

Abstract
Thrombin is a Na+-activated, allosteric serine protease that plays opposing functional roles in blood coagulation. Binding of Na+ is the major driving force behind the procoagulant, prothrombotic and signaling functions of the enzyme, but is dispensable for cleavage of the anticoagulant protein C. The anticoagulant function of thrombin is under the allosteric control of the cofactor thrombomodulin. Much has been learned on the mechanism of Na+ binding and recognition of natural substrates by thrombin. Recent structural advances have shed light on the remarkable molecular plasticity of this enzyme and the molecular underpinnings of thrombin allostery mediated by binding to exosite I and the Na+ site. This review summarized our current understanding of the molecular basis of thrombin function and allosteric regulation. The basic information emerging from recent structural, mutagenesis and kinetic investigation of this important enzyme is that thrombin exists in three forms, E*, E and E:Na+, that interconvert under the influence of ligand binding to distinct domains. The transition between the Na+-free slow from E and the Na+-bound fast form E:Na+ involves the structure of the enzyme as a whole, and so does the interconversion between the two Na+-free forms E* and E. E* is most likely an inactive form of thrombin, unable to interact with Na+ and substrate. The complexity of thrombin function and regulation has gained this enzyme pre-eminence as the prototypic allosteric serine protease. Thrombin is now looked upon as a model system for the quantitative analysis of biologically important enzymes.
1. Introduction
Blood coagulation evolved as a specialization of the complement system and immune response (Krem and Di Cera 2001), that in turn bear close evolutionary ties with developmental enzyme cascades (Krem and Di Cera 2002). Thrombin, the key enzyme of blood coagulation, is a Na+-activated allosteric serine protease (Wells and Di Cera 1992; Di Cera 2003; Di Cera et al. 2007) that diverged from the complement factors C1r, C1s or MASP2, heralding the onset of further specialization of defense mechanisms in the deuterostome lineage (Krem and Di Cera 2001; 2002). The ancestral link between clotting and immunity is reinforced by the observation that sequence homologues of fibrinogen, the terminal substrate in blood clotting and a specific substrate of thrombin, originally served in immunologic roles (Adema et al. 1997). Thrombin predated and most likely gave rise to all other vitamin K-dependent proteases (Krem and Di Cera 2001), namely factors VIIa, IXa, and Xa that define the convergence between the intrinsic and extrinsic pathways of the coagulation cascade (Gailani and Broze 1991), and activated protein C that adds negative regulation to the cascade (Esmon 2003b) and a link to the inflammatory response (Cohen 2002). Because complement evolved from developmental proteases (Krem and Di Cera 2001; 2002), as also documented by the dual role played by the Toll signaling pathway in development and host defense (Tauszig et al. 2000), it is not surprising that thrombin itself retains signatures of its descent from a growth factor. In the mouse, knock-out of the prothrombin gene results in embryonic and neonatal lethality (Sun et al. 1998; Xue et al. 1998), a wastage not seen with mouse models deficient for thrombin receptors on platelets (Coughlin 2000) or fibrinogen (Suh et al. 1995). Furthermore, thrombin is expressed in the liver, the major site of clotting factor synthesis, but also in the developing and adult rat brains (Dihanich et al. 1991). Thrombin cleaves osteopontin, a multifunctional molecule regulating chronic inflammation and vascular disease (Scatena et al. 2007). Thrombin acts nonproteolytically to induce monocyte chemotaxis (Bar-Shavit et al. 1983), has adhesive properties dependent on its RGD sequence (Bar-Shavit et al. 1991; Papaconstantinou et al. 2005) and can promote the migration of cells through the extracellular matrix, a basic activity required for embryonic development and tumor metastasis. The complexity of thrombin function and regulation, as well as the intriguing aspects of its evolutionary origin have captured the interest of many investigators over the years and earned this enzyme a most deserved pre-eminence among all clotting factors. Relevant reviews have been published recently on the structure and interactions of thrombin (Bode 2006; Davie and Kulman 2006; Di Cera et al. 2007). The present review addresses the molecular basis of thrombin procoagulant and anticoagulant activities, and especially how Na+ influences them, as well as recent structural advances on the molecular basis of thrombin allostery. We also discuss how our basic knowledge on thrombin interactions translated into the rational engineering of thrombin mutants that offer a revolutionary approach to the treatment of thrombotic emergencies.
Blood coagulation is initiated by exposure of tissue factor that forms a complex with factor VIIa and results in the generation of small quantities of factors IXa and Xa (Girard et al. 1990; Gailani and Broze 1991). The small quantities of Xa generate minute concentrations of thrombin that result in the activation of factor XI and the cofactors VIII and V. At this point, the VIIIa-IXa complex generates sufficient quantities of Xa to form the prothrombinase complex, composed of factors Va, Xa, Ca2+ and phospholipids, which leads to the explosive generation of thrombin from prothrombin (Mann et al. 2003). Thrombin is a serine protease of the chymotrypsin family (Rawlings et al. 2004), which includes enzymes involved in digestion and degradative processes, blood coagulation, cell-mediated immunity and cell death, complement, fibrinolysis, fertilization and embryonic development (Perona and Craik 1995; 1997; Page and Di Cera 2008). Once generated in the blood from its inactive precursor prothrombin, thrombin plays two important and paradoxically opposing functions (Figure 1) (Griffin 1995). It acts as a procoagulant factor when it converts fibrinogen into an insoluble fibrin clot that anchors platelets to the site of lesion and initiates processes of wound repair. This action is reinforced and amplified by activation of the transglutaminase factor XIII that covalently stabilizes the fibrin clot (Lorand et al. 1968), the inhibition of fibrinolysis via activation of TAFI (Bajzar et al. 1996), and the proteolytic activation of factors V, VIII and XI (Mann 2003; Davie and Kulman 2006). In contrast, thrombin acts as an anticoagulant through activation of protein C (Esmon 2003b). This function unfolds in vivo upon binding to thrombomodulin, a receptor on the membrane of endothelial cells. Binding of thrombomodulin suppresses the ability of thrombin to cleave fibrinogen and PAR1, but enhances >1,000-fold the specificity of the enzyme toward the zymogen protein C. The reaction is further enhanced by the presence of a specific endothelial cell protein C receptor (Esmon et al. 1999; Taylor et al. 2001). Activated protein C cleaves and inactivates factors Va and VIIIa, two essential cofactors of coagulation factors Xa and IXa that are required for thrombin generation, thereby down regulating both the amplification and progression of the coagulation cascade (Esmon 2003b). Hijacking of thrombin by thrombomodulin and activation of protein C in the microcirculation constitute the natural anticoagulant pathway that prevents massive intravascular conversion of fibrinogen into an insoluble clot upon thrombin generation (Esmon 2003b; Mann 2003). In addition, thrombin is irreversibly inhibited at the active site by the serine protease inhibitor antithrombin with the assistance of heparin (Gettins 2002; Olson and Chuang 2002) and by the thrombin-specific heparin cofactor II (Tollefsen 2006). Important cellular effects are triggered by thrombin cleavage of protease-activated receptors (PARs) (Coughlin 2000), which are members of the G-protein-coupled receptor superfamily (Brass 2003). Four PARs have been identified and share the same basic mechanism of activation: thrombin and other proteases cleave at a specific site within the extracellular N-terminus exposing a new N-terminal tethered ligand domain that binds and activates the cleaved receptor (Coughlin 2000). Thrombin activation of PAR1 (Vu et al. 1991), PAR3 (Ishihara et al. 1997; Sambrano et al. 2001) and PAR4 (Kahn et al. 1998; Xu et al. 1998; Nakanishi-Matsui et al. 2000) obeys this mechanism. PAR1 is responsible for platelet activation in humans at low thrombin concentrations and its action is reinforced by PAR4 at high enzyme concentrations (Coughlin 2000). Activation of PAR1 and PAR4 triggers platelet activation and aggregation and mediates the prothrombotic role of thrombin in the blood. PAR3 is not present on human platelets, but is widely and abundantly expressed in other cell types (O'Brien et al. 2001). In the mouse, signaling in platelets is mediated entirely by PAR4, with PAR3 facilitating PAR4 cleavage at low thrombin concentrations (Kahn et al. 1998; Nakanishi-Matsui et al. 2000). The efficiency of the coagulation cascade depends on the balance between the procoagulant and anticoagulant pathways. Thrombin is the key arbiter of this balance by virtue of its dual role and has therefore received utmost attention in structure-function studies and as a target of anticoagulant therapy (Bates and Weitz 2006).
Figure 1.

Schematic representation of the multiple roles of thrombin in the blood and how Na+ binding influences them. Upon generation from the inactive zymogen prothrombin, thrombin partitions itself between a Na+-free slow form (40% of the population of molecules in vivo) and a Na+-bound fast form (60% of the population of molecules in vivo). The fast form is responsible for the efficient cleavage of fibrinogen leading to clot formation, and activation of factors V, VIII and XI that promote the progression of the coagulation response to vascular injury. The fast form is also responsible for the activation of PAR1, PAR3 and PAR4 leading to platelet activation and cell signaling. The slow form, on the other hand, activates efficiently the anticoagulant protein C with the assistance of the cofactor thrombomodulin. Na+ binding to thrombin is the major driving force behind the procoagulant, prothrombotic and signaling roles of the enzyme in the blood, but is not required for its anticoagulant role triggered by protein C activation. The anticoagulant function of thrombin depends on the interaction with thrombomodulin.
2. Thrombin and Na+
The most striking feature of thrombin is its ability to interact with Na+ and the ensuing effects on recognition of procoagulant (fibronogen), prothrombotic (PARs) and anticoagulant (protein C) substrates (Dang et al. 1995; Dang et al. 1997a; Di Cera et al. 2007). The Na+ effect on thrombin is best understood if cast in the larger context of enzyme activation by monovalent cations (M+s) (Di Cera 2006; Page and Di Cera 2006). Regulation of activity through metal ion complexation plays a key role in many enzyme catalyzed reactions (Di Cera 2006; Page and Di Cera 2006). Over one third of known proteins are metalloproteins (Ibers and Holm 1980; Tainer et al. 1992; Castagnetto et al. 2002). Conceptual associations with protein-metal complexes tend to favor divalent metals. Examples are Fe2+ involvement in redox cycles, Ca2+ in structural stability, or Zn2+ as electrophile in an enzyme catalyzed reaction. Significance of divalent metals in protein structure and function has been reviewed in detail (Armstrong 2000; Finney and O'Halloran 2003; Rizzuto and Pozzan 2006). Indeed, many bioinorganic chemistry textbooks are devoted to the description of divalent and polyvalent ions with monovalent cations (M+s) discussed only in the context of membrane potentials. However, Na+ is the most abundant metal in human plasma, the backbone of biological fluids, and its occurrence mirrors that found in environmental liquids (Table 1) (Page and Di Cera 2006). Human health is negatively influenced by excess sodium intake that may result in hypertension and other health problems (de Wardener 2001; Frohlich and Varagic 2004; Meneton et al. 2005), but other organisms have adapted to concentrations of Na+ far exceeding that of human tolerance. Sodium chloride lies at the heart of human biology and the roots of human civilization. Abundance or absence of this simple compound has had profound effects on human health and has provided a casus belli in many important milestones in the history of man (for an excellent historical account see (Kurlansky 2002)). Several initial forms of economics were based on salt rather than metal coinage and the words “salary” and “soldier” are derived from the Latin sal for salt, as is salus, the Latin word for “health”.
Table 1.
Inorganic ion content (mmol/L) of human plasma, intracellular cytosol, and seawater.
| Plasma | Cytosol | Seawater | |
|---|---|---|---|
| Na+ | 135–146 | 25–35 | 480 |
| Cl− | 98–108 | 50–60 | 559 |
| HCO3− | 23–31 | 4–12 | 2.0 |
| Mg2+ | 0.8–1.4 | 4–20 | 54.0 |
| K+ | 3.5–5.2 | 130–145 | 10.4 |
| Ca2+ | 2.1–2.7 | <0.01 | 10.6 |
| PO42− | 0.7–1.4 | 90–110 | <0.1 |
Earliest evidence for M+ activation of enzymes was provided by Boyer (Boyer et al. 1942) with the discovery of the absolute requirement of K+ by pyruvate kinase (Kachmar and Boyer 1953). Monod demonstrated Na+-dependent catalytic rate enhancement in β-galactosidase (Cohn and Monod 1951). Following these discoveries, many enzymes were observed to display increased activity in the presence of M+ (Suelter 1970). A recent classification of M+-activated enzymes groups them based on the selectivity of the effect, as established by kinetic studies, and the mechanism of activation, as shown from structural analysis (Di Cera 2006). The effect has exquisite specificity, with Na+ or K+ being the preferred M+ (Figure 2). In general, enzymes requiring K+ such as kinases and molecular chaperones are also activated by NH4+ and Rb+, but are not activated as well or at all by the larger cation Cs+ or the smaller cations Na+ and Li+. Enzymes requiring Na+ such as β-galactosidase and clotting proteases are not activated as well by Li+, or the larger cations K+, Rb+ and Cs+. Because the concentration of Na+ and K+ is tightly controlled in vivo, M+s do not function as regulators of enzyme activity. Rather, they provide a driving force for substrate binding and catalysis by lowering energy barriers in the ground and/or transition states. Enzymes activated by M+s evolved to take advantage of the large availability of Na+ outside the cell and K+ inside the cell (Table 1) to optimize their catalytic function. Indeed, a strong correlation exists between the preference for K+ or Na+ and the intracellular or extracellular localization of such enzymes. The mechanism of M+-activation can be established from crystal structures as cofactor-like or allosteric. In the former case, the M+ anchors the substrate to the active site of the enzyme, often acting in tandem with a divalent cation like Mg2+. In such mechanism of activation, Type I, the M+ is absolutely required for catalysis. In the latter, Type II, the M+ enhances enzyme activity through conformational transitions triggered upon binding to a site where the M+ makes no direct contact with substrate. In this case, that is most relevant to our discussion on thrombin, the M+ acts as an allosteric effector and is not absolutely required for catalysis.
Figure 2.

Enzyme activity in the presence of LiCl (gray), NaCl (white), KCl (black) or RbCl (hatched) for Hsc70 (O'Brien and McKay 1995) and thrombin (Prasad et al. 2004). Values refer to s=Kcat/Km of ATP hydrolysis for Hsc70 in the presence of 150 mM salt, relative to CsCl, or the hydrolysis of H-D-Phe-Pro-Arg-p-nitroanilide by thrombin in the presence of 200 mM salt, relative to choline chloride. The preference for K+ (Hsc70) or Na+ (thrombin) is evident from the plot.
Earlier kinetic studies on the hydrolysis of chromogenic substrates by Kosow revealed that thrombin is optimally active in the presence of Na+ (Orthner and Kosow 1980), an effect originally observed in factor Xa by the same group (Orthner and Kosow 1978) and in activated protein C by Castellino (Steiner et al. 1980; Steiner and Castellino 1982; 1985a; b). Curiously, the original observation on thrombin was at odds with the accepted view at the time that salts, and Na+ in particular, actually acted as inhibitors of thrombin hydrolysis of synthetic substrates (Curragh and Elmore 1964; Workman and Lundblad 1978). In fact, the inhibitory effect of Na+ was rationalized as a direct perturbation of the catalytic H57 that would reduce nucleophilicity and effectiveness of the charge relay system (Workman and Lundblad 1978). Wells and Di Cera demonstrated that the Na+ activation of thrombin is specific and allosteric (Wells and Di Cera 1992), as expected for a Na+-activated Type II enzyme (Di Cera 2006). The property is shared with other clotting factors and proteases involved in immune response (Dang and Di Cera 1996; Krem and Di Cera 2001; Di Cera 2006). Na+ binding converts thrombin from a low activity slow (Na+-free) to a high activity fast (Na+-bound) form (Wells and Di Cera 1992). The slow and fast forms are significantly (2:3 ratio) populated under physiologic conditions because the Kd for Na+ binding is 110 mM at 37 °C (Wells and Di Cera 1992; Guinto and Di Cera 1996; Griffon and Di Stasio 2001; Prasad et al. 2003; Bah et al. 2006) and the physiologic [NaCl] (140 mM) is not sufficient for saturation. Hence, the slow-fast equilibrium in vivo is optimally poised for allosteric regulation and this is all the more significant in view of the fact that the procoagulant and anticoagulant activities of thrombin are partitioned between the fast and slow form, respectively (Figure 1) (Dang et al. 1995). Na+ binding is required for optimal cleavage of fibrinogen (Dang et al. 1995; Dang et al. 1997a; Di Cera et al. 2007) and activation of factors V (Myles et al. 2001b), VIII (Nogami et al. 2005) and XI (Yun et al. 2003) necessary for the explosive generation of thrombin in the coagulation cascade (Mann 2003; Mann et al. 2003), but is dispensable for cleavage of protein C (Figure 3) (Dang et al. 1995; Dang et al. 1997a). This proves that Na+ is the major driving force behind the procoagulant role of thrombin in the blood (Di Cera et al. 2007). Na+ binding also promotes the prothrombotic and signaling functions of the enzyme by enhancing cleavage of PAR1, PAR3 and PAR4 (Di Cera et al. 1997; Ayala et al. 2001).
Figure 3.
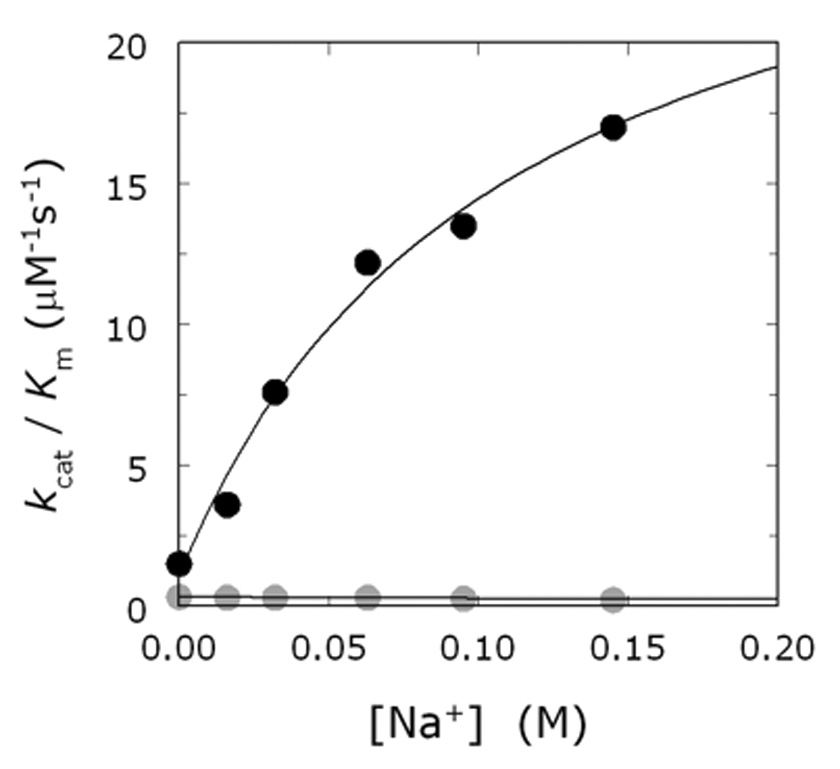
Effect of Na+ on the cleavage of fibrinogen (black circles) and protein C (gray circles) by thrombin, under experimental conditions of 5 mM Tris, 0.1% PEG, pH 7.4 at 37 °C. Values of Kcat/Km were measured as a function of [Na+] by keeping the ionic strength constant at 145 mM with choline chloride. The data for protein C were obtained in the presence of 5 mM CaCl2 and 100 nM human thrombomodulin. Note the difference in Na+ effect between the hydrolysis of the two physiologic substrates. Continuous lines were drawn according to eq 7b in the text with parameter values: s0=1.1±0.1 µM−1s−1, s1=29±3 µM−1s−1, KA=9.2±0.6 M−1 (black circles); s0=0.32±0.02 µM−1s−1, s1=0.20±0.01 µM−1s−1, KA=9.2±0.6 M−1 (gray circles). In both cases, the value of ω=1.
Due to the allosteric nature of thrombin, any effect that destabilizes Na+ binding stabilizes the slow form and produces an anticoagulant effect by prolonging the clotting time (reduced fibrinogen cleavage) and reducing platelet activation (reduced PAR1 cleavage). Several naturally occurring mutations of the prothrombin gene, like prothrombin Frankfurt (Degen et al. 1995), Salakta (Miyata et al. 1992), Greenville (Henriksen et al. 1998), Scranton (Sun et al. 2001), Copenhagen (Stanchev et al. 2006) and Saint Denis (Rouy et al. 2006) affect residues linked to Na+ binding (Pineda et al. 2004a) and are often associated with bleeding. Na+ itself can become the arbiter of the procoagulant/anticoagulant fate of thrombin. Although the Na+ concentration in the blood is tightly controlled in healthy individuals, hyponatremia (Na+<135 meq/L) (Adrogue and Madias 2000b) and hypernatremia (Na+>145 meq/L) (Adrogue and Madias 2000a) are among the most common electrolyte disorders encountered by primary care providers, nephrologists and pediatricians. Hypernatremia is often associated with venous thrombosis, especially of the cerebral vasculature (Goldman and Eckerling 1972) or of the lower extremities if secondary to diabetes (Adrogue and Madias 2000a). Infusion of hypertonic saline in healthy volunteers results in increased levels of fibrinopeptide A, a product of fibrinogen cleavage by hrombin, and a significant reduction in its generation time (Grant et al. 1985). The association of bleeding due to reduced clotting secondary to hyponatremia is more difficult to document, because one of the major causes of hyponatremia is subarachnoid emhorrage (Adrogue and Madias 2000b). However, some independent evidence of increased bleeding secondary to hyponatremia has been reported in infants (Rubin and Christian 2001). The differential effect of Na+ on fibrinogen and protein C cleavage has been the major driving force behind the rational design of anticoagulant thrombin mutants (Gibbs et al. 1995; Tsiang et al. 1996; Dang et al. 1997a; Cantwell and Di Cera 2000; Gruber et al. 2002).
3. Thrombin structure
Activated forms of blood coagulation proteases bear the chymotrypsin-like protein fold where two six-stranded β-barrels come together asymmetrically to host at their interface the residues of the catalytic triad, H57, D102 and S195 (Page and Di Cera 2008). Two residues of the triad are donated from the N-terminal β-barrel with the nucleophilic Ser and oxyanion hole generated from the C-terminal β-barrel. Thrombin is composed of two polypeptide chains of 36 (A chain) and 259 (B chain) residues that are covalently linked through a disulfide bond between residues C1 and C122 (Figure 4) (Bode et al. 1992; Bode 2006). The standard “Bode” orientation (Bode et al. 1992) puts the A chain in the back of the molecule, opposite to the front hemisphere of the B chain that hosts the entrance to the active site and all known functional epitopes of the enzyme (Pineda et al. 2004a). The A chain has received little attention in thrombin studies and is considered an appendage of the activation process from prothrombin. Previous studies have suggested that the A chain may be dispensable for function (Hageman et al. 1975; DiBella et al. 1995). However, several naturally occurring mutations of prothrombin involve residues of the A chain (Akhavan et al. 1999; Akhavan et al. 2000; Lefkowitz et al. 2000; Sun et al. 2000) and are associated with severe bleeding (Figure 5). The functional defects in prothrombins Denver (E8K and E14cK) (Lefkowitz et al. 2000), Segovia (G14mR) (Akhavan et al. 1999) and San Antonio (R15H) (Sun et al. 2000) have been attributed to perturbation of the zymogen→enzyme conversion and processing by factor Xa, resulting in severe bleeding. Such explanation is obvious for the G14mR and R15H mutations that affect the P1 (R15) and P2 (G14m) sites of recognition by factor Xa, but not for the E8K and E14cK mutations of prothrombins Denver. Other naturally mutations, like deletion of K9 or K10 (Akhavan et al. 2000), are also associated with severe bleeding. Interestingly, the defect causes impaired fibrinogen and PAR1 cleavage, reduced response to Na+ activation (De Cristofaro et al. 2004; De Cristofaro et al. 2006), and long-range perturbation of active site residues (De Cristofaro et al. 2006). The A chain is rich in charged residues that make polar interactions with partners of the B chain. Importantly, a significant number of negatively charged residues cluster toward the C-terminus, in close proximity to the Na+ site (Figure 4 and Figure 5). These charged residues may influence Na+ binding and/or allosteric transduction. The A chain is also optimally shaped to provide communication between the Na+ site and the back of the active site region (Figure 4) and could therefore influence substrate recognition and catalysis. The A chain is stabilized by the D1a-K9 and R14d-E13 ion-pairs and the R4-E8-D14-E14c ion cluster (Figure 5). Disruption of any such interactions may produce effects that ultimately influence the interaction with the B chain and its properties. The natural mutation of thrombin where K9 is deleted results in significant loss of clotting activity and Na+ effect (De Cristofaro et al. 2004). Perturbation of any residue in the ion cluster R4-E8-D14-E14c is expected to impact significantly thrombin function, also in view of the involvement of residues from the B chain. Prothrombin Denver carries two charge reversal substitutions for E8 and E14c and results in severe bleeding (Lefkowitz et al. 2000). The A chain contributes to stabilization of the B chain through numerous ionic interactions (Figure 5). There is a conspicuous polarity in the interface, with the A chain contributing several acidic residues that neutralize basic residues from the B chain. Surface interactions D1a-R206 and K14a-E23 may be energetically dispensable. On the other hand, interactions of the ion cluster with R137 and K202 are less exposed to solvent and may be quite relevant. Disruption of these interchain interactions may affect the conformation of the B chain and active site residues. Particularly interesting is the interaction of E14e with K186d and the hydroxyl group of Y184a, which could have significant consequences on Na+ binding. The van der Waals contacts of R4 with W29 and W207 are also noteworthy in view of the long-range communication existing between these Trp residues and the Na+ site, as recently documented by stopped-flow fluorescence measurements (Bah et al. 2006). A close inspection of the crystal structure reveals that W29 and W207 are members of a hydrophobic cluster stretching ~12 Å within the core of the enzyme. The two Trp residues are in van der Waals interaction with each other and R4 on one side and with V200 and P198 on the opposite side (Figure 6). P198 likely controls the backbone conformation of the entire sequence from the catalytic S195 to F199, via the highly flexible G196–G197 linker. Hence, changes originating at R4, can propagate to P198 via the W29-W207-V200-P198 hydrophobic cluster and then influence the orientation of S195 in the active site. P198 can also influence the orientation of F199, which is in van der Waals interaction with F181 near the Na+ site and Y228 near the primary specificity site around D189. Hence, mutations of R4 or other residues of the ion cluster R4-E8-D14-E14c may result in long-range effects on the active site, the primary specificity pocket and the Na+ site. The contribution of W207 and W29 to the fluorescence change induced by Na+ binding discovered recently (Bah et al. 2006) directly support the importance of this long-range communication. The hydrophobic W29-W207-V200-P198 cluster is in all likelihood a critical determinant of thrombin allostery and holds precious information on how the enzyme functions at the molecular level.
Figure 4.

Structure of thrombin bound to the active site inhibitor PPACK (stick model) and Na+ (yellow ball). The A chain (green) runs in the back of the B chain (cyan). Disulfide bonds are in orange and are numbered 1 (C1–C122), 2 (C48–C52), 3 (C168–C182), 4 (C191–C220). Relevant domains are noted. Catalytic residues (H57, D102, S195) are marked by *, and D189 is labeled. The bound Na+ is nestled between the 220-loop and the 186-loop and is within 5 Å from the side chain of D189. Numbering refers to chymotrypsin(ogen). Insertions relative to chymotrypsin are denoted by a letter in lower case following the residue number (e.g., R221a) to avoid confusion with single-site mutations. Note the position of the C-terminus of the A chain near the back of the Na+ site and the three disulfide bonds in the B chain connecting strands of the Na+ site, the primary specificity pocket and the active site.
Figure 5.
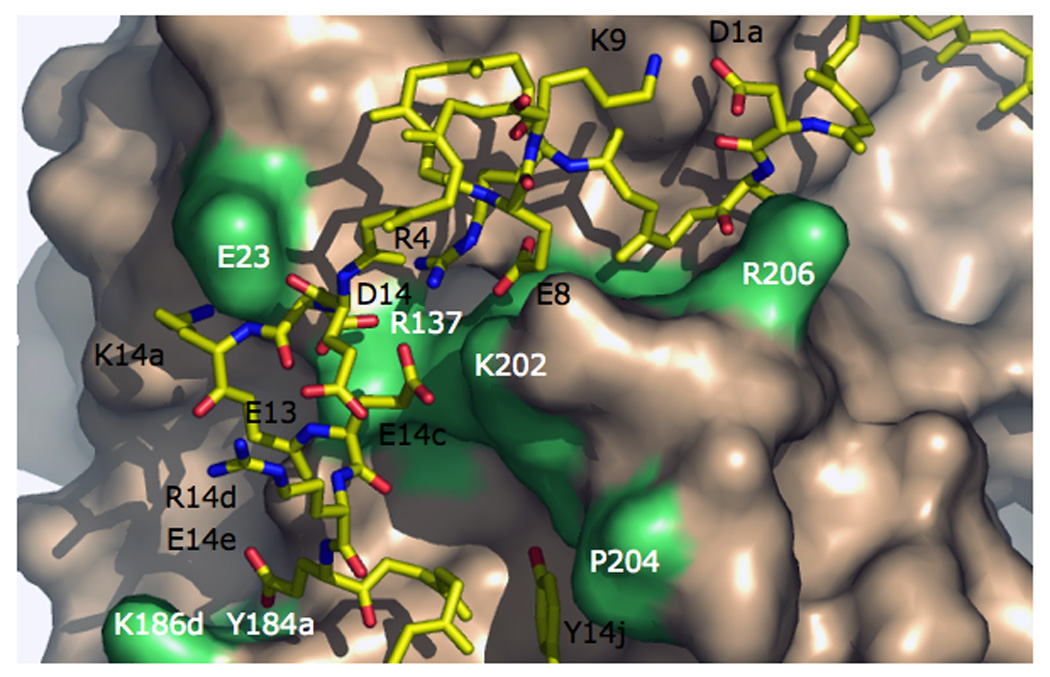
Interactions between the A chain (stick model, residues labeled in black) and B chain (wheat surface, residues labeled in white and shown in lime) of thrombin. The A chain is stabilized by the D1a-K9 and R14d-E13 ion-pairs and the R4-E8-D14-E14c ion cluster. The interaction between the A and B chain depends on the C1–C122 bond (hidden behind R206), the ionic interactions D1a-R206, E8-E14c-K202, D14-R137, K14a-E23 and E14e-K186d-Y184a, and the hydrophobic stacking Y14j-P204. Some H-bonds are omitted for clarity. The Na+ site is located below the surface of K186d and Y184a.
Figure 6.

Cross section of the thrombin structure showing the communication between R4 and important regions of the enzyme. The side chain of R4, stabilized by the polar contacts with E8 and E14c, is in van der Waals interaction with W29 (3.8 Å) and W207 (3.6 Å), which in turn are in hydrophobic contact with V200 (3.5 and 4.1 Å, respectively). V200 and W29 contact P198 (3.9 and 4.0 Å, respectively), which likely controls the backbone orientation of the entire sequence from S195 in the active site to F199 through the highly flexible G196–G197 linker. The benzene ring of F199 is in van der Waals interaction with F181 (4.0 Å) and Y228 (4.0 Å). This network of hydrophobic interactions enables R4 and the R4-E8-D14-E14c ion cluster to communicate long-range with the catalytic S195 (via P198), the primary specificity pocket (via Y228, D189 is right below it) and the Na+ site (via F181, the bound Na+ is in the cavity at the bottom right corner). The pivotal connections between R4 and the network are W29 and W207, whose conformation is under the influence of Na+ binding(Bah et al. 2006).
Trypsin-like specificity for Arg residues at P1 (Schechter and Berger 1967) is conferred to thrombin by the presence of D189 in the S1 site occupying the bottom of the catalytic pocket. Unlike trypsin, however, thrombin can efficiently cleave chymotrypsin specific substrates carrying Phe at the P1 position (Bush et al. 2006), as documented more than 40 years ago from studies on ester substrates (Martin et al. 1959; Lorand et al. 1962; Kezdy et al. 1965). This peculiar property of thrombin has obviously been overlooked when devising “general” rules for the conversion of specificity in serine proteases (Hedstrom et al. 1992; Perona and Craik 1995; Hedstrom 2002). Thrombin has a preference for small and hydrophobic side chains at P2 that pack tightly against the hydrophobic wall of the S2 site defined by residues Y60a-P60b-P60c-W60d of the 60-loop. Residues at P3 point away from the thrombin surface, whereas aromatic and hydrophobic residues at P4 tend to fold back on the thrombin surface (Cleary et al. 2002) and engage the aryl binding site defined by L99, I174 and W215 (Bode et al. 1992). The autolysis loop shapes the lower rim of access to the active site and contributes to recognition of fibrinogen (Dang et al. 1997b). The loop centered on K70 defines exosite I and is homologous to the Ca2+ binding loop of trypsin and chymotrypsin (Bartunik et al. 1989). In these proteases, Ca2+ stabilizes the fold and confers increased resistance to proteolytic digestion. In thrombin, the need for Ca2+ is eliminated by insertion of K70 in the cavity available for binding this cation. Thrombin does not bind Ca2+ up to mM concentrations (Dang et al. 1995; Yang et al. 2004). Exosite I contains several positively charged residues that give rise to an intense electrostatic field. The field provides steering and optimal pre-orientation for fibrinogen, thrombomodulin, the natural inhibitor hirudin and PAR1 to facilitate formation of a productive complex upon binding. Structural and site-directed mutagenesis data support exosite I as a binding epitope for fibrinogen (Tsiang et al. 1995; Ayala et al. 2001; Pechik et al. 2004), fibrin (Ayala et al. 2001; Pechik et al. 2006), thrombomodulin (Tsiang et al. 1995; Hall et al. 1999; Fuentes-Prior et al. 2000; Pineda et al. 2002a; Xu et al. 2005), and the thrombin receptors PAR1 (Vu et al. 1991; Ayala et al. 2001; Myles et al. 2001a) and PAR3 (Ishihara et al. 1997; Ayala et al. 2001). On the side of the enzyme opposite to exosite I, a C-terminal helix and its neighbor domains host a number of positively charged residues and define exosite II. This site is the locale for interaction with polyanionic ligands like heparin and glucosaminoglycans (Gan et al. 1994; Sheehan and Sadler 1994; Tsiang et al. 1997; Li et al. 2004; Carter et al. 2005). Heparin enhances inhibition of thrombin by antithrombin via a template mechanism in which a high affinity heparin-antithrombin complex is first formed and then docked into exosite II and the thrombin active site by electrostatic coupling (Gettins 2002; Olson and Chuang 2002; Dementiev et al. 2004; Li et al. 2004). Exosite II is also the locale for thrombin interaction with the platelet receptor GpIb (De Cristofaro et al. 2000; Ramakrishnan et al. 2001; Celikel et al. 2003; Dumas et al. 2003), the acidic moiety of the fibrinogen γ’ chain (Pineda et al. 2007) and has been involved in the binding of autoantibodies (Colwell et al. 1998).
The first X-ray structure of thrombin was solved in 1992 and revealed relevant information on the overall fold of the enzyme and especially on the arrangement of loops involved in macromolecular substrate recognition (Bode et al. 1992). However, this and many subsequent structures of thrombin completely overlooked the bound Na+, although crystals were solved at high resolution and in the presence of high concentrations of Na+. The Na+ binding site of thrombin was first identified crystallographically in 1995 from Rb+ replacement (Di Cera et al. 1995). Notably, this was also the first Na+ binding site identified in the large family of M+-activated enzymes to which thrombin belongs (Di Cera 2006). Na+ binds 16–20 Å away from residues of the catalytic triad and within 5 Å from D189 in the S1 site (Figure 7), nestled between the 220- and 186- loops and coordinated octahedrally by two carbonyl O atoms from the protein (residues R221a and K224) and four buried water molecules anchored to the side chains of D189, D221 and the backbone atoms of G223 and Y184a. The site is highly specific for Na+ that binds with significantly (>10-fold) higher affinity compared with Li+, K+ or Rb+ (Prasad et al. 2003). A Na+ binding site analogous to that of thrombin has been identified in factor Xa (Zhang and Tulinsky 1997; Scharer et al. 2005), factor VIIa (Bajaj et al. 2006) and activated protein C (Schmidt et al. 2002) and has been postulated for factor IXa by homology modeling (Schmidt et al. 2005). These enzymes are Na+-activated like thrombin based on the nature of residue 225 (Dang and Di Cera 1996). Several groups have shown that Na+ has a significant influence on the activity of factors VIIa (Dang and Di Cera 1996; Petrovan and Ruf 2000), IXa (Schmidt et al. 2005), Xa (Orthner and Kosow 1978; Monnaie et al. 2000; Rezaie and He 2000; Underwood et al. 2000; Camire 2002; Rezaie and Kittur 2004; Levigne et al. 2007) and activated protein C (Steiner et al. 1980; Steiner and Castellino 1982; 1985a; b; He and Rezaie 1999). Also, in factors IXa (Schmidt et al. 2005), Xa (Monnaie et al. 2000; Rezaie and He 2000; Underwood et al. 2000; Camire 2002; Rezaie and Kittur 2004; Levigne et al. 2007) and activated protein C (He and Rezaie 1999) the binding of Na+ influences the primary specificity pocket and is linked to the binding of Ca2+ to the 70-loop. The physiologic role of Na+ in these enzymes, however, remains unclear. Na+ affects only slightly the activity of the prothrombinase complex (Rezaie and He 2000), has a small (Schmidt et al. 2005) or negligible (Gopalakrishna and Rezaie 2006) effect on the intrinsic Xase complex and no effect on the extrinsic Xase complex (Gopalakrishna and Rezaie 2006).
Figure 7.

The Na+ binding site of thrombin. Shown are substrate (CPK, C in yellow), relevant residues (CPK, C in cyan), and Na+ (yellow sphere). Na+ binding orients the critical D189 for correct engagement of the substrate Arg side chain.
Structural biology has revealed important information on how thrombin utilizes both the active site and exosites for interaction with substrates, inhibitors and effectors. Information on how thrombin recognizes substrate at the active site has come from the structure of the enzyme in complex with the irreversible active site inhibitor H-D-Phe-Pro-Arg-CH2Cl (PPACK) (Bode et al. 1992). Arg at P1 ion-pairs to D189 in the S1 site, Pro at P2 fits snugly against P60b, P60c and W60d in the S2 site and Phe at P3, in the D-enantiomer, makes an edge-to-face interaction with W215 in the aryl binding site. The PPACK-inhibited structure reveals interactions that are relevant to recognition of natural substrates and confirms the key role played by the H-bonding network found within the active site of all trypsin-like enzymes bound to substrate (Perona and Craik 1995; 1997; Hedstrom 2002). Crucial components of this network are the bidentate ion-pair between D189 and the guanidinium group of Arg at P1, the H-bonds of the carbonyl O atom of the P1 residue with the N atoms of G193 and S195 forming the oxyanion hole, the H-bond between the N atom of the P1 residue and the carbonyl O atom of S214, and the H-bonds between the backbone O and N atoms of the P3 residue with the N and O atoms of G216. This important arrangement of H-bonds has been documented in the structures of thrombin bound to fragments of the natural substrates fibrinogen (Stubbs et al. 1992), PAR4 (Bah et al. 2007) and factor XIII (Sadasivan and Yee 2000). The structure of thrombin in complex with the potent natural inhibitor hirudin has revealed how thrombin recognizes ligands at exosite I (Rydel et al. 1991). Hirudin blocks access to the active site of thrombin using its compact N-terminal domain and binds to exosite I via its extended, acidic C-terminal domain. The mode of interaction of the C-terminal domain of hirudin has later been documented in the structures of thrombin bound to hirugen (Vijayalakshmi et al. 1994), fibrinogen (Rose and Di Cera 2002; Pechik et al. 2004; Pechik et al. 2006), PAR1 (Mathews et al. 1994), PAR3 (Bah et al. 2007), thrombomodulin (Fuentes-Prior et al. 2000) and heparin cofactor II (Baglin et al. 2002). Finally, the role of exosite II has been documented eloquently in the structures of thrombin bound to heparin (Dementiev et al. 2004; Li et al. 2004; Carter et al. 2005), the fibrinogen γ’ peptide (Pineda et al. 2007) and GpIb (Celikel et al. 2003; Dumas et al. 2003).
4. Kinetics of Na+ activation
The discovery of the Na+ effect on thrombin has provided a coherent framework to understand structure and function of the enzyme, rationalized the molecular origin of the defects associated with several naturally occurring mutations of the prothrombin gene and offered an effective strategy to engineer thrombin for optimal anticoagulant activity in vivo which may one day translate into new therapeutic tools. In view of the importance of the thrombin-Na+ interaction, below we will devote much attention to its kinetic and energetic aspects. In the study of enzyme activation by M+s, attention is often focused on the effect of M+ on the velocity of substrate hydrolysis. The activating effect of M+ binding is readily observed as an increase in reaction velocity as a function of [M+]. Specificity is detected in this assay by comparing the velocity among different M+s at the same concentration (Figure 2). Quantitative information about the energetics and mechanism of M+ requires measurements of the independent Michaelis-Menten parameters kcat and s=kcat/Km. These parameters can be derived from a plot of the velocity of product generation expressed in units of active enzyme, υ /etot, vs the substrate concentration, [S]. In this plot, the value of kcat is the asymptotic value of υ /etot as [S]→∞, and the value of s is the initial slope as [S]→0. It should be noted that Km, the concentration of substrate giving half of the maximal velocity, is not an independent parameter because its definition requires knowledge of the value of kcat. The values of s and kcat, on the other hand, can be defined independently of each other.
Scheme 1 describes Na+ activation in thrombin and is the familiar Botts-Morales scheme for the action of a modifier on substrate hydrolysis (Botts and Morales 1953; Di Cera et al. 2007). The Scheme also applies to other allosteric effectors of thrombin, like thrombomodulin (Di Cera et al. 1996) or PAR3 (Bah et al. 2007). The enzyme is assumed to exist in two forms, one free (E) and the other bound to Na+ (EM), featuring different values of kinetic rate constants for binding (k1,0, k1,1), dissociation (k−1,0, k−1,1) and hydrolysis (k2,0, k2,1) of substrate S into product P. The parameters KA=kA/k−A and are the equilibrium association constants for Na+ binding to E and ES, respectively. Detailed balance imposes a constraint among the rate constants in Scheme 1, i.e., . The analytical solution for the velocity of product formation at steady state can be found with the Hill diagram method (Hill 1977). Scheme 1 contains four species, of which only three are independent because of mass conservation. Hence, each trajectory toward a species must contain the product of three rate constants as shown in Figure 8. The sum of the trajectories toward each species defines the contribution of that species at steady state. The expression for the velocity of product formation at steady state is therefore (Page and Di Cera 2006; Di Cera et al. 2007)
| (1) |
where etot is the total concentration of active enzyme and ΣEX is the sum of the trajectories toward species EX (EX=any of the four enzyme species in scheme 1) in Figure 8. An important and often overlooked property of eq 1 is that the velocity of product formation for Scheme 1 is quadratic in [S], although the enzyme contains only a single site for S. This is a consequence of the difference in which terms are calculated for equilibrium and steady state systems (Botts and Morales 1953; Hill 1977; Di Cera 1995; Di Cera et al. 1996). The non-trivial consequence of the form of eq 1 is that, under the influence of Na+ or any allosteric effector, an enzyme like thrombin containing a single active site could in principle display an apparent cooperativity in substrate binding and hydrolysis reflected in the sigmoidal shape of the velocity of product formation as a function of [S]. Glucokinase is a relevant example of such behavior (Kamata et al. 2004). Physically, this effect can be understood as a slow isomerization of the enzyme forms E and EM that takes place after a significant amount of substrate is added to the system. By “slow” we mean an E-EM interconversion that takes place on a time scale slower than that observed in the conversion between free and substrate bound forms of the enzyme. At low substrate concentration, the EM form is low populated and the enzyme binds and hydrolyzes substrate in the low activity E form. As substrate is added, the enzyme slowly converts to the high activity EM form. When E and EM interconvert rapidly, cooperativity is not seen in the velocity curve because the ensemble of enzyme forms acts as a mixture in equilibrium. The coefficients in eq 1 can be found by application of the Hill diagram method (Hill 1977) in Figure 8 as follows (Page and Di Cera 2006)
| (2a) |
| (2b) |
| (2c) |
| (2d) |
| (2e) |
where . As with eq 1, the coefficients contain terms quadratic in [Na+]=x although the enzyme has a single binding site for Na+. The independent parameters kcat and s can be derived readily from eq 1 as
| (3a) |
| (3b) |
Scheme 1.
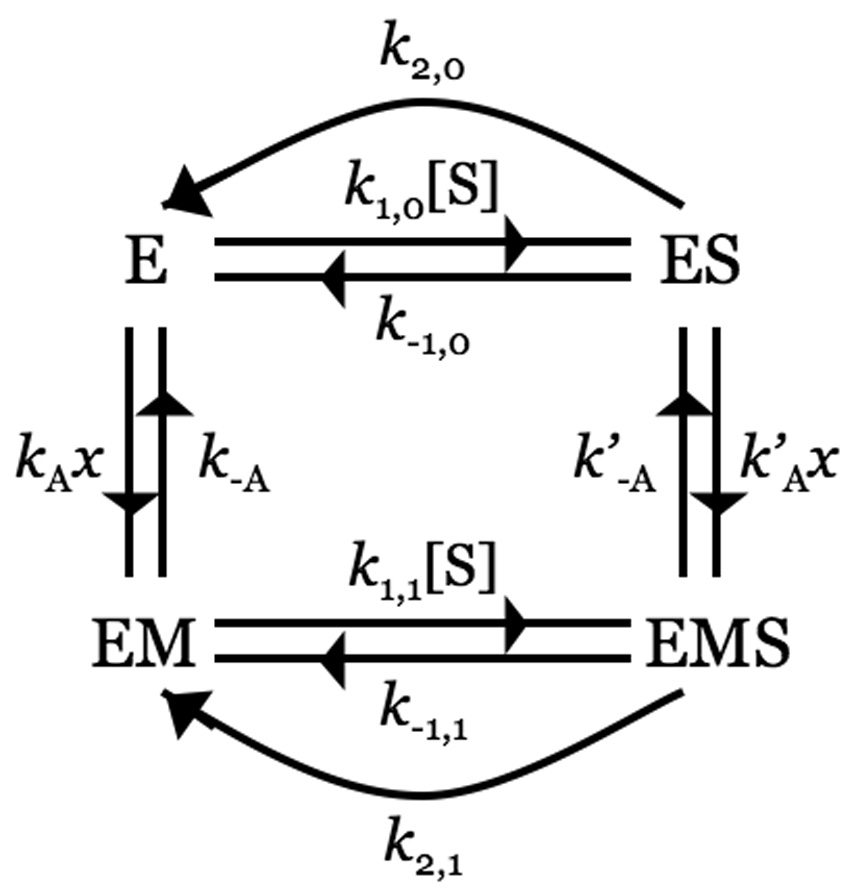
Figure 8.

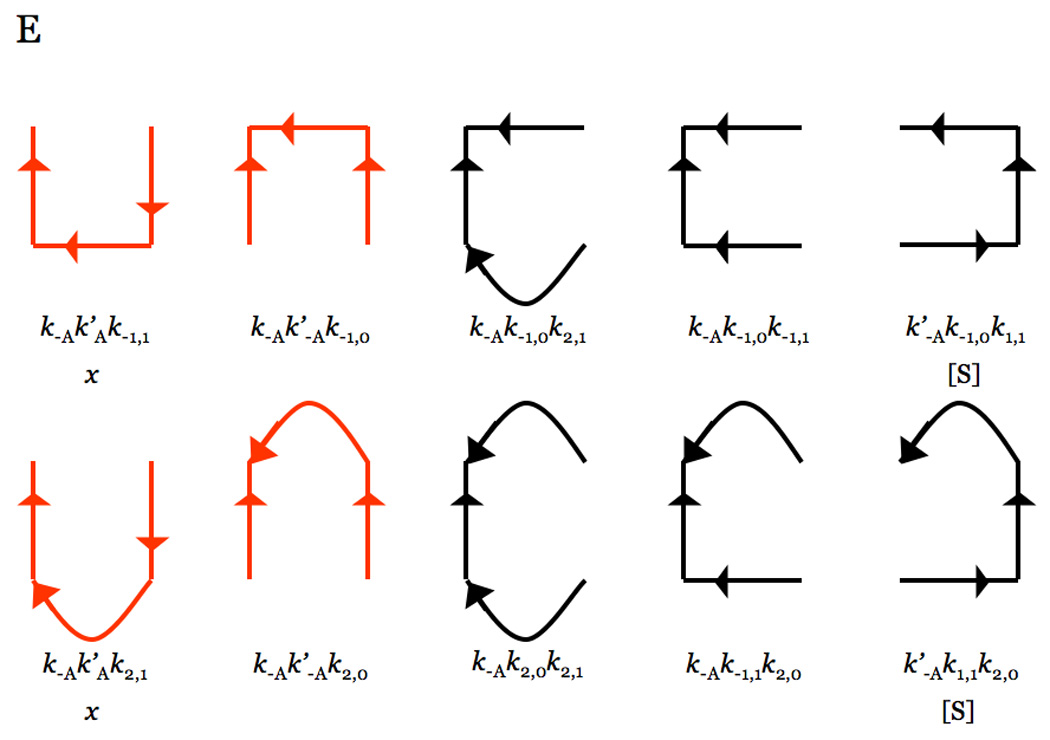
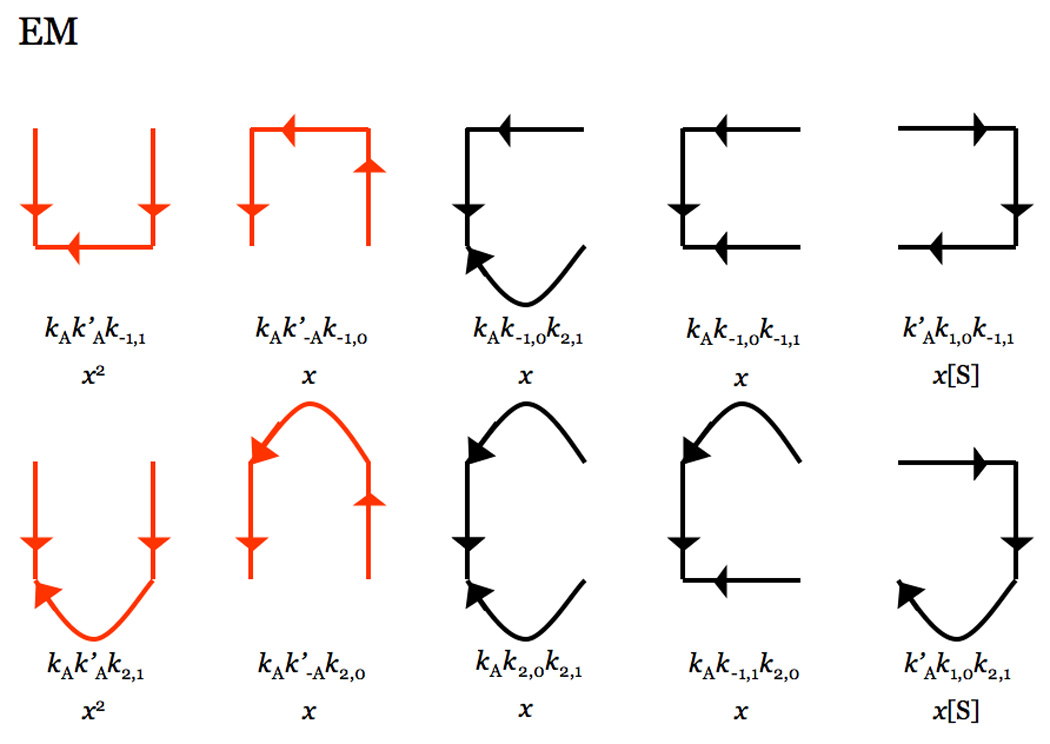

Hill diagrams depicting the trajectories toward each of the four species in the kinetic Scheme 1. Each trajectory contains the product of three rate constants in Scheme 1, because of the four species only three are independent due to mass conservation. Curved lines depict the irreversible reactions of product formation with rate constants k2,0 and k2,1 (see Scheme 1). Trajectories in red dominate under conditions where the rates of binding and dissociation of Na+ are fast compared to all other rates. Combination of all trajectories gives the expressions for the coefficients in eq 2aeq 2e.
Three independent parameters k2,0, k2,1 and K’A are resolved from measurements of kcat as a function of x, from which the binding affinity of Na+ for the ES complex can be measured directly. On the other hand, measurements of s as a function of x only resolve two parameters because of the form of eq 3b. These parameters are , obtained respectively as the values of s in the absence or presence of saturating concentrations of Na+. Resolution of KA, the important parameter measuring the binding affinity of Na+ for the free enzyme, is complicated by the additional term (ω−1)Λ that contains the independent parameters . When (ω−1)Λ makes only a small contribution to the value of s, KA can be estimated from the value of x at the midpoint of the transition of s from s0 to s1.
The velocity of product formation, eq 1, can be written in a form where its connection with the classical Michaelis-Menten equation is more readily appreciated
| (4a) |
or,
| (4b) |
The additional term, (ω−1)Θ, vanishes when ω=1 or Θ=0. The former condition was already identified by Botts and Morales and corresponds to the cases where substrate binds and dissociate rapidly from the enzyme, i.e., k−1,1>>k2,1 and k−1,0>>k2,0, or in the interesting but rather peculiar circumstance where . The expression for Θ is
| (5) |
and is a cumbersome function of [S] and rate constants of little practical utility. The value of Θ=0 is obtained when binding and dissociation of Na+ are fast compared to all other rates. In this case, the red trajectories in Figure 8 dominate the definitions of the coefficients 2a–e and eq 1 reduces to the familiar Michaelis-Menten form
| (6) |
with
| (7a) |
| (7b) |
The expression for kcat is not affected by the drastic change in the form of υ because it depends solely on the properties of the ES and EMS species. On the other hand, the form of s simplifies slightly but leaves resolution of KA difficult even in the presence of Michaelis-Menten kinetics. Measurements of this important parameter must be carried out by means of other techniques. Alternatively, the value of ω and k1,1/k1,0 must be resolved from independent measurements of the individual kinetic rate constants (Krem et al. 2002; Bobofchak et al. 2005).
Scheme 1 in its general form applies to Type II activation, i.e., when M+ acts as an allosteric effector that promotes substrate binding and catalysis by changing the conformation of the enzyme (Di Cera 2006). In this case, the steady state velocity of substrate hydrolysis should be measured accurately to confirm Michaelis-Menten kinetics. Departure from such behavior is indicative of binding and dissociation of M+ that take place on the same or slower time scale as substrate binding and dissociation. That could well be the case for other allosteric effectors of thrombin, like thrombomodulin and heparin, for which rapid equilibrium is always assumed for simplicity but remains to be proved experimentally. If the enzyme obeys Michaelis-Menten kinetics over a wide range of solution conditions, then the likely explanation is that binding and dissociation of M+ are fast compared to all other rates. This is indeed the case for Na+ binding to thrombin (Bah et al. 2006; Gianni et al. 2007). Measurements of kcat and s as a function of [Na+] reveal a significant hyperbolic increase in both parameters from finite low values, k2,0 and s0, to higher values, k2,1 and s1 (Figure 9). The midpoint of the transition in the kcat vs [Na+] plot yields K’A, the equilibrium association constant for Na+ binding to the enzyme-substrate complex. The midpoint of the transition in the s vs [Na+] plot yields an approximate measure of KA, the equilibrium association constant for Na+ binding to the free enzyme. The dependence of kcat on [Na+] unequivocally proves the existence of two alternative conformations in equilibrium, both displaying finite catalytic activity (Carrell et al. 2006). However, resolution of the important parameter KA is complicated even when the enzyme obeys Michaelis-Menten kinetics and must rely on other approaches (see below).
Figure 9.

Na+ dependence of the kinetic constants s=Kcat/Km (left) and Kcat (right) for the hydrolysis of H-D-Phe-Pro-Arg-p-nitroanilide (FPR) by thrombin. Experimental conditions are: 50 mM Tris, 0.1% PEG, pH 8.0 at 25 °C. The [Na+] was changed by keeping the ionic strength constant at 400 mM with choline chloride. The data illustrate the signatures of Type II activation with both s and Kcat showing a marked Na+ dependence and changing from low, finite values, to significantly higher values. Curves were drawn using equation 7a equation 7b, with best-fit parameter values: (data at left) s0=2.3±0.1 µM−1s−1, s1=99±3 µM−1s−1, KA=38±1 M−1; (data at right) k2,0=4.7±0.2 s−1, k2,1=78±2 s−1, KA’=45±2 M−1. Also shown is the contribution of the additional term (ω−1)Λ in eq 7b (gray line, left) calculated from the reported values of kinetic rate constants (Krem et al. 2002). This term makes at most a 3% correction at low [Na+].
It should be pointed out that the effect of Na+ depends entirely on the nature of the substrate used. Most parameters defining s and kcat pertain to properties of the enzyme-substrate complex and therefore their dependence on [Na+] is expected to change with different substrates. We have already shown the basic and physiologically important differences of the Na+ effect on the cleavage of fibrinogen and protein C (Figure 3). The origin of Na+ activation must be understood in terms of two components: the binding of Na+ to its site and the transduction of this event into changes of the catalytic properties of the enzyme. The first component is a property of the free enzyme, independent of any substrate. The second component, on the other hand, depends specifically on the particular enzyme-substrate complex under consideration. Structural changes produced by Na+ binding that result into enhanced binding and/or catalysis of fibrinogen may not be as beneficial for the binding and/or catalysis of protein C. These observations provides important clues on how Na+ binding to thrombin was fine tuned during evolution to optimize cleavage of substrates along the procoagulant/prothrombotic/signaling pathways, with no effect on the cleavage and activation of the anticoagulant protein C. We analyse the two components of Na+ activation in greater detail below.
5. Kinetic mechanism of Na+ binding: the E*, E and E:Na+ forms
Stopped-flow fluorescence measurements yield direct determinations of the parameter KA that is difficult to resolve from kinetic investigation of substrate hydrolysis at steady state and reveal the precise mechanism of binding. Na+ binding to thrombin is linked to a significant increase in intrinsic fluorescence (Wells and Di Cera 1992; Ayala and Di Cera 1994; Griffon and Di Stasio 2001; Prasad et al. 2003; De Filippis et al. 2005). The fluorescence increase has an initial rapid phase that cannot be resolved within the dead time (<0.5 ms) of the spectrometer, followed by a single exponential slow phase with a kobs that decreases as [Na+] increases (Bah et al. 2006) (Figure 10 and Figure 11). The rapid phase, resolved with a continuous-flow apparatus, yields a single exponential time course with a kobs that increases linearly with [Na+] (Figure 10 and Figure 11) (Gianni et al. 2007). The two-step mechanism of Na+ binding to thrombin solved by combination of the stopped-slow and continuous-flow measurements is then
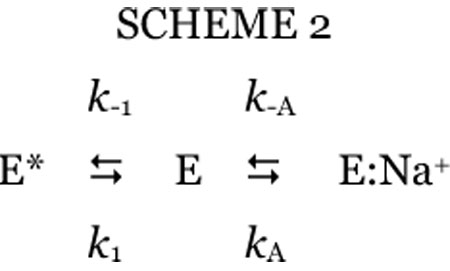 This is the mechanism recently proposed by Bah et al. (Bah et al. 2006), but validated for the binding/dissociation components of Na+ by direct resolution of the rapid phase. Thrombin exists in equilibrium between two forms, E* and E, that interconvert with kinetic rate constants k1 and k−1. Of these forms, only E can interact with Na+ with a rate constant kA to populate E:Na+, that may dissociate into the parent components with a rate constant k−A. The fast phase is due to the E-E:Na+ interconversion involving Na+ binding/dissociation, and the slow phase is due to the E-E* interconversion that precedes Na+ binding. The exact analytical solution of scheme 2 calls for two eigenvalues that give the kobs for the two exponential transitions reported in Figure 10. Separation of time scales for the E-E* (slow) and E-E:Na+ (fast) interconversions simplifies the eigenvalues to (Gianni et al. 2007)
This is the mechanism recently proposed by Bah et al. (Bah et al. 2006), but validated for the binding/dissociation components of Na+ by direct resolution of the rapid phase. Thrombin exists in equilibrium between two forms, E* and E, that interconvert with kinetic rate constants k1 and k−1. Of these forms, only E can interact with Na+ with a rate constant kA to populate E:Na+, that may dissociate into the parent components with a rate constant k−A. The fast phase is due to the E-E:Na+ interconversion involving Na+ binding/dissociation, and the slow phase is due to the E-E* interconversion that precedes Na+ binding. The exact analytical solution of scheme 2 calls for two eigenvalues that give the kobs for the two exponential transitions reported in Figure 10. Separation of time scales for the E-E* (slow) and E-E:Na+ (fast) interconversions simplifies the eigenvalues to (Gianni et al. 2007)
| (8) |
| (9) |
where KA=kA/k−A. The three species in scheme 2 portray thrombin in the Na+-free (E and E*) and Na+-bound (E:Na+) forms. The activation effect of Na+ on thrombin has very clear kinetic signatures (Figure 9) and specifically involves an increase in kcat and a decrease in Km (Orthner and Kosow 1980; Wells and Di Cera 1992; Vindigni and Di Cera 1996; Ayala and Di Cera 2000; Krem et al. 2002; Carrell et al. 2006). Such a “modifier” effect on kcat has long been known to be of diagnostic value (Botts and Morales 1953) and unequivocally proves the existence of two active forms in equilibrium, one Na+-free with low kcat and one Na+-bound with high kcat (Carrell et al. 2006; Page and Di Cera 2006). E and E:Na+ in scheme 2 are the two active forms of thrombin that account for the dependence of Kcat on [Na+] and correspond to the slow (E) and fast (E:Na+) forms originally defined by Wells and Di Cera (Wells and Di Cera 1992).
Figure 10.
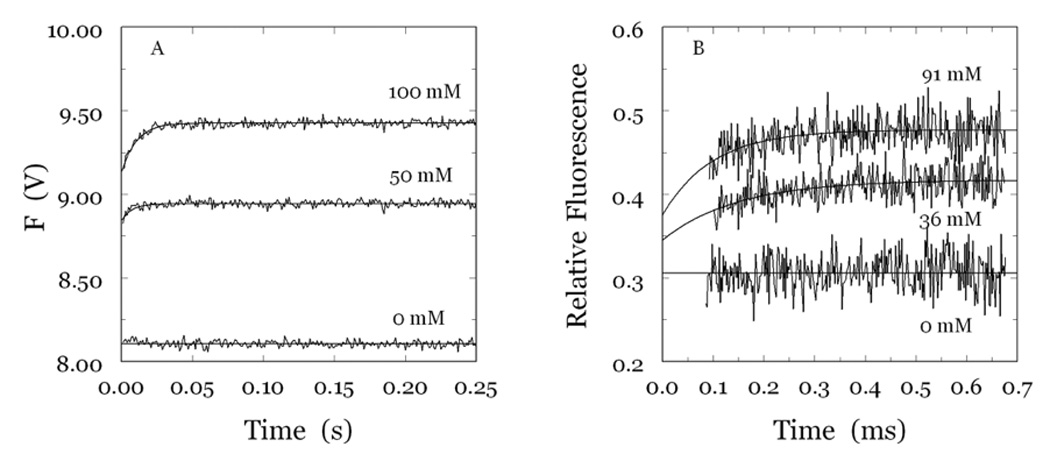
A,B. (A) Kinetic traces of Na+ binding to human thrombin in the 0–250 ms time scale. Shown are the traces obtained at 50 and 100 mM Na+ with the stopped-flow method using an Applied Photophysics SX20 spectrometer, with an excitation of 280 nm and a cutoff filter at 305 nm (Bah et al. 2006). Traces are averages of three determinations. Binding of Na+ obeys a two-step mechanism, with a fast phase completed within the dead time (<0.5 ms) of the spectrometer, followed by a single-exponential slow phase. The kobs for the slow phase decreases with increasing [Na+] (Figure 11A). (B) Kinetic traces of Na+ binding to human thrombin in the 0–700 µs time scale. Shown are the traces obtained at 36 and 91 mM Na+ with the continuous-flow method as single determinations with an exposure time of 3 s. Binding of Na+ in the 0–700 µs time scale obeys a single-exponential phase with a kobs increasing linearly with [Na+] (Figure 11B). This resolves the fast phase detected with the stopped-flow method and shown in (A). Experimental conditions for the two methods are: 5 mM Tris, 0.1% PEG8000, pH 8.0 at 25 °C. The thrombin concentration was 50 nM for the stopped-flow measurements and 40 µM for the continuous-flow measurements. The [Na+], as indicated, was changed by keeping the ionic strength constant at 400 mM with choline chloride. Continuous lines were drawn using the expression a +bexp (−kobst) with best-fit parameter values: (A) a=8.944±0.001 V, b=−0.10±0.01 V, kobs=111±9 s−1 ([Na+]=50 mM); a=9.427±0.001 V, b=−0.29±0.01 V, kobs=96±6 s−1 ([Na+]=100 mM). (B) a=0.417±0.002, b=−0.072±0.02, kobs=7±2 ms−1 ([Na+]=36 mM); a=0.477±0.002, b=−0.10±0.03, kobs=10±2 ms−1 ([Na+]=91 mM). All data were collected at least in duplicate.
Figure 11.
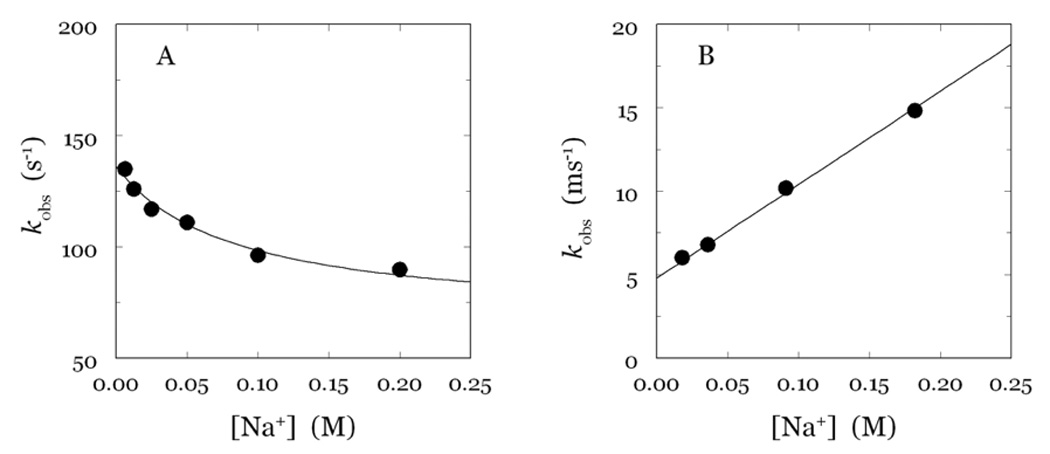
A,B. Values of kobs vs [Na+] for the slow and fast phases of fluorescence change due to Na+ binding to thrombin shown in Figure 10. Shown are the results pertaining to the stopped-flow (A) and continuous-flow (B) measurements. Note the different time scale between the two panels. Experimental conditions are given in the legend to Figure 1. Continuous lines were drawn according to eq 8 and eq 9 in the text, with best-fit parameter values: k1=67±7 s−1, k−1=69±6 s−1, kA=56,000±200 M−1s−1, k−A=4,800±200 s−1.
Precise determinations of the value of KA as a function of temperature enable dissection of the thermodynamic components of Na+ binding (Bah et al. 2006). Binding of Na+ is characterized by a large enthalpy change of −22 kcal/mol that is compensated by a large entropy loss of −64 cal/mol/K. The enthalpy change is due to formation of the six ligating interactions in the coordination shell, that also involve four buried water molecules (Figure 7) (Pineda et al. 2004a; Di Cera 2006; Page and Di Cera 2006). The entropy change reflects the uptake and ordering of water molecules within the channel embedding the primary specificity pocket and the active site linked to the occupancy of the Na+ site (Pineda et al. 2004a). As a result of the enthalpy-entropy energetic compensation, the binding affinity of Na+ is relatively weak (Kd in the mM range), as seen for many other M+-activated enzymes (Di Cera 2006; Page and Di Cera 2006). An important consequence of the large enthalpy change is that the value of KA becomes only about 10 M−1 at 37 °C, which implies that under physiologic conditions of temperature and [NaCl]=140 mM thrombin is only 60% bound to Na+. The fraction of thrombin in the procoagulant E:Na+ form is about 60%, and the anticoagulant form E accounts for about 40%. The temperature dependence also demonstrates that, of the two Na+ free forms, E* represents <1% of the population of thrombin molecules at 37 °C, which raises questions about its possible functional role in vivo. The paucity of E* forms makes scheme 1 valid under conditions of physiologic relevance. Inclusion of E* in scheme 1 is straightforward and does not affect the properties of Kcat, that depends by definition only on the properties of the enzyme forms that interact with substrate. The effect of E* on s can be determined easily under a number of conditions of interest (Carrell et al. 2006). The mechanistic significance of E* should be appreciated in the context of the effect of mutations, as recently shown for the D102N mutant (Pineda et al. 2006), or conditions that involve allosteric effectors in vivo to be identified.
6. Global effect of Na+ binding
Thrombin has a total of nine Trp residues located in the B chain (Figure 12) anywhere from 13 to 35 Å from the bound Na+ (Pineda et al. 2004a). Single-site Phe mutants of each of the nine Trp residues were used to identify fluorophores responsible for the spectral changes associated with Na+ binding (Bah et al. 2006). The 10% total increase in fluorescence observed for wild-type is retained by five Trp mutants, namely, W29F, W51F, W60dF, W96F and W237F (Figure 13). Two mutants of thrombin, W148F and W207F, experience >30% loss in total fluorescence change upon Na+ binding. On the other hand, W141F and W215F lose >70% of the total fluorescence change. The fast phase of fluorescence increase directly linked to the transition from E to E:Na+ in scheme 2 is affected in all Phe mutants, vouching for a global effect of Na+ binding on thrombin structure. The contributions of single Trp residues are not additive, lending support to the hypothesis that some of the environments in which they reside may be coupled allosterically. The coupling may ensure propagation of long-range effects originating at the Na+ site via a limited number of structural conduits. W141 and W215 make a large contribution to the fluorescence change induced by Na+ binding, and their mutation to Phe abrogates the fast phase completely (Figure 13). This implies that the environments of W141 and W215 change in the E* to E conversion, and more drastically in the conversion of E to E:Na+. The important role of W215 has been reported before (Arosio et al. 2000). This is the closest Trp residue to the bound Na+ (13 Å) and defines most of the aryl binding site involved in substrate recognition (Bode et al. 1992; Pineda et al. 2004a; Bode 2006). W141 is buried in a strategic location between the autolysis loop and exosite I and its linkage with the bound Na+, situated 23 Å away, vouches for a pivotal role in communicating changes from the Na+ site to exosite I (Figure 12). The importance of W141 is supported by recent structures of thrombin that document a flip in the indole ring in the absence of Na+ (Carrell et al. 2006) and a major involvement in the allosteric communication between exosite I and the active site in the E* to E transition (Gandhi et al. 2008). Among the other residues that contribute to the fluorescence change and the fast phase, W148 is located in the middle of the highly flexible autolysis loop, 21 Å away from the bound Na+, and is 62% exposed to solvent (Bode et al. 1992), whereas W207 is completely buried in the back of the catalytic chain, 23 Å away from the bound Na+, and in hydrophobic contact with W29 (Figure 12). The effects seen upon mutation of W207 are particularly interesting as this Trp makes numerous contacts with residues of the A chain around E8 and R4. This argues in favor of an important unanticipated role for the A chain in a linkage with the Na+ site and in channeling information from this site to other regions of the protein. Due to their proximity, W207 and W29 may function as a single fluorophore and/or quench each other. It is interesting that the Phe mutation of W29 enhances the amplitude of the fast phase, as though changes affecting W207 are better reported in the absence of W29. A similar scenario can be envisioned for W51 and W237, whose hydrophobic coupling may result in overlapping spectral effects with W51 actually hindering the full response of W237 to Na+ binding. The effects seen with the highly solvent exposed W60d and W96 suggest that these residues may be quite flexible and capable of probing different environments that reduce the fluorescence response to Na+ binding.
Figure 12.
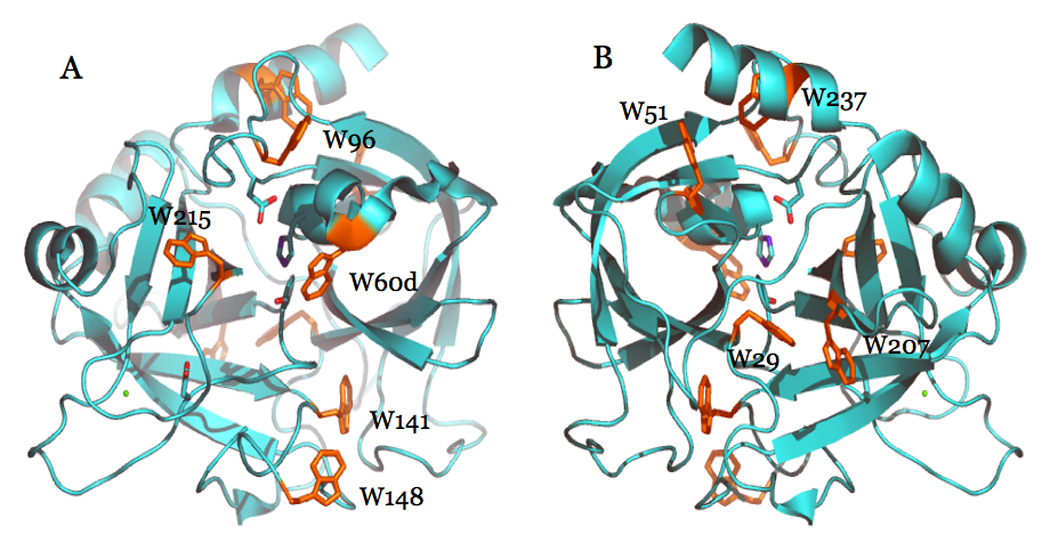
Ribbon plot of thrombin in the Na+-bound form, portraying the structure 1SG8 (Pineda et al. 2004a) with the active site in the front (A) or rotated 180° along the y axis (B). Shown are the side chains of the catalytic residues H57, D102 and S195, and the side chain of D189. Na+ is rendered as a green ball. The nine Trp residues of the enzyme are shown with their side chains in orange. The contribution of these residues to the fluorescence change induced by Na+ binding is discussed in the text. The A chain was removed for clarity.
Figure 13.
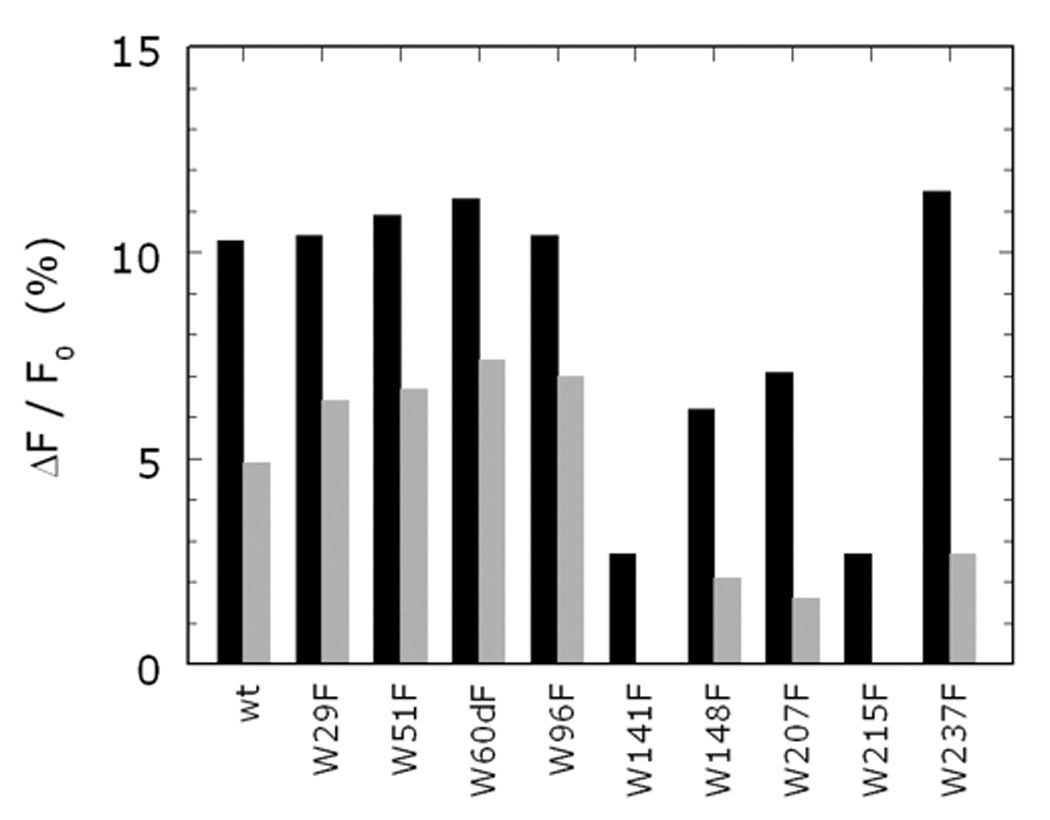
Fluorescence change induced by Na+ binding to wild-type and the Phe mutants of all nine Trp residues of human thrombin. Shown are the values of the total change in intrinsic fluorescence (black bars), or the amplitude of the fast phase (grey bars). All values are expressed as % change relative to the baseline. Experimental conditions are: 50 nM thrombin, 5 mM Tris, 0.1% PEG, pH 8.0 at 15 °C.
7. Mapping the domains energetically linked to Na+ binding
What are the domains of thrombin important for Na+ binding? Are they limited to residues in direct contact with the cation, or do they encompass more distant residues? Kinetics and site-directed mutagenesis studies of the nine Trp residues of thrombin vouch for very extensive perturbation of the enzyme structure upon Na+ binding. The extent of structural involvement is best gauged experimentally from the effect of site-specific mutations (Clackson and Wells 1995; Schreiber and Fersht 1995; Greenspan and Di Cera 1999) and the technique has been applied with enormous success in allosteric proteins like ATCase (Stevens et al. 1991) and hemoglobin (Turner et al. 1992). In the case of thrombin, much structural information can be obtained by measuring how Ala substitutions affect ligand recognition in the slow and fast forms. Different ligands have the ability to probe different domains of thrombin and can therefore report on structural changes due to Na+ binding that translate into perturbed energetics. This approach complements and expands direct measurements of Na+ binding and the effect of mutations on the thrombin-Na+ interaction.
Extensive Ala-scanning mutagenesis of thrombin has revealed an allosteric core of residues energetically linked to Na+ binding (Pineda et al. 2004a) that cluster around the Na+ site. Na+ binding is severely compromised (>30-fold decrease in affinity) upon mutation of D189, E217, D222 and Y225 that reside within 5 Å from the bound Na+. D189 assists the orientation of one of the four water molecules ligating Na+ and provides an important link between the Na+ site and the P1 residue of substrate (Prasad et al. 2004). E217 makes polar contacts with K224 and T172 that help stabilize the intervening 220-loop in the Na+ site. Mutation of K224 and T172 also affect Na+ binding, although to a lesser extent compared to E217A. Notably, the naturally occurring mutant prothrombin Scranton carries the K224T substitution and is associated with a bleeding phenotype due to reduced Na+ binding (Sun et al. 2001). The ion-pair between R187 and D222 latches the 186-loop onto the 220-loop to stabilize the Na+ site and the pore of entry of the cation to its binding site (Prasad et al. 2003). Mutation of R187 also affects Na+ binding, although to a lesser extent compared to D222A. The naturally occurring mutant prothrombin Greenville carries the R187Q substitution and is associated with a bleeding phenotype, again due to reduced Na+ binding (Henriksen et al. 1998). Y225 plays a crucial role in determining the Na+-dependent allosteric nature of serine proteases (Dang and Di Cera 1996) by allowing the correct orientation of the backbone O atom of residue 224 in the Na+ coordination shell (Guinto et al. 1999). The side chain of Y225 also secures the integrity of the water channel surrounding the primary specificity pocket required for correct substrate recognition (Guinto et al. 1999). Remarkably, the backbone of the KYG sequence around Y225 is oriented like the GYG sequence in the selectivity filter of the K+ channel (Doyle et al. 1998; Di Cera 2006; Page and Di Cera 2006).
Important information on how the active site participates in the allosteric transition has come from studies of chromogenic substrate hydrolysis (Pineda et al. 2004a). Effects of the bound Na+ are seen most dramatically on residues D189 and D221, as mutation of these residues to Ala nearly abrogates the difference between the slow and fast forms. These residues are crucial for the allosteric transduction of Na+ binding into enhanced catalytic activity. On the other hand, S214 and G223 promote substrate recognition in the slow form. Mutation of these residues produces a larger difference between the slow and fast forms compared to wild-type. The higher activity of the fast form toward FPR is due to the net balance between the favorable contribution to substrate binding and hydrolysis from D189 and D221 in the presence of Na+ and the favorable contributions from S214 and G223 in the absence of Na+. The important conclusion is that functional epitopes for substrate recognition change when Na+ binds to thrombin. In turn, this explains how the allosteric nature of thrombin can control the extent of Na+ activation in a substrate specific manner. The same approach has been used in the analysis of thrombin interaction with macromolecular ligands that probe a larger surface of the enzyme upon binding. The potent natural inhibitor hirudin (Mengwasser et al. 2005) covers 20% of the thrombin surface area (Rydel et al. 1991) and is an exceptionally sensitive probe of the conformational state of the enzyme. Hirudin also mimics the binding strategy of fibrinogen (Stubbs et al. 1992; Rose and Di Cera 2002), PAR1 (Ayala et al. 2001; Gandhi et al. 2008) and PAR3 (Bah et al. 2007). The analysis has revealed D221 and D222 in the Na+ site, along with K36, L65, T74 and R75 in exosite I and G193 in the active site as critical determinants of recognition in the fast form. The use of a more extended probe of the conformational state of the enzyme reveals long range perturbation that extends from the Na+ binding site to exosite I, in line with the results of stopped-flow measurements of Na+ binding (Bah et al. 2006). As for the interaction with FPR, preferential binding of hirudin to the fast form is contrasted by residues like E192 and S214 that stabilize binding to the slow form. Again, the functional epitopes controlling hirudin binding change upon Na+ binding. Systematic investigation of functional epitopes in the two forms of thrombin toward FPR hydrolysis and hirudin inhibition provides a paradigm that also applies to physiologic substrate recognition. Preferential cleavage of fibrinogen by the fast form is due to the balance of residues T172 and I174 that promote recognition in the fast form and K224 that promotes recognition in the slow form (Di Cera 2007; Di Cera et al. 2007). A summary of the contributions of thrombin residues to ligand recognition (FPR, hirudin and fibrinogen) in the two allosteric forms is shown in Figure 14, along with the residues energetically linked to Na+ binding. The extent of perturbation originating at the Na+ site is indeed extensive and approximates the scenario envisioned by stopped-flow fluorescence measurements. Interestingly, many Trp residues responsible for the fluorescence change due to Na+ binding are sandwiched in between the Na+ site and exosite I on the opposite poles of the enzyme defined by the two interacting β-barrels. These residues are linked to the structural conduits for allosteric communication between the two domains.
Figure 14.

Surface representation depicting the structural organization of the residues promoting ligand recognition to the fast (red) or slow (green) form of thrombin. The structure refers to 4HTC (Rydel et al. 1991) with thrombin oriented as in Figure 4. The inhibitor hirudin was removed for clarity. Shown are the residues whose Ala substitution affects FPR (Pineda et al. 2004a), hirudin (Mengwasser et al. 2005) or fibrinogen (Di Cera et al. 2007) recognition >10-fold in either form and concurrently change the difference in specificity between the slow and fast forms >3-fold. Also shown in red are residues D189, E217, D222 and Y225 in the allosteric core controlling Na+ binding to thrombin (Pineda et al. 2004a). Trp residues visible in this orientation are rendered in yellow. Note how the binding of Na+ produces long-range effects that cross the medial portion of the B chain where most of the Trp residues are located. These residues are linked to the structural conduits for allosteric communication between the two domains.
8. Structures of E*, E and E:Na+
Information gleaned from mutagenesis data and recent stopped flow analysis of Na+ binding points out a number of structural determinants that are likely involved in the transition of thrombin from the E form to the E:Na+ form. Current structural information on the molecular basis of Na+-dependent allostery accounts for many important functional differences between the E and E:Na+ forms. However, the documented structural changes are limited and do not explain the full complexity of the allosteric transition captured by functional studies (Figure 14) that vouch for a remarkable conformational transition that transduces Na+ binding into a global, long-range perturbation of the enzyme. With this caveat in mind, we will now analyze the results of recent crystallographic studies of the three forms of thrombin, E*, E and E:Na+.
An obviously important component of any informed discussion of Na+ binding to thrombin is based on the nature of the Na+-free slow form E (see scheme 2). Structural investigation of this form turned out to be even more challenging than that of the Na+-bound fast form E:Na+, because it was strictly dependent on the elimination of Na+ and active site or exosite inhibitors of the enzyme from the crystallization conditions. In fact, most of the inhibitors used in thrombin crystallization to prevent autoproteolysis bind preferentially to the Na+-bound fast form. In 2002, the thrombin mutant R77aA devoid of the autoproteolytic site in exosite I was crystallized free of Na+ and inhibitors and yielded the first structure of thrombin in the Na+-free slow form E (Pineda et al. 2002b). This structure was followed in 2004 by higher resolution structures in the Na+-free and Na+-bound forms, free or bound to the active site inhibitor PPACK (Pineda et al. 2004a) that have revealed some of the changes caused by Na+ binding. Structures of E and E:Na+ are highly similar, with r.m.s. deviations of the Cα traces of only 0.38 Å. PPACK-bound forms of thrombin are practically identical to each other (r.m.s.=0.19 Å), except for the obvious absence of Na+ in the PPACK-inhibited slow form. A small (1 Å) upward shift is observed in the 60-loop in the structure of the slow form relative to the fast form that could explain the involvement of W60d in the fluorescence change linked to E to E:Na+ transition. There are five main structural differences between the slow and fast forms of thrombin (Figure 15): 1. the R187-D222 ion-pair; 2. orientation of D189 in the primary specificity pocket; 3. conformation of E192 at the entrance of the active site; 4. position of the catalytic S195; and 5. architecture of the water network spanning >20 Å from the Na+ site to the active site. The R187-D222 ion-pair connects the 220- and 186- loops that define the Na+ site. D222 belongs to the allosteric core and the mutant D222A has drastically impaired Na+ binding, a property mirrored by the R187A mutant. In the fast form, the guanidinium N atoms of R187 are 2.7 Å and 3.1 Å from the carboxylate O atoms of D222. In the slow form, these distances become 3.3 Å and 4.8 Å respectively. Breakage of the ion-pair was observed in the low resolution structure of the slow form (Pineda et al. 2002b) and is consistent with mutagenesis data supporting a slow form type of behavior for the R187A and D222A mutants. Notably, the broken ion-pair shifts the backbone O atom of R221a that directly coordinates Na+ in the fast form and moves it into an orientation that is incompatible with Na+ binding. The structure of the thrombin mutant D221A/D222K also assumes signatures characteristic of the slow form (Pineda et al. 2004c).
Figure 15.
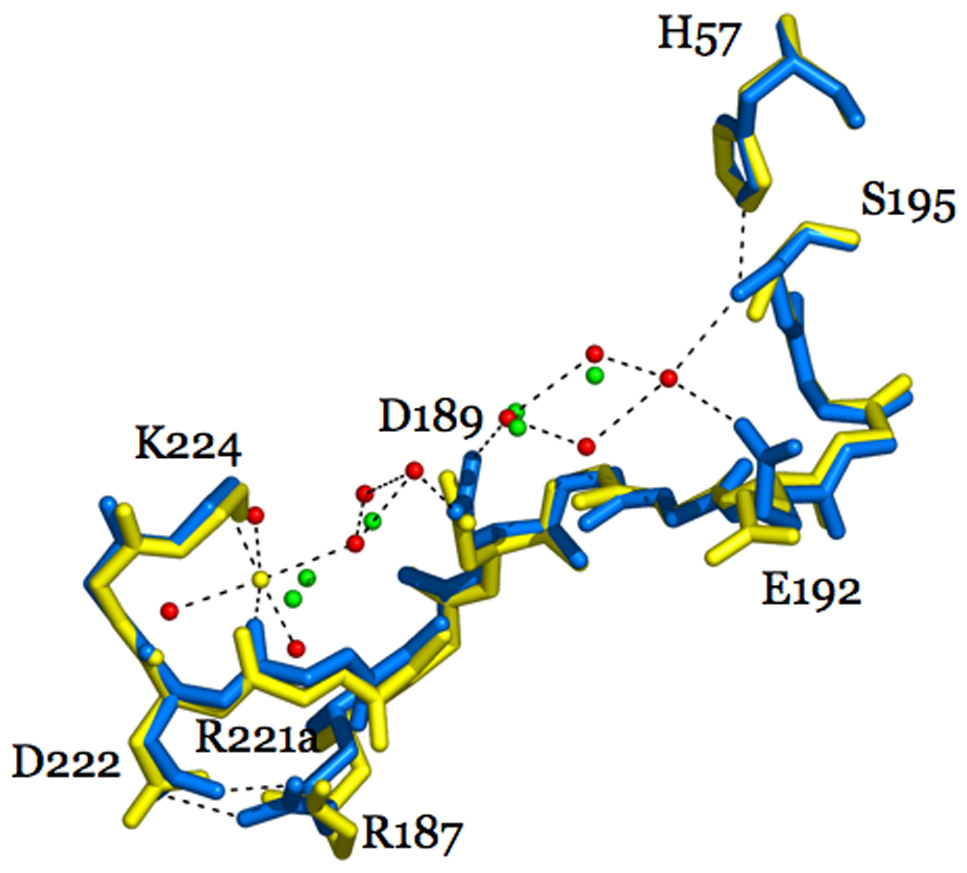
Structural changes induced by Na+ binding to thrombin depicted by the structures of the Na+-free slow (1SGI, yellow) and Na+-bound fast (1SG8, marine) forms. The main changes induced by Na+ (yellow sphere) binding are: formation of the R187-D222 ion-pair that causes a shift in the backbone O atom of R221a, reorientation of D189 that accounts for the change in substrate binding, shift of the side chain of E192, and shift in the position of the Oγatom of S195 that accounts for the change in Kcat. Also shown are the changes in the water network connecting the Na+ site to the active site S195. The water molecules in the fast form (red spheres) are organized in a network that connects Na+ to the side chain of D189 and continues on to reach the Oγatom of S195. A critical link in the network is provided by a water molecule that H-bonds to S195 and E192. This water molecule is removed in the slow form, causing a reorientation of E192. The connectivity of water molecules in the Na+-free form (green spheres) is further compromised by the lack of Na+ and proper anchoring of the side chain of D189. H-bonds are shown by broken lines and refer to the fast form.
Perturbation of the primary substrate pocket is evidenced in the E→E:Na+ transition of thrombin. D189 in the fast form is optimally oriented for electrostatic coupling with the P1 Arg residue of substrate. In the slow form, the carboxylate of D189 experiences a 30° rotation that moves the Oδ1 atom up to 1.1 Å away from its optimal coupling with the guanidinium group of Arg at the P1 position of substrate. Rearrangement of D189 upon Na+ binding enhances substrate specificity by improving the Km. The structure of the slow form supports a key role for D189 in both Na+ coordination and allosteric transduction of Na+ binding into enhanced catalytic activity (Prasad et al. 2004) consistent with results from mutagenesis data. Further conformational changes are observed near the top of the primary specificity pocket where the side chain of residue E192 moves in the slow form relative to the fast form and is positioned away from the active site region. Such movement could minimize the potential electrostatic clash with the P3 and P3’ acidic residues of protein C and could explain why the slow form of thrombin retains high activity toward this anticoagulant substrate (Dang et al. 1995; Berg et al. 1996; Rezaie and Olson 1997; Yang et al. 2004). Subtle changes at the enzyme active site also include conformational alteration of the catalytic triad. In the slow form of thrombin, the nucleophilic S195 side chain is altered. Rotation of the S195 side chain about 35° in the slow form relative to the fast form breaks the critical H-bond with the catalytic H57, which itself also shifts slightly away from S195. Integrity of the H-bond is important for catalysis (Fuhrmann et al. 2006) and the unfavorable position of S195 in the slow form may explain the lower Kcat observed in the absence of Na+. Much of the activating effect of Na+ can be explained in terms of the more favorable orientation of D189 in the S1 site, that improves Km, and of S195 in the active site, that improves kcat. It should be pointed out that the conformation of S195 in the fast form is also not optimal for substrate recognition and is intermediate to that of the slow form and the PPACK-bound forms. These changes lend support to the idea that the nucleophile of S195 can be repositioned within the active site and that Na+, and possibly other allosteric effectors of thrombin like thrombomodulin, may take advantage of this flexibility to modulate enzyme activity.
Structural changes in the slow→fast transition of thrombin do not involve E217 and Y225 in the allosteric core identified by mutagenesis. These residues appear in the same conformation in the four intermediates of thrombin free/bound to Na+ and/or PPACK. Likewise, there are no significant changes around D221, which plays a dominant role in the allosteric enhancement of catalytic activity, or around T172 and S214 (Pineda et al. 2004a). Finally, none of the Trp residues involved in the spectral changes linked to Na+ binding (Bah et al. 2006) show significant movement in the structures. It is likely that the changes undergone by these residues may be too small to detect crystallographically, or are hindered by crystal packing. The most significant structural change between the slow and fast forms, however, provides evidence of long-range communication between the Na+ site and the active site. Previous studies of thrombin in the fast form have pointed out a network of water molecules that embeds the Na+ site, the primary specificity site and the active site (Zhang and Tulinsky 1997; Krem and Di Cera 1998). This network was first discovered in the structure of thrombin inhibited at exosite I with hirugen and with the active site free (Vijayalakshmi et al. 1994). In the fast form, there is a network of eleven water molecules that connect through H-bonds the bound Na+ to the Oγ atom of S195, located >15 Å away (Figure 15). These solvent molecules act as links between the strands 215–219, 225–227 and 191–193, that define the Na+ site and the walls of the primary specificity pocket, and fine tune the function of these strands in controlling substrate docking into the active site. The network provides the long range connectivity needed to allosterically communicate information from the Na+ site to the active site S195 and to residues involved in substrate recognition, such as D189 and E192. In the slow form, only seven water molecules occupy positions in the network equivalent to those seen in the fast form and the connectivity is radically altered. Change in the number and long range ordering of water molecules linked to Na+ binding offers an explanation for the thermodynamic signatures detected experimentally. The enthalpy change is due to formation of the six ligating interactions in the Na+ coordination shell, that involve four buried water molecules (Pineda et al. 2004a; Di Cera 2006; Page and Di Cera 2006). The entropy change reflects the uptake and ordering of water molecules within the channel embedding the primary specificity pocket and the active site linked to the occupancy of the Na+ site (Pineda et al. 2004a; Bone 2006). These changes could also explain the large and negative heat capacity change measured upon Na+ binding to thrombin (Guinto and Di Cera 1996; Griffon and Di Stasio 2001; Prasad et al. 2003).
The network of water molecules embedding the primary specificity pocket and the Na+ site has a bearing on the kinetic mechanism of Na+ binding. The value of kA (scheme 2, Figure 11) is four orders of magnitude slower than the diffusion controlled limit (van Holde 2002). Although we lack data on other protein systems for comparison, we can assume that several factors contribute to slow down the kinetics of Na+ association. Without these factors, Na+ binding to thrombin would likely be too fast to measure even with the continuous-flow method. Na+ binds to its site after penetrating a pore defined by the 186- and 220- loops (Prasad et al. 2003; Page and Di Cera 2006) whose size would not admit a fully hydrated Na+. In this regard, the interaction of Na+ with thrombin has features in common with the permeation and selectivity of ion channels (Roux and MacKinnon 1999; Di Cera 2006; Page and Di Cera 2006). Dehydration of Na+ required to enter the pore is expected to slow down the kinetics of association significantly (Eisenman and Dani 1987; Roux and MacKinnon 1999). Once inside the pore, Na+ is re-hydrated into its coordination shell that involves four water molecules and the backbone oxygen atoms of K224 and R221a (Figure 7) (Pineda et al. 2004a). That in turn organizes a network of water molecules that spans the interior of the enzyme for over 15 Å up to the catalytic S195 (Pineda et al. 2004a). Ordering of this network of water molecules may require a longer time scale than the simple process of penetrating the pore, thereby contributing to the drastic reduction in the rate of binding kA measured experimentally. Combination of dehydration, penetration of the pore, and re-hydration with organization of the water network around the Na+ site could account for the significant slow down in the kinetics of Na+ association.
The available structures of the slow form E and fast form E:Na+ account for some basic functional properties of the two allosteric conformations of thrombin and explain most of the kinetic features linked to Na+ binding and allosteric activation. However, rapid kinetic measurements of Na+ binding clearly demonstrate that E is in equilibrium with a third thrombin form, E*, that is unable to interact with Na+ (scheme 2) (Bah et al. 2006; Gianni et al. 2007). The structural nature of E* is of considerable interest. E* has been suggested to portray an “inactive” slow form, unable to bind Na+ and substrate or inhibitors at the active site (Lai et al. 1997). That hypothesis has gained prominence recently in the context of several structures of inactive forms of thrombin in the Na+-free state that have appeared in the literature. These structures differ drastically from the active slow form E (Pineda et al. 2002b; Pineda et al. 2004a) and share disorder or collapse of the Na+ site and steric blockage of the active site (Carter et al. 2004; Pineda et al. 2004b; Johnson et al. 2005; Papaconstantinou et al. 2005; Carrell et al. 2006). Potential issues of concern with these structures are the presence of mutations in close proximity to the Na+ site (Carter et al. 2004; Pineda et al. 2004b) and/or substantial crystal packing interactions that bias the conformations of the active site and the Na+ site (Johnson et al. 2005; Papaconstantinou et al. 2005; Carrell et al. 2006). All existing structures portraying thrombin in inactive conformations in the absence of inhibitors and Na+ (Carter et al. 2004; Pineda et al. 2004b; Johnson et al. 2005; Papaconstantinou et al. 2005; Carrell et al. 2006) contain two molecules in the asymmetric unit, related by non-crystallographic 2-fold symmetry, that pack together through extensive contacts. The packing involves the active site and the Na+ site and calls into question the structural assignments attributed to the absence of Na+ (Pineda et al. 2006). The recently solved structure of free wild-type thrombin in an inactive conformation attributed to the lack of Na+ (Johnson et al. 2005) has two molecules in the asymmetric unit defining an intermolecular surface of interaction of ~900 Å2. The surface involves residues of the 60-, 99-, 170-, 186- and 220- loops and is the size of the interface of the complex between thrombin and the N-terminal domain of hirudin (Ayala et al. 1995). Practically, this structure of “free” thrombin is in fact bound to the second thrombin molecule in the asymmetric unit so tightly that the entire Na+ site and active site regions come under the influence of significant crystal packing. Much importance was attributed to a 3.0 Å shift in the position of E192, that occludes access to the active site (Johnson et al. 2005). However, E192 is at the interface of the two thrombin molecules in the asymmetric unit, with the Cγ atom 3.5 Å away from the Nε atom of R221a of the second molecule. The contact pushes the side chain of E192 into the active site, where it is further stabilized by two water-mediated H-bonds with a glycerol molecule at the interface. The artifactual conformation of E192 may have facilitated a flip of the N atom of G193 defining the oxyanion hole together with the N atom of the catalytic S195. Interestingly, the flip is also observed in the structure of thrombin bound to K+ (Carrell et al. 2006). Other significant packing interactions across the interface involve D221 with R221a, D222 with K60f, and K224 with D60e, all residues claimed to be involved in the allosteric transition from the inactive slow form to the active fast form (Johnson et al. 2005). There is reason to believe that the inactive conformation reported in this structure may just be an artifact of crystal packing. Similar considerations apply to the structure of the thrombin mutant E217K (Carter et al. 2004), also claimed to portray thrombin in the inactive slow form. In this case, packing interactions are even more substantial and cover a surface area >1,000 Å in the Na+ site and active site regions. The 3.4 Å contact between the Oε1 atom of E192 and the NH1 atom of R221a of the second molecule in the asymmetric unit positions E192 toward the active site. Importantly, the Nζ atom of the mutated residue K217 forms a 2.9 Å salt bridge with the Oδ2 atom of D222 in the second molecule of the asymmetric unit, contrary to the claim that this Lys side chain did not make a significant stabilizing contact in the structure (Carter et al. 2004). The inactive conformation reported in this structure may well be the result of the drastic charge reversal E217K introduced near the Na+ site and significant crystal packing. The recent structure of the thrombin mutant R77aA in the presence of KCl contains two molecules in the asymmetric unit, one with K+ bound to the Na+ site (molecule 1) and the other (molecule 2) in an inactive conformation with the Na+ site free (Papaconstantinou et al. 2005; Carrell et al. 2006). This structure contains substantial packing interactions in the asymmetric unit that affect a surface area >600 Å. The 220-loop region of molecule 2 is at the interface, and its partial disorder increasese exposure of the RGD sequence to solvent. The autolysis loop of molecule 2 is repositioned toward exosite I by its contacts with molecule 1 at the interface. C220 is in contact with R221a of molecule 1, which could explain perturbation of the 190-strand through the C191:C220 disulfide bond and closure of the access to the primary specificity pocket. Perturbation of C220 could have also caused the collapse of the 215–219 β-strand into the primary specificity pocket, as seen in the structure of the inactive W215A/E217A mutant (Pineda et al. 2004b), as well as the 190° flip in the side chain of W215 that is also observed in the structure of inactive wild-type thrombin (Johnson et al. 2005). As in the case of other structures claimed to portray thrombin in the inactive slow form (Carter et al. 2004; Johnson et al. 2005), the inactive conformation of molecule 2 in the R77aA structure in the presence of KCl (Carrell et al. 2006) may be the result of crystal packing.
A major advance in our understanding of thrombin allostery and its structural underpinnings has come from the structure of the thrombin mutant D102N in the free form (Pineda et al. 2006). This mutant was constructed, expressed and purified to inactivate thrombin with the most conservative replacement of the catalytic triad H57/D102/S195, and to produce a useful reagent for crystallization in the absence of inhibitors or in complex with physiological substrates. The available X-ray crystal structure of the rat trypsin mutant D102N in the presence of benzamidine (Sprang et al. 1987) is practically identical to that of wild-type although the mutant shows >10,000-fold reduction in catalytic activity. A very different result was observed in the case of thrombin. The structure of the thrombin mutant D102N contains a single molecule in the asymmetric unit, with minimal contacts with symmetry related molecules in the lattice (Pineda et al. 2006). The overall fold of D102N is similar to wild-type, with a backbone rms deviation of 0.77 Å compared to the Na+-bound fast form (Pineda et al. 2004a). There are no significant changes at the site of mutation that can be attributed to the D102N substitution. For example, the Nδ1 atom of N102 remains within H-bonding distance of the Nδ1 atom of the catalytic H57 (2.91 Å) and the Oγ atom of S214 (3.06 Å), as seen in the wild-type (Pineda et al. 2004a). On the other hand, the structure shows changes in the primary specificity pocket and in the Na+ binding site that are unprecedented in thrombin and the entire realm of serine proteases. These changes provide a plausible interpretation of the peculiar properties of the E* form. The 215–219 β-strand collapses into the primary specificity pocket (Figure 16). W215, whose Cα atom is 5.26 Å away from the mutated Nδ1 atom of N102, shifts 130° at the Cβ atom and causes the indole ring to pack against the hydrophobic pocket in the active site formed by W60d, Y60a, H57 and L99. In this conformation, the indole of W215 occupies the same position as the Pro ring of the active site inhibitor PPACK in the PPACK-inhibited structure of thrombin, suggesting that the collapse of W215 into the active site is driven by the favorable hydrophobic environment of the S2 site near the catalytic H57 (Bode et al. 1992). Downstream from the 215–219 β-strand, the twist in the backbone caused by the collapse of W215 moves the entire 220-loop upward and changes the orientation of several residues. The side chain of R221a, located >10 Å away from the site of mutation, rotates 95° and brings the guanidinium group in contact with D189 in the primary specificity pocket. In this conformation, the guanidinium group of R221a occupies a position analogous to that of the guanidinium group of the Arg of PPACK, or of an incoming substrate molecule. Altogether, the drastic shifts of W215 and R221a produce a conformation of thrombin that is self-inhibited by the hydrophobic engagement of the 60-loop and active site H57 by W215, and of the acidic moiety of D189 in the primary specificity pocket by R221a. Residues D221 and D222 flanking R221a shift upward, together with the entire 220-loop, and free the side chain of R187 which is ion-paired to them in the Na+-bound fast form. The acquired mobility of R187 and the upward shift of the 220-loop cause the side chain of R187 to penetrate the protein core and the Na+ site, with the guanidinium group positioned within 1 Å from where Na+ would bind. The structure of D102N portrays a conformation of thrombin that is unable to bind Na+ and ligands to the active site, in agreement with the properties of the inactive slow form E* documented by earlier kinetic studies (Lai et al. 1997).
Figure 16.
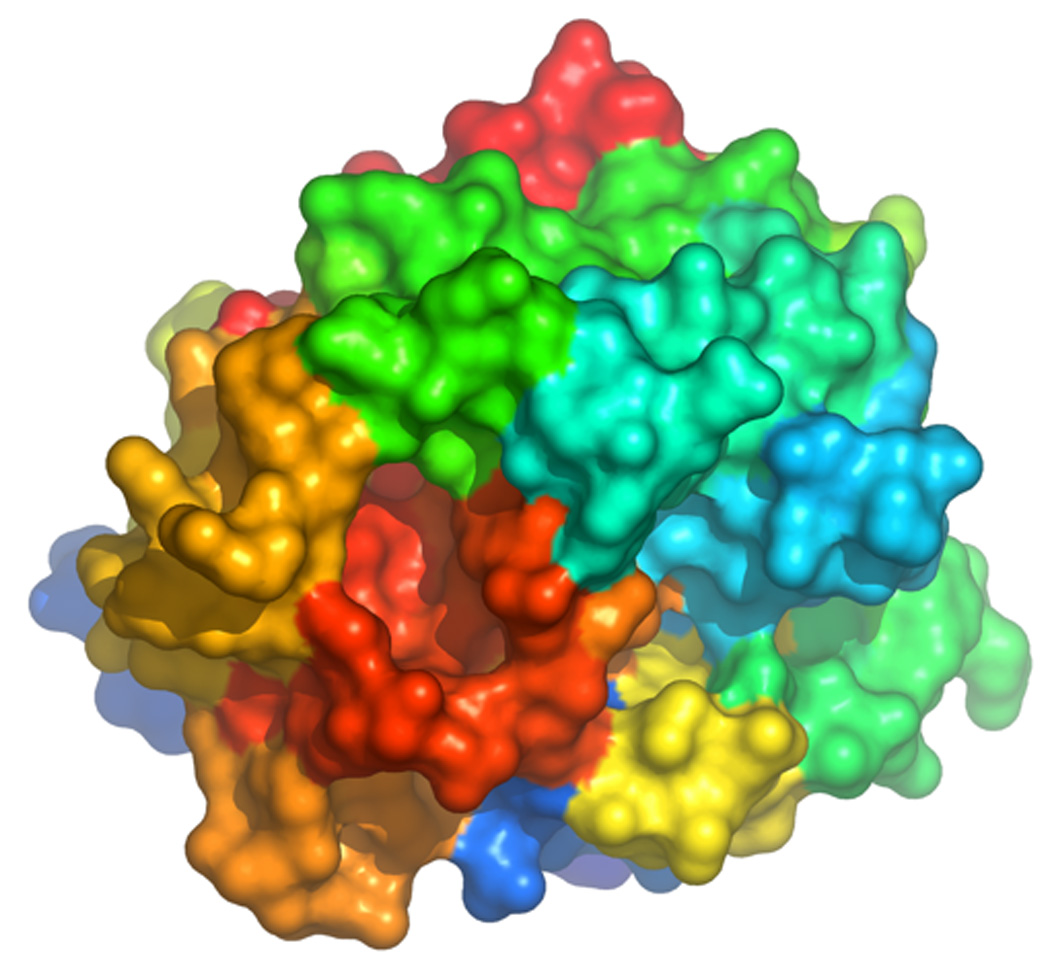
Space filling model of thrombin in the Bode orientation (active site, center; exosite I, right; exosite II, top left; Na+ site, bottom left; 60-loop, top) depicting the structure of E*. The surface is color-coded according to the spectrum (N-terminus blue, C-terminus red). Note how the collapse of the 215–219 β-strand (red) against the 60-loop (cyan) completely occludes access to the active site. Details of the conformation of E* and its conversion into E are given in Figure 24.
Because the structure of D102N contains a single molecule in the asymmetric unit, it only contains contacts with symmetry-related molecules in the lattice. These contacts do not affect the self-inhibited conformation. Minor H-bonding interactions engage K224 with E127, R77a with P204, and R126 with E97a, that are inconsequential on the burial of R221a and R187 inside the protein core. Minor van der Waals interactions involve the 60-loop from Y60a to D60e, but do not extend to W215. The structure does not feature the active site-occusion by E192 reported in recent structures of inactive thrombin (Carter et al. 2004; Johnson et al. 2005; Carrell et al. 2006), nor does it exhibit perturbation of the 60-loop (Huntington and Esmon 2003; Johnson et al. 2005) or significant exposure of the RGD sequence (Papaconstantinou et al. 2005). The N atom of G193 is flipped as in other structures of inactive thrombin (Carter et al. 2004; Johnson et al. 2005; Papaconstantinou et al. 2005; Carrell et al. 2006) and in the K+-bound form (Carrell et al. 2006), lending support to the conclusion that flexibility of the backbone in this region is an intrinsic property of thrombin. Finally, the collapse of the 215–219 β-strand into the active site is reminiscent of that observed in the structures of the mutants W215A/E217A (Pineda et al. 2004b), E217K (Carter et al. 2004) and R77aA (Carrell et al. 2006). The collapse is likely due to the intrinsic flexibility of W215 whose rearrangement causes a shift in the 216–219 backbone to avoid steric clash (Carrell et al. 2006). Moreover, the collapse of W215 into the active site observed in the structure of D102N would not be possible without an initial flip of the indole side chain as first reported in the inactive structure of the R77aA mutant (Papaconstantinou et al. 2005). Movement of W215 and its structural consequences on the 215–219 β-strand likely represent genuine properties of the E* form, together with the rearrangement of R221a and R187 never before documented in structures of thrombin or cognate serine proteases. The structural features of the D102N mutant pertain to the inactive slow form E* and are not consequences of the mutation. Stopped-flow measurements of Na+ binding to the D102N mutant yield a direct assessment of the contributions of E and E* in the equilibrium that precedes Na+ binding. These measurements demonstrate that D102N is more stabilized in the E* form compared to wild-type, therefore accounting for the higher probability of capturing the elusive E* form by X-ray crystallography using this mutant. It should be pointed out, however, that although this structure reveals drastic changes at the level of W215 and perturbation of W141, it fails to account for changes at other Trp residues that are detected by fluorescence measurements (Figure 13). The true extent of conformational perturbation accompanying Na+ binding to thrombin may ultimately defy resolution by X-ray structural biology.
9. Murine thrombin
A new twist on the role of Na+ in thrombin function has come from the recent observation that murine thrombin lacks Na+ activation, but retains high catalytic activity toward physiologic substrates because it is locked in a Na+-bound E:Na+ form (Bush et al. 2006; Marino et al. 2007). We have already pointed out how Na+ binding to human thrombin is controlled energetically by a number of residues (Pineda et al. 2004a; Mengwasser et al. 2005). Na+ binding is severely compromised (>30-fold decrease in binding affinity) upon mutation of D189, E217, D222 and Y225. The importance of these residues has been discussed. Of these four residues, E217 and Y225 are conserved in thrombin from all species sequenced to-date. D189 is Ser in the sturgeon, and the D189S mutant of human thrombin has impaired Na+ binding and substrate recognition (Prasad et al. 2004). D222 is the least conserved residue among the four. It is Ser in the sturgeon, Lys in the mouse and Asn in the rat (Banfield and MacGillivray 1992). Because of the D222K substitution, murine thrombin lacks Na+ activation, which is partially restored by the reverse K222D mutation (Bush et al. 2006).
The lack of Na+ activation in human thrombin would produce an enzyme with little catalytic activity toward fibrinogen and PAR1, incompatible with biological function (Di Cera 2003). Murine thrombin has solved this conundrum by mimicking functionally the Na+-bound fast form of the human enzyme, thereby retaining high activity toward fibrinogen, PAR1 and PAR4, and without compromising activation of protein C (Bush et al. 2006). Such molecular mimicry of Na+ activation may have turned beneficial during evolution to counter the effect of numerous mutations that in humans destabilize Na+ binding and cause bleeding (Miyata et al. 1992; Degen et al. 1995; Henriksen et al. 1998; Sun et al. 2001; Rouy et al. 2006; Stanchev et al. 2006). There are thirty-two total replacements between human and murine thrombin (Table 2), five in the A chain and twenty-seven in the catalytic B chain. The net result of the replacements is very significant: murine thrombin has a net charge difference relative to human thrombin of +6. This comes from the introduction of two positively charged residues (I24K, Q131R), the loss of one positively charged residue (K14aT) and three negatively charged residues (E127Q, D170A, D186aN), and three charge reversal substitutions (E13K, K149eE, D222K). The drastic charge difference explains some of the distinct properties of the two enzymes, most notably: the different ability to stick to ion exchange columns, the different procedures needed for activation from zymogen and especially the difficulty for murine thrombin to crystallize under solution conditions containing even minimal amounts of salts. Among the non-conservative substitutions, D186aN and D222K affect residues around the pore of entry of Na+ to its binding site (Figure 17) and account for half of the +6 charge difference between the two enzymes. The electrostatic perturbation caused by these two substitutions alone could be substantial enough to compromise or alter the response to Na+ binding in the murine enzyme. The composition and length of the 186- and 220- loops defining the pore are in fact critical for Na+ binding and selectivity over K+ (Prasad et al. 2003). There are few, but biologically relevant examples of monovalent cation activation mimicry (Di Cera 2006; Page and Di Cera 2006). Actin folds similarly to the K+-activated enzyme Hsc70 (Wilbanks and McKay 1995), but the requirement for K+ to bridge the Pα and Pβ of ADP is fulfilled by the Nζ atom of K18 (Kabsch et al. 1990). K222 in murine thrombin plays an important structural role in securing functional mimicry of Na+ activation.
Table 2.
Amino acid differences between human and murine thrombin.
| 1e | 13 | 14a | 14d | 14h | 20 | 24 | 26 | 27 | 59 | 79 | |
| Human | S | E | K | R | E | S | I | M | S | L | I |
| Murine | L | K | T | K | D | W | K | I | A | I | V |
| 90 | 106 | 126 | 127 | 129 | 129a | 131 | 145 | 149a | 149c | 149d | |
| Human | I | M | R | E | A | A | Q | K | A | V | G |
| Murine | V | L | K | Q | V | T | R | R | T | I | N |
| 149e | 150 | 170 | 184a | 186 | 186a | 186b | 186c | 222 | 236 | ||
| Human | K | G | D | Y | P | D | E | G | D | K | |
| Murine | E | I | A | F | V | N | D | T | K | R | |
Replacements in italic (9 total) indicate non-conservative charge substitutions. The thrombin chimera was constructed by replacing residues 184a, 186, 186a, 186b, 186c and 222 in human thrombin with those of the murine enzyme.
Figure 17.

(A, B, C). Surface rendering of the pore of entry to the Na+ binding site of human thrombin in the structure 1SG8 (Pineda et al. 2004a) (A), compared with the same region in murine thrombin (B) and the thrombin chimera (C). Residues lining the pore are color coded according to their physical properties (red=positively charged, blue=negatively charged, orange=hydrophobic, white=all others). In the human enzyme, the pore is defined by residue D222 in the 220-loop and the sequence PDEGKR from P186 to R187 in the 186-loop (Table 2) (A). In murine thrombin (B), residue 222 is Lys and the corresponding sequence in the 186-loop is VNDTKR (Table 2). The side chain of K222 completely occludes the pore. The side chain of N186a is glycosylated (NAG). Occlusion of the pore is also seen in the thrombin chimera (C), in which the human enzyme carries all residues around the pore as in murine thrombin. There is no glycosylation of N196a in the chimera. (D, E, F). Architecture of the pore of entry to the Na+ binding site in the same orientation as shown in the surface rendering (panels A, B, C), with relevant residues rendered in CPK model (C in yellow) and the 2Fo-Fc electron density maps contoured at the 0.7 σ level for the structures presented in this study (panels E, F). The human enzyme (D) shows the pore wide open, whereas K222 in murine thrombin (E) occludes the pore and positions the Nζ atom within H-bonding distance from K185, D186b and K186d. The backbone O atom of residue 186b is flipped relative to the position assumed in the fast form of the human enzyme. Also shown is the indole side chain of W20, which is Ser in human thrombin, as a structural signature of the murine enzyme. K222 in the thrombin chimera (F) is positioned as in the murine thrombin structure.
The structure of murine thrombin was solved at 2.2 Å resolution, free of inhibitors and salts, after many unsuccessful attempts involving the active site inhibitor PPACK and different concentrations of Na+, K+ or Li+ in the crystallization conditions (Marino et al. 2007). The conformation of murine thrombin in its free form has the active site and primary specificity pocket readily accessible to substrate. The rms deviation at the Cα atoms is only 0.42 Å relative to the fast form of the human enzyme (Pineda et al. 2004a). Because the enzyme used was free of inhibitors and in its wild-type form, autoproteolytic digestion at exosite I caused the entire segment 74–78 to be missed in the electron density map. Autoproteolytic attack of R77a followed by a second cleavage at R73 produce β-thrombin from α-thrombin in the human enzyme (Hofsteenge et al. 1988). The substantial perturbation of exosite I shifts the backbone around H71 and makes room for a flip of the indole ring of W141, as observed in the inactive monomer of the human thrombin structure solved in the presence of KCl (Papaconstantinou et al. 2005; Carrell et al. 2006). The flipped conformation is also stabilized by a rearrangement of the side chain of M32. Whether the flipped conformation of W141 is constitutive in murine thrombin remains to be established. Interest in this residue stems from its involvement in the fluorescence change linked to Na+ binding to human thrombin (Bah et al. 2006) and its key role in the alloseric communication between exosite I and the active site (Gandhi et al. 2008). The autolysis loop spanning residues 144–150 (Bode et al. 1992) is disordered, as typically seen in human thrombin even at high resolution (Pineda et al. 2004a).
The most important features of the murine thrombin structure involve the pore of entry to the Na+ binding site (Figure 17). The ion-pair between D222 and R187 is a hallmark of the Na+ bound fast form in the human enzyme (Pineda et al. 2004a; Pineda et al. 2004c) and contributes to the architecture of the pore. In murine thrombin, the presence of K222 pushes the guanidinium group of R187 away. The side chain of K222 occupies the interior of the pore and completely occludes its access from the solvent (Figure 17). The Nζ atom of K222 penetrates the pore and sits ~5 Å away from where Na+ binds in the fast form (Pineda et al. 2004a), within H-bonding distance of the backbone O atoms of K185 (2.85 Å), D186b (3.34 Å) and K186d (2.71 Å). The backbone O atom of residue 186b, recruited by the Nζ atom of K222, is flipped relative to the position assumed in the fast form of the human enzyme. Other changes of interest around the pore region include glycosylation of N186a, which is never observed in the human enzyme. W20, which is Ser in the human enzyme, is identified unequivocally from the density map and offers a unique fingerprint of the murine structure (Figure 17). The aromatic residue sits only 4.7 Å away from K186d in the pore region and 14 Å away from where Na+ binds in the human enzyme. That is about the same distance as W215 in the aryl binding site, which is a major fluorophore reporting Na+ binding to human thrombin (Arosio et al. 2000). W20 in murine thrombin could therefore interfere with the fluorescence change linked to Na+ binding that is difficult to detect even after Na+ activation is partially restored with the K222D mutation (Bush et al. 2006).
Occlusion of the Na+ pore by the side chain of K222 is linked to a conformation similar to the Na+-bound fast form of the human enzyme. That is revealed by inspection of three markers of the allosteric slow→fast transition of thrombin (Pineda et al. 2004a; Di Cera et al. 2007). E192 at the entrance of the active site, S195 in the active site and D189 in the primary specificity pocket are oriented as in the fast form (Figure 18). S195 is within H-bonding distance (3.05 Å) of the catalytic H57 to ensure efficient catalysis (Fuhrmann et al. 2006). The H-bond is not present in the slow form (Pineda et al. 2004a). D189 is optimally oriented to engage the guanidinium group of the Arg of an incoming substrate. These structural signatures of the fast form are observed in murine thrombin in the absence of bound Na+ and provide a molecular explanation for the high catalytic activity of the enzyme independent of Na+ activation. The conformations of D189 and S195 are maintained by H-bonding interactions mediated by water molecules, as in the fast form of the human enzyme (Figure 18). However, only seven water molecules are involved in these interactions, as opposed to Na+ and eleven water molecules in the fast form of human thrombin (Pineda et al. 2004a). The presence of K222 pushes R187 away and closer to D221, whose Oδ2 atom engages the guanidinium NH2 atom of R187 in a 2.55 Å ion-pair. The ion-pair positions the Oδ1 atom of D221 within 2.51 Å of water w153, which sits 3.96 Å away from the Nζ atom of K222. Water w153 is within 2.79 Å of water w51 that occupies a position equivalent (<1 Å away) to the bound Na+ in the fast form and is likewise coordinated by the backbone O atoms of R221a (2.77 Å) and K224 (2.61 Å). The H-bonding network around water w51 mimics that found around the bound Na+ in the human enzyme and establishes a connection to the Oδ2 atom of D189 via an intervening water w97. This and water w55 ensure the optimal orientation of the carboxylate of D189. S195 is fixed in its orientation by a water mediated contact with the Oε1 atom of E192, with water w63 positioned 3.19 Å away from the Oγ atom of S195 and 2.82 Å away from the the Oε1 atom of E192. Unlike the fast form of human thrombin, there is no obvious water mediated linkage between D189 and E192 that would provide connectivity from the bound Na+ to the catalytic S195 (Pineda et al. 2004a). The only two water molecules, w141 and w142, in the active site between D189 and S195 are too far away from either residue. Therefore, the conformations of the critical residues D189 and S195 in murine thrombin have been optimized independently of each other, with K222 sealing the Na+ pore and contributing indirectly to stabilization of the Na+ binding site via waters w153 and w51. The structure of murine thrombin was validated by that of a thrombin chimera where the human enzyme carries all residues around the Na+ pore of murine thrombin. The chimera lacks Na+ activation, and its crystal structure was solved at 1.75 Å resolution in the presence of PPACK and in the absence of Na+. The pore region in the chimera looks practically identical to that of the murine enzyme (Figure 17), with the side chain of K222 occupying the interior of the pore.
Figure 18.
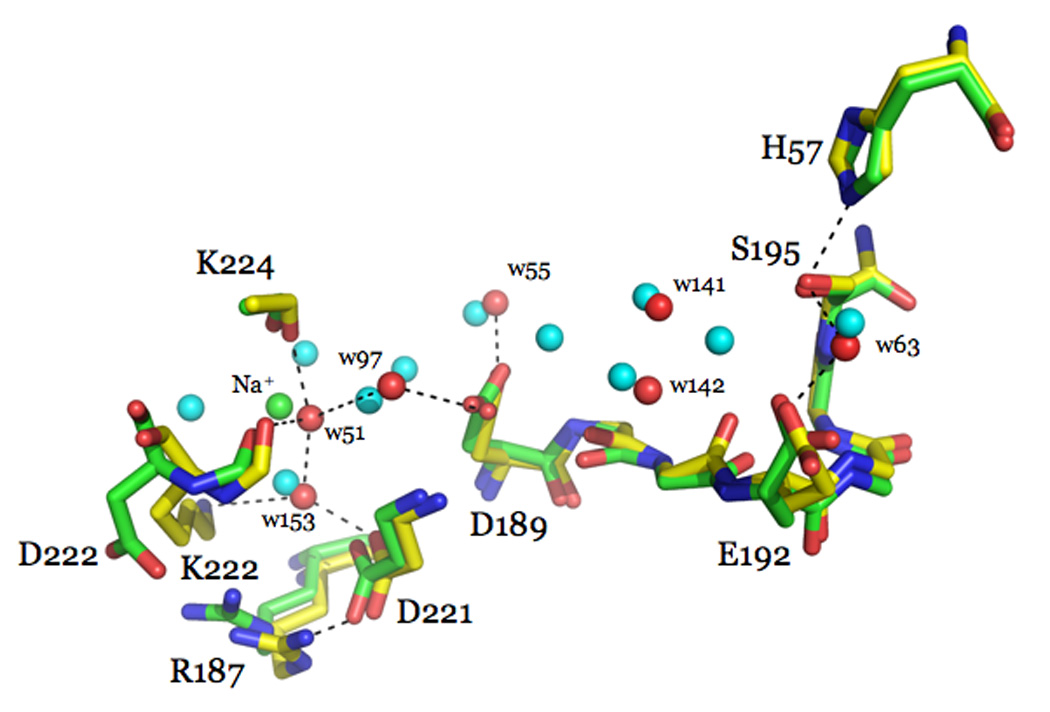
Overlay of key residues in murine thrombin (CPK, with C in yellow) and in the fast form (CPK, with C in green) of human thrombin (Pineda et al. 2004a). H-bonds (broken lines) refer to the murine thrombin structure. The presence of K222 in murine thrombin stabilizes the conformation in a fast-like form. The Oγ atom of the catalytic S195 is within H-bonding distance (3.05 Å) from H57. This H-bond is present in the fast form of the human enzyme (3.09 Å), but is broken (3.70 Å) in the Na+-free slow form (Pineda et al. 2004a). The side chain of D189 in the primary specificity pocket is oriented optimally for coordination of Arg of substrate, as seen in the fast form. The conformations of D189 and S195 are maintained by H-bonding interactions mediated by water molecules, as in the fast form of the human enzyme. However, only seven water moleluces (red balls) are present in this region of the murine thrombin structure, as opposed to Na+ (green ball) and eleven water molecules (cyan balls) present in the fast form of the human enzyme (Pineda et al. 2004a). The presence of K222 in murine thrombin pushes R187 away and closer (2.55 Å) to D221. The Nζ atom of K222 and the Oδ1 atom of D221 H-bond to water w153, which in turn stabilizes water w51 in a position equivalent (<1 Å away) to the bound Na+ in the fast form (green ball) and in contact with the backbone O atoms of R221a (2.77 Å) and K224 (2.61 Å). The H-bonding network around water w51 mimics that seen around the bound Na+ in the fast form of the human enzyme (Pineda et al. 2004a) and establishes a connection to the Oδ2 atom of D189 via water w97. The Oδ1 atom of D189 is held in place by a H-bond with water w55 (2.74 Å). S195 is fixed in its orientation by a water mediated contact with the Oε1 atom of E192, with water w63 positioned 3.19 Å away from the Oγ atom of S195 and 2.82 Å away from the the Oε1 atom of E192. The only two waters molecules, w141 and w142, between D189 and S195 are too far away from either residue. Hence, murine thrombin lacks the connectivity between the primary specificity pocket and the catalytic triad seen in the fast form of the human enzyme.
Murine thrombin has been the subject of detailed genetic analysis in the context of models of human disease (Suh et al. 1995; Sun et al. 1998; Coughlin 2000; Zhang and Ginsburg 2004). However, important differences exist in the functional interactions and regulation of thrombin between mice and humans. In addition to the basic differences uncovered in the role of PARs in platelet activation between mice and humans (Coughlin 2000; Brass 2003), thrombin in the mouse lacks Na+ activation (Bush et al. 2006), which is a hallmark feature of the human enzyme (Di Cera et al. 2007). The lack of Na+ activation in human thrombin would be inconsequential on the ability to cleave the anticoagulant protein C (Dang et al. 1995; Dang et al. 1997a; Di Cera et al. 2007), but would drastically compromise activity toward fibrinogen and the PARs (Dang et al. 1995; Dang et al. 1997a; Ayala et al. 2001; Di Cera et al. 2007) and make the enzyme unable to carry out its procoagulant, prothrombotic and signaling roles in the blood (Di Cera 2003). Murine thrombin compensates for the lack of Na+ activation by mimicking functionally the Na+-bound form of the human enzyme (Bush et al. 2006). That ensures high efficiency in the catalytic activity toward fibrinogen, PAR1 and PAR4, and does not compromise protein C activation. Previous attempts to mimic functionally the activating effect of Na+ in thrombin by replacing G184 with Lys have produced a poorly active enzyme (Roy et al. 2001). Similarly, mimicry of K+ activation in rabbit muscle pyruvate kinase (Laughlin and Reed 1997) and the ATPase domain of Hsc70 (Wilbanks and McKay 1998) have utilized Lys substitutions near the monovalent cation binding site, but in no case did the mutation result in high catalytic activity. The strategy used by murine thrombin is obviously very effective, because the enzyme shows high catalytic activity toward physiologic substrates in the absence of Na+ activation. The side chain of K222 replaces Asp in the human enzyme and occludes the pore of entry to the Na+ binding site. The same architecture is observed in the thrombin chimera where the human enzyme carries all residues of the pore in murine thrombin. The Nζ atom of K222 in murine thrombin is held in place by a H-bonding network involving three residues in the 186-loop, and is ~5 Å away from where Na+ binds in the human enzyme. It is unclear whether the Nζ atom in this position could provide effective electrostatic mimicry of the bound Na+. It is reasonable to assume, however, that the side chain of K222 could penetrate the Na+ pore further to position the Nζ atom closer to the site occupied by Na+. Nonetheless, the presence of K222 seals the entrance to the Na+ site, abrogates sensitivity to monovalent cations (Bush et al. 2006) and generates an environment within the primary specificity pocket that is optimized for substrate binding. That is accomplished through a network of water molecules that mimics the Na+ bound architecture in the proximity of D189. A water mediated contact, similar to that documented in the fast form of the human enzyme, fixes the orientation of the catalytic S195 with the assistance of the side chain of E192. As a result, both S195 and D189 assume conformations like those in the fast form of human thrombin, thereby accounting for the high catalytic activity of the murine enzyme.
Why is murine thrombin devoid of Na+ activation? The reason is most likely an evolutionary advantage to counter the effects of mutations that produce anticoagulant effects by destabilizing Na+ binding. It is known that the C-terminal domain of serine proteases dictated functional diversity during evolution (Krem et al. 1999) and is the locale preferentially targeted by naturally occurring mutations. All residues linked energetically to Na+ binding to thrombin reside in the C-terminal domain of the enzyme (Pineda et al. 2004a) and so do most of the known naturally occurring mutations of the prothrombin gene in humans (Miyata et al. 1992; Degen et al. 1995; Henriksen et al. 1998; Sun et al. 2001; Rouy et al. 2006; Stanchev et al. 2006). It would be important to establish if mutations that cause anticoagulant effects in the human enzyme behave differently in murine thrombin. The Y225P mutation selectively abolishes Na+ binding to human thrombin and compromises fibrinogen and PAR1 recognition indirectly by switching the enzyme to the anticoagulant slow form (Dang and Di Cera 1996; Dang et al. 1997a). Such mutation in murine thrombin should be inconsequential on fibrinogen and PAR1 recognition and preliminary results indicate that this is indeed the case. The W215A/E217A mutant of human thrombin features remarkable anticoagulant and antithrombotic properties in vitro and in vivo (Cantwell and Di Cera 2000; Gruber et al. 2002; Feistritzer et al. 2006; Gruber et al. 2006). The anticoagulant effect of the mutation has been explained structurally in terms of the total collapse of the locale for fibrinogen and PAR1 recognition (Pineda et al. 2004b). The murine W215A/E217A mutant has largely (>1,000-fold) compromised fibrinogen cleavage, but the reduction is almost 20-fold less pronounced than in the human variant (Bush et al. 2006). Cleavage of PAR4 is compromised 600-fold. Notably, cleavage of PAR1 is reduced only 60-fold as opposed to >1,000-fold in the human enzyme. On the other hand, cleavage of protein C is reduced <7-fold, just as for the human variant. Mutation of W215 and E217 affects the activity of murine thrombin toward fibrinogen, PAR1 and PAR4 because these residues make direct contacts with substrate (Dang et al. 1997a; Arosio et al. 2000; Ayala et al. 2001). However, unlike the human enzyme, murine thrombin is spared an additional ~20-fold drop in activity caused by stabilization of the Na+-free slow form upon the Ala replacement of W215 and E217 (Pineda et al. 2004a). Because of the constitutive Na+ activation in murine thrombin, the W215A/E217A mutation is significantly less anticoagulant in the mouse than in humans.
10. Thrombin interaction with protein C
Two well documented pathways of allosteric regulation exist in thrombin: one involves the Na+ site and the other involves exosite I. As we have seen, binding of Na+ to thrombin enhances activity toward procoagulant and prothrombotic substrates like fibrinogen and PARs (Di Cera 2003; Di Cera et al. 2007), whereas binding of thrombomodulin to exosite I enhances activity toward the anticoagulant protein C (Esmon and Mather 1998; Esmon 2003b). A significant linkage also exists between the two allosteric sites (Ayala and Di Cera 1994; Lai et al. 1997; Kroh et al. 2007). Thrombin is the only enzyme in the blood capable of activating protein C (Di Cera 2003). Activated protein C is a natural anticoagulant that inactivates factors Va and VIIIa with the assistance of protein S, thereby promoting the down-regulation of the coagulation cascade (Esmon 2003b). In addition to its anticoagulant role, protein C has recently emerged as a regulator of inflammatory response and as an anti-apoptotic agent (Griffin et al. 2002; Esmon 2003a; Feistritzer et al. 2006). This has in turn expanded the roles of thrombin in blood coagulation and established an intriguing link between thrombosis and inflammation via the protein C pathway.
The interaction of thrombin with protein C has been studied in considerable detail. Under physiologic concentrations of Ca2+ thrombin has only marginal affinity for protein C in the absence of thrombomodulin (Esmon et al. 1983; Wu et al. 1991; Rezaie and Esmon 1992; Dang et al. 1995; Yang et al. 2004). The presence of thrombomodulin increases the Kcat/Km of thrombin for protein C >1,000-fold (Esmon et al. 1983; Wu et al. 1991; Rezaie and Esmon 1992; Dang et al. 1995; Yang et al. 2004). In the absence of Ca2+, thrombomodulin has only a modest effect on protein C activation by thrombin and the reaction proceeds with a Kcat/Km comparable to that observed under physiologic conditions of Ca2+ and thrombomodulin (Esmon et al. 1983; Wu et al. 1991; Rezaie and Esmon 1992; Dang et al. 1995; Yang et al. 2004). Because Ca2+ has no effect on thrombin structure and function (Dang et al. 1995), but changes drastically the conformation of the activation peptide of protein C (Yang et al. 2004), it is reasonable to assume that an important component of thrombomodulin action is to allosterically affect protein C to relieve the inhibitory effect of Ca2+ binding on the conformation of the domain containing the scissile bond. It is unlikely, however, that such an allosteric effect would explain >1,000-fold change in kcat/Km. Recent work from the group of Rezaie has shown that engineering a disulfide bond in the Ca2+ binding loop of the protease domain of protein C removes the Ca2+ effect and produces a protein C derivative that is activated 50-fold better by thrombin in the absence of thrombomodulin (Yang et al. 2006). The thrombin mutant R35E activates this mutant protein C without thrombomodulin with a Kcat/Km comparable to that measured using wild-type thrombin in the presence of wild-type protein C and thrombomodulin (Yang et al. 2006). This shows that the mechanism of thrombomodulin enhancement of protein C activation encompasses at least two components: a change in the conformation of thrombin around R35, and a change in the Ca2+ binding loop of protein C.
A key observation on the kinetic effect of thrombomodulin on protein C activation by thrombin reveals that the 1,000-fold increase in Kcat/Km arises from a 10-fold decrease in Km and a 100-fold increase in Kcat (Esmon et al. 1983). Let us consider the basic kinetic scheme for substrate hydrolysis by serine proteases
 The substrate S interacts with the enzyme E to form a complex with a rate constant k1. The complex can dissociate back into the parent species with a rate constant k−1, or turn over the substrate to give the product P and regenerate the free enzyme E. The rate of product formation is given by k2. The Michaelis-Menten parameters associated with the kinetic scheme 3 are
The substrate S interacts with the enzyme E to form a complex with a rate constant k1. The complex can dissociate back into the parent species with a rate constant k−1, or turn over the substrate to give the product P and regenerate the free enzyme E. The rate of product formation is given by k2. The Michaelis-Menten parameters associated with the kinetic scheme 3 are
| (10) |
| (11) |
From the definition of Km in eq 10, it follows that in order for this parameter to decrease 10-fold when the value of Kcat increases 100-fold, the value of k1 must increase at least 1,000-fold when thrombomodulin binds to thrombin. In other words, the most striking effect of thrombomodulin is expected to influence the rate at which substrate and enzyme come together to form a productive complex. How is this remarkable feat accomplished? Several hypotheses on the action of thrombomodulin have been presented in the literature. Some groups favor the idea that thrombomodulin changes the conformation of thrombin to enable efficient protein C activation. Support to this proposal comes from the observation that binding of thrombomodulin to thrombin alters the spectral properties of probes tethered to the active site of the enzyme (Ye et al. 1991). Furthermore, several mutations of thrombin have been found to enhance protein C activation in the absence, but not in the presence of thrombomodulin (Le Bonniec and Esmon 1991; Rezaie and Yang 2003). These findings suggest that thrombomodulin changes thrombin structure in a way that can be mimicked by ad hoc amino acid substitutions. However, changes in spectral probes are qualitative in nature and may bear little on the mechanism of protein C activation by thrombin. As to the thrombin mutations that increase protein C activation in the absence of thrombomodulin, the effects are small and nowhere close to the large (>1,000-fold) enhancement induced by thrombomodulin. Furthermore, these effects are reproduced entirely by mutations of protein C affecting domains interacting with thrombin (Richardson et al. 1992; Grinnell et al. 1994), thereby raising the alternative possibility that thrombomodulin may affect the structure of protein C rather than thrombin. In contrast to previous claims of a significant effect of thrombomodulin on the active site of thrombin (Ye et al. 1991), analysis of the hydrolysis of several chromogenic substrates by thrombin shows little dependence on thrombomodulin binding (Vindigni et al. 1997). More importantly, the effect is reproduced both qualitatively and quantitatively by hirugen binding to exosite I, the major determinant of thrombomodulin recognition by thrombin (Pineda et al. 2002a), even though hirugen has no effect on the activation of protein C.
Critical to the understanding of the molecular mechanism of protein C activation is information on the value of k1 in the kinetic scheme 3. That value can be obtained from the temperature dependence of the parameter s=Kcat/Km (Vindigni and Di Cera 1996; Ayala and Di Cera 2000; Krem et al. 2002; Pineda et al. 2004a; Pineda et al. 2004c; Bobofchak et al. 2005; Xu et al. 2005), i.e.,
| (12) |
where R is the gas constant, T the absolute temperature, and α=k2/k−1 defines the “stickiness” of substrate as the ratio between k2 (the rate of acylation) and k−1 (the rate of dissociation of the enzyme-substrate complex into the parent species). The Arrhenius terms E1 and Eα=E−1–E2 define respectively the activation energy for substrate binding to the active site of the enzyme and the difference between the activation energies for substrate dissociation and acylation. The values of k1 and α refer to the reference temperature T0=298.15 K. Measurements of s as a function of temperature resolve k1, α, E1 and Eαin eq 12, provided the plot shows curvature. The curvature is indicative of a change in the rate-limiting step for substrate hydrolysis due to the shift from k2>>k−1 at low temperatures, to k−1>>k2 at high temperatures. The shift is caused by the drastic difference in activation energies for substrate acylation and dissociation, when E−1>>E2 or Eα>>0. The rate constant k1 is limited by diffusion (van Holde 2002) or conformational rearrangements that facilitate docking. Its energetic balance is determined by the Arrhenius energy of activation E1. Typically, diffusion-limited enzyme-substrate encounters feature k1 values in excess of 107 M−1s−1 linked to small activation energies (E1 < 10 kcal/mol) (van Holde 2002). Values of E1 that exceed 20 kcal/mol are conducive to structural rearrangements of the enzyme-substrate complex to facilitate productive complex formation and are usually linked to slower (<107 M−1s−1) values of k1. Information on k1 and E1 is therefore quite valuable in establishing mechanisms of enzyme-substrate interaction. In the case of thrombin interacting with protein C, these values can be used to establish the effect of thrombomodulin on how protein C docks on the thrombin active site to enable productive binding and activation. Measurements of k1 confirm that thrombomodulin enhances diffusion of protein C into the active site of thrombin >1,000-fold with a small but significant (4 kcal/mol) decrease in the energy of activation E1 (Xu et al. 2005). The larger value of E1 in the absence of thrombomodulin indicates substantial structural strains linked to formation of the thrombin-protein C complex. The strains are partially relieved by the presence of cofactor. The main function of thrombomodulin is to facilitate diffusion of protein C into the thrombin active site. An ancillary function is to induce changes in the molecular surface of recognition between thrombin and protein C.
Extensive Ala-scanning mutagenesis of thrombin has revealed the functional epitopes for protein C recognition in the absence or presence of thrombomodulin (Figure 19) (Xu et al. 2005). There are significant differences in the structure of the epitopes that underscore molecular changes in the thrombin-protein C interface due to cofactor binding. In the absence of thrombomodulin, there are several residues of thrombin critically involved in protein C recognition. These residues cluster around the Na+ site (T172, T184a, K224, Y225, G226), and define the primary specificity pocket (D189), the oxyanion hole (G193) and portions of the aryl binding site in the 60-loop (Y60a). A striking property of the epitope is that it contains residues that once mutated to Ala produce a small but significant enhancement of protein C activation. These residues are R35 and P37 in the 30-loop and F60h at the distal end of the 60-loop. Interestingly, these residues lie close to each other (blue in Figure 19) and define a patch on the surface of thrombin next to the active site where docking of protein C is unfavorable in the wild-type. The important role of R35 has been emphasized (Hall et al. 1999; Rezaie and Yang 2003; Yang et al. 2006), together with the unfavorable contribution of other residues in the 30-loop like S36a, Q38 and E39 (Hall et al. 1999). The spatial contiguity of F60h, R35 and P37 suggests that a domain of protein C experiences unfavorable contacts with this region upon binding to thrombin. Indeed, Grinnell et al. (Grinnell et al. 1994) have identified a cluster of positively charged residues in the primed sites of protein C (K174, R177 and R178) that once mutated to Glu produces a mutant that is activated 16-fold faster by thrombin in the absence but not in the presence of thrombomodulin. They suggested that the cluster experiences electrostatic repulsion with the positively charged region of exosite I of thrombin. Given that exosite I makes no contribution to protein C recognition (Figure 19), it is likely that some of these positively charged residues of protein C clash with R35 and the 30-loop of thrombin. The scenario changes significantly when thrombomodulin binds to thrombin. Recognition of protein C by thrombin involves a smaller set of residues compared to the absence of cofactor. Practically only D189 in the primary specificity pocket and Y60a in the aryl binding site make a dominant contribution to binding. The critical role played by the oxyanion hole (G193) and the Na+ site (T172, Y184a, K224, Y225, G226) is diminished when thrombomodulin binds. The unfavorable contribution from residues in the 30-loop and the distal portion of the 60-loop disappears in the presence of thrombomodulin. Because binding of thrombomodulin depends little on residues located in the 30-loop, the 60-loop, the oxyanion hole and the Na+ site (Pineda et al. 2002a; Xu et al. 2005), the change in the epitope is not due to hijacking of these domains by the cofactor. The drastic difference in the epitopes is due to the mode of interaction of protein C with thrombin and proves that the thrombin-protein C interface changes when thrombomodulin is bound: the interface is reduced to the primary specificity pocket and the aryl binding site around Tyr-60a in the presence of cofactor.
Figure 19.

Space filling model of thrombin in the Bode orientation (active site, center; exosite I, right; exosite II, top left; Na+ site, bottom left; 60-loop, top) depicting the structural organization of the epitopes recognizing protein C in the absence (left) or presence (right) of thrombomodulin. Residues affected by Ala replacement are color coded based on the log change in the value of s=Kcat/Km for protein C activation: blue, −1.5/−0.5 units (1/30- to 1/3- fold change in s); cyan, −0.5/0.5 units (1/3- to 3- fold change in s); green, 0.5/1.5 units (3- to 30- fold change in s); yellow, 1.5/2.5 units (30- to 300-fold chage in s); red, >2.5 units (>300-fold change in s). Residues not subject to Ala-scanning mutagenesis are in gray. Crucial residues are labeled. The epitope in the absence of thrombomodulin is split into one domain providing favorable interactions (residues in yellow and red: visible are Y60a, T172, D189, G193 and K224) and a second domain providing steric/electrostatic hindrance (residues in blue: from top to bottom, F60h, R35 and P37). In the presence of thrombomodulin, the shape of the epitope changes drastically and only Y60a and D189 make significant contribution to protein C recognition. The region of unfavorable contributions to recognition (residues in blue at left) disappears.
Thrombomodulin promotes diffusion of protein C into the thrombin active site, which is indicated eloquently by the drastic change in the value of k1 when thrombomodulin binds. Additional contributions come from conformational rearrangements of either thrombin or protein C as evidenced by the small, but significant decrease in the value of E1. The effect of thrombomodulin on the molecular surface of recognition between thrombin and protein C are felt mainly at the Na+ site region and at the 30-loop. Are these predictions consistent with structural investigation of the thrombin-protein C interaction? The structure of thrombin bound to a fragment of thrombomodulin at exosite I failed to reveal significant conformational changes in the active site or other regions (Fuentes-Prior et al. 2000). Such changes might have been obliterated by the presence of the active site inhibitor used in the crystallization. A number of peptides targeting exosite I influence allosterically the active site of thrombin, bring about significant changes in activity and even substrate specificity (Liu et al. 1991; Parry et al. 1993; Ayala and Di Cera 1994; Lai et al. 1997; Vindigni et al. 1997). One of these peptides, hirugen, is derived from the C-terminal fragment of hirudin. The structure of thrombin bound to hirugen was solved with the active site free (Vijayalakshmi et al. 1994), but again failed to reveal any significant conformational changes as for the thrombomodulin-bound structure (Fuentes-Prior et al. 2000). We are therefore faced with a conundrum. Although a plethora of functional data vouch for extensive alterations of thrombin upon thrombomodulin or hirugen binding to exosite I, structural investigation fails to document any significant conformational change when thrombin is bound to exosite I. Solution to this puzzle has come unexpectedly from studies of how thrombin recognizes the receptors PAR1, PAR3 and PAR4.
11. Thrombin interaction with the PARs
The prothrombotic role of thrombin depends mainly on cleavage of protease-activated receptors (PARs), that are members of the G-protein-coupled receptor superfamily (Coughlin 2000; Brass 2003). Four PARs have been cloned and they all share the same basic mechanism of activation: thrombin, and other proteases derived from the circulation and inflammatory cells, cleave at a specific site within the extracellular N-terminus to expose a new N-terminal tethered ligand domain, which binds intramolecularly and activates the cleaved receptor (Coughlin 2000). Thrombin activates PAR1 (Vu et al. 1991), PAR3 (Ishihara et al. 1997; Sambrano et al. 2001) and PAR4 (Kahn et al. 1998; Xu et al. 1998; Nakanishi-Matsui et al. 2000) in this manner. Human platelets express PAR1 and PAR4 that mediate signaling and aggregation at low and high thrombin concentrations, respectively, and with distinct temporal characteristics (Shapiro et al. 2000). By contrast, murine platelets express PAR3 and PAR4. Upon cleavage by thrombin, PAR3, rather than itself mediating transmembrane signaling, functions as a cofactor that supports cleavage and activation of PAR4 at low thrombin concentrations (Kahn et al. 1998; Nakanishi-Matsui et al. 2000). On endothelial cells, PAR3 assists activation of PAR1 by promoting receptor heterodimerization (McLaughlin et al. 2007).
We have already illustrated the role of exosite I, located 20–25 Å away from the active site moiety, as a binding epitope for fibrinogen (Tsiang et al. 1995; Ayala et al. 2001; Pechik et al. 2004; Pechik et al. 2006), thrombomodulin (Tsiang et al. 1995; Hall et al. 1999; Fuentes-Prior et al. 2000; Pineda et al. 2002a; Xu et al. 2005), and the thrombin receptors PAR1 (Vu et al. 1991; Mathews et al. 1994; Ayala et al. 2001; Myles et al. 2001a) and PAR3 (Ishihara et al. 1997; Ayala et al. 2001). The potent natural inhibitor hirudin also targets exosite I via its extended, acidic C-terminal domain (Rydel et al. 1991). An hirudin-like acidic domain is present in PAR1 and PAR3 immediately downstream of the tethered ligand domain and has been invoked in the ability of these receptors to engage exosite I of thrombin (Vu et al. 1991; Kahn et al. 1998). Recognition of PAR4 is less dependent on exosite binding and relies almost entirely on the active site moiety (Ayala et al. 2001), especially through a pair of Pro residues at the P2 and P4 positions (Cleary et al. 2002; Jacques and Kuliopulos 2003). In addition, PAR4 constructs carrying an hirudin-like acidic domain fail to produce enhanced binding or catalytic processing (Jacques and Kuliopulos 2003), suggesting that PAR4 folds into a conformation unable to bind to exosite I. The cofactor function of PAR3 on PAR4 cleavage and activation by thrombin in murine platelets (Kahn et al. 1998; Nakanishi-Matsui et al. 2000) implies binding of both PAR4 and the cleaved form of PAR3 to thrombin to form a ternary complex. After cleavage of PAR3, thrombin would remain bound to the receptor via exosite I. This complex would then engage PAR4 for proteolytic activation optimized by an allosteric effect of PAR3 bound to exosite I on the active site moiety. A similar mechanism likely exists in human platelets where PAR4 can be co-factored by other receptors (Kahn et al. 1998). The cofactor function of PAR3 on PAR4 cleavage and activation has many features in common with the interaction of thrombin with protein C that is cofactored by thrombomodulin. Structural studies of thrombin-PAR interactions were therefore undertaken with the expectation that they might reveal important details on the allosteric mechanism underlying binding to exosite I.
Murine thrombin inactivated with the single site mutation S195A was crystallized in complex with the extracellular fragment of murine PAR3 (Bah et al. 2007), 38SFNGGPQNTFEEFPLSDIE56, corresponding to the sequence downstream of the cleavage site at K37 and containing the hirudin-like motif 47FEEFP51 predicted to bind to exosite I (Nakanishi-Matsui et al. 2000; Jacques and Kuliopulos 2003). Although the structure was solved at relatively high resolution (2.0 Å), only the sequence 44QNTFEEFPLSDIE56 of the extracellular fragment of PAR3 could be detected in the electron density map. The PAR3 fragment engages exosite I of murine thrombin in a number of well-defined interactions starting with F47 (Figure 20). The three residues 44QNT46 fold away from the thrombin surface, as seen in the NMR structure of a cleaved fragment of PAR1 (Seeley et al. 2003). The aromatic ring of F47 is edge-to-face with the aromatic ring of F34 of thrombin. On the opposite side of F34 lies the side chain of F50, also oriented perpendicular to the benzene ring of the thrombin residue. F50 is at the center of a hydrophobic cluster that includes L52 and I55 of PAR3 in van der Waals interactions. The cluster docks on an extended hydrophobic patch of exosite I comprising F34, L65, Y76 and I82. In addition to the substantial hydrophobic contacts at the thrombin-PAR3 interface, the complex is stabilized by electrostatic and polar interactions. The backbone N atom of G48 engages the backbone O atom of T74 of thrombin and the Oε1 atom forms an ion-pair with the Nε atom of R75 and an H-bond with the backbone N atom of Y76 of thrombin. D54 uses its carboxylate oxygens to contact R77a of thrombin via the NH2 and Nε atoms. Finally, E56 engages the Nζ atom of K110 of thrombin via its C-terminal carboxylate atoms and H-bonds to the O atom of I82 via its N atom. Overall, the surface of interaction between thrombin and PAR3 is composed of a hydrophobic patch surrounded by a periphery of polar/electrostatic contacts. The interactions involve the hirudin-like motif 47FEEFP51 predicted to bind to exosite I (Nakanishi-Matsui et al. 2000; Jacques and Kuliopulos 2003), but also the sequence 52LSDIE56 that internally stabilizes the fold of the bound PAR3 and provides additional polar contacts with the thrombin surface.
Figure 20.
A–D. Structures of murine thrombin in complex with fragments of murine PAR3 (A,C) and PAR4 (B,D). (A,B) Thrombin is rendered in surface representation (wheat) with the residues <4 Å from the bound fragment of PAR3 or PAR4 (stick model) colored in light blue. The orientation is centered on exosite I (A) or the active site (B). The orientation in (A) is obtained from (B) by ~90° rotation along the y-axis. Electron density maps (green mesh) are contoured at 1.0 (B) or 0.7 (D) σ. (C,D) Details of the molecular contacts at the thrombin-PAR interface, with hydrophobic regions of the thrombin epitope colored in orange and polar regions colored in light blue. H-bonds are depicted as broken lines. Residues involved in contacts <4 Å are listed in Table 1 and are labeled in black for thrombin and red for PAR. (C) The cleaved fragment of PAR3 engages exosite I through polar and hydrophobic interactions. (D) The fragment of PAR4 comprising the scissile bond makes extensive interactions with the active site moiety of the enzyme utilizing the P56 and P58 clamp at the P2 and P4 positions. After exiting the active site, the fragment folds into a short helical turn and is deflected away from exosite I and to the autolysis loop right below the active site region.
Comparison with the structure of the murine thrombin-PAR3 complex (Bah et al. 2007) with that of murine thrombin in the free form (Marino et al. 2007) reveals an enzyme conformation that is largely unmodified except for one notable feature. The presence of PAR3 bound to exosite I causes a rearrangement of the 60-loop lining the upper rim of the active site entrance (Figure 21). In the free enzyme, and in human thrombin (Bode et al. 1992; Pineda et al. 2004a), the indole ring of W60d partially occludes access to the active site and restricts specificity toward physiologic substrates and inhibitors (Bode 2006). When PAR3 binds to exosite I, the 60-loop shifts 3.8 Å upward and causes a 180° flip of W60d that projects the indole ring into the solvent and opens up the active site fully. Interestingly, cleaved PAR3 binds to exosite I of murine thrombin in a conformation almost identical to that determined for hirugen bound to exosite I of the human enzyme (Vijayalakshmi et al. 1994). The conformation and molecular contacts of the hirudin-like motif 47FEEFP51 are practically identical to those of the sequence 56FEEIP60 of hirugen. Yet, binding of hirugen has minimal effects on the position of the 60-loop of thrombin and W60d retains an orientation that partially occludes the active site cleft and screens the catalytic H57 from solvent (Vijayalakshmi et al. 1994). On the other hand, the structure of murine thrombin bound to cleaved PAR3 at exosite I reveals the basis of the allosteric communication between exosite I and the active site cleft. Key to this allosteric effect is the ability of W60d to move like a flap and regulate substrate diffusion into the active site. The structural flexibility documented for this and other Trp residues of thrombin is in agreement with recent spectroscopic measurements (Bah et al. 2006).
Figure 21.

Allosteric effect induced by binding of the cleaved fragment of murine PAR3 (stick model in gold) to exosite I of murine thrombin (ribbon model in light green) on the conformation of the 60-loop (blue) and the position of W60d. Comparison with the free structure of murine thrombin (ribbon model in wheat, with 60-loop and W60d in red) shows a significant upward shift of the 60aYPPWDK60f region of the 60-loop and a 180° flip of the indole ring of W60d. The change produces complete aperture of the active site to facilitate substrate diffusion. The allosteric communication between exosite I and the 60-loop documented in the thrombin-PAR3 structure may be relevant to the interaction of thrombin with other exosite I ligands, like thrombomodulin.
The murine thrombin mutant S195A was also crystallized in complex with the extracellular fragment of murine PAR4 (Bah et al. 2007), 51KSSDKPNPR↓GYPGKFCANDSDTLELPASSQA81 (↓=site of cleavage), containing the cleavage site at R59. Although the structure was solved at low resolution (3.5 Å), twenty-six residues of the fragment from K51 to P76 could be traced in the electron density map. Overall, the thrombin conformation is similar to that seen in the thrombin-PAR3 complex (rmsd 0.52 Å), but W60d retains its canonical orientation with the indole ring pointing down over the active site (Figure 20) in contact with PAR4. Of great importance are the contacts made by PAR4 with the active site of thrombin. R59 penetrates the primary specificity pocket and engages several residues. The guanidinium group makes a strong bidentate ion-pair with the carboxylate oxygens of D189 and three H-bonds with the backbone O atoms of G216, E219 and A190. The backbone O atom of R59 makes a short H-bond with the N atom of G193 in the oxyanion hole and the backbone N atom H-bonds to the backbone O atom of S214. These are the canonical interactions expected for Arg of substrate in the specificity pocket of a trypsin-like protease (Perona and Craik 1995; Hedstrom 2002; Page et al. 2005). Extensive interactions are also present away from the primary specificity pocket. P58 at the P2 position of substrate makes numerous van der Waals contacts with the catalytic H57, Y60a and W60d in the 60-loop. Its backbone O atom could experience electrostatic repulsion from the Oε2 atom of E192 that is 3.4 Å away. The P3 residue N57 points away from the thrombin surface with its side chain, as expected for an amino acid in the L-enantiomer at this position (Bode et al. 1992), but its backbone N and O atoms H-bond to the O and N atoms of G216 again in compliance with the canonical antiparallel mode of interaction of substrate with the active site of serine proteases (Perona and Craik 1995; Hedstrom 2002; Page et al. 2005). The P4 residue P56 makes numerous van der Waals interactions with the aryl binding site composed of L99, I174 and W215 with which it makes a strong edge-to-face interaction similar to that seen for the H-D-Phe residue of the inhibitor PPACK (Bode et al. 1992; Pineda et al. 2004a). Together, P56 and P58 produce a clamp that latches PAR4 onto the active site of thrombin utilizing a large hydrophobic surface from the aryl binding site on one side and the 60-loop on the opposite side of the catalytic H57. The conformation of the 56PNPR59 sequence of PAR4 is practically identical to that predicted by NMR studies of the human PAR4 sequence 44PAPR47 (Cleary et al. 2002) and supports the critical role of this Pro pair in thrombin recognition uncovered by functional studies (Jacques and Kuliopulos 2003). The P5 residue K55 leaves the active site moiety, although a slight conformational change could put it in electrostatic coupling with E97a of thrombin. Upstream of K55, D54 at P6 makes a long H-bond with the guanidinium group of R173 before the rest of the PAR4 peptide leaves the thrombin surface.
Downstream from the scissile bond, the P1’ residue G60 initiates a turn followed by a short helical segment stabilized by a H-bond between the O atom of P62 and the N atom of C66. The helix turn is an intrinsic property of PAR4 because the conformation of the fragment in this region is not influenced by any packing interactions with symmetry related molecules. Without P62, the fragment of PAR4 would likely pursue the 30-loop and exosite I of thrombin. This suggests obvious future experiments of mutagenesis with PAR4. The helix turn stabilizes and redirects PAR4 toward the autolysis loop right below the entrance to the active site. The mode of interaction of PAR4 with thrombin calls for numerous interactions with the active site and the aryl binding site, leaving exosite I and the 30-loop free. Cleavage of PAR4 does not require binding to exosite I of thrombin, in agreement with earlier mutagenesis studies (Nakanishi-Matsui et al. 2000; Jacques and Kuliopulos 2003). The structured helix of PAR4 makes it difficult to predict whether introduction of an hirudin-like sequence downstream of the scissile bond would confer affinity toward exosite I. Contrasting results have been reported in this connection (Nakanishi-Matsui et al. 2000; Jacques and Kuliopulos 2003). If the helix remains rigid, the backbone downstream from C66 should be directed toward the autolysis loop and not exosite I. However, insertion of a properly spaced hirudin-like sequence could endow PAR4 with exosite I binding by unraveling the helix turn. Finally, the presence of the short helical turn includes residues of the tethered ligand domain 60GYPGKF65 (Kahn et al. 1998; Xu et al. 1998; Nakanishi-Matsui et al. 2000) and suggests that the activation peptide may bind to the receptor utilizing a structured conformation rather than an extended sheet. The strategy used by PAR4 to contact the active site of thrombin in a way that avoids interaction with exosite I is highly relevant to the interaction of thrombin with protein C, for which no structural information is currently available. The obvious conclusion to be drawn from the structures of murine thrombin bound to PAR3 and PAR4 is that the cleaved form of PAR3 bound to exosite I does not interfere with binding of the intact PAR4 to thrombin. The cleaved PAR3 and intact PAR4 fragments bind to thrombin without overlap and can generate a ternary complex where PAR3 functions as a cofactor that allosterically optimizes PAR4 cleavage (Figure 22).
Figure 22.

Putative ternary complex of thrombin, cleaved PAR3 and PAR4 derived from an overlay of the crystal structures of the murine thrombin-PAR4 and thrombin-PAR3 complexes. Thrombin (green) refers to the thrombin-PAR4 complex. Binding of cleaved PAR3 (gold) to exosite I does not interfere with binding of PAR4 (red) to the active site of the enzyme. Cleaved PAR3 may therefore act as a cofactor of PAR4 cleavage by thrombin.
The role of exosite I as a locale for cofactor binding to thrombin is well documented. A number of peptides targeting exosite I influence allosterically the active site of thrombin, bring about significant changes in activity and even substrate specificity (Liu et al. 1991; Parry et al. 1993; Ayala and Di Cera 1994; Lai et al. 1997; Vindigni et al. 1997). On the surface of platelets, the cofactor role of PAR3 may exploit the allosteric communication between exosite I and the active site, or simply manifest itself as a favorable anchoring of thrombin that reduces the cost of diffusion when PAR4 is targeted for cleavage. The structure of thrombin bound to PAR3 reveals a snapshot of the mechanism for the allosteric communication in terms of a shift in the indole ring of W60d and upward movement of the entire 60-loop that open up the active site cleft. That facilitates substrate diffusion into the active site and enhances Kcat/Km values for hydrolysis (Ayala and Di Cera 1994). A similar mechanism may exist upon thrombomodulin binding to thrombin, because the large change in specificity elicited toward protein C is due, as we have seen, to enhanced substrate diffusion into the active site (Xu et al. 2005). The structure of murine thrombin bound to cleaved PAR3 refutes a long held assumption on the rigidity of the 60-loop (Bode et al. 1992), which was first called into question by the structure of the thrombin mutant E192Q bound to the Kunitz inhibitor BPTI that forced its way into the active site of the enzyme (van de Locht et al. 1997). This documented a significant departure from the modest mobililty seen in previous structures (Vijayalakshmi et al. 1994; Malkowski et al. 1997). The structure of the thrombin-PAR3 complex offers convincing evidence that the position of W60d and the 60-loop can be modified allosterically by binding to exosite I when the active site of thrombin is free. A recent structure of thrombin bound to PAR1 provides crystallographic evidence of a much larger conformational change experienced by thrombin when exosite I is bound to a ligand (Gandhi et al. 2008). The findings reveal the full extent of structural flexibility accessible to this important allosteric enzyme and a precise pathway of long-range communication.
Human thrombin inactivated with the single site mutation D102N was crystallized in complex with the extracellular fragment of human PAR1 (Gandhi et al. 2008), 42SFLLRNPNDKYEPFWEDEEKN62, corresponding to the sequence downstream from the cleavage site at R41 (Vu et al. 1991) and containing the hirudin-like motif 52YEPFWE57 predicted to bind to exosite I (Vu et al. 1991; Coughlin 2000). Although the structure was solved at a resolution of 2.2 Å, only the sequence 49NDKYEPFWE57 of the extracellular fragment of PAR1 could be traced in the electron density map, leaving the tethered ligand domain 42SFLLRN47 and the acidic tail of the fragment unresolved. A similar problem was discussed for the cleaved fragment of murine PAR3 bound to exosite I of murine thrombin (Bah et al. 2007). The tethered ligand domain presumably folds away from the thrombin surface after cleavage at R41 and its conformation becomes disordered, as documented by NMR studies (Seeley et al. 2003). As seen for the binding epitope of PAR3 (Bah et al. 2007), the PAR1 fragment engages exosite I of thrombin in a number of polar and hydrophobic interactions starting with D50 whose Oδ1 atom H-bonds to the NH1 and NH2 atoms of R73 and the Nζ atom of K149e of thrombin (Figure 23). Other polar interactions involve the O and OH atoms of Y52 of PAR1 with the Nε2 atom of N38, the NH1 atom of R73 and the N atom of L40 of thrombin. The Oε1 and N atoms of E53 H-bond to the N atom of Y76 and the O atom of T74 of thrombin, respectively. Finally, a weak H-bond couples the N atom of W56 to the OH atom of Y76 of thrombin. The hydrophobic interactions are substantial and involve F34, L65 and I82 that accommodate in a cluster Y52, F55 and W56 of PAR1. F34 is edge-to-face with respect to Y52 on one side and F55 on the other side. F55 is in van der Waals contact with the entire hydrophobic cluster in exosite I and is also engaged in a cation-π interaction by R67 of thrombin. W56 interacts with I82 of thrombin at the boundary of the binding epitope. The contacts documented by the crystal structure are in agreement with the results of Ala scanning mutagenesis of thrombin (Ayala et al. 2001). In particular, mutagenesis studies have pointed out the peculiar contribution of R67 to PAR1 recognition. The guanidinium group of R67 is within 4.2 Å from the benzene ring of F55 of PAR1, in optimal cation-π interaction, but not so relative to F50 of PAR3 (Bah et al. 2007). That could explain why the R67A mutation is far more deleterious for PAR1 binding relative to PAR3 (Ayala et al. 2001). Ala scanning mutagenesis of PAR1 have singled out Y52, E53 and F55 as important determinants of recognition by exosite I (Vu et al. 1991), in agreement with the results of the crystal structure. However, the important role of D50 of PAR1 in the interaction with thrombin has been missed by previous mutagenesis studies (Vu et al. 1991; Ayala et al. 2001).
Figure 23.

A–B. Structure of the human thrombin mutant D102N in complex with the extracellular fragment of human PAR1. (A) Thrombin is rendered in surface representation (wheat) with residues <4 Å from the bound fragment of PAR1 (stick model) colored in light blue. The orientation is centered on the 30-loop that separates exosite I on the right from the active site cleft on the left. The 60-loop occupies the upper rim of the active site. The electron density 2F0-Fc map (green mesh) is contoured at 1.0 σ. (B) Details of the molecular contacts at the thrombin-PAR1 interface, with hydrophobic regions of the thrombin epitope colored in orange and polar regions colored in light blue. H-bonds are depicted as broken lines. Residues involved in contacts <4 Å are listed in Table 1 and are labeled in black for thrombin and red for PAR1. The extracellular fragment of PAR1 engages exosite I through polar and hydrophobic interactions.
A previous structure of thrombin bound to a fragment of PAR1 revealed a non-productive binding mode bridging two thrombin molecules in the crystal lattice without any significant conformational change in the enzyme (Mathews et al. 1994). The structure of the D102N-PAR1 complex shows the fragment of PAR1 recognizing exosite I basically in the same conformation as reported previously (Mathews et al. 1994). However, the structure documents a large conformational change that propagates from F34 and R73 in exosite I, to W215 in the aryl binding site and R221a in the 220-loop located up to 28 Å away on the opposite side of the active site relative to exosite I. The conformational change is revealed by comparison of the structure of D102N bound to the fragment of PAR1 at exosite I with that of the free mutant solved recently (Pineda et al. 2006). The free mutant D102N folds in a self-inhibited conformation with the active site occluded (Figure 16 and Figure 24). Binding of PAR1 to exosite I restores an active conformation utilizing a long-range allosteric communication between exosite I and the active site.
Figure 24.
Allosteric effect induced by binding of the extracellular fragment of PAR1 (stick model in gold) to exosite I of thrombin (ribbon model in light green) on the conformation of the 215–219 β-strand and the 220-loop (blue). The position of W215 and R221a is indicated as a stick model. Thrombin is shown in the standard Bode orientation (Bode et al. 1992) with the active site cleft in the middle and exosite I to the right. Comparison with the free structure of thrombin (ribbon model in wheat, with the 215–219 β-strand and the 220-loop, W215 and R221a in red) shows a drastic rearrangement that pushes the 215–219 β-strand back >6 Å. W215 and R221a relocate >9 Å to restore access to the active site and primary specificity pocket that was obliterated in the free form‥ The allosteric communication between exosite I and the 215–219 β-strand and 220-loop spans almost 30 Å across the thrombin molecule (see also Figure 25) and reveals a possible mechanism for the conversion of thrombin from its inactive form E* into the active form E.
The allosteric communication is triggered by a change in the conformation of F34 and R73 in exosite I. The Cε2 atom of F34 is 4.3 Å away from the S atom of M32 in the free form of D102N. Upon binding of PAR1 to exosite I, the phenyl ring of F34 rotates to optimize its interaction with F55 and Y52 of PAR1 and pulls the Cε atom of M32 along causing a 5.6 Å shift (Figure 25). The indole ring of W141 switches places with M32 and causes a >1 Å concerted translation of the entire 141–146 β-strand. Contributing to this shift is the repositioning of R73 for engagement of D50 and Y52 of PAR1 that pushes Q151 toward N143 causing electrostatic clash between the side chain O atoms coming within 2.96 Å. P152 pushes G142 back contributing to the realignment of the 141–146 and 191–193 β-strands that become stabilized by H-bonds between the backbone N atom of N143 with the Oε1 atom of Q151 on one side and the backbone O atom of E192 on the other side (Figure 25). The latter H-bond flips back the peptide bond with G193 into the position seen in the wild-type and reconstitutes the oxyanion hole with the correct orientation of the backbone N atom. As the 141–146 β-strand is pushed back, the backbone O atom of N143 collides with the S atom of C220. As a result, the C191:C220 disulfide bond twists and relocates 4.29 Å deeper inside the protein causing a major rearrangement in the backbone of the 215–219 β-strand and the 220-loop. The shift in the 141–146 β-strand initiated at W141 and amplified by R73 propagates to the hinge region of the flexible autolysis loop and orders the side chain of E146 for a strong ion-pair with R221a in the 220-loop, whose guanidinium group moves >9 Å from the interior of the primary specificity pocket to the surface of the protein. The entire 215–219 β-strand moves back >6 Å and W215 re-establishes its stacking interaction with F227 moving >10 Å away from the catalytic H57, thereby restoring access to the active site. D221 restores its important ion-pair interaction with R187 whose guanidinium group moves away from the Na+ binding pocket.
Figure 25.
Molecular basis of the allosteric communication between exosite I and the 215–219 β-strand and 220-loop spanning almost 30 Å across the thrombin molecule (see also Figure 24). The extracellular fragment of PAR1 is rendered as a stick model with C atoms in yellow and the thrombin residues in the PAR1 bound form are rendered as stick models with C atoms in green. Residues of the free form of thrombin are rendered as stick models uniformly colored in light green. Relevant H-bonds are indicated as broken lines. The allosteric communication initiates with a rotation of the benzene ring of F34 and a shift in the side chain of R73 (labeled in black, as all other thrombin residues) in exosite I due to binding of PAR1 via F55, Y52 and D50 (labeleled in red). The changes propagate to the 141–146 β-strand via M32 and Q151. In turn, that re-establishes H-bond connections with the 191–193 β-strand, restores the oxyanion hole and the orientation/location of the C191:C220 disulfide bond, which relocates the entire 215–219 β-strand and 220-loop in their canonical positions to free access to the active site and the primary specificity pocket. W215 folds back almost 10 Å into the aryl binding site, relinquishing its hydrophobic interaction with the catalytic H57 (the catalytic D102 and S195 are also shown for completeness). R221a leaves the interior of the primary specificity pocket where it binds to D189 (not shown) and moves >9 Å to the surface to engage E146 in a strong bidentate ion-pair. E146 is disordered in the free structure. Additional changes involving the 186-loop restoring access to the Na+ binding site are omitted for clarity. The allosteric communication documented in the structure of the thrombin-PAR1 complex relative to the free form of the enzyme is testimony to the flexibility of the thrombin fold and prove that the various forms of the enzyme (E*, E and E:Na+) interconvert under the influence of ligand binding to distinct domains.
The peculiar self-inhibited conformation documented originally for D102N (Pineda et al. 2006) and pertaining to the E* form can therefore convert into a catalytically active conformation through a structural transition that can be traced to a set of residues organized in four layers (Figure 25). A first layer directly in contact with ligands recognizing exosite I (F34 and R73), a second layer of “transducing” residues connecting to the 141–146 β-strand (M32 and Q151), a third layer comprising the interactions between the 141–146 and 191–193 β-strands (W141, N143 and E192) and a final layer where such interactions are transmitted to the 215–219 β-strand and the 220-loop via the C191:C220 disulfide bond and E146. The conformational change documented in the structure of the thrombin mutant D102N bound to a fragment of PAR1 at exosite I differs markedly from that uncovered for PAR3 binding, which opens the question as to the true extent of conformational perturbation induced by binding to exosite I. Consideration of the distinct conformational states that thrombin assumes when free and bound to Na+ rationalizes the differences. The structural changes in the 60-loop documented upon PAR3 binding to exosite I of murine thrombin (Bah et al. 2007) pertain to a conformation of the enzyme that is stabilized in the highly active E:Na+ form (Bush et al. 2006; Marino et al. 2007). As we have seen, murine thrombin is constitutively stabilized in the E:Na+ form by the D222K replacement in the Na+ site, where K222 provides molecular mimicry of the bound Na+ (Marino et al. 2007). On the other hand, the larger structural effects caused by binding of PAR1 to exosite I in the D102N mutant of human thrombin pertain to the E* conformation of the enzyme that is severely compromised for both substrate and Na+ binding. Binding to exosite I in the E:Na+ form would cause a shift in the 60-loop to open the active site fully. Binding to exosite I in the E* form would cause a larger conformational transition with a long-range communication that can be traced from exosite I to the opposite side of the molecule almost 30 Å away. As a result of this drastic conformational change induced allosterically by binding to exosite I, access to the active site is fully restored and the self-inhibited E* conformation of D102N is converted into the active form E.
12. Dissociating procoagulant and anticoagulant activities
The multifunctional nature of thrombin has long motivated interest in dissociating its procoagulant and anticoagulant activities (Griffin 1995). Thrombin mutants with anticoagulant activity help rationalize the phenotypes of several naturally occurring mutations and could eventually provide new tools for pharmacological intervention (Bates and Weitz 2006). The group of Sadler first reported that mutation of R75 to Glu in exosite I had normal fibrinogen clotting activity but only 7% activity toward protein C in the presence of thrombomodulin, whereas mutation of K60f to Glu in the 60-loop had 2.5-fold increased protein C activation and 17% activity toward fibrinogen (Wu et al. 1991). More pronounced anticoagulant effects were reported by the group of Leung with the E217A and E217K mutations, that significantly shifted thrombin specificity toward protein C relative to wild type (Gibbs et al. 1995; Tsiang et al. 1996). This group also provided the first proof of principle on the efficacy of such anticoagulant thrombin mutants in vivo (Gibbs et al. 1995; Tsiang et al. 1996) and pioneered systematic Ala scanning mutagenesis of the epitopes recognizing fibrinogen and protein C.
A comprehensive library of Ala mutants was recently utilized to map the epitopes recognizing fibrinogen (Di Cera et al. 2007), protein C (Xu et al. 2005) and thrombomodulin (Xu et al. 2005) in order to identify residues that contribute differentially to the procoagulant and anticoagulant functions of thrombin. Residues important for fibrinogen recognition are distributed over the entire surface of contact between enzyme and substrate (Rose and Di Cera 2002), and involve the 60-loop, exosite I, the primary specificity pocket, the aryl binding site, and the Na+ binding site (Di Cera et al. 2007). The surface of recognition between thrombin and protein C changes significantly upon thrombomodulin binding and is reduced mainly to the primary specificity pocket and portions of the 60-loop in the presence of cofactor (Xu et al. 2005). This makes it possible to identify residues that are significantly more important for fibrinogen recognition than protein C activation and to engineer thrombin mutants with enhanced anticoagulant activity in three steps: 1. identify residues that contribute differentially to fibrinogen and protein C recognition; 2. select among these residues those that make independent contributions to substrate recognition; 3. construct multiple mutations involving these residues to achieve additivity. Targets can be set for optimal anticoagulant activity in vivo by demanding safety and potency of the constructs. For a mutant to be safe, activity toward fibrinogen should be <0.1% of that of wild type. Potency demands activity toward protein C to be >10% of that of wild type. These targets define a region in Figure 26 where “ideal” anticoagulant thrombin mutants should map for selection. None of the 80 Ala mutants reported in Figure 26 map in that region, but W215A and E217A stand out. Because W215 and E217 participate in fibrinogen recognition via distinct interactions (Stubbs et al. 1992), the double mutant W215A/E217A (WE) was constructed (Cantwell and Di Cera 2000) hoping for additivity of mutational effects (Wells 1990) that would boost the anticoagulant activity relative to the single mutations. Additivity was indeed found and WE mapped in the desired section in Figure 26 (Cantwell and Di Cera 2000). The crystal structure of WE in the absence of inhibitors shows a conformation with the active site occluded by a collapse of the 215–217 strand, whereas the PPACK-bound form is similar to that of wild-type (Pineda et al. 2004b). The mutant is practically inactive toward synthetic and natural substrates, and only recovers activity in the presence of thrombomodulin and protein C (Pineda et al. 2004b). Hence, W215A/E217A is presumably stabilized in the E* form and converts to the E form upon binding of thrombomodulin following a molecular transition similar to that documented for the interaction of the D102N mutant with PAR1. These are most desirable properties for a recombinant thrombin to be used in vivo as an anticoagulant.
Figure 26.
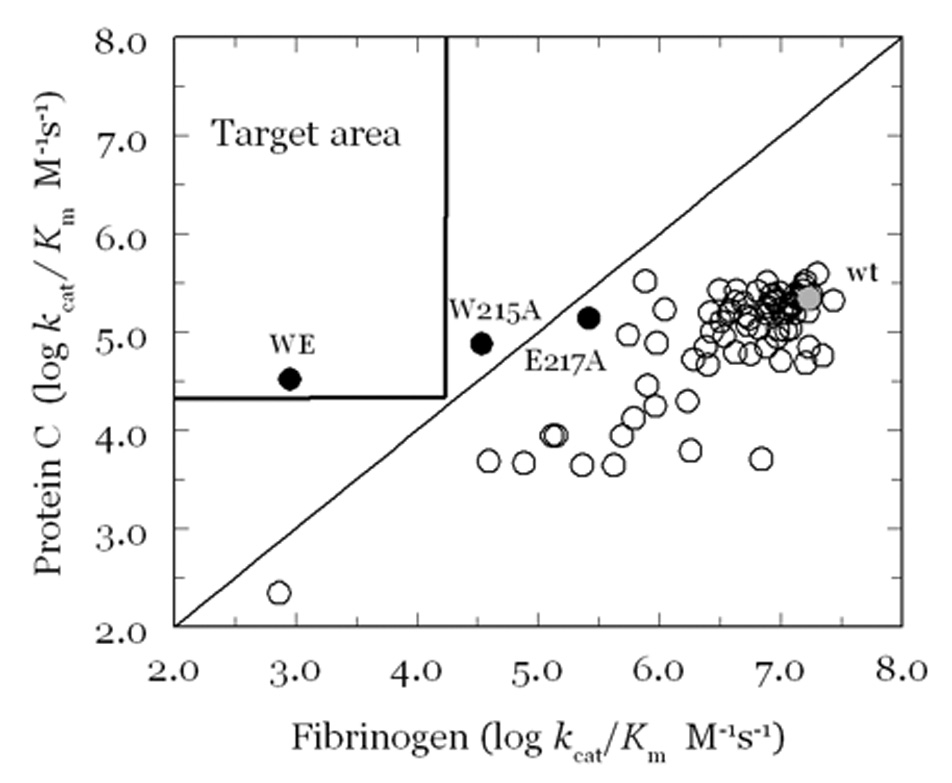
Specificity constants Kcat/Km for the hydrolysis of fibrinogen and protein C, in the presence of 10 nM thrombomodulin and 5 mM CaCl2, under experimental conditions of 5 mM Tris, 0.1% PEG, 145 mM NaCl, pH 7.4 at 37 °C. Plotted are the wild-type (gray) and 80 Ala mutants of thrombin. Safety and potency of an anticoagulant thrombin mutant for in vivo applications demand <0.1% activity toward fibrinogen and >10% activity toward protein C compared to wild-type. These boundaries define a target region in the plot where mutations should map. W215A and E217A are the most promising single Ala mutants. Combination of the two mutations in the W215A/E217A produces additivity and a construct with the required anticoagulant profile.
13. WE: a prototypic anticoagulant/antithrombotic thrombin
Systemic administration of activated protein C (APC) has antithrombotic and antiinflammatory effects (Taylor et al. 1987; Gruber et al. 1989; Gruber et al. 1990) that are now utilized in the treatment of severe sepsis (Bernard et al. 2001). Since infused thrombin activates protein C and activated protein C is antithrombotic, thrombin infusion could act, in theory, as an antithrombotic agent. The thrombin analog WE was tested in a wide dose range for safety and efficacy in a baboon model of acute vascular graft thrombosis (Gruber et al. 2002; Gruber et al. 2007). In this model, a permanent surgically implanted arteriovenous shunt is temporarily extended with a thrombogenic vascular graft segment. Acute thrombus formation is visualized and quantified in real time using gamma camera imaging the deposition of radiolabeled platelets in the graft. WE was initially administered as an intravenous bolus in order to evaluate pharmacokinetics. The lowest bolus dose of WE tested, 11 µg/kg, reduced platelet accumulation by 80% one hour after the beginning of thrombosis, and was at least as effective as the direct administration of 40-fold more (0.45 mg/kg bolus) APC. Baboons treated with WE at doses as high as 200 µg/kg showed no clinical or laboratory signs of thrombosis, hemorrhage, or organ failure. No procoagulant activity could be detected for up to one week in baboon plasma obtained following bolus WE administration. Meanwhile, rapid systemic anticoagulation was observed which dissipated with the biological half life of circulating APC as determined in previous experiments in baboons. Higher doses of WE (>20 µg/kg) had pronounced anticoagulant effect, triggered by a burst in the levels of circulating endogenous APC following injection of WE (Gruber et al. 2006). High dose WE infusion exhausted the cofactors of protein C activation before consumption of the substrate reserve and the process was rapidly down regulated >90% following a WE overdose. The exact molecular or cellular mechanism of this self-limiting pharmacological process is not yet known, but failure of protein C activation upon WE overdose can be overcome by cofactor supplementation using soluble recombinant thrombomodulin (Gruber et al. 2006). These results suggest that the thrombin-thrombomodulin complex is efficiently and rapidly inhibited in vivo and that thrombomodulin does not recycle rapidly once WE (and possibly thrombin) is bound to it. The true significance of this surprising finding is that pharmacological protein C activation by WE appears to be intrinsically safe compared to all other accepted methods of anticoagulation, that are ultimately fatal when overdosed. Studies with low dose WE infusions revealed that pharmacological levels of APC and marginal systemic anticoagulation could be maintained by continuous WE infusion for at least 5 hr without substantial decrease in protein C levels. Since WE has potent antithrombotic effects even at very low doses that do not induce systemic anticoagulation (Gruber et al. 2007), natural protein C reserves and production could keep up with the consumption of protein C during sustained antithrombotic WE treatment.
The hemostatic safety of continuous WE infusion in comparison to equi-efficacious doses of low molecular weight heparin infusion for the prevention of acute vascular graft thrombus propagation in the baboon model were also evaluated. Based on previously established anticoagulant effect and antithrombotic efficacy of circulating APC in the baboon model (Gruber et al. 1989; Gruber et al. 1990) we originally predicted that, in systemic blood samples, an APTT prolongation of at least 1.5-fold over baseline, and APC levels at least 20-fold over baseline, sustained for at least 40 min, would result in a significant antithrombotic benefit. The results exceeded our expectations and we found that WE had potent antithrombotic effect even at a dose (2.1 µg/kg/70 min) when the APTT was not demonstrably affected (Gruber et al. 2007). This indicated that WE was a very potent antithrombotic agent in primates (Gruber et al. 2007). Low doses of WE (2.1, 4.2, or 8.3 µg/kg/70 min) outperformed higher doses of exogenous APC (28 and 222 µg/kg/70 min) and were as efficient as interventional doses of intravenous enoxaparin (325 to 2600 µg/kg/70 min) in preventing the propagation of thrombi. The lowest dose of WE tested still increased circulating APC levels by approximately 5-fold (to about 20 ng/mL), and we do not yet know whether even lower doses of WE that may not detectably increase APC levels would also be efficacious. The comparably effective plasma concentration of exogenous (recombinant) APC exceeded the endogenous APC level following low dose WE infusion by several-fold. The fact that WE is very effective at such small doses strongly argues for limited receptor-mediated mechanism. The considerably higher antithrombotic efficacy of WE compared to exogenous APC suggests that APC generated by WE on the surface of endothelial cells may remain transiently bound to the receptor (Esmon et al. 1999; Taylor et al. 2001) and elicit additional effects by signaling via PAR1 (Feistritzer et al. 2006; Gruber et al. 2007). Although ex vivo data suggest that the signaling effect is dependent on generation of APC on the cell surface, one may speculate that WE could exhibit direct (PAR1-mediated) cytoprotective signaling in the presence of EPCR-bound protein C (Bae et al. 2007a; Bae et al. 2007b). WE may be retained on the cell surface in the vicinity of PAR1 by receptors other than thrombomodulin, similar to the case of its interaction with platelet GpIb which appears to be active only under shear flow conditions (Berny et al. 2008).
The molar ratio of the equi-efficacious doses of WE and enoxaparin exceeded 1:1000. However, this extraordinary potency would be ultimately and clinically irrelevant if the safety profile of WE were not substantially different from other anticoagulants. The striking finding was that these potent antithrombotic doses of WE infusion did not impair primary hemostasis, an outcome never before seen in baboons that received comparably effective doses of commercially available antithrombotic agents (Gruber et al. 2007). Interestingly, the comparably antithrombotic, and reasonably low doses of WT thrombin, 40 µg/kg/hr (Hanson et al. 1993) and WE, about 2 µg/kg/hr (Gruber et al. 2007) appear to be at least one order of magnitude apart to the advantage of WE, despite the 90% reduced activity of WE towards protein C. One of the possible explanations for this discrepancy could be the propensity of wild-type thrombin to bind to and activate abundant prothrombotic substrates (e.g., fibrinogen, PAR1) in the blood flow, leaving only a fraction of the dose to diffuse across the boundary layer of flow to surfaces. Meanwhile, binding and interaction of WE with fibrinogen in blood is dramatically reduced, the rate of such interactions as fibrinogen cleavage and inhibition by antithrombin are delayed by several orders of magnitude. The delay leaves more time for the administered enzyme to reach surfaces and become available for interaction with or consumption by transmembrane cofactors and receptors, such as thrombomodulin and PAR1. This can thus lead to effective activation of the protein C-dependent pathways on solid surfaces such as the endothelium. Altogether, this makes WE thrombin an agent that targets thrombosis and help explain the very high antithrombotic efficacy of this enzyme.
Acknowledgments
I am indebted to Dr Andras Gruber for his assistance in writing the section on WE, and to some current members of my laboratory (Alaji Bah, Leslie Bush-Pelc, Chris Carrell, Zhiwei Chen, Prafull Gandhi, Francesca Marino, Michael Page, Matthew Papacostantinou) who have contributed significantly to the work presented in this review. This work was supported in part by NIH research grants HL49413, HL58141 and HL73813.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adema CM, Hertel LA, Miller RD, Loker ES. A family of fibrinogen-related proteins that precipitates parasite-derived molecules is produced by an invertebrate after infection. Proc Natl Acad Sci U S A. 1997;94:8691–8696. doi: 10.1073/pnas.94.16.8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrogue HJ, Madias NE. Hypernatremia. N Engl J Med. 2000a;342:1493–1499. doi: 10.1056/NEJM200005183422006. [DOI] [PubMed] [Google Scholar]
- Adrogue HJ, Madias NE. Hyponatremia. N Engl J Med. 2000b;342:1581–1589. doi: 10.1056/NEJM200005253422107. [DOI] [PubMed] [Google Scholar]
- Akhavan S, Mannucci PM, Lak M, Mancuso G, Mazzucconi MG, Rocino A, Jenkins PV, Perkins SJ. Identification and three-dimensional structural analysis of nine novel mutations in patients with prothrombin deficiency. Thromb Haemost. 2000;84:989–997. [PubMed] [Google Scholar]
- Akhavan S, Rocha E, Zeinali S, Mannucci PM. Gly319 --> arg substitution in the dysfunctional prothrombin Segovia. Br J Haematol. 1999;105:667–669. doi: 10.1046/j.1365-2141.1999.01423.x. [DOI] [PubMed] [Google Scholar]
- Armstrong RN. Mechanistic diversity in a metalloenzyme superfamily. Biochemistry. 2000;39:13625–13632. doi: 10.1021/bi001814v. [DOI] [PubMed] [Google Scholar]
- Arosio D, Ayala YM, Di Cera E. Mutation of W215 compromises thrombin cleavage of fibrinogen, but not of PAR-1 or protein C. Biochemistry. 2000;39:8095–8101. doi: 10.1021/bi0006215. [DOI] [PubMed] [Google Scholar]
- Ayala Y, Di Cera E. Molecular recognition by thrombin. Role of the slow⤍fast transition, site-specific ion binding energetics and thermodynamic mapping of structural components. J Mol Biol. 1994;235:733–746. doi: 10.1006/jmbi.1994.1024. [DOI] [PubMed] [Google Scholar]
- Ayala YM, Cantwell AM, Rose T, Bush LA, Arosio D, Di Cera E. Molecular mapping of thrombin-receptor interactions. Proteins. 2001;45:107–116. doi: 10.1002/prot.1130. [DOI] [PubMed] [Google Scholar]
- Ayala YM, Di Cera E. A simple method for the determination of individual rate constants for substrate hydrolysis by serine proteases. Protein Sci. 2000;9:1589–1593. doi: 10.1110/ps.9.8.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala YM, Vindigni A, Nayal M, Spolar RS, Record MT, Jr, Di Cera E. Thermodynamic investigation of hirudin binding to the slow and fast forms of thrombin: evidence for folding transitions in the inhibitor and protease coupled to binding. J Mol Biol. 1995;253:787–798. doi: 10.1006/jmbi.1995.0591. [DOI] [PubMed] [Google Scholar]
- Bae JS, Yang L, Manithody C, Rezaie AR. The ligand occupancy of endothelial protein C receptor switches the protease-activated receptor 1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood. 2007a;110:3909–3916. doi: 10.1182/blood-2007-06-096651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae JS, Yang L, Rezaie AR. Receptors of the protein C activation and activated protein C signaling pathways are colocalized in lipid rafts of endothelial cells. Proc Natl Acad Sci U S A. 2007b;104:2867–2872. doi: 10.1073/pnas.0611493104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baglin TP, Carrell RW, Church FC, Esmon CT, Huntington JA. Crystal structures of native and thrombin-complexed heparin cofactor II reveal a multistep allosteric mechanism. Proc Natl Acad Sci U S A. 2002;99:11079–11084. doi: 10.1073/pnas.162232399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bah A, Chen Z, Bush-Pelc LA, Mathews FS, Di Cera E. Crystal structures of murine thrombin in complex with the extracellular fragments of murine protease-activated receptors PAR3 and PAR4. Proc Natl Acad Sci USA. 2007;104:11603–11608. doi: 10.1073/pnas.0704409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bah A, Garvey LC, Ge J, Di Cera E. Rapid kinetics of Na+ binding to thrombin. J Biol Chem. 2006;281:40049–40056. doi: 10.1074/jbc.M608600200. [DOI] [PubMed] [Google Scholar]
- Bajaj SP, Schmidt AE, Agah S, Bajaj MS, Padmanabhan K. High Resolution Structures of p-Aminobenzamidine- and Benzamidine-VIIa/Soluble Tissue Factor: UNPREDICTED CONFORMATION OF THE 192–193 PEPTIDE BOND AND MAPPING OF Ca2+, Mg2+, Na+, AND Zn2+ SITES IN FACTOR VIIa. J. Biol. Chem. 2006;281:24873–24888. doi: 10.1074/jbc.M509971200. [DOI] [PubMed] [Google Scholar]
- Bajzar L, Morser J, Nesheim M. TAFI, or plasma procarboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem. 1996;271:16603–16608. doi: 10.1074/jbc.271.28.16603. [DOI] [PubMed] [Google Scholar]
- Banfield DK, MacGillivray RT. Partial characterization of vertebrate prothrombin cDNAs: amplification and sequence analysis of the B chain of thrombin from nine different species. Proc Natl Acad Sci U S A. 1992;89:2779–2783. doi: 10.1073/pnas.89.7.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Shavit R, Kahn A, Wilner GD, Fenton JW., 2nd Monocyte chemotaxis: stimulation by specific exosite region in thrombin. Science. 1983;220:728–731. doi: 10.1126/science.6836310. [DOI] [PubMed] [Google Scholar]
- Bar-Shavit R, Sabbah V, Lampugnani MG, Marchisio PC, Fenton JW, 2nd, Vlodavsky I, Dejana E. An Arg-Gly-Asp sequence within thrombin promotes endothelial cell adhesion. J Cell Biol. 1991;112:335–344. doi: 10.1083/jcb.112.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartunik HD, Summers LJ, Bartsch HH. Crystal structure of bovine beta-trypsin at 1.5 A resolution in a crystal form with low molecular packing density. Active site geometry, ion pairs and solvent structure. J Mol Biol. 1989;210:813–828. doi: 10.1016/0022-2836(89)90110-1. [DOI] [PubMed] [Google Scholar]
- Bates SM, Weitz JI. The status of new anticoagulants. Br J Haematol. 2006;134:3–19. doi: 10.1111/j.1365-2141.2006.06134.x. [DOI] [PubMed] [Google Scholar]
- Berg DT, Wiley MR, Grinnell BW. Enhanced protein C activation and inhibition of fibrinogen cleavage by a thrombin modulator. Science. 1996;273:1389–1391. doi: 10.1126/science.273.5280.1389. [DOI] [PubMed] [Google Scholar]
- Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, et al. Recombinant human protein C Worldwide Evaluation in Severe Sepsis (PROWESS) study group. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- Berny MA, White TC, Tucker EI, Bush-Pelc LA, Di Cera E, Gruber A, McCarty OJ. Thrombin mutant W215A/E217A acts as a platelet GpIb antagonist. Arterioscler Thromb Vasc Biol. 2008 doi: 10.1161/ATVBAHA.107.156273. in press. [DOI] [PubMed] [Google Scholar]
- Bobofchak KM, Pineda AO, Mathews FS, Di Cera E. Energetic and structural consequences of perturbing Gly-193 in the oxyanion hole of serine proteases. J Biol Chem. 2005;280:25644–25650. doi: 10.1074/jbc.M503499200. [DOI] [PubMed] [Google Scholar]
- Bode W. Structure and interaction modes of thrombin. Blood Cells Mol Dis. 2006;36:122–130. doi: 10.1016/j.bcmd.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Bode W, Turk D, Karshikov A. The refined 1.9-A X-ray crystal structure of D-Phe-Pro-Arg chloromethylketone-inhibited human alpha-thrombin: structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Protein Sci. 1992;1:426–471. doi: 10.1002/pro.5560010402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bone S. Dielectric studies of water clusters in cyclodextrins: Relevance to the transition between slow and fast forms of thrombin. J Phys Chem B Condens Matter Mater Surf Interfaces Biophys. 2006;110:20609–20614. doi: 10.1021/jp063811j. [DOI] [PubMed] [Google Scholar]
- Botts J, Morales M. Analytical description of the effects of modifiers and of multivalency upon the steady state catalyzed reaction rate. Trans Faraday Soc. 1953;49:696–707. [Google Scholar]
- Boyer PD, Lardy HA, Phillips PH. The role of potassium in muscle phosphorylations. J Biol Chem. 1942;146:673–681. [Google Scholar]
- Brass LF. Thrombin and platelet activation. Chest. 2003;124:18S–25S. doi: 10.1378/chest.124.3_suppl.18s. [DOI] [PubMed] [Google Scholar]
- Bush LA, Nelson RW, Di Cera E. Murine thrombin lacks Na+ activation but retains high catalytic activity. J Biol Chem. 2006;281:7183–7188. doi: 10.1074/jbc.M512082200. [DOI] [PubMed] [Google Scholar]
- Camire RM. Prothrombinase assembly and S1 site occupation restore the catalytic activity of FXa impaired by mutation at the sodium-binding site. J Biol Chem. 2002;277:37863–37870. doi: 10.1074/jbc.M203692200. [DOI] [PubMed] [Google Scholar]
- Cantwell AM, Di Cera E. Rational design of a potent anticoagulant thrombin. J Biol Chem. 2000;275:39827–39830. doi: 10.1074/jbc.C000751200. [DOI] [PubMed] [Google Scholar]
- Carrell CJ, Bush LA, Mathews FS, Di Cera E. High resolution crystal structures of free thrombin in the presence of K+ reveal the basis of monovalent cation selectivity and an inactive slow form. Biophys Chem. 2006;121:177–184. doi: 10.1016/j.bpc.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Carter WJ, Cama E, Huntington JA. Crystal structure of thrombin bound to heparin. J Biol Chem. 2005;280:2745–2749. doi: 10.1074/jbc.M411606200. [DOI] [PubMed] [Google Scholar]
- Carter WJ, Myles T, Gibbs CS, Leung LL, Huntington JA. Crystal structure of anticoagulant thrombin variant E217K provides insights into thrombin allostery. J Biol Chem. 2004;279:26387–26394. doi: 10.1074/jbc.M402364200. [DOI] [PubMed] [Google Scholar]
- Castagnetto JM, Hennessy SW, Roberts VA, Getzoff ED, Tainer JA, Pique ME. MDB: The Metalloprotein Database and Browser at The Scripps Research Institute. Nucleic Acids Research. 2002;30:379–382. doi: 10.1093/nar/30.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celikel R, McClintock RA, Roberts JR, Mendolicchio GL, Ware J, Varughese KI, Ruggeri ZM. Modulation of alpha-thrombin function by distinct interactions with platelet glycoprotein Ibalpha. Science. 2003;301:218–221. doi: 10.1126/science.1084183. [DOI] [PubMed] [Google Scholar]
- Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- Cleary DB, Trumbo TA, Maurer MC. Protease-activated receptor 4-like peptides bind to thrombin through an optimized interaction with the enzyme active site surface. Arch Biochem Biophys. 2002;403:179–188. doi: 10.1016/s0003-9861(02)00220-5. [DOI] [PubMed] [Google Scholar]
- Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- Cohn M, Monod J. [Purification and properties of the beta-galactosidase (lactase) of Escherichia coli.] Biochim Biophys Acta. 1951;7:153–174. doi: 10.1016/0006-3002(51)90013-3. [DOI] [PubMed] [Google Scholar]
- Colwell NS, Blinder MA, Tsiang M, Gibbs CS, Bock PE, Tollefsen DM. Allosteric effects of a monoclonal antibody against thrombin exosite II. Biochemistry. 1998;37:15057–15065. doi: 10.1021/bi980925f. [DOI] [PubMed] [Google Scholar]
- Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- Curragh EF, Elmore DT. Kinetics and mechanism of catalytic of proteolytic enzymes. Biochem J. 1964;93:163–171. doi: 10.1042/bj0930163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang OD, Vindigni A, Di Cera E. An allosteric switch controls the procoagulant and anticoagulant activities of thrombin. Proc Natl Acad Sci U S A. 1995;92:5977–5981. doi: 10.1073/pnas.92.13.5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang QD, Di Cera E. Residue 225 determines the Na(+)-induced allosteric regulation of catalytic activity in serine proteases. Proc Natl Acad Sci U S A. 1996;93:10653–10656. doi: 10.1073/pnas.93.20.10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang QD, Guinto ER, Di Cera E. Rational engineering of activity and specificity in a serine protease. Nat Biotechnol. 1997a;15:146–149. doi: 10.1038/nbt0297-146. [DOI] [PubMed] [Google Scholar]
- Dang QD, Sabetta M, Di Cera E. Selective loss of fibrinogen clotting in a loop-less thrombin. J Biol Chem. 1997b;272:19649–19651. doi: 10.1074/jbc.272.32.19649. [DOI] [PubMed] [Google Scholar]
- Davie EW, Kulman JD. An overview of the structure and function of thrombin. Semin Thromb Hemost. 2006;32 Suppl 1:3–15. doi: 10.1055/s-2006-939550. [DOI] [PubMed] [Google Scholar]
- De Cristofaro R, Akhavan S, Altomare C, Carotti A, Peyvandi F, Mannucci PM. A Natural Prothrombin Mutant Reveals an Unexpected Influence of A-chain Structure on the Activity of Human {alpha}-Thrombin. J. Biol. Chem. 2004;279:13035–13043. doi: 10.1074/jbc.M312430200. [DOI] [PubMed] [Google Scholar]
- De Cristofaro R, Carotti A, Akhavan S, Palla R, Peyvandi F, Altomare C, Mannucci PM. The natural mutation by deletion of Lys9 in the thrombin A-chain affects the pKa value of catalytic residues, the overall enzyme's stability and conformational transitions linked to Na+ binding. Febs J. 2006;273:159–169. doi: 10.1111/j.1742-4658.2005.05052.x. [DOI] [PubMed] [Google Scholar]
- De Cristofaro R, De Candia E, Rutella S, Weitz JI. The Asp(272)-Glu(282) region of platelet glycoprotein Ibalpha interacts with the heparin-binding site of alpha-thrombin and protects the enzyme from the heparin-catalyzed inhibition by antithrombin III. J Biol Chem. 2000;275:3887–3895. doi: 10.1074/jbc.275.6.3887. [DOI] [PubMed] [Google Scholar]
- De Filippis V, De Dea E, Lucatello F, Frasson R. Effect of Na+ binding on the conformation, stability and molecular recognition properties of thrombin. Biochem J. 2005;390:485–492. doi: 10.1042/BJ20050252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wardener HE. The hypothalamus and hypertension. Physiol Rev. 2001;81:1599–1658. doi: 10.1152/physrev.2001.81.4.1599. [DOI] [PubMed] [Google Scholar]
- Degen SJ, McDowell SA, Sparks LM, Scharrer I. Prothrombin Frankfurt: a dysfunctional prothrombin characterized by substitution of Glu-466 by Ala. Thromb Haemost. 1995;73:203–209. [PubMed] [Google Scholar]
- Dementiev A, Petitou M, Herbert JM, Gettins PG. The ternary complex of antithrombin-anhydrothrombin-heparin reveals the basis of inhibitor specificity. Nat Struct Mol Biol. 2004;11:863–867. doi: 10.1038/nsmb810. [DOI] [PubMed] [Google Scholar]
- Di Cera E. Thermodynamic Theory of Site-Specific Binding Processes in Biological Macromolecules. Cambridge, UK: Cambridge University Press; 1995. [Google Scholar]
- Di Cera E. Thrombin interactions. Chest. 2003;124:11S–17S. doi: 10.1378/chest.124.3_suppl.11s. [DOI] [PubMed] [Google Scholar]
- Di Cera E. A structural perspective on enzymes activated by monovalent cations. J Biol Chem. 2006;281:1305–1308. doi: 10.1074/jbc.R500023200. [DOI] [PubMed] [Google Scholar]
- Di Cera E. Thrombin as procoagulant and anticoagulant. J Thromb Haemost. 2007;5:196–202. doi: 10.1111/j.1538-7836.2007.02485.x. [DOI] [PubMed] [Google Scholar]
- Di Cera E, Dang QD, Ayala YM. Molecular mechanisms of thrombin function. Cell Mol Life Sci. 1997;53:701–730. doi: 10.1007/s000180050091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cera E, Guinto ER, Vindigni A, Dang QD, Ayala YM, Wuyi M, Tulinsky A. The Na+ binding site of thrombin. J Biol Chem. 1995;270:22089–22092. doi: 10.1074/jbc.270.38.22089. [DOI] [PubMed] [Google Scholar]
- Di Cera E, Hopfner KP, Dang QD. Theory of allosteric effects in serine proteases. Biophys J. 1996;70:174–181. doi: 10.1016/S0006-3495(96)79558-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cera E, Page MJ, Bah A, Bush-Pelc LA, Garvey LC. Thrombin allostery. Phys Chem Chem Phys. 2007;9:1292–1306. doi: 10.1039/b616819a. [DOI] [PubMed] [Google Scholar]
- DiBella EE, Maurer MC, Scheraga HA. Expression and folding of recombinant bovine prethrombin-2 and its activation to thrombin. J Biol Chem. 1995;270:163–169. doi: 10.1074/jbc.270.1.163. [DOI] [PubMed] [Google Scholar]
- Dihanich M, Kaser M, Reinhard E, Cunningham D, Monard D. Prothrombin mRNA is expressed by cells of the nervous system. Neuron. 1991;6:575–581. doi: 10.1016/0896-6273(91)90060-d. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Dumas JJ, Kumar R, Seehra J, Somers WS, Mosyak L. Crystal structure of the GpIbalpha-thrombin complex essential for platelet aggregation. Science. 2003;301:222–226. doi: 10.1126/science.1083917. [DOI] [PubMed] [Google Scholar]
- Eisenman G, Dani JA. An introduction to molecular architecture and permeability of ion channels. Annu Rev Biophys Biophys Chem. 1987;16:205–226. doi: 10.1146/annurev.bb.16.060187.001225. [DOI] [PubMed] [Google Scholar]
- Esmon CT. Inflammation and thrombosis. J Thromb Haemost. 2003a;1:1343–1348. doi: 10.1046/j.1538-7836.2003.00261.x. [DOI] [PubMed] [Google Scholar]
- Esmon CT. The protein C pathway. Chest. 2003b;124:26S–32S. doi: 10.1378/chest.124.3_suppl.26s. [DOI] [PubMed] [Google Scholar]
- Esmon CT, Mather T. Switching serine protease specificity. Nat Struct Biol. 1998;5:933–937. doi: 10.1038/2906. [DOI] [PubMed] [Google Scholar]
- Esmon CT, Xu J, Gu JM, Qu D, Laszik Z, Ferrell G, Stearns-Kurosawa DJ, Kurosawa S, Taylor FB, Jr, Esmon NL. Endothelial protein C receptor. Thromb Haemost. 1999;82:251–258. [PubMed] [Google Scholar]
- Esmon NL, DeBault LE, Esmon CT. Proteolytic formation and properties of gamma-carboxyglutamic acid-domainless protein C. J Biol Chem. 1983;258:5548–5553. [PubMed] [Google Scholar]
- Feistritzer C, Schuepbach RA, Mosnier LO, Bush LA, Di Cera E, Griffin JH, Riewald M. Protective signaling by activated protein C is mechanistically linked to protein C activation on endothelial cells. J Biol Chem. 2006;281:20077–20084. doi: 10.1074/jbc.M600506200. [DOI] [PubMed] [Google Scholar]
- Finney LA, O'Halloran TV. Transition metal speciation in the cell: insights from the chemistry of metal ion receptors. Science. 2003;300:931–936. doi: 10.1126/science.1085049. [DOI] [PubMed] [Google Scholar]
- Frohlich ED, Varagic J. The role of sodium in hypertension is more complex than simply elevating arterial pressure. Nat Clin Pract Cardiovasc Med. 2004;1:24–30. doi: 10.1038/ncpcardio0025. [DOI] [PubMed] [Google Scholar]
- Fuentes-Prior P, Iwanaga Y, Huber R, Pagila R, Rumennik G, Seto M, Morser J, Light DR, Bode W. Structural basis for the anticoagulant activity of the thrombin-thrombomodulin complex. Nature. 2000;404:518–525. doi: 10.1038/35006683. [DOI] [PubMed] [Google Scholar]
- Fuhrmann CN, Daugherty MD, Agard DA. Subangstrom crystallography reveals that short ionic hydrogen bonds, and not a His-Asp low-barrier hydrogen bond, stabilize the transition state in serine protease catalysis. J. Am. Chem. Soc. 2006;128:9086–9102. doi: 10.1021/ja057721o. [DOI] [PubMed] [Google Scholar]
- Gailani D, Broze GJ., Jr Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–912. doi: 10.1126/science.1652157. [DOI] [PubMed] [Google Scholar]
- Gan ZR, Li Y, Chen Z, Lewis SD, Shafer JA. Identification of basic amino acid residues in thrombin essential for heparin-catalyzed inactivation by antithrombin III. J Biol Chem. 1994;269:1301–1305. [PubMed] [Google Scholar]
- Gandhi PS, Chen Z, Mathews FS, Di Cera E. Structural identification of the pathway of long-range communication in an allosteric enzyme. Proc Natl Acad Sci USA. 2008 doi: 10.1073/pnas.0710894105. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gettins PG. Serpin structure, mechanism, and function. Chem Rev. 2002;102:4751–4804. doi: 10.1021/cr010170+. [DOI] [PubMed] [Google Scholar]
- Gianni S, Ivarsson Y, Bah A, Bush-Pelc LA, Di Cera E. Mechanism of Na+ binding to thrombin resolved by ultra-rapid kinetics. Biophys Chem. 2007;131:111–114. doi: 10.1016/j.bpc.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs CS, Coutre SE, Tsiang M, Li WX, Jain AK, Dunn KE, Law VS, Mao CT, Matsumura SY, Mejza SJ, et al. Conversion of thrombin into an anticoagulant by protein engineering. Nature. 1995;378:413–416. doi: 10.1038/378413a0. [DOI] [PubMed] [Google Scholar]
- Girard TJ, MacPhail LA, Likert KM, Novotny WF, Miletich JP, Broze GJ., Jr Inhibition of factor VIIa-tissue factor coagulation activity by a hybrid protein. Science. 1990;248:1421–1424. doi: 10.1126/science.1972598. [DOI] [PubMed] [Google Scholar]
- Goldman JA, Eckerling B. Intracranial dural sinus thrombosis following intrauterine instillation of hypertonic saline. Am J Obstet Gynecol. 1972;112:1132–1133. doi: 10.1016/0002-9378(72)90193-7. [DOI] [PubMed] [Google Scholar]
- Gopalakrishna K, Rezaie AR. The influence of sodium ion binding on factor IXa activity. Thromb Haemost. 2006;95:936–941. doi: 10.1160/TH06-03-0156. [DOI] [PubMed] [Google Scholar]
- Grant PJ, Tate GM, Hughes JR, Davies JA, Prentice CR. Does hypernatraemia promote thrombosis? Thromb Res. 1985;40:393–399. doi: 10.1016/0049-3848(85)90274-9. [DOI] [PubMed] [Google Scholar]
- Greenspan NS, Di Cera E. Defining epitopes: It's not as easy as it seems. Nat Biotechnol. 1999;17:936–937. doi: 10.1038/13590. [DOI] [PubMed] [Google Scholar]
- Griffin JH. Blood coagulation. The thrombin paradox. Nature. 1995;378:337–338. doi: 10.1038/378337a0. [DOI] [PubMed] [Google Scholar]
- Griffin JH, Zlokovic B, Fernandez JA. Activated protein C: potential therapy for severe sepsis, thrombosis, and stroke. Semin Hematol. 2002;39:197–205. doi: 10.1053/shem.2002.34093. [DOI] [PubMed] [Google Scholar]
- Griffon N, Di Stasio E. Thermodynamics of Na+ binding to coagulation serine proteases. Biophys Chem. 2001;90:89–96. doi: 10.1016/s0301-4622(01)00129-6. [DOI] [PubMed] [Google Scholar]
- Grinnell BW, Gerlitz B, Berg DT. Identification of a region in protein C involved in thrombomodulin-stimulated activation by thrombin: potential repulsion at anion-binding site I in thrombin. Biochem J. 1994;303(Pt 3):929–933. doi: 10.1042/bj3030929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber A, Cantwell AM, Di Cera E, Hanson SR. The thrombin mutant W215A/E217A shows safe and potent anticoagulant and antithrombotic effects in vivo. J Biol Chem. 2002;277:27581–27584. doi: 10.1074/jbc.C200237200. [DOI] [PubMed] [Google Scholar]
- Gruber A, Fernandez JA, Bush L, Marzec U, Griffin JH, Hanson SR, Di Cera E. Limited generation of activated protein C during infusion of the protein C activator thrombin analog W215A/E217A in primates. J Thromb Haemost. 2006;4:392–397. doi: 10.1111/j.1538-7836.2006.01760.x. [DOI] [PubMed] [Google Scholar]
- Gruber A, Griffin JH, Harker LA, Hanson SR. Inhibition of platelet-dependent thrombus formation by human activated protein C in a primate model. Blood. 1989;73:639–642. [PubMed] [Google Scholar]
- Gruber A, Hanson SR, Kelly AB, Yan BS, Bang N, Griffin JH, Harker LA. Inhibition of thrombus formation by activated recombinant protein C in a primate model of arterial thrombosis. Circulation. 1990;82:578–585. doi: 10.1161/01.cir.82.2.578. [DOI] [PubMed] [Google Scholar]
- Gruber A, Marzec UM, Bush L, Di Cera E, Fernandez JA, Berny MA, Tucker EI, McCarty OJ, Griffin JH, Hanson SR. Relative antithrombotic and antihemostatic effects of protein C activator versus low molecular weight heparin in primates. Blood. 2007;109:3733–3740. doi: 10.1182/blood-2006-07-035147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinto ER, Caccia S, Rose T, Futterer K, Waksman G, Di Cera E. Unexpected crucial role of residue 225 in serine proteases. Proc Natl Acad Sci U S A. 1999;96:1852–1857. doi: 10.1073/pnas.96.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinto ER, Di Cera E. Large heat capacity change in a protein-monovalent cation interaction. Biochemistry. 1996;35:8800–8804. doi: 10.1021/bi9608828. [DOI] [PubMed] [Google Scholar]
- Hageman TC, Endres GF, Scheraga HA. Mechanism of action of thrombin on fibrinogen. On the role of the A chain of bovine thrombin in specificity and in differentiating between thrombin and trypsin. Arch Biochem Biophys. 1975;171:327–336. doi: 10.1016/0003-9861(75)90039-9. [DOI] [PubMed] [Google Scholar]
- Hall SW, Nagashima M, Zhao L, Morser J, Leung LL. Thrombin interacts with thrombomodulin, protein C, and thrombin-activatable fibrinolysis inhibitor via specific and distinct domains. J Biol Chem. 1999;274:25510–25516. doi: 10.1074/jbc.274.36.25510. [DOI] [PubMed] [Google Scholar]
- Hanson SR, Griffin JH, Harker LA, Kelly AB, Esmon CT, Gruber A. Antithrombotic effects of thrombin-induced activation of endogenous protein C in primates. J Clin Invest. 1993;92:2003–2012. doi: 10.1172/JCI116795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Rezaie AR. Identification and characterization of the sodium-binding site of activated protein C. J Biol Chem. 1999;274:4970–4976. doi: 10.1074/jbc.274.8.4970. [DOI] [PubMed] [Google Scholar]
- Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102:4501–4524. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- Hedstrom L, Szilagyi L, Rutter WJ. Converting trypsin to chymotrypsin: the role of surface loops. Science. 1992;255:1249–1253. doi: 10.1126/science.1546324. [DOI] [PubMed] [Google Scholar]
- Henriksen RA, Dunham CK, Miller LD, Casey JT, Menke JB, Knupp CL, Usala SJ. Prothrombin Greenville, Arg517⤍Gln, identified in an individual heterozygous for dysprothrombinemia. Blood. 1998;91:2026–2031. [PubMed] [Google Scholar]
- Hill TL. Free Energy Transduction in Biology. New York, NY: Academic Press; 1977. [Google Scholar]
- Hofsteenge J, Braun PJ, Stone SR. Enzymatic properties of proteolytic derivatives of human alpha-thrombin. Biochemistry. 1988;27:2144–2151. doi: 10.1021/bi00406a049. [DOI] [PubMed] [Google Scholar]
- Huntington JA, Esmon CT. The molecular basis of thrombin allostery revealed by a 1.8 a structure of the "Slow" form. Structure (Camb) 2003;11:469–479. doi: 10.1016/s0969-2126(03)00049-2. [DOI] [PubMed] [Google Scholar]
- Ibers JA, Holm RH. Modeling coordination sites in metallobiomolecules. Science. 1980;209:223–235. doi: 10.1126/science.7384796. [DOI] [PubMed] [Google Scholar]
- Ishihara H, Connolly AJ, Zeng D, Kahn ML, Zheng YW, Timmons C, Tram T, Coughlin SR. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature. 1997;386:502–506. doi: 10.1038/386502a0. [DOI] [PubMed] [Google Scholar]
- Jacques SL, Kuliopulos A. Protease-activated receptor-4 uses dual prolines and an anionic retention motif for thrombin recognition and cleavage. Biochem J. 2003;376:733–740. doi: 10.1042/BJ20030954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DJ, Adams TE, Li W, Huntington JA. Crystal structure of wild-type human thrombin in the Na +-free state. Biochem J. 2005;392:21–28. doi: 10.1042/BJ20051217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Mannherz HG, Suck D, Pai EF, Holmes KC. Atomic structure of the actin:DNase I complex. Nature. 1990;347:37–44. doi: 10.1038/347037a0. [DOI] [PubMed] [Google Scholar]
- Kachmar JF, Boyer PD. Kinetic analysis of enzyme reactions. II. The potassium activation and calcium inhibition of pyruvic phosphoferase. J Biol Chem. 1953;200:669–682. [PubMed] [Google Scholar]
- Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, Farese RV, Jr, Tam C, Coughlin SR. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–694. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- Kamata K, Mitsuya M, Nishimura T, Eiki J, Nagata Y. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure. 2004;12:429–438. doi: 10.1016/j.str.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Kezdy FJ, Lorand L, Miller KD. Titration of active centers in thrombin solutions. Standardization of the enzyme. Biochemistry. 1965;4:2302–2308. [Google Scholar]
- Krem MM, Di Cera E. Conserved water molecules in the specificity pocket of serine proteases and the molecular mechanism of Na+ binding. Proteins. 1998;30:34–42. doi: 10.1002/(sici)1097-0134(19980101)30:1<34::aid-prot3>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Krem MM, Di Cera E. Molecular markers of serine protease evolution. Embo J. 2001;20:3036–3045. doi: 10.1093/emboj/20.12.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krem MM, Di Cera E. Evolution of enzyme cascades from embryonic development to blood coagulation. Trends Biochem Sci. 2002;27:67–74. doi: 10.1016/s0968-0004(01)02007-2. [DOI] [PubMed] [Google Scholar]
- Krem MM, Prasad S, Di Cera E. Ser(214) is crucial for substrate binding to serine proteases. J Biol Chem. 2002;277:40260–40264. doi: 10.1074/jbc.M206173200. [DOI] [PubMed] [Google Scholar]
- Krem MM, Rose T, Di Cera E. The C-terminal sequence encodes function in serine proteases. J Biol Chem. 1999;274:28063–28066. doi: 10.1074/jbc.274.40.28063. [DOI] [PubMed] [Google Scholar]
- Kroh HK, Tans G, Nicolaes GAF, Rosing J, Bock PE. Expression of allosteric linkage between the sodium ion binding site and exosite I of thrombin during prothrombin activation. J Biol Chem. 2007;282:16095–16104. doi: 10.1074/jbc.M610577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurlansky M. Salt: A World History. Toronto, Canada: Random House Publishing Limited; 2002. p. 484. [Google Scholar]
- Lai MT, Di Cera E, Shafer JA. Kinetic pathway for the slow to fast transition of thrombin. Evidence of linked ligand binding at structurally distinct domains. J Biol Chem. 1997;272:30275–30282. doi: 10.1074/jbc.272.48.30275. [DOI] [PubMed] [Google Scholar]
- Laughlin LT, Reed GH. The monovalent cation requirement of rabbit muscle pyruvate kinase is eliminated by substitution of lysine for glutamate 117. Arch Biochem Biophys. 1997;348:262–267. doi: 10.1006/abbi.1997.0448. [DOI] [PubMed] [Google Scholar]
- Le Bonniec BF, Esmon CT. Glu-192----Gln substitution in thrombin mimics the catalytic switch induced by thrombomodulin. Proc Natl Acad Sci U S A. 1991;88:7371–7375. doi: 10.1073/pnas.88.16.7371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz JB, Haver T, Clarke S, Jacobson L, Weller A, Nuss R, Manco-Johnson M, Hathaway WE. The prothrombin Denver patient has two different prothrombin point mutations resulting in Glu-300⤍Lys and Glu-309⤍Lys substitutions. Br J Haematol. 2000;108:182–187. doi: 10.1046/j.1365-2141.2000.01810.x. [DOI] [PubMed] [Google Scholar]
- Levigne S, Thiec F, Cherel S, Irving JA, Fribourg C, Christophe OD. Role of the alpha-helix 163–170 in factor Xa catalytic activity. J Biol Chem. 2007;282:31569–31579. doi: 10.1074/jbc.M704837200. [DOI] [PubMed] [Google Scholar]
- Li W, Johnson DJ, Esmon CT, Huntington JA. Structure of the antithrombin-thrombin-heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat Struct Mol Biol. 2004;11:857–862. doi: 10.1038/nsmb811. [DOI] [PubMed] [Google Scholar]
- Liu LW, Vu TK, Esmon CT, Coughlin SR. The region of the thrombin receptor resembling hirudin binds to thrombin and alters enzyme specificity. J Biol Chem. 1991;266:16977–16980. [PubMed] [Google Scholar]
- Lorand L, Brannen WT, Jr, Rule NG. Thrombin-catalyzed hydrolysis of p-nitrophenyl esters. Arch Biochem Biophys. 1962;96:147–151. doi: 10.1016/0003-9861(62)90463-0. [DOI] [PubMed] [Google Scholar]
- Lorand L, Downey J, Gotoh T, Jacobsen A, Tokura S. The transpeptidase system which crosslinks fibrin by gamma-glutamyle-episilon-lysine bonds. Biochem Biophys Res Commun. 1968;31:222–230. doi: 10.1016/0006-291x(68)90734-1. [DOI] [PubMed] [Google Scholar]
- Malkowski MG, Martin PD, Guzik JC, Edwards BFP. The co-crystal structure of unliganded bovine {alpha}-thrombin and prethrombin-2: Movement of the Tyr-Pro-Pro-Trp segment and active site residues upon ligand binding. Protein Sci. 1997;6:1438–1448. doi: 10.1002/pro.5560060708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann KG. Thrombin formation. Chest. 2003;124:4S–10S. doi: 10.1378/chest.124.3_suppl.4s. [DOI] [PubMed] [Google Scholar]
- Mann KG, Butenas S, Brummel K. The dynamics of thrombin formation. Arterioscler Thromb Vasc Biol. 2003;23:17–25. doi: 10.1161/01.atv.0000046238.23903.fc. [DOI] [PubMed] [Google Scholar]
- Marino F, Chen ZW, Ergenekan C, Bush-Pelc LA, Mathews FS, Di Cera E. Structural basis of Na+ activation mimicry in murine thrombin. J Biol Chem. 2007;282:16355–16361. doi: 10.1074/jbc.M701323200. [DOI] [PubMed] [Google Scholar]
- Martin CJ, Golubow J, Axelrod AE. The hydrolysis of carbobenzoxy-L-tyrosine p-nitrophenyl ester by various enzymes. J Biol Chem. 1959;234:1718–1725. [PubMed] [Google Scholar]
- Mathews II, Padmanabhan KP, Ganesh V, Tulinsky A, Ishii M, Chen J, Turck CW, Coughlin SR, Fenton JW., 2nd Crystallographic structures of thrombin complexed with thrombin receptor peptides: existence of expected and novel binding modes. Biochemistry. 1994;33:3266–3279. doi: 10.1021/bi00177a018. [DOI] [PubMed] [Google Scholar]
- McLaughlin JN, Patterson MM, Malik AB. Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad Sci USA. 2007;104:5662–5667. doi: 10.1073/pnas.0700763104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meneton P, Jeunemaitre X, de Wardener HE, MacGregor GA. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol Rev. 2005;85:679–715. doi: 10.1152/physrev.00056.2003. [DOI] [PubMed] [Google Scholar]
- Mengwasser KE, Bush LA, Shih P, Cantwell AM, Di Cera E. Hirudin binding reveals key determinants of thrombin allostery. J Biol Chem. 2005;280:23997–27003. doi: 10.1074/jbc.M502678200. [DOI] [PubMed] [Google Scholar]
- Miyata T, Aruga R, Umeyama H, Bezeaud A, Guillin MC, Iwanaga S. Prothrombin Salakta: substitution of glutamic acid-466 by alanine reduces the fibrinogen clotting activity and the esterase activity. Biochemistry. 1992;31:7457–7462. doi: 10.1021/bi00148a005. [DOI] [PubMed] [Google Scholar]
- Monnaie D, Arosio D, Griffon N, Rose T, Rezaie AR, Di Cera E. Identification of a binding site for quaternary amines in factor Xa. Biochemistry. 2000;39:5349–5354. doi: 10.1021/bi9926781. [DOI] [PubMed] [Google Scholar]
- Myles T, Le Bonniec BF, Stone SR. The dual role of thrombin's anion-binding exosite-I in the recognition and cleavage of the protease-activated receptor 1. Eur J Biochem. 2001a;268:70–77. doi: 10.1046/j.1432-1327.2001.01844.x. [DOI] [PubMed] [Google Scholar]
- Myles T, Yun TH, Hall SW, Leung LL. An extensive interaction interface between thrombin and factor V is required for factor V activation. J Biol Chem. 2001b;276:25143–25149. doi: 10.1074/jbc.M011324200. [DOI] [PubMed] [Google Scholar]
- Nakanishi-Matsui M, Zheng YW, Sulciner DJ, Weiss EJ, Ludeman MJ, Coughlin SR. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. 2000;404:609–613. doi: 10.1038/35007085. [DOI] [PubMed] [Google Scholar]
- Nogami K, Zhou Q, Myles T, Leung LL, Wakabayashi H, Fay PJ. Exosite-interactive regions in the A1 and A2 domains of factor VIII facilitate thrombin-catalyzed cleavage of heavy chain. J Biol Chem. 2005;280:18476–18487. doi: 10.1074/jbc.M412778200. [DOI] [PubMed] [Google Scholar]
- O'Brien MC, McKay DB. How potassium affects the activity of the molecular chaperone Hsc70. I. Potassium is required for optimal ATPase activity. J Biol Chem. 1995;270:2247–2250. doi: 10.1074/jbc.270.5.2247. [DOI] [PubMed] [Google Scholar]
- O'Brien PJ, Molino M, Kahn M, Brass LF. Protease activated receptors: theme and variations. Oncogene. 2001;20:1570–1581. doi: 10.1038/sj.onc.1204194. [DOI] [PubMed] [Google Scholar]
- Olson ST, Chuang YJ. Heparin activates antithrombin anticoagulant function by generating new interaction sites (exosites) for blood clotting proteinases. Trends Cardiovasc Med. 2002;12:331–338. doi: 10.1016/s1050-1738(02)00183-4. [DOI] [PubMed] [Google Scholar]
- Orthner CL, Kosow DP. The effect of metal ions on the amidolytic acitivity of human factor Xa (activated Stuart-Prower factor) Arch Biochem Biophys. 1978;185:400–406. doi: 10.1016/0003-9861(78)90182-0. [DOI] [PubMed] [Google Scholar]
- Orthner CL, Kosow DP. Evidence that human alpha-thrombin is a monovalent cation-activated enzyme. Arch Biochem Biophys. 1980;202:63–75. doi: 10.1016/0003-9861(80)90406-3. [DOI] [PubMed] [Google Scholar]
- Page MJ, Di Cera E. Role of Na+ and K+ in enzyme function. Physiol Rev. 2006;86:1049–1092. doi: 10.1152/physrev.00008.2006. [DOI] [PubMed] [Google Scholar]
- Page MJ, Di Cera E. Serine peptidases: classification, structure and function. Cell Mol Life Sci. 2008 doi: 10.1007/s00018-008-7565-9. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page MJ, MacGillivray RTA, Di Cera E. Determinants of specificity in coagulation proteases. J Thromb Haemost. 2005;3:2401–2408. doi: 10.1111/j.1538-7836.2005.01456.x. [DOI] [PubMed] [Google Scholar]
- Papaconstantinou ME, Carrell CJ, Pineda AO, Bobofchak KM, Mathews FS, Flordellis CS, Maragoudakis ME, Tsopanoglou NE, Di Cera E. Thrombin functions through its RGD sequence in a non-canonical conformation. J Biol Chem. 2005;280:29393–29396. doi: 10.1074/jbc.C500248200. [DOI] [PubMed] [Google Scholar]
- Parry MA, Stone SR, Hofsteenge J, Jackman MP. Evidence for common structural changes in thrombin induced by active-site or exosite binding. Biochem J. 1993;290(Pt 3):665–670. doi: 10.1042/bj2900665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechik I, Madrazo J, Mosesson MW, Hernandez I, Gilliland GL, Medved L. Crystal structure of the complex between thrombin and the central "E" region of fibrin. Proc Natl Acad Sci U S A. 2004;101:2718–2723. doi: 10.1073/pnas.0303440101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechik I, Yakovlev S, Mosesson MW, Gilliland GL, Medved L. Structural basis for sequential cleavage of fibrinopeptides upon fibrin assembly. Biochemistry. 2006;45:3588–3597. doi: 10.1021/bi0525369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perona JJ, Craik CS. Structural basis of substrate specificity in the serine proteases. Protein Sci. 1995;4:337–360. doi: 10.1002/pro.5560040301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perona JJ, Craik CS. Evolutionary divergence of substrate specificity within the chymotrypsin-like serine protease fold. J Biol Chem. 1997;272:29987–29990. doi: 10.1074/jbc.272.48.29987. [DOI] [PubMed] [Google Scholar]
- Petrovan RJ, Ruf W. Role of residue Phe225 in the cofactor-mediated, allosteric regulation of the serine protease coagulation factor VIIa. Biochemistry. 2000;39:14457–14463. doi: 10.1021/bi0009486. [DOI] [PubMed] [Google Scholar]
- Pineda AO, Cantwell AM, Bush LA, Rose T, Di Cera E. The thrombin epitope recognizing thrombomodulin is a highly cooperative hot spot in exosite I. J Biol Chem. 2002a;277:32015–32019. doi: 10.1074/jbc.M205009200. [DOI] [PubMed] [Google Scholar]
- Pineda AO, Carrell CJ, Bush LA, Prasad S, Caccia S, Chen ZW, Mathews FS, Di Cera E. Molecular dissection of Na+ binding to thrombin. J Biol Chem. 2004a;279:31842–31853. doi: 10.1074/jbc.M401756200. [DOI] [PubMed] [Google Scholar]
- Pineda AO, Chen ZW, Bah A, Garvey LC, Mathews FS, Di Cera E. Crystal structure of thrombin in a self-inhibited conformation. J Biol Chem. 2006;281:32922–32928. doi: 10.1074/jbc.M605530200. [DOI] [PubMed] [Google Scholar]
- Pineda AO, Chen ZW, Caccia S, Cantwell AM, Savvides SN, Waksman G, Mathews FS, Di Cera E. The anticoagulant thrombin mutant W215A/E217A has a collapsed primary specificity pocket. J Biol Chem. 2004b;279:39824–39828. doi: 10.1074/jbc.M407272200. [DOI] [PubMed] [Google Scholar]
- Pineda AO, Chen ZW, Marino F, Mathews FS, Mosesson MW, Di Cera E. Crystal structure of thrombin in complex with fibrinogen gamma' peptide. Biophys Chem. 2007;125:556–559. doi: 10.1016/j.bpc.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Pineda AO, Savvides SN, Waksman G, Di Cera E. Crystal structure of the anticoagulant slow form of thrombin. J Biol Chem. 2002b;277:40177–40180. doi: 10.1074/jbc.C200465200. [DOI] [PubMed] [Google Scholar]
- Pineda AO, Zhang E, Guinto ER, Savvides SN, Tulinsky A, Di Cera E. Crystal structure of the thrombin mutant D221A/D222K: the Asp222:Arg187 ion-pair stabilizes the fast form. Biophys Chem. 2004c;112:253–256. doi: 10.1016/j.bpc.2004.07.027. [DOI] [PubMed] [Google Scholar]
- Prasad S, Cantwell AM, Bush LA, Shih P, Xu H, Di Cera E. Residue Asp-189 controls both substrate binding and the monovalent cation specificity of thrombin. J Biol Chem. 2004;279:10103–10108. doi: 10.1074/jbc.M312614200. [DOI] [PubMed] [Google Scholar]
- Prasad S, Wright KJ, Roy DB, Bush LA, Cantwell AM, Di Cera E. Redesigning the monovalent cation specificity of an enzyme. Proc Natl Acad Sci U S A. 2003;100:13785–13790. doi: 10.1073/pnas.2333109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan V, DeGuzman F, Bao M, Hall SW, Leung LL, Phillips DR. A thrombin receptor function for platelet glycoprotein Ib-IX unmasked by cleavage of glycoprotein V. Proc Natl Acad Sci U S A. 2001;98:1823–1828. doi: 10.1073/pnas.98.4.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Tolle DP, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2004;32(Database issue):D160–D164. doi: 10.1093/nar/gkh071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaie AR, Esmon CT. The function of calcium in protein C activation by thrombin and the thrombin-thrombomodulin complex can be distinguished by mutational analysis of protein C derivatives. J Biol Chem. 1992;267:26104–26109. [PubMed] [Google Scholar]
- Rezaie AR, He X. Sodium binding site of factor Xa: role of sodium in the prothrombinase complex. Biochemistry. 2000;39:1817–1825. doi: 10.1021/bi992006a. [DOI] [PubMed] [Google Scholar]
- Rezaie AR, Kittur FS. The critical role of the 185–189-loop in the factor Xa interaction with Na+ and factor Va in the prothrombinase complex. J Biol Chem. 2004;279:48262–48269. doi: 10.1074/jbc.M409964200. [DOI] [PubMed] [Google Scholar]
- Rezaie AR, Olson ST. Contribution of lysine 60f to S1' specificity of thrombin. Biochemistry. 1997;36:1026–1033. doi: 10.1021/bi9620823. [DOI] [PubMed] [Google Scholar]
- Rezaie AR, Yang L. Thrombomodulin allosterically modulates the activity of the anticoagulant thrombin. Proc Natl Acad Sci U S A. 2003;100:12051–12056. doi: 10.1073/pnas.2135346100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson MA, Gerlitz B, Grinnell BW. Enhancing protein C interaction with thrombin results in a clot-activated anticoagulant. Nature. 1992;360:261–264. doi: 10.1038/360261a0. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- Rose T, Di Cera E. Three-dimensional modeling of thrombin-fibrinogen interaction. J Biol Chem. 2002;277:18875–18880. doi: 10.1074/jbc.M110977200. [DOI] [PubMed] [Google Scholar]
- Roux B, MacKinnon R. The cavity and pore helices in the KcsA K+ channel: electrostatic stabilization of monovalent cations. Science. 1999;285:100–102. doi: 10.1126/science.285.5424.100. [DOI] [PubMed] [Google Scholar]
- Rouy S, Vidaud D, Alessandri JL, Dautzenberg MD, Venisse L, Guillin MC, Bezeaud A. Prothrombin Saint-Denis: a natural variant with a point mutation resulting in Asp to Glu substitution at position 552 in prothrombin. Br J Haematol. 2006;132:770–773. doi: 10.1111/j.1365-2141.2005.05920.x. [DOI] [PubMed] [Google Scholar]
- Roy DB, Rose T, Di Cera E. Replacement of thrombin residue G184 with Lys or Arg fails to mimic Na+ binding. Proteins. 2001;43:315–318. doi: 10.1002/prot.1042. [DOI] [PubMed] [Google Scholar]
- Rubin D, Christian C. Retinal hemorrhages in infants with hyponatremic seizures. Pediatr Emerg Care. 2001;17:313–314. doi: 10.1097/00006565-200108000-00028. [DOI] [PubMed] [Google Scholar]
- Rydel TJ, Tulinsky A, Bode W, Huber R. Refined structure of the hirudin-thrombin complex. J Mol Biol. 1991;221:583–601. doi: 10.1016/0022-2836(91)80074-5. [DOI] [PubMed] [Google Scholar]
- Sadasivan C, Yee VC. Interaction of the factor XIII activation peptide with alpha - thrombin. Crystal structure of its enzyme-substrate analog complex. J Biol Chem. 2000;275:36942–36948. doi: 10.1074/jbc.M006076200. [DOI] [PubMed] [Google Scholar]
- Sambrano GR, Weiss EJ, Zheng YW, Huang W, Coughlin SR. Role of thrombin signalling in platelets in haemostasis and thrombosis. Nature. 2001;413:74–78. doi: 10.1038/35092573. [DOI] [PubMed] [Google Scholar]
- Scatena M, Liaw L, Giachelli CM. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol. 2007;27:2302–2309. doi: 10.1161/ATVBAHA.107.144824. [DOI] [PubMed] [Google Scholar]
- Scharer K, Morgenthaler M, Paulini R, Obst-Sander U, Banner DW, Schlatter D, Benz J, Stihle M, Diederich F. Quantification of cation-pi interactions in protein-ligand complexes: crystal-structure analysis of Factor Xa bound to a quaternary ammonium ion ligand. Angew Chem Int Ed Engl. 2005;44:4400–4404. doi: 10.1002/anie.200500883. [DOI] [PubMed] [Google Scholar]
- Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- Schmidt AE, Padmanabhan K, Underwood MC, Bode W, Mather T, Bajaj SP. Thermodynamic linkage between the S1 site, the Na+ site, and the Ca2+ site in the protease domain of human activated protein C (APC). Sodium ion in the APC crystal structure is coordinated to four carbonyl groups from two separate loops. J Biol Chem. 2002;277:28987–28995. doi: 10.1074/jbc.M201892200. [DOI] [PubMed] [Google Scholar]
- Schmidt AE, Stewart JE, Mathur A, Krishnaswamy S, Bajaj SP. Na+ site in blood coagulation factor IXa: effect on catalysis and factor VIIIa binding. J Mol Biol. 2005;350:78–91. doi: 10.1016/j.jmb.2005.04.052. [DOI] [PubMed] [Google Scholar]
- Schreiber G, Fersht AR. Energetics of protein-protein interactions: analysis of the barnase-barstar interface by single mutations and double mutant cycles. J Mol Biol. 1995;248:478–486. doi: 10.1016/s0022-2836(95)80064-6. [DOI] [PubMed] [Google Scholar]
- Seeley S, Covic L, Jacques SL, Sudmeier J, Baleja JD, Kuliopulos A. Structural Basis for Thrombin Activation of a Protease-Activated Receptor: Inhibition of Intramolecular Liganding. Chemistry & Biology. 2003;10:1033–1041. doi: 10.1016/j.chembiol.2003.10.014. [DOI] [PubMed] [Google Scholar]
- Shapiro MJ, Weiss EJ, Faruqi TR, Coughlin SR. Protease-activated receptors 1 and 4 are shut off with distinct kinetics after activation by thrombin. J Biol Chem. 2000;275:25216–25221. doi: 10.1074/jbc.M004589200. [DOI] [PubMed] [Google Scholar]
- Sheehan JP, Sadler JE. Molecular mapping of the heparin-binding exosite of thrombin. Proc Natl Acad Sci U S A. 1994;91:5518–5522. doi: 10.1073/pnas.91.12.5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprang S, Standing T, Fletterick RJ, Stroud RM, Finer-Moore J, Xuong NH, Hamlin R, Rutter WJ, Craik CS. The three-dimensional structure of Asn102 mutant of trypsin: role of Asp102 in serine protease catalysis. Science. 1987;237:905–909. doi: 10.1126/science.3112942. [DOI] [PubMed] [Google Scholar]
- Stanchev h, Philips M, Villoutreix BO, Aksglaede L, Lethagen S, Thorsen S. Prothrombin deficiency caused by compound heterozigosity for two novel mutations in the prothrombin gene associated with a bleeding tendency. Thromb Haemost. 2006;95:195–198. [PubMed] [Google Scholar]
- Steiner SA, Amphlett GW, Castellino FJ. Stimulation of the amidase and esterase activity of activated bovine plasma protein C by monovalent cations. Biochem Biophys Res Commun. 1980;94:340–347. doi: 10.1016/s0006-291x(80)80226-9. [DOI] [PubMed] [Google Scholar]
- Steiner SA, Castellino FJ. Kinetic studies of the role of monovalent cations in the amidolytic activity of activated bovine plasma protein C. Biochemistry. 1982;21:4609–4614. doi: 10.1021/bi00262a015. [DOI] [PubMed] [Google Scholar]
- Steiner SA, Castellino FJ. Effect of monovalent cations on the pre-steady-state kinetic parameters of the plasma protease bovine activated protein C. Biochemistry. 1985a;24:1136–1141. doi: 10.1021/bi00326a011. [DOI] [PubMed] [Google Scholar]
- Steiner SA, Castellino FJ. Kinetic mechanism for stimulation by monovalent cations of the amidase activity of the plasma protease bovine activated protein C. Biochemistry. 1985b;24:609–617. doi: 10.1021/bi00324a011. [DOI] [PubMed] [Google Scholar]
- Stevens RC, Chook YM, Cho CY, Lipscomb WN, Kantrowitz ER. Escherichia coli aspartate carbamoyltransferase: the probing of crystal structure analysis via site-specific mutagenesis. Protein Eng. 1991;4:391–408. doi: 10.1093/protein/4.4.391. [DOI] [PubMed] [Google Scholar]
- Stubbs MT, Oschkinat H, Mayr I, Huber R, Angliker H, Stone SR, Bode W. The interaction of thrombin with fibrinogen. A structural basis for its specificity. Eur J Biochem. 1992;206:187–195. doi: 10.1111/j.1432-1033.1992.tb16916.x. [DOI] [PubMed] [Google Scholar]
- Suelter CH. Enzymes activated by monovalent cations. Science. 1970;168:789–795. doi: 10.1126/science.168.3933.789. [DOI] [PubMed] [Google Scholar]
- Suh TT, Holmback K, Jensen NJ, Daugherty CC, Small K, Simon DI, Potter S, Degen JL. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 1995;9:2020–2033. doi: 10.1101/gad.9.16.2020. [DOI] [PubMed] [Google Scholar]
- Sun WY, Burkart MC, Holahan JR, Degen SJ. Prothrombin San Antonio: a single amino acid substitution at a factor Xa activation site (Arg320 to His) results in dysprothrombinemia. Blood. 2000;95:711–714. [PubMed] [Google Scholar]
- Sun WY, Smirnow D, Jenkins ML, Degen SJ. Prothrombin Scranton: substitution of an amino acid residue involved in the binding of Na+ (LYS-556 to THR) leads to dysprothrombinemia. Thromb Haemost. 2001;85:651–654. [PubMed] [Google Scholar]
- Sun WY, Witte DP, Degen JL, Colbert MC, Burkart MC, Holmback K, Xiao Q, Bugge TH, Degen SJ. Prothrombin deficiency results in embryonic and neonatal lethality in mice. Proc Natl Acad Sci U S A. 1998;95:7597–7602. doi: 10.1073/pnas.95.13.7597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tainer JA, Roberts VA, Getzoff ED. Protein metal-binding sites. Current Opinion in Biotechnology. 1992;3:378–387. doi: 10.1016/0958-1669(92)90166-g. [DOI] [PubMed] [Google Scholar]
- Tauszig S, Jouanguy E, Hoffmann JA, Imler JL. Toll-related receptors and the control of antimicrobial peptide expression in Drosophila. Proc Natl Acad Sci U S A. 2000;97:10520–10525. doi: 10.1073/pnas.180130797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor FB, Chang A, Esmon CT, D'Angelo A, Vigano-D'Angelo S, Blick KE. Protein C prevents the coagulopathic and lethal effects of Escherichia coli infusion in the baboon. J Clin Invest. 1987;79:918–925. doi: 10.1172/JCI112902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor FB, Jr, Peer GT, Lockhart MS, Ferrell G, Esmon CT. Endothelial cell protein C receptor plays an important role in protein C activation in vivo. Blood. 2001;97:1685–1688. doi: 10.1182/blood.v97.6.1685. [DOI] [PubMed] [Google Scholar]
- Tollefsen DM. Heparin Cofactor II Modulates the Response to Vascular Injury. Arterioscler Thromb Vasc Biol. 2006 doi: 10.1161/01.ATV.0000256471.22437.88. [DOI] [PubMed] [Google Scholar]
- Tsiang M, Jain AK, Dunn KE, Rojas ME, Leung LL, Gibbs CS. Functional mapping of the surface residues of human thrombin. J Biol Chem. 1995;270:16854–16863. doi: 10.1074/jbc.270.28.16854. [DOI] [PubMed] [Google Scholar]
- Tsiang M, Jain AK, Gibbs CS. Functional requirements for inhibition of thrombin by antithrombin III in the presence and absence of heparin. J Biol Chem. 1997;272:12024–12029. doi: 10.1074/jbc.272.18.12024. [DOI] [PubMed] [Google Scholar]
- Tsiang M, Paborsky LR, Li WX, Jain AK, Mao CT, Dunn KE, Lee DW, Matsumura SY, Matteucci MD, Coutre SE, et al. Protein engineering thrombin for optimal specificity and potency of anticoagulant activity in vivo. Biochemistry. 1996;35:16449–16457. doi: 10.1021/bi9616108. [DOI] [PubMed] [Google Scholar]
- Turner GJ, Galacteros F, Doyle ML, Hedlund B, Pettigrew DW, Turner BW, Smith FR, Moo-Penn W, Rucknagel DL, Ackers GK. Mutagenic dissection of hemoglobin cooperativity: effects of amino acid alteration on subunit assembly of oxy and deoxy tetramers. Proteins. 1992;14:333–350. doi: 10.1002/prot.340140303. [DOI] [PubMed] [Google Scholar]
- Underwood MC, Zhong D, Mathur A, Heyduk T, Bajaj SP. Thermodynamic linkage between the S1 site, the Na+ site, and the Ca2+ site in the protease domain of human coagulation factor xa. Studies on catalytic efficiency and inhibitor binding. J Biol Chem. 2000;275:36876–36884. doi: 10.1074/jbc.M001386200. [DOI] [PubMed] [Google Scholar]
- van de Locht A, Bode W, Huber R, Le Bonniec BF, Stone SR, Esmon CT, Stubbs MT. The thrombin E192Q-BPTI complex reveals gross structural rearrangements: implications for the interaction with antithrombin and thrombomodulin. Embo J. 1997;16:2977–2984. doi: 10.1093/emboj/16.11.2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Holde KE. A hypothesis concerning diffusion-limited protein-ligand interactions. Biophys Chem. 2002;101–102:249–254. doi: 10.1016/s0301-4622(02)00176-x. [DOI] [PubMed] [Google Scholar]
- Vijayalakshmi J, Padmanabhan KP, Mann KG, Tulinsky A. The isomorphous structures of prethrombin2, hirugen-, and PPACK-thrombin: changes accompanying activation and exosite binding to thrombin. Protein Sci. 1994;3:2254–2271. doi: 10.1002/pro.5560031211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vindigni A, Di Cera E. Release of fibrinopeptides by the slow and fast forms of thrombin. Biochemistry. 1996;35:4417–4426. doi: 10.1021/bi952834d. [DOI] [PubMed] [Google Scholar]
- Vindigni A, White CE, Komives EA, Di Cera E. Energetics of thrombin-thrombomodulin interaction. Biochemistry. 1997;36:6674–6681. doi: 10.1021/bi962766a. [DOI] [PubMed] [Google Scholar]
- Vu TK, Wheaton VI, Hung DT, Charo I, Coughlin SR. Domains specifying thrombin-receptor interaction. Nature. 1991;353:674–677. doi: 10.1038/353674a0. [DOI] [PubMed] [Google Scholar]
- Wells CM, Di Cera E. Thrombin is a Na(+)-activated enzyme. Biochemistry. 1992;31:11721–11730. doi: 10.1021/bi00162a008. [DOI] [PubMed] [Google Scholar]
- Wells JA. Additivity of mutational effects in proteins. Biochemistry. 1990;29:8509–8517. doi: 10.1021/bi00489a001. [DOI] [PubMed] [Google Scholar]
- Wilbanks SM, McKay DB. How potassium affects the activity of the molecular chaperone Hsc70. II. Potassium binds specifically in the ATPase active site. J Biol Chem. 1995;270:2251–2257. doi: 10.1074/jbc.270.5.2251. [DOI] [PubMed] [Google Scholar]
- Wilbanks SM, McKay DB. Structural replacement of active site monovalent cations by the epsilon-amino group of lysine in the ATPase fragment of bovine Hsc70. Biochemistry. 1998;37:7456–7462. doi: 10.1021/bi973046m. [DOI] [PubMed] [Google Scholar]
- Workman EF, Lundblad RL. The effect of monovalent cations on the catalytic activity of thrombin. Arch Biochem Biophys. 1978;185:544–548. doi: 10.1016/0003-9861(78)90199-6. [DOI] [PubMed] [Google Scholar]
- Wu QY, Sheehan JP, Tsiang M, Lentz SR, Birktoft JJ, Sadler JE. Single amino acid substitutions dissociate fibrinogen-clotting and thrombomodulin-binding activities of human thrombin. Proc Natl Acad Sci U S A. 1991;88:6775–6779. doi: 10.1073/pnas.88.15.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Bush LA, Pineda AO, Caccia S, Di Cera E. Thrombomodulin changes the molecular surface of interaction and the rate of complex formation between thrombin and protein C. J Biol Chem. 2005;280:7956–7961. doi: 10.1074/jbc.M412869200. [DOI] [PubMed] [Google Scholar]
- Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, Gilbert T, Davie EW, Foster DC. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci U S A. 1998;95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Wu Q, Westfield LA, Tuley EA, Lu D, Zhang Q, Shim K, Zheng X, Sadler JE. Incomplete embryonic lethality and fatal neonatal hemorrhage caused by prothrombin deficiency in mice. Proc Natl Acad Sci U S A. 1998;95:7603–7607. doi: 10.1073/pnas.95.13.7603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Manithody C, Rezaie AR. Activation of protein C by the thrombin-thrombomodulin complex: cooperative roles of Arg-35 of thrombin and Arg-67 of protein C. Proc Natl Acad Sci U S A. 2006;103:879–884. doi: 10.1073/pnas.0507700103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Prasad S, Di Cera E, Rezaie AR. The conformation of the activation peptide of protein C is influenced by Ca2+ and Na+ binding. J Biol Chem. 2004;279:38519–38524. doi: 10.1074/jbc.M407304200. [DOI] [PubMed] [Google Scholar]
- Ye J, Esmon NL, Esmon CT, Johnson AE. The active site of thrombin is altered upon binding to thrombomodulin. Two distinct structural changes are detected by fluorescence, but only one correlates with protein C activation. J Biol Chem. 1991;266:23016–23021. [PubMed] [Google Scholar]
- Yun TH, Baglia FA, Myles T, Navaneetham D, Lopez JA, Walsh PN, Leung LL. Thrombin activation of factor XI on activated platelets requires the interaction of factor XI and platelet glycoprotein Ib alpha with thrombin anion-binding exosites I and II, respectively. J Biol Chem. 2003;278:48112–48119. doi: 10.1074/jbc.M306925200. [DOI] [PubMed] [Google Scholar]
- Zhang B, Ginsburg D. Familial multiple coagulation factor deficiencies: new biologic insight from rare genetic bleeding disorders. J Thromb Haemost. 2004;2:1564–1572. doi: 10.1111/j.1538-7836.2004.00857.x. [DOI] [PubMed] [Google Scholar]
- Zhang E, Tulinsky A. The molecular environment of the Na+ binding site of thrombin. Biophys Chem. 1997;63:185–200. doi: 10.1016/s0301-4622(96)02227-2. [DOI] [PubMed] [Google Scholar]