

Abstract
Bone mass is maintained constant in vertebrates through bone remodeling (BR). BR is characterized by osteoclastic resorption of preexisting bone followed by de novo bone formation by osteoblasts. This sequence of events and the fact that bone mass remains constant in physiological situation lead to the assumption that resorption and formation are regulated by each other during BR. Recent evidence shows that cells of the osteoblastic lineage are involved in osteoclast differentiation. However, the existence of a functional link between the two activities, formation and resorption, has never been shown in vivo. To define the role of bone formation in the control of bone resorption, we generated an inducible osteoblast ablation mouse model. These mice developed a reversible osteopenia. Functional analyses showed that in the absence of bone formation, bone resorption continued to occur normally, leading to an osteoporosis of controllable severity, whose appearance could be prevented by an antiresorptive agent. This study establishes that bone formation and/or bone mass do not control the extent of bone resorption in vivo.
Vertebrates constantly renew bone through remodeling. This process is characterized by two successive phases: resorption of preexisting bone by osteoclasts followed by de novo bone formation by the osteoblasts (1). Physiologically, resorption and formation are balanced to maintain a stable bone mass. Generally, it has been assumed that bone formation and bone resorption functions are linked mechanistically during bone remodeling (BR; refs. 2–4). One implication of this model is that the absence of or a functional defect in one cell type would greatly decrease the function of the other cell type to maintain a constant bone mass. Although osteoblast progenitors secrete a factor required for osteoclast differentiation (5), a requirement of the bone-formation function to regulate the bone-resorption function has neither been established nor ruled out in vivo.
EXPERIMENTAL PROCEDURES
Generation of Transgenic (tg) Mice.
We generated tg mice that bear a fusion gene composed of a 1.3-kb fragment of the mouse osteocalcin gene 2 (OG2) promoter fused to the 1.5-kb fragment of the herpes simplex virus thymidine kinase (Hsv-Tk) gene from position +2 relative to the transcription start site to 236 bp downstream of the polyadenylation site. tg founders were generated by pronuclear injection according to standard techniques (6). This study evaluated two lines of tg animals. Their genotype was determined by Southern blotting (7) with the Hsv-Tk gene as a probe. RNA extraction and Northern blotting were performed according to standard procedures (7). Serum osteocalcin and deoxypyridinoline crosslink levels were evaluated with kits and reagents from Biomedical Technologies (Stoughton, MA) and Biometra (Tampa, FL), respectively.
Treatment of Mice.
Ganciclovir (GCV) treatment (150 μg/kg per day injected i.p.) of wild-type (wt) and tg mice lasted 4 weeks or 8 weeks. Alendronate treatment lasted 4 weeks (35 mg/kg every third day). Mice were analyzed at the end of the treatment periods.
Histologic and Histomorphometric Analyses.
Bone specimens were processed as described (8). Histological analyses were performed on undecalcified sections stained with toluidine blue. Double labeling has been described (9); tetracycline injection was followed by the same dose of calcein 10 days later, and animals were killed 2 days later. Static and dynamic histomorphometries were performed according to protocols (10) with the OsteoMeasure Analysis System (Osteometrics, Atlanta). Statistical differences between groups were assessed by Student’s t test.
RESULTS
To determine whether bone resorption is influenced by bone formation in vivo, tg mice were generated with a 1.3-kb fragment of the mouse OG2 promoter, which was used to drive expression of Hsv-Tk (refs. 11 and 12; Fig. 1A). OG2, an osteoblast-specific gene, is expressed only in differentiated osteoblasts but not in osteoblast progenitors (13–15). Because dividing cells expressing Hsv-Tk die on treatment with GCV (11, 12), expression of Hsv-Tk in dividing osteoblasts should allow inducible ablation of differentiated osteoblasts in vivo without affecting the pool of progenitor cells. For each experiment, identical results were obtained with the progenies of two independent lines. Northern blot analysis showed the bone-specific expression of the transgene (Fig. 1A). In situ hybridization experiments showed that Hsv-Tk was expressed in osteoblasts (data not shown). To assess the efficacy of the GCV treatment, primary calvarial cell cultures [which include osteoblasts (16, 17)] of tg mice were maintained in the absence or presence of GCV. A 4-day GCV treatment of these calvarial cell cultures greatly decreased osteocalcin expression, and after an 8-day treatment, its expression was abolished (Fig. 1B) indicating that Hsv-Tk was expressed in dividing osteoblasts that were killed by GCV.
Figure 1.

Expression and activity of the OG2-Tk transgene. (A) Schematic representation of the transgene (Upper) and Northern blot analysis of its expression (Lower). (B) In vitro osteoblast ablation by GCV in calvarial cell culture of tg mice. Northern blot analysis shows osteocalcin (OC) expression in cells cultured without GCV for 4 days (−) and in the presence of 25 μM GCV for 4 days (4d) and 8 days (8d). Equivalent amounts of intact RNA were run in each lane as indicated by hybridization to an 18S RNA probe. (C) The graph shows in vivo osteoblast ablation after GCV treatment: measurement of circulating osteocalcin (OC) in wt animals (dotted line, n = 6) or tg animals (solid line, n = 6) before (6 weeks old), at the end of (10 weeks old), or 4 weeks after the GCV treatment period (14 weeks old).
To assess the consequences of osteoblast ablation in vivo, 6-week-old wt and tg mice, in which most but not all skeletal growth has occurred, were treated with GCV for 4 weeks. Blood tests showed a dramatic decrease in the level of circulating osteocalcin, an osteoblast-specific gene product (refs. 13–15; Fig. 1C), whereas levels of phosphate and calcium were not altered (data not shown). In tg mice, this treatment led to an arrest of skeletal growth and to the development of kyphosis (Fig. 2Ab), a feature frequently observed in osteopenic conditions (18), suggesting that bone formation was affected. Radiological examination of long bones and vertebrae of these animals showed thinner cortices and decreased bone density compared with wt littermates (Fig. 2Af). Histological analysis showed bone surfaces denuded of osteoblasts and a near absence of trabecular bone (Fig. 2Aj). During longitudinal bone growth, osteoblasts deposit bone matrix at the surface of persistent calcified cartilage septa. Because of the absence of osteoblasts, this bone-matrix deposition does not occur in tg mice, and therefore longitudinal bone growth does not take place. Biomechanical study showed that failure load, an indicator of bone strength, was significantly reduced in GCV-treated tg mice (data not shown). Wt mice treated with GCV exhibited none of these features (Fig. 2A c, g, and k).
Figure 2.
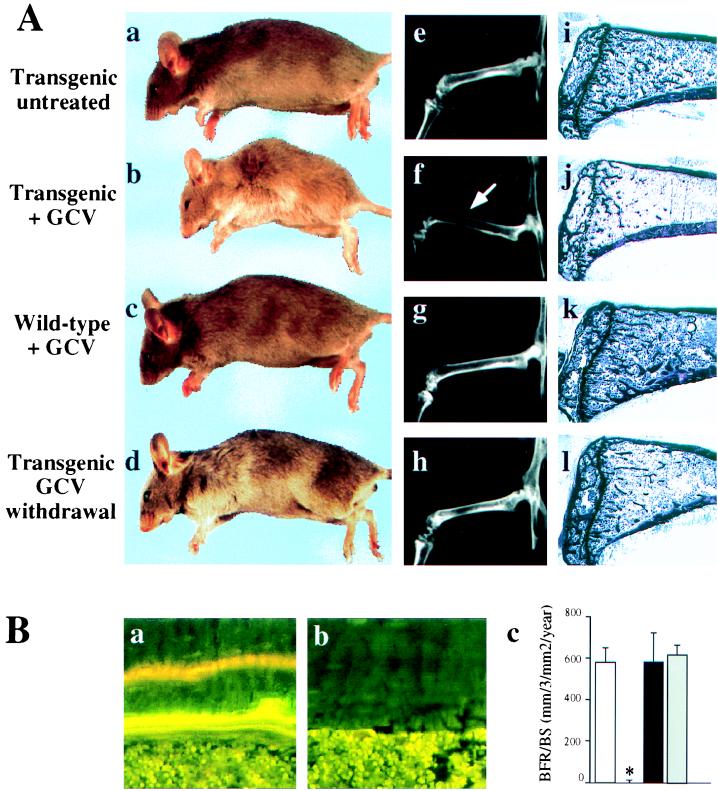
Bone-formation parameters in GCV-treated animals. (A) Morphological, radiological, and histological comparison of 10-week-old tg mice untreated (a, e, and i) or treated daily for 4 weeks with GCV (b, f, and j), 10 week-old wt mice treated daily for 4 weeks with GCV (c, g, and k), or 14 week-old treated tg mice 4 weeks after GCV withdrawal (d, h, and l). Note the thinning of the cortices (arrow) and the lucent aspect of the bones on the x-rays (f) as well as the loss of the trabecular bone (j) in GCV-treated tg mice. These abnormalities were reversible after GCV withdrawal (compare f with h and j with l). (B) Dynamic histomorphometric analysis after tetracycline/calcein double labeling. Fluorescent micrographs of the two labeled mineralization fronts in representative section at the mid diaphysis of the tibia of wt (a) or tg mice (b) at the end of a 4-week GCV treatment period. (c) Measurement of the bone formation rate. Open bar, 10-week-old untreated tg mice; hatched bar, 10-week-old tg mice GCV-treated for 4 weeks; closed bar, 10-week-old wt mice GCV-treated for 4 weeks; gray bar, 14-week-old tg mice 4 weeks after GCV withdrawal. The asterisk indicates a statistically significant difference between wt and tg mice (P < 0.001; n = 4).
The bone-loss phenotype of the GCV-treated tg animals suggested that bone resorption was not hampered and led us to perform an histomorphometric analysis at the end of the treatment period. This analysis showed a nearly complete absence of cells with morphologic features of osteoblasts (data not shown). Absence of detectable double labeling with tetracycline/calcein, a marker of new bone formation (10), showed the concomitant complete arrest of bone-matrix deposition (Fig. 2B a and b). Consistent with this observation, the rate of bone formation was nil (Fig. 2Bc). In every tg animal on withdrawal of GCV, there was a complete reversal of the morphologic, histologic, and histomorphometric abnormalities within 4 weeks (Fig. 2A d, h, and l; Fig 2Bc).
Two other important findings of the histomorphometric analysis were that the number of osteoclasts was unchanged and that trabecular bone volume was decreased in long bones and vertebrae (Fig. 3A). This latter finding, reminiscent of what is observed in osteoporosis (18, 19), could be explained only if bone resorption was still occurring. Thus, we studied osteoclastic activity in these mice. Measurement of urinary pyridinoline crosslinks, the breakdown product of collagen, is a reliable, although correlative, indicator of bone resorption (20, 21). No difference in urine levels of pyridinoline crosslinks was detected between untreated mice and GCV-treated mice (Fig. 3B), suggesting that the absence of bone formation did not affect the level of bone resorption activity.
Figure 3.
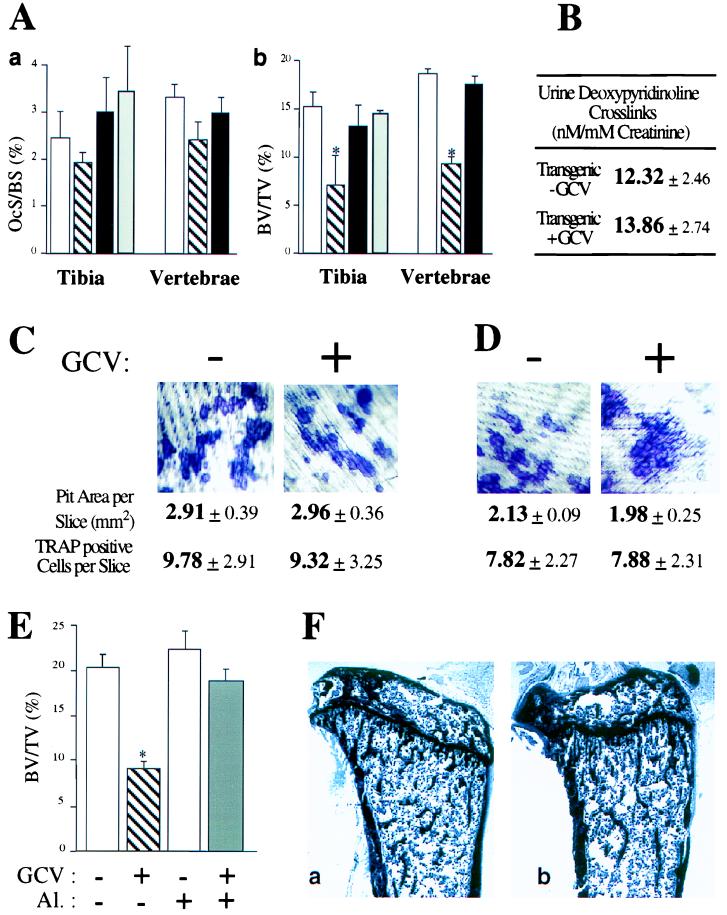
Bone resorption parameters in GCV-treated animals. (A) Measurement of osteoclast surface (a) and bone volumes (b) in tibia and vertebrae. Open bars, 10-week-old tg mice untreated; hatched bars, 10-week-old tg mice GCV-treated for 4 weeks; closed bars, 10-week-old wt mice GCV-treated for 4 weeks; gray bars, 14-week-old tg mice 4 weeks after GCV withdrawal. Asterisks indicate statistically significant differences following GCV treatment (P < 0.005; n = 4). (B) Urine deoxypyridinoline crosslinks levels in 10-week-old tg mice untreated or GCV-treated for 4 weeks. (C) Coculture of bone marrow cells (BMCs) and calvarial cells derived from tg mice treated in the absence (−) or presence (+) of GCV for 7 days. (D) Similar coculture assay of BMCs derived from 10-week-old tg mice treated for 4 weeks with GCV. (E) Measurement of bone volume after GCV and/or alendronate treatment of tg animals. Open bar, 10-week-old untreated mice; hatched bar, 10-week-old mice GCV-treated for 4 weeks; light gray bar, 10-week-old mice treated with alendronate (Al.) for 4 weeks; dark gray bar, 10-week-old mice treated with GCV and alendronate for 4 weeks. (F) Similar structural appearance of tibia of 10-week-old mouse untreated (a) or treated with GCV and alendronate for 4 weeks (b).
To study osteoclast function directly, we first used an in vitro coculture assay of BMCs and osteoblasts derived from calvaria (22). In this assay, osteoclast differentiation is measured by the appearance of multinucleated cells staining positive for tartrate-resistant acid phosphatase (TRAP), a marker of the osteoclast, and bone resorption is measured by the appearance of resorption pits on dentin slices (22). A 7-day GCV treatment of cocultured BMCs and calvarial cells derived from tg mice did not inhibit pit formation (Fig. 3C). To rule out the existence of any osteoblast contamination in the bone marrow preparation during this short-term GCV treatment, we repeated this experiment with cells derived from tg mice treated with GCV for 4 weeks. BMCs derived from GCV-treated and untreated animals differentiated equally well into TRAP-positive cells able to form resorption pits (Fig. 3D).
Next, we asked whether we could prevent bone loss in GCV-treated animals by inhibiting osteoclast function. For that purpose we used alendronate, an inhibitor of osteoclast function (23). We treated 6-week-old tg mice for 4 weeks with GCV alone, alendronate alone, or a combination of both drugs, before histomorphometric analyses. As expected, the GCV-treated animals developed a severe bone loss, and their bone volumes were decreased (Fig. 3E). In contrast, treatment of tg animals with both GCV and alendronate prevented bone loss, and the bone volumes of these animals remained normal (Fig. 3E). These results show that the osteoporotic syndrome occurring in the GCV-treated tg animals was caused by active osteoclastic bone resorption that could be prevented with a classical antiresorptive agent. Interestingly, in the GCV- and alendronate-treated animals, there was no active BR, because both formation and resorption were inhibited. Nevertheless, these animals were healthy, and their bones appeared structurally normal (Fig. 3F), therefore raising the question of the requirement of BR in absence of stress.
For these tg mice to be a valuable model of osteoporosis, the severity of the bone formation defect should be amenable to modulation, and bone resorption should be maintained for even longer periods of time. To test whether these two requirements could be met, we used older animals that have a slower rate of BR. We treated 12-week-old tg mice for either 4 or 8 weeks with GCV. After 4 weeks of GCV treatment, these mice developed an osteopenic phenotype characterized by a moderate decrease of the bone formation rate (Fig. 4). As was the case in younger animals, the osteoclast number was not affected by the GCV treatment, and bone volume was decreased, indicating that bone resorption was still occurring (Fig. 4B). This phenotype, reminiscent of a mild osteoporosis, was reversible at the arrest of the treatment (data not shown). When the GCV treatment was prolonged for 8 weeks, the osteoblast number was more severely decreased, and bone formation was no longer detectable (Fig. 4B). After this long GCV treatment, the number of osteoclasts had still not decreased, and trabecular bone volume was reduced further, indicating that bone resorption had still not stopped (Fig. 4B). Thus, this animal model can be used to mimic osteoporosis of variable severity marked by continuing bone resorption in the face of little or no bone formation.
Figure 4.

Induction of osteoporosis of variable severity. Tg animals aged 12 weeks were GCV-treated for 4–8 weeks. (A) Histological analysis of tibiae of 16-week-old mice untreated (a); 16-week-old mice GCV-treated for 4 weeks (b); 20-week-old mice GCV-treated for 8 weeks (c). Note the progressive loss of trabecular bone. (B) Static and dynamic histomorphometric analyses. Measurement of bone formation rate (a) and bone volume (b). Open bars, 16-week-old animal untreated; hatched bars, 16-week-old animals GCV-treated for 4 weeks; closed bars, 20-week-old animals GCV-treated for 8 weeks. For each parameter similar values were observed in 16-week-old and 20-week-old untreated animals. Asterisks indicate statistically significant differences between wt and tg mice (P < 0.01; n = 4).
DISCUSSION
This study shows unequivocally that despite a complete arrest of bone formation and a dramatic decrease in bone mass, bone resorption continues unaffected in mice.
The requirement for an osteoclast differentiation factor synthesized by osteoblast progenitors and the maintenance of a near constant bone mass through many cycles of BR lead to the assumption that the two activities, formation and resorption, control each other. Several in vivo biological and medical arguments have challenged this hypothesis, at least partly. First, in c-fos-deficient mice that have no osteoclasts, histologic evidence suggests that bone formation is not arrested, even though there is no bone resorption (E. Wagner, personal communication). Second, c-src-deficient mice have nonfunctional osteoclasts and develop an osteopetrosis. This phenotype worsens over a 6-month period because of a persistence of bone formation (M.A. and R.B., unpublished data). Third, tg mice overexpressing osteoprotegerin, a secreted protein recently identified, have a marked decrease in trabecular osteoclast number (24). Their osteopetrotic phenotype worsens over an 8- to 10-week period, suggesting that bone formation was not arrested during this long period, although bone resorption was arrested (24). Fourth, several human conditions regrouped under the name of osteosclerosis are marked by an increase in bone formation, without any evidence of a decrease in bone resorption (25, 26). In some cases, the functional manifestations of the disease were not identified before adulthood. Taken together, these observations and this study’s confirmation that one can arrest bone formation without altering bone resorption in the OG2-Tk mice indicates that there is no obligatory crossregulation between the two activities. Although we cannot rule out the possibility that osteoblast progenitors, which are not affected by GCV treatment, could express signals yet to be identified that would regulate osteoclast function, our data suggest that a coupling mechanism has to work at the level of differentiation and not function.
If there is no obligatory crosscontrol of the two activities, how is bone mass maintained constant? One possibility is that the bone matrix contains cytokines, produced locally or brought in by the general circulation, that would control bone formation and bone resorption. Such cytokines, if they exist, have yet to be identified. Alternatively, BR could be controlled, like many other physiological processes, by hormone-like factors. It is tempting to speculate by analogy with what has been learned in the last few years about the control of appetite and fat deposition for instance (27) that similar mechanisms may regulate BR. These hypotheses warrant further investigation. Beyond these conceptual challenges, the OG2-Tk mouse model provides a tool to study bone resorption in vivo and to test new therapeutic avenues for bone loss conditions.
Acknowledgments
We thank A. Ulmann for his early support, R. Behringer, H. Kronenberg, G. Rodan, N. Takahashi, T. Suda, and I. Swartz for reagents and suggestions during this study, F. Glorieux, R. St. Arnaud, and P. Soriano for critical reading of the manuscript, and Dr. G. Delling for his support. Animal care was in accordance with institutional guidelines. This work was supported by Hoechst Marion Roussel, the March of Dimes Foundation, and National Institutes of Health grants to G.K. and R.B.
ABBREVIATIONS
- BMC
bone marrow cell
- BR
bone remodeling
- GCV
ganciclovir
- tg
transgenic
- wt
wild type
Footnotes
A Commentary on this article begins on page 13361.
References
- 1.Frost H M. Calcif Tissue Res. 1969;3:211–237. doi: 10.1007/BF02058664. [DOI] [PubMed] [Google Scholar]
- 2.Hattner R, Epker B N, Frost H M. Nature (London) 1965;206:489–490. doi: 10.1038/206489a0. [DOI] [PubMed] [Google Scholar]
- 3.Rodan G A, Martin T J. Calcif Tissue Int. 1981;33:349–351. doi: 10.1007/BF02409454. [DOI] [PubMed] [Google Scholar]
- 4.Rodan G A. In: Osteoporosis. Marcus R, Feldman D, Kelsey J, editors. San Diego: Academic; 1996. pp. 290–301. [Google Scholar]
- 5.Lacey D L, Timms E, Tan H L, Kelley M J, Dunstan C R, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, et al. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 6.Bonnerot C, Nicolas J F. Methods Enzymol. 1993;225:747–802. doi: 10.1016/0076-6879(93)25031-v. [DOI] [PubMed] [Google Scholar]
- 7.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current Protocols in Molecular Biology. New York: Wiley; 1995. [Google Scholar]
- 8.Hahn M, Vogel M, Delling G. Virchows Arch A Pathol Anat Histopathol. 1991;418:1–7. doi: 10.1007/BF01600238. [DOI] [PubMed] [Google Scholar]
- 9.Vignery A, Baron R. Anat Rec. 1980;196:191–200. doi: 10.1002/ar.1091960210. [DOI] [PubMed] [Google Scholar]
- 10.Parfitt A M, Drezner M K, Glorieux F H, Kanis J A, Malluche H, Meunier P J, Ott S M, Recker R R. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 11.Borrelli E, Heyman R A, Hisi M, Evans R M. Proc Natl Acad Sci USA. 1988;85:7572–7576. doi: 10.1073/pnas.85.20.7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borrelli E, Heyman R A, Arias C, Sawchenko P E, Evans R M. Nature (London) 1989;339:538–541. doi: 10.1038/339538a0. [DOI] [PubMed] [Google Scholar]
- 13.Desbois C, Hogue D A, Karsenty G. J Biol Chem. 1994;269:1183–1190. [PubMed] [Google Scholar]
- 14.Aubin J E, Liu F. In: Principles of Bone Biology. Bilizikian J P, Raisz L G, Rodan R A, editors. San Diego: Academic; 1996. pp. 51–68. [Google Scholar]
- 15.Hauschka P V, Lian J B, Cole D, Gundberg C. Physiol Rev. 1989;69:990–1047. doi: 10.1152/physrev.1989.69.3.990. [DOI] [PubMed] [Google Scholar]
- 16.Peck W A, Birge S I, Fedah S A. Science. 1964;146:1476–1477. doi: 10.1126/science.146.3650.1476. [DOI] [PubMed] [Google Scholar]
- 17.Aubin J, Hursche J N M, Meritels J M, Sodek J. J Cell Biol. 1982;92:452–461. doi: 10.1083/jcb.92.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delmas P D. Endocrinol Metab Clin North Am. 1990;19:1–18. [PubMed] [Google Scholar]
- 19.Parfitt A M, Riggs B L, Melton L J. Osteoporosis: Etiology, Diagnosis and Management. New York: Raven; 1988. [Google Scholar]
- 20.Eyre D R. Acta Orthop Scand. 1987;66:166–170. [PubMed] [Google Scholar]
- 21.Eyre D R, Dickson I R, Van Ness K P. Biochem J. 1988;252:495–500. doi: 10.1042/bj2520495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takahashi N, Akatsu T, Udagawa N, Sasaki T, Yamaguchi A, Moseley J M, Martin T J, Suda T. Endocrinology. 1988;123:2600–2602. doi: 10.1210/endo-123-5-2600. [DOI] [PubMed] [Google Scholar]
- 23.Rodan G A, Fleisch H A. J Clin Invest. 1996;97:2692–2696. doi: 10.1172/JCI118722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whyte M P. In: Metabolic Bone Disease. Avioli L V, Krane S M, editors. San Diego: Academic; 1998. pp. 697–738. [Google Scholar]
- 25.Lajeunesse D, Busque L, Menard P, Brunette M G, Bonny Y. J Clin Invest. 1996;98:1835–1842. doi: 10.1172/JCI118984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chua S C, Jr, Chung W K, Wu-Peng X S, Zhang Y, Liu S M, Tartaglia L, Leibel R L. Science. 1996;271:994–996. doi: 10.1126/science.271.5251.994. [DOI] [PubMed] [Google Scholar]
- 27.Collins S, Kuhn C M, Petro A E, Swick A G, Chrunyk B A, Surwit R S. Nature (London) 1996;380:677. doi: 10.1038/380677a0. (lett.). [DOI] [PubMed] [Google Scholar]