparent phenotypic changes (Shadel et al. 2000; Li et al. 2004); however, footprinting studies indicate some degree of protection in these loops (Fig. 2A). The likely explanation of the observed protection of some parts of the terminal loops in stems eP19, ymP5, and ymP7 is involvement of these regions in protein-mediated tertiary interactions in the holoenzyme. Indeed, analysis of the available crystal structures of the eubacterial RNase P RNA demonstrates multiple examples of tertiary interactions involving terminal regions of helical stems. Some of these tertiary interactions may be important for structural stability and/or high level of activity of the enzyme, but their disruption does not necessarily result in a complete inactivation of the enzyme and can be compensated for in vivo. The eubacterial RNase P RNA can provide a good example of an important tertiary interaction that can be eliminated without complete loss of activity. The “mini-RNase P,” which has stems P13, P14 removed (these stems are involved in tertiary interactions stabilizing the CR-II/CR-III region in eubacterial RNase P) (Krasilnikov et al. 2004), still demonstrated activity, albeit substantially reduced (Waugh et al. 1989).
Our footprinting analysis has identified the regions of eukaryotic RNase P and RNase MRP RNAs affected by binding of the protein components in the holoenzyme. The patterns of protection suggest a role for the proteins in stabilizing the fold of the RNA component and indicate a high degree of similarity between the putative catalytic domains of the two evolutionarily related ribonucleoprotein complexes.
MATERIALS AND METHODS
RNases MRP and P were purified from yeast strain YSW1 using tandem affinity purification (TAP-tag method) (Riqaut et al. 1999) generally following a purification protocol described by Salinas et al. (2005) with minor modifications. Sixteen liters of yeast (S. cerevisiae strain YSW1 with TAP fusion cassette attached to the carboxyl terminus of RNase MRP/P protein component Pop4) (Salinas et al. 2005) were grown at 30°C with vigorous aeration on YPD media (1% [w/v] yeast extract, 2% [w/v] peptone, 2% [w/v] dextrose) to about 2 × 108 cells/mL. The culture was cooled on ice, the cells (about 150 g) were harvested by centrifugation at 4000g (4°C), washed with water, and resuspended in a buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM KCl, 10% glycerol, 1 mM PMSF, and 1 mM EDTA. The cells were disrupted on ice using a Beadbeater (Biospec), then Tween 20 was added to 0.1% (v/v), and the extract was clarified by centrifugation at 17,000g for 10 min (4°C) followed by ultracentrifugation at 100,000g for 3 h (4°C). The clarified extract was mixed with 3 mL rabbit IgG agarose (Sigma) and incubated at 4°C for 5 h with light agitation. The IgG agarose was washed six times with 5 volumes of the buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM KCl, 10% glycerol, 1 mM PMSF, 1 mM EDTA, 0.1% (v/v) Tween 20 (Buffer A) and resuspended in 2 mL of the same buffer. Then 300 units of tobacco etch virus protease were added and the sample was incubated for 12 h at 4°C with light agitation. The resin was separated by centrifugation at 500g for 10 min (4°C) and the supernatant collected; the resin was additionally washed twice with 5 mL of Buffer A and the supernatants combined. CaCl2 was added to 4 mM, and the sample was incubated with 0.5 mL of calmodulin affinity resin (Amersham) for 3 h at 4°C with light agitation. The resin was washed six times with 10 mL of Buffer A supplemented with 4 mM CaCl2 and 1 mM Na-imidazole (pH 7.4). After the final wash, the resin was resuspended in 10 mL of Buffer A supplemented with 20 mM EGTA and 10 mM Na-imidazole (pH 7.4), and the RNases MRP/P were eluted for 2 h at 4°C with light agitation and concentrated in a Centricon-YM100 concentrator (Amicon). After purification the enzymes (Fig. 3) were immediately used or transferred into buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM KCl, 0.1 mM EDTA, and 50% (v/v) glycerol and stored at −20°C.
FIGURE 3.
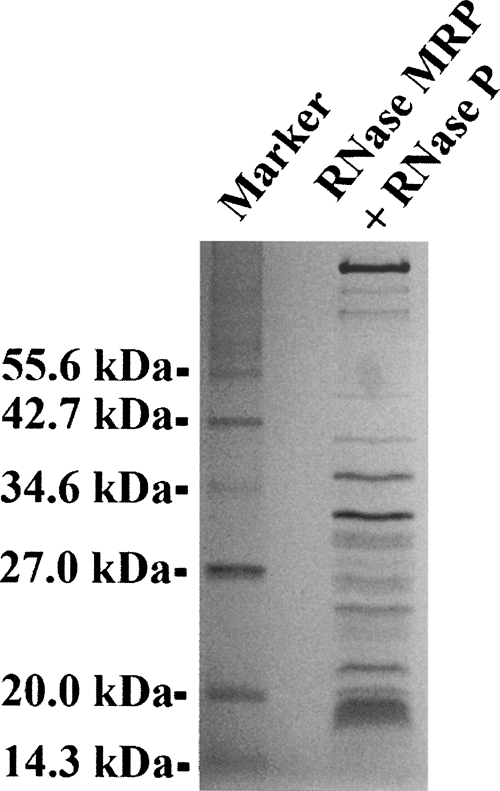
The protein components of RNase MRP/ RNase P preparations. This is a 15% SDS-polyacrylamide gel stained with Coomassie blue.
To analyze the RNA composition of the samples, RNA was extracted with phenol-chloroform, fractionated on a 6% denaturing polyacrylamide gel (8 M urea), stained with ethidium bromide, and quantified using a phosphorimager (Molecular Dynamics). To confirm the identities of the bands, RNA was sequenced by primer extension with SuperScript reverse transcriptase (Invitrogen) using the primers used in the footprinting experiments (below). To identify the protein components, purified samples were fractionated on a 12% SDS-polyacrylamide gel and stained with Coomassie blue, then the protein bands were excised from the gel, subjected to overnight in-gel digestion with trypsin, and analyzed by a capillary liquid chromatography-tandem mass spectrometry. To test for RNase MRP activity of the RNase MRP/RNase P mix, an in vitro transcribed fragment of yeast rRNA ITS1 containing the A3 site was used as a substrate; supS1 transcript (Drainas et al. 1989) was used as a substrate to test for RNase P activity. The yeast ITS1 fragment was produced by in vitro transcription with SP6 RNA polymerase using the plasmid pJA110 (Gill et al. 2004) as a template; supS1 was produced by in vitro transcription with T7 RNA polymerase using a corresponding synthetic DNA oligonucleotide (see sequence below) as a template. The transcripts were gel purified using 6% denaturing (8 M urea) polyacrylamide gels. The digestion (5 pmol of the substrate and 0.0005 to 0.1 pmol of RNase MRP/P) was performed for 30 min at 30°C in a buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM KCl, 5 mM MgCl2, and 5 mM DTT. The products of the digestion were analyzed using ethidium bromide stained 6% denaturing (8 M urea) polyacrylamide gels. The total molar concentration of RNases MRP/P was roughly estimated based on the sample's absorption at 260 nm using a NanoDrop ND-1000 spectrophotometer; the relatively small contribution of the protein moiety to the absorption at 260 nm was disregarded in the estimation of concentration. To obtain an estimation of the total molar concentration, the weighted average of the molecular weights of RNase MRP RNA (112 kDa) and RNase P RNA (120 kDa) was used.
For footprinting assays, the samples were transferred into buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM KCl, and 5 mM MgCl2, and divided into two parts. The first part was used in the assays directly, while the second part was first deproteinated with Proteinase K and then used in the assays as RNA-only controls. For deproteination the sample containing ∼100 pmol of RNases MRP/P was treated with 50 μg of Proteinase K (Fermentas) for 15 min at 25°C, then Proteinase K was inactivated by addition of 1 mM PMSF. The completeness of the deproteinization was monitored using silver-stained SDS-polyacrylamide gels. The amount of Proteinase K used in the deproteination was 10-fold higher that the minimal amount required for a complete digestion of RNase MRP/RNase P proteins in the sample. The use of RNase P RNA deproteinated with Proteinase K as a control in footprinting experiments has been previously reported (Tranguch et al. 1994).
The Fe-EDTA footprinting assays were performed following a well-established procedure (Pan 1995). The modifications were performed in a buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM KCl, 5 mM MgCl2, 5 mM DTT, 1 mM ascorbic acid, 1 mM (NH4)2Fe(SO4)2, and 1 mM Na-EDTA (pH 8.0) for 90 min at 30°C. The reactions were stopped by addition of thiourea to 10 mM and ethanol to 5%. The chemical modifications with dimethyl sulfate (DMS; Sigma) were performed for 20 min at 25°C in a buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM KCl, 5 mM MgCl2, and 0.05% to 0.5% DMS; the reactions were stopped by addition of 2-mercaptoethanol to 900 mM. The chemical modifications with CMCT (Sigma) were performed for 60 min at 25°C in a buffer containing 100 mM K-Borate (pH 7.8), 100 mM KCl, 5 mM MgCl2, and 0.5%–5% CMCT; the reactions were stopped by phenol extraction. The chemical modifications with kethoxal (Sigma) were performed for 20 min at 25°C in a buffer containing 50 mM Na-HEPES (pH 8.0), 100 mM KCl, 5 mM MgCl2, and 0.25 mM to 4 mM kethoxal; the reactions were stopped by addition of potassium borate to 0.25 M followed by phenol extraction. The enzymatic probing with RNase A and RNase V1 was performed in a buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM KCl, and 5 mM MgCl2 with varying concentrations of RNases for 10 min at 4°C for RNase A (Sigma) or 15 min at 30°C for RNase V1 (Ambion). The reactions were stopped by phenol extraction.
The results of the chemical and enzymatic probing were visualized by primer extension with reverse transcriptase followed by gel electrophoresis. Following the chemical/enzymatic treatment, the RNA (0.25 μg of RNase MRP and RNase P RNA mix per reaction, or about 1 pmol of each RNA) was extracted by phenol, ethanol precipitated, and dissolved in a buffer containing 5 mM Tris-HCl (pH 8.0), 10 mM KCl, and 0.1 mM EDTA. The DNA oligonucleotide primers complementary to RNase MRP or RNase P RNA were added in a 100-fold molar excess (100 pmol) over the template RNA and the primer annealing was performed by incubation for 3 min at 85°C followed by immediate cooling on ice for 15 min. To guarantee a reliable and redundant coverage of the whole length of RNase MRP and RNase P RNAs, seven different primers were used for each RNA (RTP1B, RTP1AL, RTP15A, RTP2A, RTP25, RTP3B, and RTP4 for RNase MRP RNA and PRTP1K, PRTP1B, PRTP15, PRTP2A, PRTP25A, PRTP3, and PRTP4 for RNase P RNA; see sequences below). Following primer annealing, the primer extension was performed in two steps: 32P labeling of the product and the primer extension itself. Radioactive labeling was performed in 50 mM Tris-HCl (pH 8.0), 75 mM KCl, 5 mM MgCl2, 20 mM DTT, and 1 unit of RNase inhibitor (RNaseIn; Ambion) with 50 units of the reverse transcriptase (SuperScript II; Invitrogen) in the presence of 0.5 mM each of dGTP, dTTP, and dATP, and 1 pmol of α-32P dCTP for dCTP labeling (or 0.5 mM each of dGTP, dTTP, and dCTP, and 1 pmol of α-32P dATP for dATP labeling). To optimize the incorporation of the label in primer extension reactions, labeling with α-32P dATP was used with the following primers: RTP1AL, RTP1B, RTP15A, RTP2A, RTP3B, RTP4, PRTP1K, PRTP15, PRTP2A, PRTP25A, and PRTP4. Labeling with α-32P dCTP was used with the following primers: RTP25, PRTP1B, and PRTP3. The labeling was performed for 5 min at 42°C, and cold dCTP (for 32P-dCTP labeling) or dATP (for 32P-dATP labeling) was added to 0.5 mM, and primer extension was continued for 10 min at 50°C. The reactions were stopped by addition of EDTA to 20 mM, and the samples were fractionated on 6% denaturing (8 M urea) polyacrylamide gels. The radioactive bands were visualized and quantified using a phosphorimager (Molecular Dynamics).
Primers used in primer extension reactions for the RNase MRP RNA:
RTP1AL (complementary to nucleotides 324–340): 5′-GGGTGAATCCATGGACC-3′;
RTP1B (complementary to nucleotides 316–340): 5′-GGTGAATCCATGGACCAAGAATAG-3′;
RTP15A (complementary to nucleotides 243–276): 5′-CAATCGTCATAACTATGGTTTAGATTCCATACAA-3′;
RTP2A (complementary to nucleotides 213–248): 5′-ATACAAACGGAATCCAGTACCGAAGAAAACACCAGT-3′;
RTP25 (complementary to nucleotides 168–201): 5′-GTGGTACCAGGTCAAGAAGCAGAATACCCAAGGG-3′;
RTP3B (complementary to nucleotides 133–174): 5′-CCAAGGGCATCCTCCTTAATGAGTTGATTTAAACAATTTAAA-3′;
RTP4 (complementary to nucleotides 91–126): 5′-AAATCTCATTACGTTTTCCGCATACGAATTGGTGGG-3′.
Primers used in primer extension reactions for the RNase P RNA:
PRTP1K (complementary to nucleotides 359–369): 5′-GCTGGAACAGC-3′;
PRTP1B (complementary to nucleotides 343–369): 5′-GCTGGAACAGCAGCAGT AATCGGTATC-3′;
PRTP15 (complementary to nucleotides 284–315): 5′-TTGCACTCAACAGACCTTGACGCTCTCGCCGT-3′;
PRTP2A (complementary to nucleotides 260–290): 5′-ACGCCGTAGCGGGCGACAAGTCAAACGGAAC-3′;
PRTP25A (complementary to nucleotides 200–230): 5′-CTAGGCCGAACTCCGTGAATTTCTGATAACA-3′;
PRTP3 (complementary to nucleotides 112–138): 5′-AAAGCGACATTAACCCGGAGGACAAGG-3′; and
PRTP4 (complementary to nucleotides 95–130): 5′-ATTAACCCGGAGGACAAGGCTCCACTGTGTTCCACC-3′.
DNA oligonucleotide used as a template for Sup1 transcript: 5′-CGACACCAGCAGGATTTGAACCTGCGCGCGGAGACCGCAACAGATTTAGAGTCTGTCCCTTTAACCACTCAGGCATAGTGTCCTGGACGATATTACTTTAGCTTGTATTCC TATAGTGAGTCGTATTA-3′.
The secondary structure diagrams (Fig. 2) are generally based on works by Frank et al. (2000), Li et al. (2002), and Walker and Avis (2004), with the following corrections. To accommodate the results of our footprinting assays, in RNase MRP (Fig. 2A) the top and the bottom of the stem P2 are relocated with corresponding changes in the adjacent stems P1 and eP19; U200 is shown bulged, and the putative stem P7 (Walker and Avis 2004) is not shown. Also, A85 is shown bulged and the P4 stem is shown to include A84 to reflect crystallographic data obtained for bacterial RNase P (Kazantsev et al. 2005); similar changes are made to the P4 stem of RNase P (Fig. 2B).
The accessibility of the nucleotides of eubacterial RNases P was estimated for structures of the S domains of RNase P from B. subtilis (PDB ID 1NBS), T. thermophilus (PDB ID 1U9S), RNase P RNAs from T. maritima (PDB ID 2E2E), and Bacillus stearothermophilus (PDB ID 2A64) using AREAIMOL (Bailey 1994).
ACKNOWLEDGMENTS
We thank Paul Babitzke, Phil Bevilacqua, and Alex Yakhnin for valuable suggestions. This work was supported by an American Heart Association Scientist Development Grant (AHA 0730131N) and by a Beckman Young Investigator Award to A.S.K. and NIH Grant GM063798 to M.E.S.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.1106408.
REFERENCES
- Altman S., Kirsebom L. Ribonuclease P. In: Gesteland R.F., et al., editors. The RNA world. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1999. pp. 351–380. [Google Scholar]
- Aspinall T.V., Gordon J.M.B., Bennett H.J., Karahalios P., Bukowski J.P., Walker S.C., Engelke D.R., Avis J.M. Interactions between subunits of Saccharomyces cerevisiae RNase MRP support a conserved eukaryotic RNase P/MRP architecture. Nucleic Acids Res. 2007;35:6439–6450. doi: 10.1093/nar/gkm553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey S. The CCP4 suite—programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Buck A.H., Kazantsev A.V., Dalby A.B., Pace N.R. Structural perspective on the activation of RNase P RNA by protein. Nat. Struct. Mol. Biol. 2005;12:958–964. doi: 10.1038/nsmb1004. [DOI] [PubMed] [Google Scholar]
- Cai T., Schmitt M.E. Characterization of ribonuclease MRP function. Methods Enzymol. 2001;342:135–142. doi: 10.1016/s0076-6879(01)42541-9. [DOI] [PubMed] [Google Scholar]
- Chamberlain J.R., Lee Y., Lane W.S., Engelke D.R. Purification and characterization of the nuclear RNase P holoenzyme complex reveals extensive subunit overlap with RNase MRP. Genes & Dev. 1998;12:1678–1690. doi: 10.1101/gad.12.11.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D.D., Clayton D.A. A novel endoribonuclease cleaves at a priming site of mouse mitochondrial DNA replication. EMBO J. 1987;6:409–417. doi: 10.1002/j.1460-2075.1987.tb04770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drainas D., Zimmerly S., Willis I., Soll D. Substrate structural requirements of Schizosaccharomyces pombe RNase P. FEBS Lett. 1989;251:84–88. doi: 10.1016/0014-5793(89)81433-4. [DOI] [PubMed] [Google Scholar]
- Ehresmann C., Baudin F., Mougel M., Romby P., Ebel J.-P., Ehresmann B. Probing the structure of RNAs in solution. Nucleic Acids Res. 1987;15:9109–9128. doi: 10.1093/nar/15.22.9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster A.C., Altman S. Similar cage-shaped structures for the RNA components of all ribonuclease P and ribonuclease MRP enzymes. Cell. 1990;62:407–409. doi: 10.1016/0092-8674(90)90003-w. [DOI] [PubMed] [Google Scholar]
- Frank D.N., Adamidi C., Ehringer M.A., Pitulle C., Pace N.R. Phylogenetic-comparative analysis of the eukaryal ribonuclease P RNA. RNA. 2000;6:1895–1904. doi: 10.1017/s1355838200001461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill T., Cai T., Aulds J., Wierzbicki S., Schmitt M.E. RNase MRP cleaves the CLB2 mRNA to promote cell cycle progression: Novel method of mRNA degradation. Mol. Cell. Biol. 2004;24:945–953. doi: 10.1128/MCB.24.3.945-953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalan V. Uniformity amid diversity in RNase P. Proc. Natl. Acad. Sci. 2007;104:2031–2032. doi: 10.1073/pnas.0611193104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrier-Takada C., Altman S. Catalytic activity of an RNA molecule prepared by transcription in vitro. Science. 1984;223:285–286. doi: 10.1126/science.6199841. [DOI] [PubMed] [Google Scholar]
- Guerrier-Takada C., Gardiner K., Marsh T., Pace N., Altman S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35:849–857. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- Haas E.S., Armbruster D.W., Vucson B.M., Daniels C.J., Brown J.W. Comparative analysis of ribonuclease P RNA structure in Archaea. Nucleic Acids Res. 1996;24:1252–1259. doi: 10.1093/nar/24.7.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry Y., Wood H., Morrissey J.P., Petfalski E., Kearsey S., Tollervey D. The 5′ end of yeast 5.8S rRNA is generated by exonucleases from an upstream cleavage site. EMBO J. 1994;13:2452–2463. doi: 10.1002/j.1460-2075.1994.tb06530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrous N. Human ribonuclease P: Subunits, function, and intranuclear localization. RNA. 2002;8:1–7. doi: 10.1017/s1355838202011184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrous N., Wolenski J.S., Wesolowski D., Lee C., Altman S. Localization in the nucleolus and coiled bodies of protein subunits of the ribonucleoprotein ribonuclease P. J. Cell Biol. 1999;146:559–571. doi: 10.1083/jcb.146.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwan R., Bennett J.L., Clayton D.A. Nuclear RNase MRP processes RNA at multiple discrete sites—interaction with an upstream G-box is required for subsequent downstream cleavages. Genes & Dev. 1991;5:1264–1276. doi: 10.1101/gad.5.7.1264. [DOI] [PubMed] [Google Scholar]
- Kazantsev A.V., Pace N.R. Bacterial RNase P: A new view of an ancient enzyme. Nat. Rev. Microbiol. 2006;4:729–740. doi: 10.1038/nrmicro1491. [DOI] [PubMed] [Google Scholar]
- Kazantsev A.V., Krivenko A.A., Harrington D.J., Holbrook S.R., Adams P.D., Pace N.R. Crystal structure of a bacterial ribonuclease P RNA. Proc. Natl. Acad. Sci. 2005;102:13392–13397. doi: 10.1073/pnas.0506662102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikovska E., Svard S.G., Kirsebom L.A. Eukaryotic RNase P RNA mediates cleavage in the absence of protein. Proc. Natl. Acad. Sci. 2007;104:2062–2067. doi: 10.1073/pnas.0607326104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsebom L.A. RNase P RNA mediated cleavage: Substrate recognition and catalysis. Biochimie. 2007;89:1183–1194. doi: 10.1016/j.biochi.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Kiss T., Filipowicz W. Evidence against a mitochondrial location of the 7-2/MRP RNA in mammalian cells. Cell. 1992;70:11–16. doi: 10.1016/0092-8674(92)90528-k. [DOI] [PubMed] [Google Scholar]
- Krasilnikov A.S., Mondragon A. On the occurrence of the T-loop RNA folding motif in large RNA molecules. RNA. 2004;9:640–643. doi: 10.1261/rna.2202703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasilnikov A.S., Yang X.J., Pan T., Mondragon A. Crystal structure of the specificity domain of ribonuclease P. Nature. 2003;421:760–764. doi: 10.1038/nature01386. [DOI] [PubMed] [Google Scholar]
- Krasilnikov A.S., Xiao Y.H., Pan T., Mondragon A. Basis for structural diversity in homologous RNAs. Science. 2004;306:104–107. doi: 10.1126/science.1101489. [DOI] [PubMed] [Google Scholar]
- Li X., Frank D.N., Pace N.R., Zengel J.M., Lindahl L. Phylogenetic analysis of the structure of RNase MRP in yeast. RNA. 2002;8:740–751. doi: 10.1017/s1355838202022082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Zaman S., Langdon Y., Zengel J.M., Lindahl L. Identification of a functional core in the RNA component of RNase MRP of budding yeasts. Nucleic Acids Res. 2004;32:3703–3711. doi: 10.1093/nar/gkh689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl L., Fretz S., Epps N., Zengel J.M. Functional equivalence of hairpins in the RNA subunits of RNase MRP and RNase P in Saccharomyces cerevisiae . RNA. 2000;6:653–658. doi: 10.1017/s1355838200992574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria A., Pan T. Domain structure of the ribozyme from eubacterial ribonuclease P. RNA. 1996;2:551–563. [PMC free article] [PubMed] [Google Scholar]
- Lygerou Z., Allmang C., Tollervey D., Seraphin B. Accurate processing of a eukaryotic precursor ribosomal RNA by ribonuclease MRP in vitro . Science. 1996;272:268–270. doi: 10.1126/science.272.5259.268. [DOI] [PubMed] [Google Scholar]
- Marquez S.M., Chen J.L., Evans D., Pace N.R. Structure and function of eukaryotic ribonuclease P RNA. Mol. Cell. 2006;24:445–456. doi: 10.1016/j.molcel.2006.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan T. Higher-order folding and domain analysis of the ribozyme from Bacillus subtilis ribonuclease P. Biochemistry. 1995;34:902–909. doi: 10.1021/bi00003a024. [DOI] [PubMed] [Google Scholar]
- Pannucci J.A., Haas E.S., Hall T.A., Harris J.K., Brown J.W. RNase P RNAs from some Archaea are catalytically active. Proc. Natl. Acad. Sci. 1999;96:7803–7808. doi: 10.1073/pnas.96.14.7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perederina A., Esakova O., Schmitt M.E., Krasilnikov A.S. Specific binding of a Pop6/Pop7 heterodimer to the P3 stem of the yeast RNase MRP and RNase P RNAs. RNA. 2007;13:1648–1655. doi: 10.1261/rna.654407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinelli P., Rosenblad M.A., Samuelson T. Identification and analysis of ribonuclease P and MRP RNA in a broad range of eukaryotes. Nucleic Acids Res. 2005;33:4485–4495. doi: 10.1093/nar/gki756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers T., Noller H.F. Hydroxyl radical footprinting of ribosomal proteins on 16S rRNA. RNA. 1995;1:194–209. [PMC free article] [PubMed] [Google Scholar]
- Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Seraphin B. The tandem-affinity purification (TAP) method: A general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- Reich C., Olsen G.J., Pace B., Pace N.R. Role of the protein moiety of ribonuclease P, a ribonucleoprotein enzyme. Science. 1988;239:178–181. doi: 10.1126/science.3122322. [DOI] [PubMed] [Google Scholar]
- Reiner R., Ben-Asouli Y., Krilovetzky I., Jarrous N. A role for the catalytic ribonucleoprotein RNase P in RNA polymerase III transcription. Genes & Dev. 2006;20:1621–1635. doi: 10.1101/gad.386706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridanpaa M., van Eenennaam H., Pelin K., Chadwick R., Johnson C., Yuan B., van Venrooij W., Pruijn G., Salmela R., Rockas S., et al. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, cartilage-hair hypoplasia. Cell. 2001;104:195–203. doi: 10.1016/s0092-8674(01)00205-7. [DOI] [PubMed] [Google Scholar]
- Riqaut G., Shevchenko A., Rutz B., Wilm M., Mann M., Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- Salinas K., Wierzbicki S., Zhou L., Schmitt M.E. Characterization and purification of Saccharomyces cerevisiae RNase MRP reveals a new unique protein component. J. Biol. Chem. 2005;280:11352–11360. doi: 10.1074/jbc.M409568200. [DOI] [PubMed] [Google Scholar]
- Schmitt M.E., Clayton D.A. Nuclear RNase MRP is required for correct processing of pre-5.8S ribosomal RNA in Saccharomyces cerevisiae . Mol. Cell. Biol. 1993;13:7935–7941. doi: 10.1128/mcb.13.12.7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt M.E., Clayton D.A. Characterization of a unique protein component of yeast RNase MRP—an RNA-binding protein with a zinc-cluster domain. Genes & Dev. 1994;8:2617–2628. doi: 10.1101/gad.8.21.2617. [DOI] [PubMed] [Google Scholar]
- Shadel G.S., Buckenmeyer G.A., Clayton D.A., Schmitt M.E. Mutational analysis of the RNA component of Saccharomyces cerevisiae RNase MRP reveals distinct nuclear phenotypes. Gene. 2000;245:175–184. doi: 10.1016/s0378-1119(00)00013-5. [DOI] [PubMed] [Google Scholar]
- Smith J.K., Hsieh J., Fierke C.A. Importance of RNA–protein interactions in bacterial ribonuclease P structure and catalysis. Biopolymers. 2007;87:329–338. doi: 10.1002/bip.20846. [DOI] [PubMed] [Google Scholar]
- Srisawat C., Engelke D.R. Streptavidin aptamers: Affinity tags for the study of RNAs and ribonucleoproteins. RNA. 2001;7:632–641. doi: 10.1017/s135583820100245x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srisawat C., Houser-Scott F., Bertrand E., Xiao S., Singer R.H., Engelke D.R. An active precursor in assembly of yeast nuclear ribonuclease P. RNA. 2002;8:1348–1360. doi: 10.1017/s1355838202027048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark B.C., Kole R., Bowman E.J., Altman S. Ribonuclease P: An enzyme with an essential RNA component. Proc. Natl. Acad. Sci. 1978;75:3717–3721. doi: 10.1073/pnas.75.8.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Larios A., Swinger K.K., Krasilnikov A.S., Pan T., Mondragon A. Crystal structure of the RNA component of bacterial ribonuclease P. Nature. 2005;437:584–587. doi: 10.1038/nature04074. [DOI] [PubMed] [Google Scholar]
- Torres-Larios A., Swinger K.K., Pan T., Mondragon A. Structure of ribonuclease P—a universal ribozyme. Curr. Opin. Struct. Biol. 2006;16:327–335. doi: 10.1016/j.sbi.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Tranguch A.J., Kindelberger D.W., Rohlman C.E., Lee J.Y., Engelke D.R. Structure-sensitive RNA footprinting of yeast nuclear ribonuclease P. Biochemistry. 1994;33:1778–1787. doi: 10.1021/bi00173a022. [DOI] [PubMed] [Google Scholar]
- True H.L., Celander D.W. Protein components contribute to active site architecture for eukaryotic ribonuclease P. J. Biol. Chem. 1998;273:7193–7196. doi: 10.1074/jbc.273.13.7193. [DOI] [PubMed] [Google Scholar]
- Tsai H.-Y., Pulukkunat D.K., Woznick W.K., Gopalan V. Functional reconstitution and characterization of Pyrococcus furiosus RNase P. Proc. Natl. Acad. Sci. 2006;103:16147–16152. doi: 10.1073/pnas.0608000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eenennaam H., van der Heijden A., Janssen R.J.R.J., van Venrooij W.J., Pruijn G.J.M. Basic domains target protein subunits of the RNase MRP complex to the nucleolus independently of complex association. Mol. Biol. Cell. 2001;12:3680–3689. doi: 10.1091/mbc.12.11.3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker S.C., Avis J.M. A conserved element in the yeast RNase MRP RNA subunit can participate in a long-range base-pairing interaction. J. Mol. Biol. 2004;341:375–388. doi: 10.1016/j.jmb.2004.05.076. [DOI] [PubMed] [Google Scholar]
- Waugh D.S., Green C.J., Pace N.R. The design and catalytic properties of a simplified ribonuclease P RNA. Science. 1989;244:1569–1571. doi: 10.1126/science.2472671. [DOI] [PubMed] [Google Scholar]
- Welting T.J.M., van Venrooij W.J., Pruijn G.J.M. Mutual interactions between subunits of the human RNase MRP ribonucleoprotein complex. Nucleic Acids Res. 2004;32:2138–2146. doi: 10.1093/nar/gkh539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welting T.J.M., Peters F.M.A., Hensen S.M.M., van Doorn N.L., Kikkert B.J., Raats J.M.H., van Venrooij W.J., Pruijn G.J.M. Heterodimerization regulates RNase MRP/RNase P association, localization, and expression of Rpp20 and Rpp25. RNA. 2007;13:65–75. doi: 10.1261/rna.237807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Stribinskis V., Ramos K.S., Li Y. Sequence analysis of RNase MRP reveals its origination from eukaryotic RNase P RNA. RNA. 2006;12:699–706. doi: 10.1261/rna.2284906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziehler W.A., Morris J., Scott F.H., Millikin C., Engelke D.R. An essential protein-binding domain of nuclear RNase P RNA. RNA. 2001;7:565–575. doi: 10.1017/s1355838201001996. [DOI] [PMC free article] [PubMed] [Google Scholar]