

Abstract
We have investigated geometries and excitation energies of bovine rhodopsin and some of its mutants by hybrid quantum mechanical/molecular mechanical (QM/MM) calculations in ONIOM scheme, employing B3LYP and BLYP density functionals as well as DFTB method for the QM part and AMBER force field for the MM part. QM/MM geometries of the protonated Schiff-base 11-cis-retinal with B3LYP and DFTB are very similar to each other. TD-B3LYP/MM excitation energy calculations reproduce the experimental absorption maximum of 500 nm in the presence of native rhodopsin environment and predict spectral shifts due to mutations within 10 nm, whereas TD-BLYP/MM excitation energies have red-shift error of at least 50 nm. In the wild-type rhodopsin, Glu113 shifts the first excitation energy to blue and accounts for most of the shift found. Other amino acids individually contribute to the first excitation energy but their net effect is small. The electronic polarization effect is essential for reproducing experimental bond length alternation along the polyene chain in protonated Schiff-base retinal, which correlates with the computed first excitation energy. It also corrects the excitation energies and spectral shifts in mutants, more effectively for deprotonated Schiff-base retinal than for the protonated form. The protonation state and conformation of mutated residues affect electronic spectrum significantly. The present QM/MM calculations estimate not only the experimental excitation energies but also the source of spectral shifts in mutants.
I. Introduction
The visual pigment rhodopsin (Rh) is a G-protein-coupled receptor that is responsible for light/dark vision in vertebrate eye.1–3 It contains an 11-cis-retinal chromophore attached covalently to a lysine residue (Lys296) via a protonated Schiff-base (PSB) linkage. It is biologically activated by the light-induced 11-cis to all-trans isomerization of PSB retinal (PSBR) that triggers a sequence of events resulting in the excitation of the visual nerve and perception of light in the brain. The chromophore in the protein environment of bovine Rh absorbs at 500 nm,1–3 while the absorption occurs at about 450 nm in organic solvents4 and 610 nm in vacuo.5
For understanding how protein environment regulates absorption maxima (spectral tuning) of the retinal chromophore, many experimental and theoretical studies have been performed. The mutagenesis experiments highlight the importance of some amino acids, but the molecular basis of spectral tuning in various retinal proteins is not clear. Hybrid quantum mechanical/molecular mechanical (QM/MM) calculations can take the entire protein into account and thus are viable in understanding the visual event. In practice, however, computation of the wave length of absorption maxima for retinal proteins is a challenging problem.6–28
For all-trans retinal (Schiff-base terminal = −NH2/−NHCH3) in the gas phase, CASPT2 (at CASSCF geometry = 534/545 nm)22 and TD-B3LYP (this study at B3LYP geometry = 501/509 nm) excitation energies with the 6−31G* basis set agree within 35 nm but differ significantly from an another CASPT2 result with an ANO basis set (at DFTB and MP2 geometries = 606 nm).21,23 The later reproduce the experimental result of 610 nm.5 In a CASPT2/AMBER study on bovine Rh, the improvement of the 6−31G* basis set to ANO-S corrects the first excitation energy by 30 nm.24 Hence, the difference of 60 nm between two CASPT2 results can be attributed partly to the differences in the basis set (6−31G* vs ANO) and partly to geometry (CASSCF vs DFTB or MP2). The errors in the computed gas-phase excitation energy of all-trans retinal (experiment = 610 nm) are quite variable by using basis sets at similar sizes: −100 nm (TD-B3LYP/6−31G* at B3LYP geometry),6−65 nm (CASPT2/6−31G* at CASSCF geometry),22 −36 nm (OM2-MRCI at DFTB geometry),8 57 nm (SORCI/SV(P) at DFTB geometry),8 and more than 200 nm (SACCI/D95(d) at B3LYP geometry).25
Gas-phase calculations7,9 on an 11-cis-retinal model (without β-ionone ring) optimized in the protein matrix of bovine Rh show that (a) incorporation of dynamical correlation in the calculations corrects the excitation energies significantly; (b) CASPT2/6−31G* (at CASSCF geometry), SORCI/SV(P) (at DFTB geometry), and OM2-MRCI (at DFTB geometry) calculations yield comparable excitation energies to each other; and (c) the excitation energies calculated at these three levels deviate from the experimental result (500 nm) by 55−80 nm in the absence of β-ionone ring and protein environment.
The computed gas-phase excitation energy of the full 11-cis-retinal that is optimized in the protein matrix of bovine Rh is also quite variable, analogous to all-trans retinal in the gas phase (see above): 643 nm (CASPT2/ANO at DFTB geometry),21 545 nm (CASPT2/6−31G* at CASSCF geometry),24 616 nm (aug-MCQDPT2/cc-pVDZ at PBE0 geometry),26 912 nm (SACCI/D95(d) at B3LYP geometry),25,27 and 490 nm (TD-B3LYP/D95(d) at B3LYP geometry).27 When Glu113 is included in the gas-phase model, the excitation energy is blue-shifted by ca. 150 and 210 nm at CASPT2/(ANO or 6−31G*)21,24 and aug-MCQDPT2 (cc-pVDZ)26 levels, respectively. The remaining protein environment of bovine Rh shifts the first excitation energy by ca. 15/20/−7 nm at CASPT2/ANO level using NPA/Mulliken/CHARMM charges for the protein environment21 and 110 nm at aug-MCQDPT2 level using effective fragment potential (EFP) method.26 In contrast to these variable contributions, the resulting CASPT2/MM (502/507/495 nm21) and aug-MCQDPT2/EFP (515 nm)26 excitation energies are very close to the experimental value (500 nm) and the results of other QM/MM studies including only retinal in the QM region: 479/489 nm at CASPT2/AMBER (6−31G*/ANO-S)10,24 and 481 nm at TD-B3LYP/AMBER (6−31G*)12 levels. Similarly, TD-B3LYP/AMBER and SACCI/AMBER excitation energies with D95(d) basis set are 508 (492) nm and 601 (506) nm in the absence (presence) of Glu113 in the QM region.27 Hence, although several QM/MM excitation energy calculations reproduce the experimental absorption maximum for bovine Rh, gas-phase results and QM/MM interaction energies are strongly dependent on the chosen QM method and the environmental fixed protein charges. This suggests that protein environment fixes some amount of error in the gas phase or most QM/MM excitation energy calculations favor from lucky error cancelations.
Absorption maximum (569 nm) of all-trans retinal in bacteriorhodopsin (bR) is underestimated by ca. 200 nm at TD-HF/AMBER level.6 Dynamical correlation corrects the QM/MM excitation energy significantly. However, it is still underestimated by ca. 40 nm at SORCI/CHARMM8 and 80 nm at TD-B3LYP/AMBER,6,27 OM2-MRCI/CHARMM,8 and OM2-CIS/CHARMM8 levels. Although the experimental result is overestimated by 100 nm in a SACCI/AMBER study that includes only the retinal in the QM region, it is reproduced within 15 nm (556 nm) when the QM region is extended to Asp85 and the neighboring water molecule W402, indicating electron transfer from Asp85 to retinal.27 Asp85 blue-shifts the first excitation energy by ca. 100 nm also at SORCI/CHARMM and OM2-MRCI/CHARMM levels but without its inclusion in the QM region.8 Almost all QM/MM studies incorporate polarization of the QM part by environmental residues but ignore polarization of the protein environment by QM charges. At a semiempirical level, environmental polarization red-shifts the first excitation energy of bR by ca. 80 nm.28 If this semiempirical estimation is applied to TD-B3LYP/AMBER and OM2/CHARMM results, the experimental excitation energy is reproduced. However, the amount of environmental polarization contribution to the first excitation energy may be quite different at these levels. All computational trends summarized here for the QM/MM studies of bR are the same as those of sensory rhodopsin II.8,25,27
Correlated ab initio calculations that require large QM regions and active spaces for retinal proteins are very expensive. The full 11-cis-retinal is already very large for single-reference coupled cluster calculations. Density functional theory (DFT) calculations with B3LYP functional give comparable QM/MM excitation energies with CASPT210,24 and seem the cost-effective reliable method of choice to explore molecular basis of spectral tuning of retinal proteins. We here present a systematic hybrid density functional QM/MM study in the ONIOM scheme for geometries and excitation energies of Rh to enhance our understanding on the spectral tuning mechanism.
II. Computational Details
A. Setup of the System
We started from the best-resolved X-ray structure available for bovine Rh (pdb code: 1U19, resolution = 2.2 Å), which has no missing amino acid.29 We included only the protein chain A and the corresponding water chain of the crystallographic dimer. Nonamino acid residues at the protein surface (membrane lipids) as well as Hg and Zn ions were omitted. A water molecule was put in the place of surface ion Zn963 for matching its interaction with His278. Met1 was acetylated as in the X-ray structure and C terminal was used for Ala348.
The results of PROPKA30 and PROCHECK31 programs were used, in combination with visual inspection, to assign the protonation states of all titratable residues. Asp83, Glu122, Glu181, and Glu249 were protonated (neutralized). FTIR and UV–vis spectral studies of site-directed mutants of the first three residues indicate that they are in fact neutral.32,33 We assigned His152 and His211 to be protonated at the ε nitrogen only and His195 and His278 to be protonated at the δ nitrogen only. The other histidines (65 and 100) were fully protonated at both nitrogen atoms. Sulfur atoms in Cys110 and Cys187 were not protonated as they formed a disulfide bond. Standard protonation states were used for all other amino acids. The overall charge of the system after adding missing hydrogen atoms (total of 5632 atoms) is +1 due to protonated Schiff-base (PSB) linkage between the 11-cis-retinal and Lys296, compensated by the negatively charged Glu113 counterion.33–35
Although β-ionone ring orientation is 6-s-cis at the C6–C7 bond of the polyene chain of the chromophore in all X-ray studies,29,36–38 NMR studies give conflicting results: 6-s-trans in refs 39 and 40; 6-s-cis in refs 41–43; 6-s-cis (6-s-trans) with a 74% (26%) component in ref 44. Low-level semiempirical MNDO-CI45 and DFTB46 calculations estimate 6-s-trans geometry more stable. Later B3LYP/MM studies12 show that the protein pocket tolerates both conformations, with energetic preference to 6-s-cis conformation. NMR parameters calculated at B3LYP/MM level are also more consistent with experiment for the 6-s-cis conformation.12 These results suggest that 6-strans conformation will appear only very rarely during protein dynamics. Hence, we have considered only 6-s-cis conformation in this study.
B. Force Field Calculations
All MM calculations [pure MM or hybrid ONIOM(QM:MM)] were performed with AMBER all-atom force field and TIP3P water model.47 All available force field parameters, charges and types of each atom in each amino acid were taken from AMBER library.47 As AMBER force field is derived with RESP charges calculated at HF/6−31G* level, we also used RESP charges calculated at the same level for protonated and deprotonated SB retinals that are not defined in AMBER library. As there is no AMBER charge derived for side chain bound Lys in AMBER library and only part of it is included in the model part (see below), the charges of its Cδ and Cε atoms along with those of attached hydrogens were modified slightly. The nonstandard AMBER charges and parameters used in this study are as given in Supporting Information.
The entire Rh system was first optimized at pure AMBER force field level to remove close contacts. For mutants, after mutating residues on this AMBER geometry of Rh, the systems were again optimized at AMBER level. The resulting geometries were used as the initial geometry of ONIOM optimizations. During pure AMBER (as well as ONIOM) geometry optimizations, no restraints were applied. The positions of backbone atoms in the seven transmembrane α-helices of Rh as well as the environment of ions ignored were well conserved with AMBER optimization (see Figure 1). However, in the absence of crystal packing effects and solvent layer, surface residues that connect the helices showed large variations compared with experiment. As those surface residues are significantly away than the chromophore, they are not expected to affect the computed electronic spectra. During ONIOM geometry optimizations, no significant conformational change was observed compared with AMBER geometry.
Figure 1.
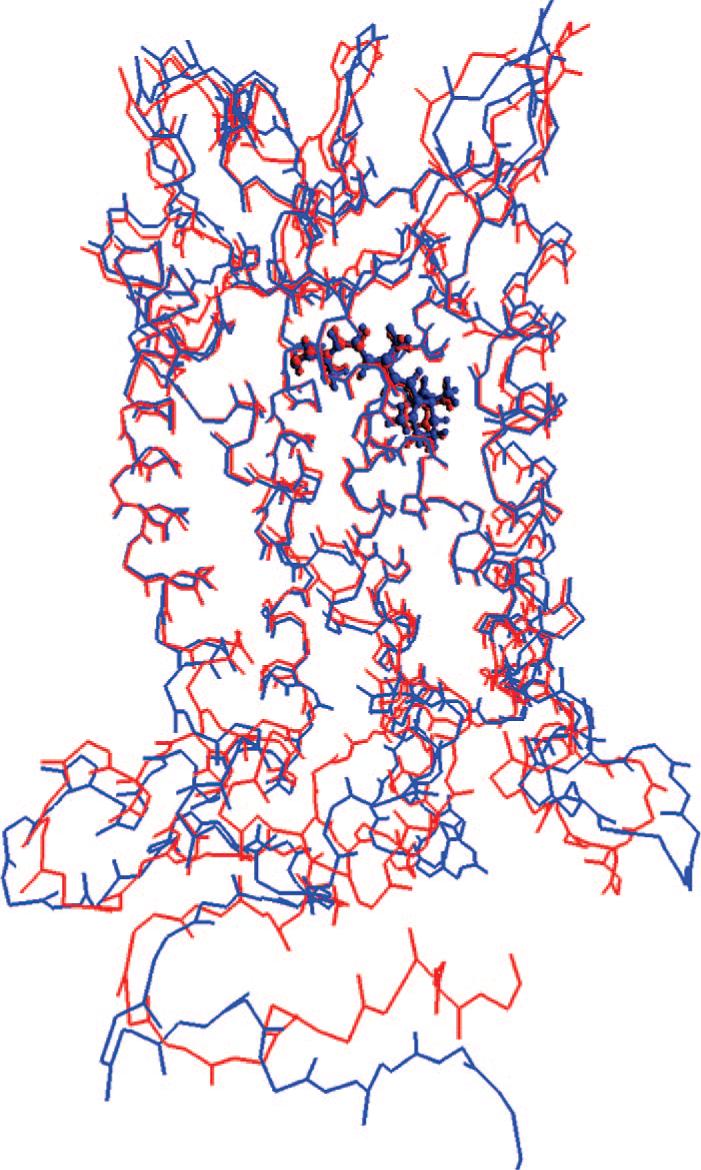
Backbone atom positions in the X-ray structure of Rh (pdb code: 1U19, in blue) and its AMBER-optimized geometry (in red). The 11-cis-retinal is shown with the ball and stick model.
There are two water molecules near the chromophore,29,36 one of which bridges backbone and carboxylic side chain oxygens of Glu113, and the other bridges backbone oxygen of Cys187 and carboxylic side chain oxygen of Glu181. These water molecules were not resolved at lower resolution X-ray structures.37,38 They were shown previously to be crucial for the stabilization of the 11-cis-retinal and for a recently proposed proton-transfer mechanism in photobleaching reaction.13,48 The positions of these water molecules were well conserved during both AMBER and ONIOM geometry optimizations.
C. ONIOM Hybrid Calculations
QM/MM calculations were performed with the use of two-layer ONIOM(QM:MM) scheme,49–51 in which the interface between QM and MM region is treated by hydrogen link atoms52 and the total energy of the system (EONIOM) is obtained from three independent calculations:
where EMM,real is the MM energy of the entire system, called real system in ONIOM terminology; EQM,model is the QM energy of a part of real system that has main chemical interest, called model part; and EMM,model is the MM energy of the model part.
In this study, electrostatic interactions between two layers were calculated using both mechanical and electronic embedding schemes.50 In the mechanical embedding (ME) scheme,49–52 they were calculated at the MM level. In the electronic embedding (EE) scheme, 52 the electrostatic interactions between the two layers are present in all three energy terms (see above).49 Thus, the electrostatic interactions terms included at the MM energies (EMM,real and EMM,model) cancel out, leaving only the interaction energy term that also includes polarization of the model part wave function by the surrounding charges included in EQM,model.
When performing ONIOM-EE geometry optimization, the interaction term of QM model part and the surrounding were calculated using ESP charges of the model part obtained at each step of the optimization. Our test calculations on Rh show that geometries and excitation energies are almost the same when Mulliken charges are used for the calculation of QM/MM interaction term instead of ESP charges. The changes in the charge distribution of the model part upon electronic excitation are automatically taken into account in ONIOM-EE excitation energies, as QM/MM interaction term is present in the QM Hamiltonian. In standard ONIOM-ME calculations, the electrostatic interaction energy is calculated by fixed MM charges, which do not reflect electrostatic energy change upon electronic excitation as the same fixed charges are used for ground and excited states artificially. Instead, we have performed separate calculations for the ground (S0) and excited (S1 and S2) states to include electrostatic energy change upon electronic excitations in the ONIOM-ME electronic excitation energies by using Mulliken charges of the respective state of the model part.
The model part comprises the full 11-cis-retinal chromophore along with covalently bound N (NH) moiety of Lys296 for deprotonated (protonated) SB linkage, resulting in total of 50 (51) atoms including the link atom (see Figure 2). When the model part is extended to include the Cε atom of Lys296 and the attached hydrogens, the first excitation energy red-shifts only 10 nm. Further extension of QM/MM boundary to the complete Lys296 does not affect the first excitation energy.
Figure 2.
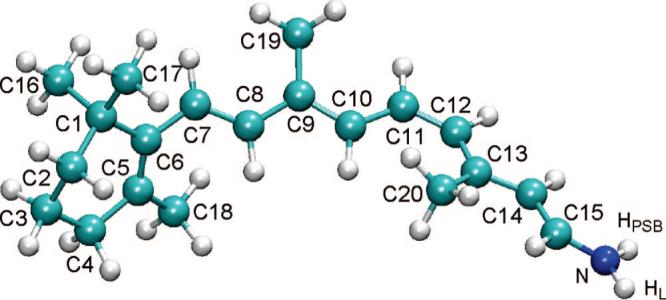
The model part of the ONIOM calculations with hydrogen link atom HL. HPSB is not present for deprotonated SBR.
The ONIOM calculations were performed for the QM part with BLYP53,54 and B3LYP54,55 density functionals to be representative of the class of generalized gradient and hybrid functionals, respectively. Geometry optimizations with the “self-consistent charge density-functional tight-binding” (abbreviated as SCC-DFTB or DFTB)56 were also performed for comparing its geometries obtained with BLYP and B3LYP functionals as well as excitation energies computed on top of them.
In this study, we performed ONIOM-ME and ONIOM-EE ground-state singlet geometry optimizations (QM= BLYP, B3LYP and DFTB) starting from the pure AMBER-optimized geometries and then calculated the vertical excitation energies with time-dependent (TD)-DFT employing BLYP and B3LYP functionals on top of the ONIOM-optimized geometries. The level of an ONIOM(QM:MM) calculation will be abbreviated in the form “QM level-embedding scheme”, e.g., B3LYP-EE, DFTB-ME, etc., for ONIOM(B3LYP/6−31G*:AMBER)-EE, ONIOM(DFTB:AMBER)-ME, etc., respectively. Calculations in the gas phase (without protein environment, in order to assess the protein effects) are labeled like B3LYP-none. Labels before // is used for the method for vertical excitation energy calculation and labels after // refer to the method for ground-state geometry optimization. For instance, B3LYP-EE//B3LYP-ME refers to a TD-B3LYP-EE singlet excitation energy calculation at the B3LYP-ME optimized ground-state geometry.
All calculations were performed with 6−31G* basis set57 using development versions of Gaussian03 program package.58 D95(d) and correlation consistent cc-pVTZ basis sets red-shift the TD-B3LYP/6−31G* excitation energy only by 5 and 8 nm, respectively. SACCI excitation energies are also affected less than 5 nm when the D95(d) basis set is improved to cc-pVDZ.59 The improvement of the 6−31G* basis set to ANO-S red-shifts the first excitation energy of bovine Rh by 30 nm in a previous CASPT2/AMBER study.24 DFT calculations converge much more quickly to the basis set limit than correlated ab initio treatments. Hence, the error in the present results due to the use of the 6−31G* basis set should be smaller than 30 nm (3 kcal/mol).
D. Mutation Types
The environment of the 11-cis-retinal in bovine Rh is as shown in Figure 3. We have investigated mutation of the highlighted residues in Figure 3 to assess the spectral tuning mechanism. The changes in the side chains with the investigated mutations are shown in Figure 4.
Figure 3.
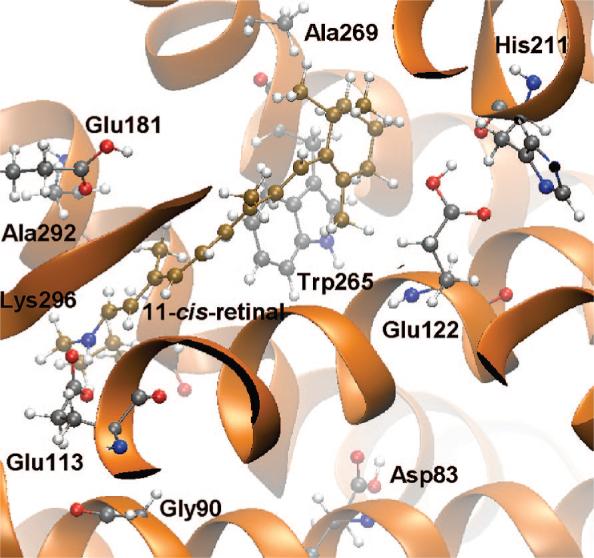
Environment of the 11-cis-retinal in Rh. The 11-cis-retinal and the attached Lys296 are shown in brown.
Figure 4.

Changes in the side chains of amino acids with mutations and experimental absorption maxima.
III. Results
A. Geometry of the 11-cis-Retinal
At present, there are no reliable parameters that can be used in fitting the experimental diffraction data of the 11-cis-retinal with twisted and extended π-system whose delocalized charge interacts with a carboxylate group (Glu113) in the standard X-ray refinement softwares.22 Hence, the MM-fitted C—C bond lengths along the polyene chain of the chromophore in the available X-ray structures, which show a clear single/double bond alternation pattern (see the zigzags in Figure 5a), differ from each other as large as 0.1 Å.29,36–39 The deviations in the chromophore structures are more significant for the MM-fitted bond angles (see Figure 6a) and dihedral angles (see Supporting Information) along the polyene chain.29,36–39 This highlights the importance of QM/MM calculations for revealing a more reliable chromophore structure in the protein environment.
Figure 5.
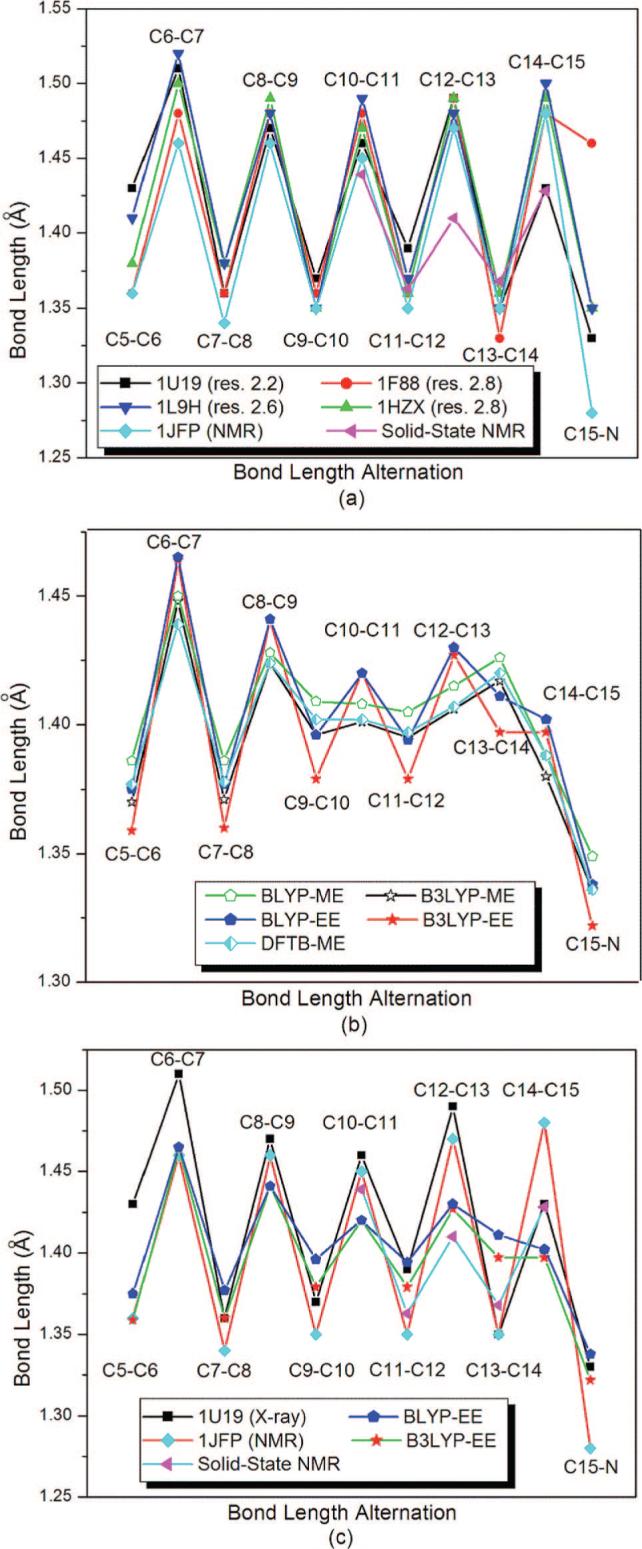
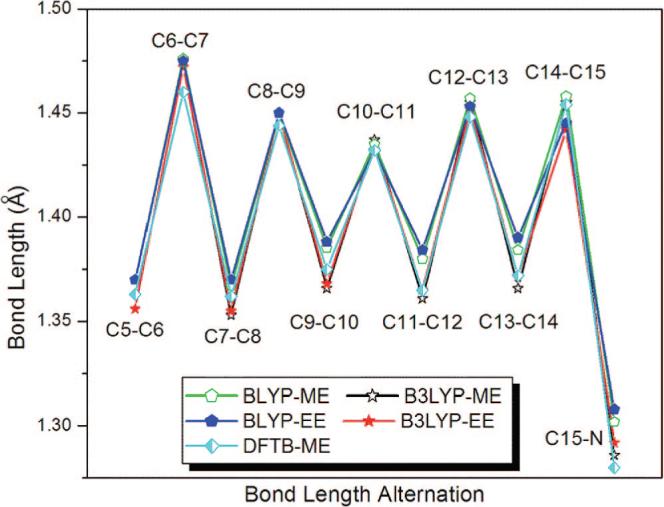
Bond length alternation along the polyene chain of the PSB 11-cis-retinal chromophore of Rh. Comparison of (a) the MM-fitted values in the X-ray structures (pdb codes: 1U19,29 1L9H,36 1F88,37 and 1HZX;38 chain A), an NMR structure with pdb code of 1JFP,39 and the high resolution double-quantum solid-state NMR data62 for the C10–C15 moiety including vibrational correction that have confidence level of ± 0.025 Å, (b) the results of present ONIOM calculations, and (c) NMR experiments and DFT-EE results.
Figure 6.
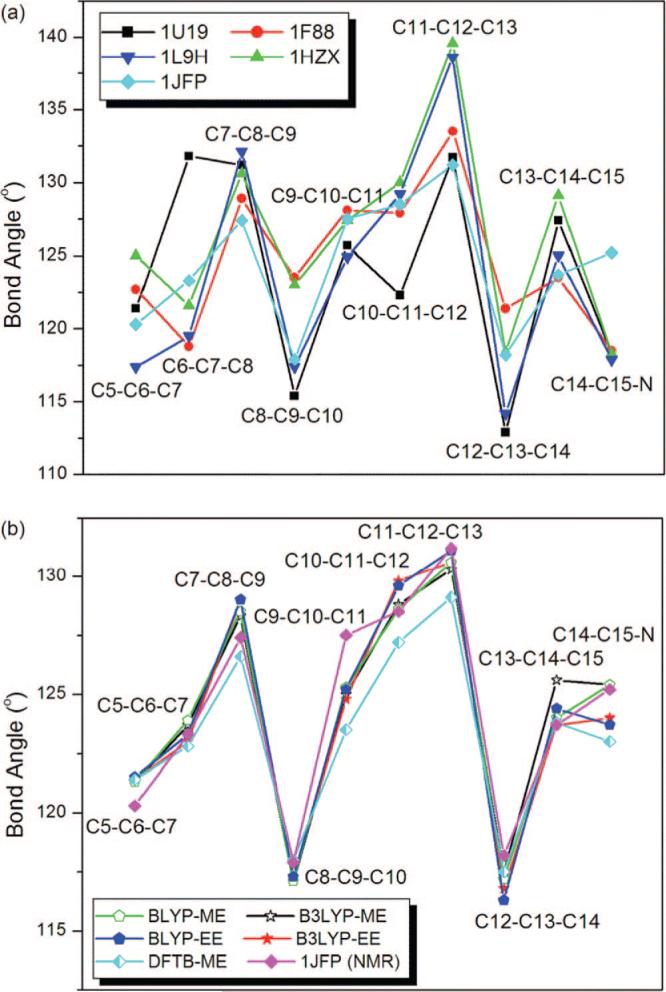
Bond angles along the polyene chain of the PSB 11-cis-retinal chromophore of Rh. Comparison of (a) the MM-fitted values in the X-ray structures and the NMR values and (b) the NMR experiment and the results of present ONIOM calculations.
For Rh with PSBR, ONIOM QM-ME geometries with DFTB and B3LYP are nearly identical (see Figures 5 and 6b for bond lengths and angles). On the other hand, BLYP bond lengths show deviations around 0.02 Å from the B3LYP ones. Gas-phase model studies for PSBR using polyene chains with different lengths and an NH2 terminal show that B3LYP geometries are almost the same as CASPT2 ones (less than 0.01 Å deviation in bond lengths).26 Hence, B3LYP should perform better than BLYP for the retinal geometry. All computed bond angles of PSBR are reasonably in agreement with each other and with an NMR structure (pdb code: 1JFP, see Figure 6b). The MM-fitted bond angles in the X-ray structures are not consistent to each other (see Figure 6a) and with the computed ones.
ONIOM QM-ME calculations do not show any obvious bond length alternation (BLA) pattern for C9–N moiety (see Figure 5b), analogous to previous gas-phase studies.61 The agreement between the gas-phase model61 and ONIOM QM-ME studies in BLA pattern can be assigned to the fact that the QM wave function, and thus the electronic structure, is not polarized in ME calculations. BLA is recovered when electronic polarization of the chromophore by the environmental point charges is included in the calculations (QM-EE) except C13–C15 moiety (see below). Although QM-EE C–C bond lengths along the polyene chain agree with the MM-fitted bond lengths in the best resolved X-ray structure (pdb code: 1U19)29 and an earlier NMR (pdb code: 1JFP)40 structure better than the MM-fitted bond lengths in the lower resolution X-ray structures36–38 (see Figure 5c), BLA pattern obtained in ONIOM QM-EE calculations is significantly less pronounced compared with that in the experimental structures (see Figure 5c), in agreement with the previous same level calculations.12,27,29,46 Recent high resolution double-quantum solid-state NMR experiments62 for C10–C15 moiety give vibrationally corrected bond lengths which demonstrate significantly less pronounced BLA than those in the other experimental structures (see Figures 5a and c). These new bond lengths agree with the present ONIOM QM-EE results (see Figure 5c) within the experimental confidence level (±0.025Å).62
To investigate polyene chain structure for the SB 11-cis-retinal in Rh, we moved the Schiff-base hydrogen (HPSB in Figure 2) to one of the carboxylic oxygen of Glu113 (OE2).63 In this case, the chromophore becomes neutral and its polyene chain demonstrates a clear BLA pattern (see Figure 7). An analogous BLA pattern was seen when both retinal and Glu113 were taken deprotonated (see Supporting Information).
Figure 7.
Bond length alternation along the polyene chain of the 11-cis-retinal chromophore of Rh after transferring the Schiff-base hydrogen to Glu113 (deprotonated SBR and protonated Glu113).63
The generality of the computational trends given in this section was further confirmed (see Supporting Information) on the E122Q and E113Q mutants with both PSBR (deprotonated Glu113) and SBR (with both protonated and deprotonated Glu113).
B. Electronic Structure of the PSB 11-cis-Retinal
Mulliken charges computed with both ME and EE schemes show that total charge (+1) of the PSB 11-cis-retinal is shared mainly by the C15−NH2 moiety and the carbon atoms that have one methyl group attached (C13, C9, and C5), indicating that the bonds formed by these carbon atoms have dominant single-bond characters. For example, Mulliken charges of the ground-state singlet (S0) computed at our best level (B3LYP-EE) for those chromophore atoms are as follows: NH2, 0.17; C5, 0.14; C9, 0.20; C13, 0.21; C15H, 0.33. Location of the positive charge densities on the atoms adjacent to the methyl groups suggests that strong electron donating capability of the methyl groups results in electron densities pushed toward the C15–N moiety, leaving positive charges on those atoms.
The preceding analysis suggests that the ground-state wave function of the PSB 11-cis-retinal is composed of essentially linear combination of wave functions that belong to five resonance structures (|a>, |b>, |c>, |d>, and |e>, see Figure 8) analogous to all-trans PSBR in the gas phase.61 In the absence of electronic polarization effect of the environmental residues of the chromophore (gas phase and ME calculations), |c> and |e> contribute to the ground-state wave function significantly in addition to |a> and |b>, destroying single/double bond alternation pattern (see Figure 5b). Polarizing effect of the protein environment helps to stabilize electron density on the methyl groups, resulting in smaller contributions from |c> and |d>. Hence, BLA pattern becomes more obvious in the EE calculations than the ME ones. C13–C14 bond tends to single (double) bond in ME (EE) calculations as |c> and |d> contribution is larger (smaller).
Figure 8.
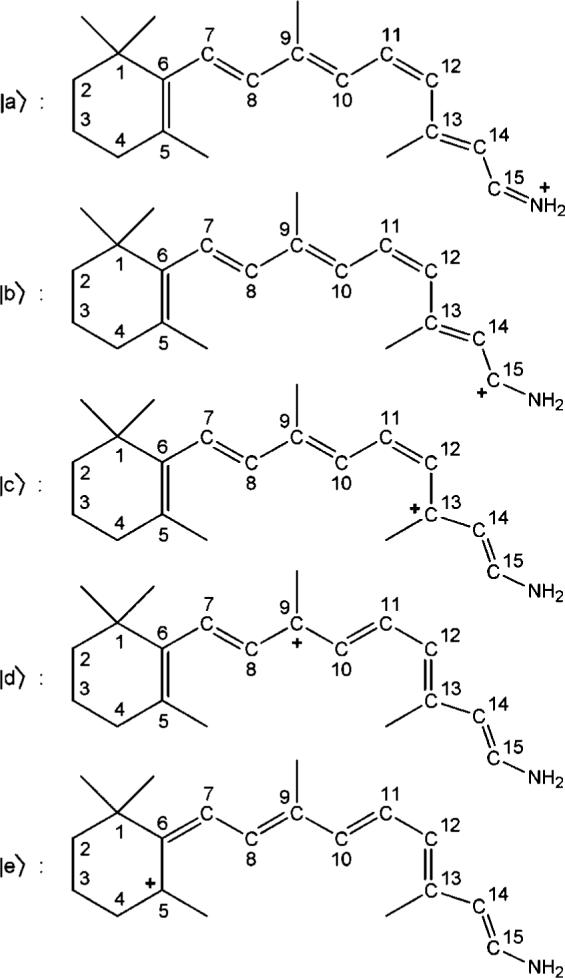
The most important resonance structures of the PSB 11-cis-retinal in Rh
The positive Mulliken charge on the C5–C6 bond in the first excited singlet state (S1) increases by 0.05 with a concomitant decrease of the charges along the polyene chain compared with the ground state (S0) charges, indicating a larger contribution from |e> in the S1 state. The positive Mulliken charge on the NH2 moiety (β-ionone ring) decreases (increases) by 0.12 going from S0 to the second excited singlet state (S2). This indicates that C15–N bond is mainly covalent in the S2 state with a small contribution from |a>.
C. Excitation Energies and Spectral Tuning
In the following, we mainly discuss ONIOM(TD-DFT:AMBER) vertical excitation energy from S0 to S1 in bovine Rh and some of its mutants, which corresponds to the energy of the absorption maximum for the longest wavelength in the electronic absorption spectrum. Unless stated otherwise, we refer to the “S0 → S1 vertical excitation energy” when saying “excitation energy” or “absorption maximum”. Vertical excitation energies to the next excited singlet state (S0 → S2) are not discussed much but included in the Supporting Information. For Rh and its mutants, we calculated oscillator strength (f) of the S0 → S1 (S0 → S2) excitation as 0.7−1.4 (0.3−0.6), suggesting that the S1 state has a larger ionic character than the S2 state64 and both are absorbing states (see Supporting Information for the full list of f values). We always label the bright states as S1 and S2. When QM region is extended to Glu113, some dark states appear lower in energy than the bright states.
1. The Effects of Geometry and HF Admixture
To assess the effects of geometry and exact exchange on the excitation energies for Rh, ONIOM geometry optimizations and the subsequent ONIOM TD-DFT:MM calculations were performed with B3LYP (20% HF exchange) and BLYP (no HF exchange) density functionals along with DFTB (geometry optimization only) method. We also performed TD-DFT calculations on the isolated QM part of ONIOM-optimized geometries to assess the influence of protein environment on excitation energies. Although some experiments indicate that the 11-cis-retinal has a PSB linkage to Rh,1–3 the protons have not been resolved in the X-ray structures. Also, the structures of retinal and Glu113 have not yet been determined with high resolutions. Hence, we have calculated vertical excitation energies for deprotonated SBR with both protonated and deprotonated Glu113, as well. This allows us to assess if the excitation energy range for SBR is compatible with that for PSBR and the behavior of the computational methods with SBR. The results are given in Table 1 for Rh with PSBR and in Table 2 for Rh with deprotonated SBR and protonated Glu113.63 The results for deprotonated SBR and Glu113 are given in Supporting Information and will be briefly discussed here and in the following sections.
TABLE 1.
Vertical S0 → S1 Excitation Energies (in nm) Calculated at Gas Phase (Labeled none) and Different ONIOM(ME or EE) Levels at Different Ground State Optimized Geometries for the Wild-Type Bovine Rhodopsin with PSBR
TABLE 2.
Vertical S0 → S1 Excitation Energies (in nm) Calculated at Gas Phase (Labeled none) and Different ONIOM (ME or EE) Levels at Different Ground State Optimized Geometries for the Wild-Type Bovine Rhodopsin with SBR and Protonated Glu113 at the OE2 Atom63
| ground state geometry |
|||
|---|---|---|---|
| excitation | B3LYP-ME | BLYP-ME | DFTB-ME |
| B3LYP-nonea | 400 | 420 | 416 |
| B3LYP-ME | 399 | 418 | 415 |
| B3LYP-EE | 412 (429b) | 430 (447c) | 427 |
| BLYP-nonea | 464 | 483 | 472 |
| BLYP-ME | 459 | 475 | 470 |
| BLYP-EE | 481 | 497 (513d) | 488 |
Gas-phase TD-DFT result of the isolated model QM part.
B3LYP-EE//B3LYP-EE.
B3LYP-EE//BLYP-EE.
BLYP-EE//BLYP-EE.
Our preferred ONIOM calculations are the ones with EE scheme since their results include both electrostatic and electronic polarization effects of the protein environment on the chromophore. ONIOM-EE calculations for Rh with PSBR (see Table 1) estimate the first excitation energy around 500 nm with TD-B3LYP (reproducing the experiments1–3) and 550 nm with TD-BLYP. This suggests that for retinal proteins with PSBR a density functional with 20% HF admixture (B3LYP) gives a more balanced description of electronic absorption spectrum than that with no HF admixture (BLYP, red-shift error of ca. 50 nm). Although ME and EE geometries give almost the same first excitation energy for PSBR with B3LYP, EE correction over ME geometries with BLYP worsens the agreement between experiment (ca. 500 nm) and computations slightly (ca. 550 nm → 572 nm). The EE geometry optimization with BLYP density affects the ONIOM-EE excitation energies also for SBR with deprotonated Glu113 (ca. 480 nm → 455 nm, see Supporting Information), although they are not affected much when using B3LYP. The charges (ESP or Mulliken) derived from BLYP density thus seem less appropriate for EE geometry optimizations.
When using geometries optimized with BLYP, the error in TD-B3LYP excitation energies of Rh with PSBR is very small (around 10 nm; see the first three rows of the first two columns in Table 1). Excitation energies calculated on the geometries optimized with B3LYP and DFTB (see the first and last columns of Table 1) are almost the same for Rh with PSBR since these geometries are very close to each other (see Figure 5b). Hence, DFTB can be used in ONIOM calculations as a cost-effective geometry optimization method for calculating TD-B3LYP-ME or EE excitations of PSBR.
For Rh with SBR (see Table 2 and Supporting Information), excitation energies computed with TD-BLYP are significantly red-shifted than those with TD-B3LYP (at least 60 nm), analogous to PSBR case. Excitation energies computed on geometries optimized with BLYP and DFTB are very similar to each other and deviate from those with B3LYP by ca. 20 nm, arising from 0.02 Å difference in the average bond lengths.
The summarized trends in the first excitation energies of Rh above for PSBR and SBR are the same as those obtained on the E122Q and E113Q (see Supporting Information) mutants. In the following, we will discuss the protein effect on the first excitation energy computed with B3LYP since its results for Rh are more consistent with experiments and CASPT2 results.10,24
2. The Effects of Protein Environment
Here we discuss the effects the protein environment on the excitation energies and spectral shifts systematically for wild-type (WT) Rh and its several mutants. In order to decompose the overall effects of protein into various contributions, we have performed a series of calculations, as shown in Table 3. At first, we use the optimized geometry at the B3LYP-ME level. A single point calculation using B3LYP for only the model QM part (gas phase calculation labeled as none in Table 3) gives a reference excitation energy of the chromophore. The B3LYP-ME excitation energy at this geometry, relative to the reference excitation energy above, gives the electrostatic contribution of protein environment. The B3LYP-EE excitation energy includes, in addition to the electrostatic effects, the effects of change of charge density distribution within the chromophore caused by the protein environment, or the electronic polarization effects. The polarization effects also change the geometries of the chromophore and the protein, resulting in the B3LYP-EE optimized geometry. The geometrical effect is calculated as the energy difference between B3LYP-EE//B3LYP-EE and B3LYP-EE//B3LYP-ME and is the measure of effect of geometry change due to electronic polarization of the chromophore. The B3LYP-EE//B3LYP-EE energy is our final target energy and includes all the effects: electrostatic, electronic polarization, and geometrical. Thus, this series of calculations allows us to elucidate the source of spectral shifts.
TABLE 3.
Vertical S0 → S1 Excitation Energies (in nm) Calculated Using B3LYP at Gas Phase (Labeled none) and at Different ONIOM (ME or EE) Levels for the Wild-Type Rh and Some of its Mutants along with the Shifts in Excitation Energies (in nm, in Parentheses) with Respect to the Wild-Type Rh
| retinal protonation | B3LYP-none//B3LYP-ME | B3LYP-ME//B3LYP-ME | B3LYP-EE//B3LYP-ME | B3LYP-none//B3LYP-EE | B3LYP-EE//B3LYP-EE | experiment | |
|---|---|---|---|---|---|---|---|
| WT | PSBR | 535 (0) | 504 (0) | 505 (0) | 560 (0) | 503 (0) | 500 (0)1-3 |
| WT | SBR | 400 (−135)a | 399 (−105) | 412 (−93) | 408 (−152) | 429 (−74) | |
| 399 (−136)b | 366 (−138) | 382 (−123) | 396 (−164) | 380 (−123) | |||
| E122Q | PSBR | 533 (−2)c | 503 (−1) | 497 (−8) | 566 (6) | 492 (−11) | 480 (−20)33-35 |
| 544 (9)d | 509 (5) | 512 (7) | 566 (6) | 514 (11) | |||
| E122Q | SBR | 402 (−133)a,c | 395 (−109) | 406 (−99) | 408 (−152) | 418 (−85) | |
| 399 (−136)b,c | 362 (−142) | 382 (−123) | 397 (−163) | 379 (−124) | |||
| E113Q | PSBR | 536 (1)e | 521 (17) | 526 (21) | 536 (−24) | 530 (27) | 496 (−4)32,34,66 |
| E113Q | SBR | 398 (−137)e | 390 (−114) | 397 (−108) | 398 (−162) | 409 (−94) | 384 (−116)32,34,66 |
| 399 (−136)f | 396 (−108) | 408 (−97) | 407 (−153) | 425 (−78) | |||
| 391 (−144)g | 389 (−115) | 397 (−108) | 395 (−165) | 422 (−81) | |||
| E113D | PSBR | 533 (−2) | 504 (0) | 505 (0) | 557 (−3) | 499 (−4) | 510 (10)67 |
| G90D | PSBR | 544 (9)h | 493 (−11) | 490 (−15) | 587 (27) | 475 (−28) | 483 (−17)68 |
| 544 (9)i | 501 (−3) | 503 (−2) | 574 (14) | 502 (−1) | |||
| A292S | PSBR | 542 (7)j | 500 (−4) | 503 (−2) | 571 (11) | 501 (−2) | 491 (−9)68 |
| 537 (2)k | 496 (−8) | 499 (−6) | 567 (7) | 500 (−3) | |||
| 542 (7)l | 502 (−2) | 504 (−1) | 570 (10) | 507 (4) | |||
| A269T | PSBR | 542 (7)m | 502 (−2) | 504 (−1) | 568 (8) | 506 (3) | 514 (14)70 |
| 543 (8)n | 499 (−5) | 502 (−3) | 571 (11) | 506 (3) | |||
| 547 (12)o | 500 (−4) | 504 (−1) | 574 (14) | 503 (0) | |||
| 542 (7)p | 500 (−4) | 502 (−3) | 568 (8) | 498 (−5) | |||
| D83N | PSBR | 534 (−1) | 501 (−3) | 503 (−2) | 570 (10) | 502 (−1) | 495 (−5)33 |
| W265Y | PSBR | 536 (1) | 495 (−9) | 503 (−2) | 574 (14) | 497 (−6) | 485 (−15)69 |
| H211C | PSBR | 543 (8) | 499 (−5) | 501 (−4) | 573 (13) | 501 (−2) | 495 (−5)71 |
| E181Q | PSBR | 541 (6)q | 499 (−5) | 501 (−4) | 571 (11) | 499 (−4) | 502 (2)72 |
| 543 (8)r | 500 (−4) | 502 (−3) | 570 (10) | 500 (−3) |
The computational results in the row were obtained with
Protonated Glu113.
Deprotonated Glu113.
Q112 whose NH2 moiety is oriented toward one of the methyl groups attached to C1 atom of the chromophore.
Q112 whose O=C-NH2 plane is rotated by ca. 90° compared with that in footnote c.
Q113 whose side chain NH2 and carbonyl O moieties are oriented toward its backbone carbonyl O and Schiff-base nitrogen, respectively.
Q113 whose side chain NH2 moiety is oriented to form a H-bond with Schiff-base nitrogen.
Q113 whose side chain NH2 and carbonyl O moieties are oriented toward Gly90 and Schiff-base nitrogen, respectively.
Deprotonated Asp90.
Protonated Asp90.
Side chain -OH unit of S292 that forms a H-bond with carbonyl O of Met288.
Side chain -OH unit of S292 that forms a H-bond with a water molecule.
Side chain -OH unit of S292 that is oriented toward -OH unit of Ser186, a higher energy conformer.
Side chain methyl moiety of T269 that is oriented toward Leu266 side chain and side chain -OH unit that is oriented toward its backbone carbonyl O and becomes closer to a methyl group attached to C1 atom of the chromophore (ca. 2.6 Å).
Side chain methyl moiety of T269 that is oriented toward C1 (one of methyl groups attached to it) and C2 atoms of the chromophore and side chain -OH unit as in footnote m.
Side chain methyl moiety of T269 as in footnote n and side chain -OH unit that forms a H-bond with Leu266.
Side chain methyl moiety of T269 as in footnote n and side chain -OH unit that forms a H-bond with Trp265.
OE2 atom of E181 that is replaced with NH2 moiety.
OE1 atom of E181 that is replaced with NH2 moiety.
(a) Wild-Type Rh with PSBR
For the wild-type (WT) Rh, vertical excitation energies of PSBR computed at our target level [B3LYP-EE//B3LYP-EE: S0 → S1 (503 nm); S0 → S2 (381 nm)] reproduces the experimental results [S0 → S1 (500 ± 2 nm); S0 → S2 (380 ± 2 nm)]1–3 and agree with the results of other QM/MM studies within 20 nm (see Introduction).10,12,21,24,26,27
The change in the 11-cis-retinal geometry due to protein electrostatics does not affect the first excitation energy much (gas-phase result: at gas-phase geometry, 527 nm; at ME geometry, 535 nm). Electrostatic effect of Rh protein environment blue-shifts the excitation energy (ME, 504 nm) by ca. 30 nm (ca. 3 kcal/mol). The change in the retinal geometry, e.g., in BLA, induced by electronic polarization effect of the protein environment red-shifts the first excitation energy in the gas phase calculation by 25 nm (gas phase: 535 nm → 560 nm). However, this effect is counterbalanced with the effect of the change in the H-bond distance between N[H] atom of PSBR and one of the carboxylic oxygen atoms of Glu113 (ME = 2.81 [1.79] Å; EE = 2.73 [1.70] Å) in the WT Rh. The excitation energy of the WT Rh in protein environment does not change by going from ME//ME (504 nm) through EE//ME (503 nm) to EE//EE (503 nm), namely the net effect of polarization is nearly zero both in the presence and absence of its influence on geometry in the WT Rh. The full protein effect thus happens to be equal to the electrostatic contribution only. As mentioned above, this is due to cancelation of the effects of changes of retinal geometry and H-bonding, and this cancelation does not occur in all mutants, such as G90D (to be discussed below).
The decrease in the H-bond distance between PSBR and Glu113 with EE correction suggests that negatively charged Glu113 helps localizing more positive charge on the C15–N moiety of PSBR, which decreases the contributions of resonance structures |c> and |d> in the ground-state wave function and, thus recovers experimental BLA pattern. Analogous decreases in this H-bond with EE optimizations occur for all studied mutant structures with PSBR (see Supporting Information).
Calculations at the EE//ME and EE//EE levels but turning off the charge of Glu113 forming a H-bond with PSBR, e.g., zeroing the interaction between the chromophore and Glu113, give the excitation energies of 535 and 569 nm, respectively, which are almost the same as that calculated for the model part in the gas phase (none//ME = 535 nm and none//EE = 560 nm). In addition, when Glu113 is included in the model QM part (without further geometry relaxation), EE//ME and EE//EE excitation energies (499 and 500 nm) are almost the same as those calculated on the gas phase model (none//ME = 500 and none//EE = 500 nm). Hence, the effects of amino acids other than Glu113 seem to have canceled out with no or little net effect on the first excitation energies. The above results with Glu113 in the model QM part are also very close to those with Glu113 in the MM part (503−505 nm, see Table 3), validating the placement of Glu113 in the MM part in the ONIOM calculations.
The results of the analysis of the energy contributions are significantly different among the present study and various previous QM/MM studies (see also Introduction). Blue-shifts of the first excitation energy of the 11-cis-retinal caused by Glu113 in the gas-phase CASPT2 (basis set: ANO and 6−31G*; ca. 150 nm)10,21,24 and aug-MCQDPT2 (basis set: cc-pVDZ; ca. 200 nm)26 calculations are more than two times larger than that of the present study (none//EE without/with Glu113 = 560/500 nm, e.g., 60 nm shift). When Glu113 is in the QM region, the remaining protein environment shifts the first excitation energy to red by ca. 100 nm at aug-MCQDPT2/EFP and to blue by ca. 100 nm in SACCI/AMBER level,26,27 while its effect is small in the previous CASPT2 (ANO basis set, ± 20 nm depending on the charge model of the protein environment)21 and present (almost no net effect) studies. When Glu113 is in the MM region, protein environment blue-shifts the gas-phase excitation energies by ca. 310 nm (SACCI/AMBER),27 65 nm (CASPT2/AMBER),10,24 and 57 nm (present TD-B3LYP/AMBER study = 503 vs 560 nm; see Table 3). The source of this diversity of the calculated effects of Glu113 is not clear at the present time and requires further careful investigation.
When Glu113 is included in the QM part of ONIOM-EE calculations, there appears another bright singlet transition (500 nm with f = 0.5; transition occurs mainly from p orbitals of carboxylate oxygens of Glu 113 to a retinal orbital), which is almost degenerate to the singlet transition mainly between retinal orbitals (499 nm with f = 0.9; the same as S1 when Glu113 is not in the QM region), in agreement with CASPT2 calculations on model systems that do not include β-ionone ring.9,10 When Glu113 is in the QM part, the S2 state in the absence of Glu113 in the QM region appears as the sixth root without any change in its excitation energy and oscillator strength as a result of intervening of silent charge transfer transitions from Glu113. The S2 state does not couple with Glu113 orbitals.
(b) Putative Wild-Type Rh with SBR
As seen in Table 3, the protein contributions to SBR excitation energies are significantly different than those to PSBR. We at first look at SBR with protonated Glu113. The first excitation energy is not affected much by the electrostatic effects of protein environment (none//ME = 400 to ME//ME = 399 nm in Table 3), while the second excitation energy is blue-shifted (see Supporting Information). The polarization effect by protein environment red-shifts the first excitation energy (ME//ME = 399 to EE//ME = 412 nm) by 13 nm. The polarization geometrical effect shifts this to red further by 17 nm (EE//EE = 429 nm), one-half of which arises from the BLA change and the other half is a result of the decrease in the H-bond length (0.16 Å) between Schiff-base nitrogen (SBR-N) and protonated Glu113. One may recall in PSBR that the effect of polarization both in presence and absence of its influence on geometry was nearly zero as a result of counterbalancing of the changes in the chromophore geometry and the strength of the H-bond between PSBR and deprotonated Glu113.
When both SBR-N and Glu113 are deprotonated (see Table 3), the electrostatic interaction blue-shifts the first (second) excitation energy by 33 (15) nm. However, electronic polarization red-shifts the first excitation energy by 16 nm. The repulsive interaction between SBR-N and carboxylic oxygens of Glu113 becomes more pronounced when ME geometries are refined with EE, which increase the N–O distance by ca. 0.4 Å. This change in Glu113 side chain conformation does not influence the EE//EE first excitation energies much since SBR-N and carboxylate O of Glu113 are already well separated in ME geometries (3.4 Å). As Glu113 is far away than the retinal, electronic polarization does not cause any significant change on the retinal geometry. When a H-bonding water is placed between deprotonated SBR and Glu113, excitation energies become the same as the SBR system with protonated Glu113. In conclusion, the absorption maximum is at around 430 (380) nm when nitrogen atom of SBR has a H-bonding (repulsive) interaction with a nearby residue (see Table 3 for WT and E122Q).
(c) E122Q Mutant
When protonated Glu122 (E122) residue in Rh is replaced by Gln (Q), the experimental absorption maximum blue-shifts from 500 to 480 nm (shift = −20 nm).33–35 Gas-phase QM results for the isolated model systems from ME optimized geometries of the WT Rh and of its E122Q mutant are almost the same (535 vs 533 nm), indicating that mutational change in electrostatic energy does not affect the chromophore geometry significantly. ONIOM electrostatic interaction blue-shifts the first excitation energy by 30 nm (Table 3, none//ME = 533 to ME//ME = 503 nm) as in the WT Rh. Electronic polarization blue-shifts by 6 nm (EE//ME = 497 nm), and geometry effect adds 5 nm (EE/EE = 492 nm), resulting in a net polarization energy and geometry effect of ca. 10 nm for E122Q. Thus, electrostatic contributions to the first excitation energy (ca. 30 nm) are the same in the WT Rh and its E122Q mutant, and spectral shift in E122Q compared with the WT Rh is a result of electronic polarization of the chromophore. Calculated (−11 nm) and experimental (−20 nm) mutation shifts agree within 10 nm, which corresponds to ca. 1 kcal/mol.
The effect of BLA change with E122Q mutation (6 nm) is counterbalanced with the effect of the change in the H-bond between PSBR and Glu113. If the charge of each atoms on the residue 122 of the WT Rh and its E122Q mutant is turned off, EE//EE excitation energies are shifted by −3 nm (503 → 500 nm) and +7 nm (492 → 499 nm), respectively, reproducing the calculated mutation shit of −11 nm. This indicates that the net mutation shift arises from the polarization effects of residue 122.
In E122Q mutant, the carboxylic OH moiety of protonated E122 (AMBER charge = −0.19) is replaced by an NH2 group (AMBER charge = −0.09). Thus, the negative charge on the mutated part is halved. The H atoms in the NH2 moiety of Q122 are oriented toward the H atoms of one of the methyl group attached to the C1 atom of PSBR (see Figure 2 for the atom labeling), with separation of at least 2.4 Å. The electron withdrawing force from the methyl to the SBR-N is decreased as the negative charge around that methyl group decreases. In this situation, polyene chain possesses more positive charge and thus increased BLA pattern (more stretching). The energy gradient on S1 surface at the Franck–Condon point was previously shown to correlate with the stretching coordinate for the retinal chromophores.65 Hence, the vertical excitation energy is blue-shifted when the structure features an increased BLA. As the side chain terminal of deprotonated Asp (D) possesses more negative charge than Gln (Q), the absorption maximum of E122D (475 nm)34 is more blue-shifted than that of E122Q (480 nm).
When the O=C−NH2 plane of Gln122 in E122Q is rotated by ca. 90°, it triggers a number of changes in the side chain orientations of some of its environmental amino acids and Q122 becomes more distant to the PSBR. In this case, our best calculation (EE//EE = 514 nm, see Table 3) predicts red-shift in the excitation energy, contrary to the experiment. This emphasizes the importance of proper side chain alignments in mutation studies.
(d) E113Q Mutant
Experiments show that absorption spectrum of the E113Q mutant (Glu113 → Gln) is pH dependent (496 nm at pH ) 5.5, 384 nm at pH = 8.2 in the dark state).32,34,66 Excitation energies computed for the bare chromophore taken from the ME geometries of the E113Q mutant (both PSBR and SBR) are almost the same as those of the WT Rh and its E122Q mutant (see Table 3), indicating that spectral shifts of PSBR and SBR in E113Q are not related to the changes in the chromophore geometry due to protein electrostatics.
We have found three stable Q113 side chain conformations in E113Q with SBR (see Table 3). In these structures, the excitation energy is shifted to blue by the electrostatic effect (less than 10 nm) and to red by the polarization (20−30 nm), more than half of which is the result of geometry relaxation effect in EE. The excitation energies for these structures at the EE//EE level are 409−425 nm (experiment = 384 nm, see Table 3). In E113Q with PSBR, electrostatic and electronic polarizing effect of protein environment shifts the first excitation energy to blue by 15 and red by 9 nm, respectively, bringing it to 530 nm at the EE/EE level (experiment = 496 nm). Hence, the computed first excitation energy is red-shifted at least 25 nm for both SBR (409 nm vs 384 nm) and PSBR (530 nm vs 496 nm) compared with the experiment.
We have examined several possibilities that may be the reason for the difference between experimental and calculated excitation energies for E113Q. (a) As discussed above, results of all levels of previous calculations for the contributions of Glu113 to the first excitation energy are very much different to each other. The computed excitation energies of Glu113 mutant may thus suffer from the same unknown difficulties in evaluating the effects of Glu133 in WT Rh. We note however that the present EE//EE as well as the previous CASPT2/AMBER24 calculations underestimate the experimental absorption maximum of E113D67 by only 10 nm. (b) When EE optimizations are performed with Mulliken charges rather than the ESP charges, the EE excitation energy is affected only by 1 nm. (c) Inclusion of Q113 in the model QM part does not affect the first excitation energy. (d) There is a water molecule that forms a H-bond with Glu113 side chain in the WT Rh and stays nearby the NH2 moiety of Q113 in E113Q. When this water molecule is removed from E113Q, the calculated excitation energy is not affected. (e) A water molecule can be fitted to the space above SBR-N for the cases given footnotes f and g of Table 3. SBR-N has a H-bond with this added water molecule for both cases, and the calculated excitation energy (409 nm) is now the same as that for the case given footnote e of Table 3, e.g., in the presence of a H-bond between SBR-N and Q113. (f) SBR excitation energy in the WT Rh is shifted to blue by deprotonated Glu113 carboxylate O atoms. To account for the effect of such a negative charge, we added a Cl− atom near the SBR-N for cases given footnotes f and g in Table 3. However, there is no stabilizing residue of this negative charge near SBR. Thus, it moves away SBR during geometry optimizations. A positive charge near SBR would shift the excitation energies to the red, as seen for SBR with protonated Glu113 and in the case of (e) with a H-bonding water, which cannot be the source of the difference in experiment and present calculations. (g) We suppose that the E113Q mutant has an analogous active site to the WT Rh. However, acidity and thus protonation states of some residues may change with E113Q mutation. This may trigger significant changes in the protein structure. In the absence of crystal structure of E113Q, it is difficult to model such a structure from Rh coordinates. Appearance of an NH2 moiety (Q113) near the SBR-N red-shifts excitation energies and thus this group may be away from the N terminal of the 11-cis-retinal in the true E113Q structure. From the experience on the putative WT and E122Q structures with SBR, it seems necessary to have a deprotonated carboxylic unit near the SBR-N to have excitation energies around 380 nm. Absorption difference spectral measurements detect many photoproducts of E113Q, which are assigned in the experimental study by supposing that phototransduction mechanisms are the same in the WT Rh and E113Q mutant.66 Excitation energies calculated at our best EE//EE level for E113Q with Q113 side chain (retinal = 11-cis), whose NH2 and O moieties are oriented respectively to Gly90 and N atom of the chromophore (see Table 3, 409 nm for SBR and 530 nm for PSBR), agree perfectly with those belong to one of the E113 photoproducts assigned to bathorhodopsin in the experiment [408 nm at pH = 8.2 (SBR) and 530 nm at pH = 5.5 (PSBR)].66 These computational results indicate that retinal in the dark state of E113Q may be different than 11-cis as the cavity around 11-cis-retinal increases with Glu113 mutation and assignations done previously66 to E113Q and its photolysis products may need to be revised.
(e) Several Other Mutants
To further assess reliability of our methodology and how certain amino acids affect absorption spectra, we have calculated first vertical excitation energies (experimental values in parentheses) of G90D (483 nm),68 A292S (491 nm),69 A269T (514 nm),70 D83N (495 nm),33 W265Y (485 nm),69 H211C (495 nm),71 and E181Q (502 nm72 or 510 nm73) mutants (see Table 3).
A292S (Ala292 → Ser) and A269T (Ala269 → Thr) mutants have several energetically close-lying (within 2 kcal/mol) side-chain conformations for the mutated residue (Ser or Thr), and each of them may contribute to the absorption spectra. Experimental excitation energies are shifted to blue by 9 nm (red by 14 nm) for the A292S (A269T) mutant, relative to the WT Rh. In agreement with experiment, we have found more conformations that shift the excitation energies to the blue (red) for the A292S (A269T) mutant. The mechanism for the blue- and red-shifts can be rationalized in terms of bond strength change as explained for E122Q above. For example, side-chain-OH moiety of T269 in A269T for conformations given with m and n footnotes in Table 3 is very close to one of the methyl groups attached to C1 atom of the chromophore. Negative charge of –OH moiety pushes electrons from β-ionone to polyene chain. Hence, electron densities along the polyene chain are increased, corresponding to decreased BLA pattern. As a result, the vertical excitation energies shift to red (see above for the relationship between BLA pattern and S1 surface).
Excitation energy of G90D (Gly90 → Asp) is blue-shifted by 17 nm experimentally (see Table 3).66 When D90 in the G90D mutant is deprotonated, the water molecule that bridges backbone and side-chain carboxylic moieties of Glu113 in the WT Rh moves to bridge carboxyl groups of Glu13 and Asp90 in the G90D mutant, which decreases the strength of the H-bond between PSBR-N and Glu113. When Glu113 moves away farther than the chromophore, the –CNH-moiety of the chromophore possesses less electron density, resulting in BLA increase. Hence, excitation energies shift to blue in agreement with experiment.66 When D90 in the G90D mutant is taken protonated, it forms a H-bond with Glu113, which decreases the strength of the H-bond between PSBR-N and one of the carboxylic O atoms of Glu113 and moves the other carboxylic O atom of Glu113 toward PSBR-N. Hence, the excitation energy with protonated D90 is almost unshifted, excluding the possibility of protonated D90.
There are two different experimental results for the E181Q mutant of bovine Rh, one of which63 shows almost no spectral shift and the other73 finds red-shift of 10 nm. Our results are more consistent with the first. However, it should be noted that the difference between two experimental results is within the computational error bar. We have found single conformation for the D83N, W265Y, and H211C mutants and good agreement for the excitation energies and spectral shifts at our final EE/EE level with experiment (see Table 3).
As a result, B3LYP-EE//B3LYP-EE calculations reproduces experimental excitation energies of bovine Rh and its mutants within 10 nm with correct spectral shift sign as long as orientation and protonation state of the side chain of the mutated residue are properly defined, except E113 mutants (see Table 3). In the absence of protein environment, neither experimental absorption maximum nor its shift can be explainable for the mutants, which emphasizes the importance of electrostatic and electronic polarizing effects of protein environment on the chromophore orbitals.
3. BLA Dependence of the First Excitation Energy
Vibrational frequencies and, thus, the changes in bond lengths along polyene chain of the chromophore are known to be a sensitive probe for spectral tuning in visual pigments.8,74–77 Average BLA, defined as the difference between the average single and double bond lengths along the polyene chain (C5–N), shows a linear relationship with the excitation energy (see Figure 9, geometries and excitation energies computed at B3LYP-EE and TD-B3LYP-EE levels, data points given in Supporting Information), as shown previously for bacteriorhodopsin, phoborhodopsin, and its mutants.8 Data points that show significant deviation from the linear correlation belong to the WT Rh and its E122Q mutant with SBR and deprotonated Glu113 and E113Q with PSBR, whose calculated excitation energies are also not consistent with experiment. Hence, the correlation is destroyed for the putative case with SBR and deprotonated Glu113, in which the separation between SBR and Glu113 is very large (see Supporting Information). This indicates that presence of a H-bond interaction between Schiff-base nitrogen and Glu113 side-chain is necessary for the linear correlation and thus important for the spectral tuning.
Figure 9.

Correlation between the calculated average BLA (B3LYP-EE geometry) and the first excitation energy (B3LYP-EE//B3LYP-EE) for Rh and its mutants.
IV. Discussion
The calculation of accurate absorption energies for retinal proteins is a great challenge. First of all, the initial coordinates may affect the calculated protein structure and thus the computed excitation energies in QM/MM studies. Previously, two water molecules near the chromophore, which are not resolved at some lower resolutions, were shown to be crucial for the stabilization of the chromophore.13,47 The differences in the H-bond network of one of these water molecules was also shown to affect the first excitation energy by 10 nm in a CASPT2/AMBER study.24
The protonation state of titratable residues (Asp, Glu, and His) must be determined during the system setup on the X-ray coordinates since side chains of these residues will adjust themselves to the assigned protonation state during geometry optimization, resulting in conformations differing from those in the X-ray structures in case of wrong assignment. Our calculations show that side chain orientation of the mutated residues and their protonation states affect excitation energies considerably (see Table 3). Our calculations reveal that the first excitation energy of Rh and its mutants will be around 500 (400) nm for PSBR (SBR). The large difference of ca. 100 nm in the computed excitation energies with PSBR and SBR clearly excludes the possibility of deprotonated SB 11-cis-retinal in WT Rh.
Second, an appropriate QM method for calculating geometries and excitation energies must be used. The computed gas-phase excitation energies are quite method dependent (see Introduction). However, the protein contributions to QM wave functions draw the excitation energies to the experimental range in protein matrix generally. Hence, either protein environment fixes some amount of errors in the computational methods, or QM/MM calculations favor from lucky error cancelations. B3LYP and CASPT2 methods show the same computational trends in the protein environment.
Third, a single conformation may not be enough as a representative structure. The inexpensive DFTB, whose PSBR geometries are almost identical to those of B3LYP, allows for long proper QM/MM MD simulations to sample the conformational space of PSBR. Hence, TD-B3LYP/MM excitation energies can be calculated on the snapshots of DFTB/MM trajectory of PSBR to obtain a realistic absorption spectrum.
B3LYP-EE//B3LYP-EE excitation energies of the mutants are consistent with experiment within 10 nm (except E113Q) and show generally the correct shift sign as long as side chains of the mutated residues are properly aligned and protonated. The agreement between calculated and experimental excitation energies of the mutants may be improved by solvating the system and sampling the conformational space using DFTB. As the error in excitation energies is already small and MD simulations will bring no new insight to present study in that sense, we do not attempt to perform any MD study here.
Fourth, interaction of the chromophore with the protein environment must be treated properly. In the mechanical embedding (ME) scheme, it is calculated with classical point-charge model (MM) and thus includes only the electrostatic effect. In the electronic embedding (EE) scheme, it includes also the effect of electronic polarization of the chromophore as it is calculated at the QM level. Although the calculations with the EE scheme includes both electrostatic and electronic polarization effects of the protein environment and thus give the best results, we have performed the calculations with both ME and EE schemes and for the isolated model QM part from ME geometries (the bare chromophore in Table 4), which allow us to decompose the protein effects on the excitation energy into its components for revealing the source of spectral shifts (see Table 4). We decompose the computed first excitation energies only for the WT Rh and its E122Q and E113Q mutants into its contributions in Table 4. Analogous analyses can be performed for the other mutants considered by using the data in Table 3.
TABLE 4.
The Effects of the Protein Environment (Electrostatic and Electronic Polarization Contribution without and with its Effect on Geometry) and of Protonation State of the 11-cis-retinal (PSBR → SBR) on the First Excitation Energy (in nm) at ONIOM(TD-B3LYP:MM) Level at ONIOM(B3LYP:MM) Optimized Ground State Geometries for the WT Rh and its E122Q and E113Q Mutants
| B3LYP-ME geometry |
B3LYP-EE geometry |
|||||
|---|---|---|---|---|---|---|
| the bare chromophorea + | electrostaticsb + | electronic polarizationc + | geometry relaxation due to polarizationd = | the resulting first excitation energye (spectral shiftf) | ||
| WT | PSBR | 535 | −31 | +1 | −2 | 503 (0) |
| SBRg | 400 | −1 | +13 | +17 | 429 (−74) | |
| SBRh | 399 | −33 | +16 | −2 | 380 (−123) | |
| E122Qi | PSBR | 533 | −30 | −6 | −5 | 492 (0) |
| SBRg | 402 | −7 | +11 | +12 | 418 (−74) | |
| SBRh | 399 | −37 | +20 | −3 | 379 (−113) | |
| E113Qj | PSBR | 536 | −15 | +5 | +4 | 530 (0) |
| SBR | 398 | −8 | +7 | +12 | 409 (−121) | |
The B3LYP-none first excitation energy for the isolated QM model part from B3LYP-ME optimized geometry.
The difference in the first excitation energies between B3LYPME//B3LYP-ME and B3LYP-none//B3LYP-ME.
The difference in the first excitation energies between B3LYP-EE//B3LYP-ME and B3LYP-ME//B3LYP-ME.
The difference in the first excitation energies between B3LYP-EE//B3LYP-EE and B3LYP-EE//B3LYP-ME.
The B3LYP-EE//B3LYP-EE first excitation energy.
Spectral shift in this table corresponds to the effect of deprotonation of the chromophore (PSBR → SBR) on the first excitation energy in the WT Rh or the mutants.
With protonated Glu113.
With deprotonated Glu113.
With Gln122 whose side-chain NH2 moiety is oriented toward one of the methyl groups attached to C1 atom of the chromophore.
With Gln113 whose side-chain NH2 (carbonyl O) moiety is oriented toward its backbone carbonyl O (Schiff-base N).
Electrostatics of protein environment (the second numerical column in Table 4) blue-shift the first excitation energy of PSBR by 30−40 nm for the WT Rh and its mutants except for E113Q (Glu113 → Gln). ONIOM calculations with charges turned off show that electrostatic contribution to the first excitation energy comes mainly from Glu113. Gln113 in the E113Q mutant has a smaller red-shift electrostatic contribution of 15 nm to the first excitation energy of PSBR. Although electronic polarization without and with its effect on the geometry (the third and fourth numerical columns in Table 4, respectively) does not contribute much to the first excitation energy of the WT Rh with PSBR, it contributes to the first excitation energies in the mutants to some extent. For example, the computed spectral shift of −11 nm in the E122Q mutant with PSBR relative to the WT Rh is totally due to the electronic polarization, half of which comes from its geometry relaxation effect (from ONIOM-ME geometry to ONIOM-EE geometry). Polarization effects bring also significant correction of 9 nm to the first excitation energy of the E113Q mutant with PSBR, being comparable with the electrostatic contribution of −15 nm (see Table 4). Electronic polarization effect of the protein environment is more pronounced for SBR than that for PSBR. For example, it contributes to the first excitation energy of putative SBR analogues of the WT Rh and its E122Q mutant with protonated (deprotonated) Glu113 and of the E113Q mutant with SBR by ca. 25 (15) nm and 19 nm, respectively, whereas electrostatic contribution is around −4(−35) and −8 nm. Hence, both electrostatic and electronic polarization effects are important for the absolute values of excitation energies and to some extent for spectral shifts in the mutants.
Geometry relaxation from ME geometries due to electronic polarization (the fourth numerical column in Table 4) brings significant correction to the first excitation energies in most cases. For SBR with protonated Glu113, the EE geometry effect does not change the chromophore geometry much (see Figure 7), but it changes the computed excitation energies. This effect comes from the H-bond between SBR-N and Glu113 side chain (Gln113 side chain in E113Q) strengthened with EE optimization. In the case of SBR with deprotonated Glu113, the distance between SBR-N and Glu113 side chain is notably different between B3LYP-ME (3.4 Å) and B3LYP-EE (3.8 Å) geometries. The change in this distance with EE geometry optimization does not influence the excitation energies much (see the fourth numerical column in Table 4) as Glu113 is already significantly away than SBR-N. Electronic polarization corrects the ME geometry of PSBR notably, contrary to that of SBR. For the WT Rh, the effect of this correction on the first excitation energy is counterbalanced with the effect of the increase in the H-bond strength between NH unit of PSBR and Glu113 carboxylic oxygen.
Our Mulliken charge analysis shows that total positive charge of the PSBR is mainly distributed, in addition to C15−NH2 moiety, to carbon atoms that have a methyl substituent, the situation similar to all-trans PSBR in the gas phase.61 The protein environment modulates electron-donating ability of the methyl groups and thus the charge distribution within the chromophore (electronic polarization). When more (less) electron drawing group is placed near these methyl groups, polyene chain of PSBR possesses less (more) positive charge and, thus, decreased (increased) BLA pattern, which corresponds to less (more) stretching. As S1 surface gradient at the Franck–Condon point correlates with stretching coordinate of the retinal,65 the first excitation energy is shifted to red (blue) with the decreased (increased) BLA.
Finally, solvation of the system as well as the polarization of the protein environment by QM charges,18,19,28,79 in addition to polarization of the chromophore by the protein environment, may affect the computed QM/MM excitation energies. We have shown that side chain orientation of some specific residues results in excitation energies that are not consistent with experiment. Solvent molecules may help keeping the side chains in the preferred alignments during protein dynamics. We have shown that the main counterion in Rh is Glu113 and its inclusion in the model QM part does not influence computed excitation energies much. Hence, we do not expect notable effect of environmental polarization on excitation energies.
V. Conclusions
We have investigated geometries and excitation energies of Rh and some of its mutants by hybrid ONIOM(QM:MM) calculations. The present QM/MM calculations enhance our understanding on spectral tuning mechanism in visual pigments and its relation with geometry. The main conclusions drawn in this study can be summarized as follows.
(a) B3LYP-EE//B3LYP-EE calculations reproduce experimental absorption maxima for Rh and predict spectral shifts within 10 nm for its mutants except for E113Q. The calculated excitation energies for E113Q with the 11-cis-retinal agree with the experimental absorption maxima assigned to E113Q with the bathorhodopsin by comparing absorption spectra of Rh and its E113Q mutant.66 The chromophore structures may thus be different in Rh and its E113Q mutant, and further experimental studies should be performed for determining the chromophore structure in E113Q.
(b) The geometries of the DFTB method are almost identical to those of B3LYP for PSBR, which makes it less expensive to obtain appropriate geometries for TD-B3LYP excitation energy calculations. TD-B3LYP excitation energies of SBR at DFTB geometries are not as appropriate as in PSBR. Calculations with BLYP have a red-shift error of at least 50 nm and thus are not appropriate for electronic spectra calculations.
(c) Glu113 acts as a counterion and shifts the first excitation energy to blue significantly. The other amino acids have no net effect on excitation energies of the WT Rh but their individual effects emerge with mutations.
(d) Electronic polarization of the chromophore by protein environment is essential for spectral tuning even if its effect on geometry is not considered, especially in the case of SBR. Electronic polarization of the chromophore by protein environment affects geometry of charged (+1) PSBR significantly. The changes in PSBR geometry due to electronic polarization are associated with a change in the strength of a H-bond between the PSBR and Glu113, which counterbalances their effects on excitation energies in WT Rh. Electronic polarization effect is less pronounced on the SBR chromophore geometry. However, the changes in Glu113 coordinates with electronic polarization affect the first excitation energy of SBR with protonated Glu113. When both retinal and Glu113 are deprotonated, they are too far away to affect the excitation energy upon electronic embedding correction on the geometries.
(e) The average BLA along polyene chain of the chromophore correlates with the computed first excitation energy. If a group with more negative charge is placed near the methyl groups, more electron is drawn to polyene chain that results in decreased BLA and thus red-shift in excitation energies, or vice versa. In conclusion, the excitation energies are very sensitive to the environment of methyl groups on the chromophore in addition to that of its –CN(H)– moiety. This also implies the importance of the side chain alignment of the mutated residues around the active site.
Acknowledgment
We thank Thom Vreven and Guishan Zheng for helpful discussions. This work was supported by a grant from the National Institutes of Health (1R01EY016400).
Footnotes
Supporting Information Available: Nonstandard AMBER settings, computed geometry parameters of the chromophore, and the first and second vertical excitation energies along with their oscillator strengths. This information is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References and Notes
- 1.Vikram RR, Oprian DD. Annu. Rev. Biophys. Biomol. Struct. 1996;25:287–314. doi: 10.1146/annurev.bb.25.060196.001443. [DOI] [PubMed] [Google Scholar]
- 2.Sakmar TP, Menon ST, Marin EP, Awad ES. Annu. Rev. Biophys. Biomol. Struct. 2002;31:443–484. doi: 10.1146/annurev.biophys.31.082901.134348. [DOI] [PubMed] [Google Scholar]
- 3.Filipek S, Stenkamp RE, Teller DC, Palczewski K. Annu. Rev. Physiol. 2003;65:851–879. doi: 10.1146/annurev.physiol.65.092101.142611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Logunov SL, Song L, El-Sayed MA. J. Phys. Chem. 1996;100:18586–18591. [Google Scholar]
- 5.Andersen LH, Nielsen IB, Kristensen MB, El Ghazaly MOA, Haacke S, Nielsen MB, Petersen MA. J. Am. Chem. Soc. 2005;127:12347–12350. doi: 10.1021/ja051638j. [DOI] [PubMed] [Google Scholar]
- 6.Vreven T, Morokuma K. Theor. Chem. Acc. 2003;109:125–132. [Google Scholar]
- 7.Wanko M, Hoffmann M, Strodel P, Koslowski A, Thiel W, Neese F, Frauenheim T, Elstner M. J. Phys. Chem. B. 2005;109:3606–3615. doi: 10.1021/jp0463060. [DOI] [PubMed] [Google Scholar]
- 8.Hoffmann M, Wanko M, Strodel P, König PH, Frauenheim T, Schulten K, Thiel W, Tajkhorshid E, Elstner M. J. Am. Chem. Soc. 2006;128:10808–10818. doi: 10.1021/ja062082i. [DOI] [PubMed] [Google Scholar]
- 9.Ferre N, Olivucci M. J. Am. Chem. Soc. 2003;125:6868–6869. doi: 10.1021/ja035087d. [DOI] [PubMed] [Google Scholar]
- 10.Andruniow T, Ferre N, Olivucci M. Proc. Natl. Acad. Sci. U.S.A. 2004;101:17908–17913. doi: 10.1073/pnas.0407997101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferre N, Cembran A, Garavelli M, Olivucci M. Theor. Chem. Acc. 2004;112:335–341. [Google Scholar]
- 12.Gascon JA, Sproviero EM, Batista VS. Acc. Chem. Res. 2006;39:184–193. doi: 10.1021/ar050027t. [DOI] [PubMed] [Google Scholar]
- 13.Gascon JA, Batista VS. Biophys. J. 2004;87:2931–2941. doi: 10.1529/biophysj.104.048264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tachikawa H, Kawabata H. J. Photochem. Photobiol. B: Biol. 2005;79:191–195. doi: 10.1016/j.jphotobiol.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 15.Blomgren F, Larsson S. J. Comput. Chem. 2005;26:738–742. doi: 10.1002/jcc.20210. [DOI] [PubMed] [Google Scholar]
- 16.Hufen J, Sugihara M, Buss V. J. Phys. Chem. B. 2004;108:20419–20426. [Google Scholar]
- 17.Kazutomo K, Yamato T. Chem. Phys. Lett. 2006;430:386–390. [Google Scholar]
- 18.Ren L, Martin CH, Wise KJ, Gillespie NB, Luecke H, Lanyi JK, Spudich JL, Birge RR. Biochemistry. 2001;40:13906–13914. doi: 10.1021/bi0116487. [DOI] [PubMed] [Google Scholar]
- 19.Kusnetzow A, Dukkipati A, Babu KR, Singh D, Vought BW, Knox BE, Birge RR. Biochemistry. 2001;40:7832–7844. doi: 10.1021/bi010387y. [DOI] [PubMed] [Google Scholar]
- 20.Rajamani R, Gao J. J. Comput. Chem. 2002;23:96–105. doi: 10.1002/jcc.1159. [DOI] [PubMed] [Google Scholar]
- 21.Sekharan S, Sugihara M, Buss V. Angew. Chem., Int. Ed. 2007;46:269–271. doi: 10.1002/anie.200603306. [DOI] [PubMed] [Google Scholar]
- 22.Cembran A, Gonzalez-Luque R, Altoe P, Merchan M, Bernardi F, Olivucci M, Garavelli M. J. Phys. Chem. A. 2005;109:6597–6605. doi: 10.1021/jp052068c. [DOI] [PubMed] [Google Scholar]
- 23.Sekharan S, Weingart O, Buss V. Biophys. J. 2006;91:L07–L09. doi: 10.1529/biophysj.106.087122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coto PB, Strambi A, Ferre N, Olivucci M. Proc. Natl. Acad. Sci. U.S.A. 2006;103:17154–17159. doi: 10.1073/pnas.0604048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujimoto K, Hasegawa J-Y, Hayashi S, Kato S, Nakatsuji H. Chem. Phys. Lett. 2005;414:239–242. [Google Scholar]
- 26.Bravaya K, Bochenkova A, Granowsky A, Nemukhin A. J. Am. Chem. Soc. 2007;129:13035–13042. doi: 10.1021/ja0732126. [DOI] [PubMed] [Google Scholar]
- 27.Fujimoto K, Hayashi S, Hasegawa J-Y, Nakatsuji H. J. Chem. Theory Comput. 2007;3:605–618. doi: 10.1021/ct6002687. [DOI] [PubMed] [Google Scholar]
- 28.Houjou H, Inoue Y, Sakurai M. J. Phys. Chem. B. 2001;105:867–879. [Google Scholar]
- 29.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. J. Mol. Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Robertson AD, Jensen JH. Proteins: Struct., Funct., Bioinf. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 31.a Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]; b Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 32.Yan ECY, Kazmi MA, De S, Chang BSW, Seibert C, Marin EP, Mathies RA, Sakmar TP. Biochemistry. 2002;41:3620–3637. doi: 10.1021/bi0160011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fahmy K, Jäger F, Beck M, Zvyaga TA, Sakmar TP, Siebert F. Proc. Natl. Acad. Sci. U.S.A. 1993;90:10206–10210. doi: 10.1073/pnas.90.21.10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakmar TP, Franke RR, Khorana HG. Proc. Natl. Acad. Sci. U.S.A. 1989;86:8309–8313. doi: 10.1073/pnas.86.21.8309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhukovsky EA, Oprian DD. Science. 1989;246:928–930. doi: 10.1126/science.2573154. [DOI] [PubMed] [Google Scholar]
- 36.Okada T, Fujiyoshi Y, Silow M, Navarro J, Landau EM, Shichida Y. Proc. Natl. Acad. Sci. U.S.A. 2002;99:5982–5987. doi: 10.1073/pnas.082666399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 38.Teller DC, Okada T, Behnke CA, Palczewski K, Stenkamp RE. Biochemistry. 2001;40:7761–7772. doi: 10.1021/bi0155091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yeagle PL, Choi G, Albert AD. Biochemistry. 2001;40:11932–11937. doi: 10.1021/bi015543f. [DOI] [PubMed] [Google Scholar]
- 40.Gröbner G, Burnett IJ, Glaubitz C, Choi G, Mason AJ, Watts A. Nature. 2000;405:810–813. doi: 10.1038/35015604. [DOI] [PubMed] [Google Scholar]
- 41.Mathies RA, Lugtenburg J. In: Handbook of Biological Physics. Stavenga DG, DeGrip WJ, Pugh EN Jr., editors. Vol. 3. Amsterdam; North-Holland: 2000. pp. 55–90. Chapter 2. [Google Scholar]
- 42.Creemers AFL, Kiihne S, Bovee-Geurts PHM, DeGrip WJ, Lugtenburg J, de Groot HJM. Proc. Natl. Acad. Sci. U.S.A. 2002;99:9101–9106. doi: 10.1073/pnas.112677599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith SO, Courtin J, de Groot H, Gebhard R, Lugtenburg J. Biochemistry. 1991;30:7409–7415. doi: 10.1021/bi00244a007. [DOI] [PubMed] [Google Scholar]
- 44.Spooner PJR, Sharples JM, Verhoeven MA, Lugtenburg J, Glaubitz C, Watts A. Biochemistry. 2002;41:7549–7555. doi: 10.1021/bi020007o. [DOI] [PubMed] [Google Scholar]
- 45.Singh D, Hudson BS, Middleton C, Birge RR. Biochemistry. 2001;40:4201–4204. doi: 10.1021/bi001911o. [DOI] [PubMed] [Google Scholar]
- 46.Sugihara M, Buss V, Entel P, Elstner M, Frauenheim T. Biochemistry. 2002;41:15259–15266. doi: 10.1021/bi020533f. [DOI] [PubMed] [Google Scholar]
- 47.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr., Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 48.Birge RR, Knox BE. Proc. Natl. Acad. Sci. U.S.A. 2003;100:9105–9107. doi: 10.1073/pnas.1733801100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maseras F, Morokuma K. J. Comput. Chem. 1995;16:1170–1179. [Google Scholar]
- 50.Dapprich S, Komáromi I, Byun S, Morokuma K, Frisch MJ. J. Mol. Struct.: THEOCHEM. 1999;461:1–21. [Google Scholar]
- 51.Vreven T, Byun KS, Komáromi I, Dapprich S, Montgomery JA, Jr., Morokuma K, Frisch MJ. J. Chem. Theory Comput. 2006;2:815–826. doi: 10.1021/ct050289g. [DOI] [PubMed] [Google Scholar]
- 52.Bakowies D, Thiel W. J. Phys. Chem. 1996;100:10580–10594. [Google Scholar]
- 53.Becke AD. Phys. Rev. A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 54.Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 55.Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 56.Elstner M, Porezag D, Jungnickel G, Elsner J, Haugk M, Frauenheim T, Suhai S, Seifert G. Phys. Rev. B. 1998;58:7260–7268. [Google Scholar]
- 57.a Ditchfield R, Hehre WJ, Pople JA. J. Chem. Phys. 1971;54:724–728. [Google Scholar]; b Hehre WJ, Ditchfield R, Pople JA. J. Chem. Phys. 1972;56:2257–2261. [Google Scholar]; c Hariharan PC, Pople JA. Theor. Chim. Acta. 1973;28:213–222. [Google Scholar]; d Clark T, Chandrasekhar J, Spitznagel GW, Schleyer P. v. R. J. Comput. Chem. 1983;4:294–301. [Google Scholar]
- 58.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr., Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian03 development version, Revisions F.01 for DFTB geometry optimizations and D.01 for all other calculations. Gaussian, Inc.; Wallingford, CT: 2007. [Google Scholar]
- 59.Fujimoto K, Hasegawa J-Y, Hayashi S, Nakatsuji H. Chem. Phys. Lett. 2006;432:252–256. [Google Scholar]
- 60.Page CS, Olivucci M. J. Comput. Chem. 2003;24:298–309. doi: 10.1002/jcc.10145. [DOI] [PubMed] [Google Scholar]
- 61.Froese RDJ, Komaromi I, Byun KS, Morokuma K. Chem. Phys. Lett. 1997;272:335–340. [Google Scholar]
- 62.Carravetta M, Zhao X, Johannessen OG, Lai WC, Verhoeven MA, Bovee-Geurts PHM, Verdegem PJE, Kiihne S, Luthman H, de Groot HJM, de-Grip WJ, Lugtenburg J, Levitt MH. J. Am. Chem. Soc. 2004;126:3948–3953. doi: 10.1021/ja039390q. [DOI] [PubMed] [Google Scholar]
- 63.For Rh with SBR and protonated Glu113, geometry parameters of the chromophore and excitation energies computed are not affected much by the choice of side chain oxygen atoms of Glu113 (OE1 and OE2) for protonation. However, the structure with protonated OE2 is ca. 2 kcal/mol more stable. Hence, we present the results with protonated OE2 throughout the paper
- 64.Gonzalez-Luque R, Garavelli M, Bernardi M, Merchan M, Robb MA, Olivucci M. Proc. Natl. Acad. Sci. U.S.A. 2000;97:9379–9384. doi: 10.1073/pnas.97.17.9379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garavelli M, Negri F, Olivucci M. J. Am. Chem. Soc. 1999;121:1023–1029. [Google Scholar]
- 66.Lewis JW, Szundi I, Fu W-Y, Sakmar TP, Kliger DS. Biochemistry. 2000;39:599–606. doi: 10.1021/bi991860z. [DOI] [PubMed] [Google Scholar]
- 67.Sakmar TP, Franke RR, Khorana HG. Proc. Natl. Acad. Sci. U.S.A. 1991;88:3079–3083. doi: 10.1073/pnas.88.8.3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rao VR, Cohen GB, Oprian DD. Nature. 1994;367:639–642. doi: 10.1038/367639a0. [DOI] [PubMed] [Google Scholar]
- 69.Lin SW, Kochendoerfer GG, Carroll KS, Wang D, Mathies RA, Sakmar TP. J. Biol. Chem. 1998;273:24583–24591. doi: 10.1074/jbc.273.38.24583. [DOI] [PubMed] [Google Scholar]
- 70.Chan T, Lee M, Sakmar TP. J. Biol. Chem. 1992;267:9478–9480. [PubMed] [Google Scholar]
- 71.Nathans J. Biochemistry. 1990;29:937–942. doi: 10.1021/bi00456a013. [DOI] [PubMed] [Google Scholar]
- 72.Nathans J. Biochemistry. 1990;29:9746–9752. doi: 10.1021/bi00493a034. [DOI] [PubMed] [Google Scholar]
- 73.Terakita A, Yamashita T, Shichida Y. Proc. Natl. Acad. Sci. U.S.A. 2000;97:14263–14267. doi: 10.1073/pnas.260349597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kochendoerfer GG, Wang D, Oprian DD, Mathies RA. Biochemistry. 1997;36:6577–6587. doi: 10.1021/bi970322o. [DOI] [PubMed] [Google Scholar]
- 75.Kochendoerfer GG, Lin SW, Sakmar TP, Mathies RA. Trends Biochem. Sci. 1999;24:300–305. doi: 10.1016/s0968-0004(99)01432-2. [DOI] [PubMed] [Google Scholar]
- 76.Baasov T, Sheves M. J. Am. Chem. Soc. 1985;107:7524–7533. [Google Scholar]
- 77.Lin SW, Imamoto Y, Fukada Y, Shichida Y, Yoshizawa T, Mathies RA. Biochemistry. 1994;33:2151–2160. doi: 10.1021/bi00174a023. [DOI] [PubMed] [Google Scholar]
- 78.Heyde ME, Gill D, Kilponen RG, Rimai L. J. Am. Chem. Soc. 1979;93:6776–6780. doi: 10.1021/ja00754a012. [DOI] [PubMed] [Google Scholar]
- 79.Warshel A, Chu ZT. J. Phys. Chem. B. 2001;105:9857–9871. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.