

Summary
The human APOBEC3G protein restricts the replication of Vif-deficient HIV-1 by deaminating nascent viral cDNA cytosines to uracils, leading to viral genomic strand G-to-A hypermutations [1–4]. However, the HIV-1 Vif protein triggers APOBEC3G degradation, helping explain why this innate defense does not protect patients [5]. The APOBEC3G-Vif interaction is a promising therapeutic target, but the benefit of enabling HIV-1 restriction in patients is unlikely to be known until Vif antagonists are developed. As a necessary prelude to such studies, cell-based HIV-1 evolution experiments were done to ask whether APOBEC3G can provide a long-term block to Vif-deficient virus replication and, if so, whether HIV-1 variants would emerge that resist restriction. APOBEC3G-expressing T cells were infected with Vif-deficient HIV-1. Virus infectivity was suppressed in 45/48 cultures for over 5 weeks, but replication was eventually detected in 3 cultures. Virus growth characteristics and sequencing demonstrated that these isolates were still Vif-deficient. Rather, these viruses had acquired a promoter mutation and a Vpr null mutation. Resistance occurred by a novel tolerance mechanism in which the resistant viruses packaged less APOBEC3G and accumulated fewer hypermutations. These data support the development of anti-retrovirals that antagonize Vif and thereby enable endogenous APOBEC3G to suppress HIV-1 replication.
Results
APOBEC3G has been shown to decrease the infectivity of Vif-deficient HIV-1 by 10- to several 100-fold (e.g., [2–4, 6–9]). However, in almost all reported experiments there was clearly a population of viruses that appeared unrestricted. These apparent escapees may be attributable to low levels of virus replication in the presence of APOBEC3G, to a failure of APOBEC3G to express in some cells and/or to assays operating near resolution limits. Regardless of the explanations, the overall and long-term potency of this important antiviral protein is unknown.
To address these questions and gain further insights into the mechanism of HIV-1 restriction, we used a long-term continuous cell culture system to ask whether Vif-deficient viruses could evolve resistance to APOBEC3G. The human T cell line CEM-SS was transfected stably with an APOBEC3G expression construct or a vector control. APOBEC3G-expressing clones restricted the short-term spreading infection of Vif-deficient HIV-1IIIB, but not that of the Vif-proficient virus (Figure 1A & Table S1; originally shown in [3]). In contrast, vector control clones supported the replication of both viruses. It is noteworthy that the CEM-SS clones selected for these experiments expressed near-physiologic APOBEC3G levels, similar to those in the parental line CEM (non-permissive for Vif-deficient virus replication), the unrelated T cell line H9 or activated CD4-positive primary T lymphocytes (Figures 1B & S1).
Figure 1. Selecting Vif-Deficient HIV-1 Isolates that Resist APOBEC3G.
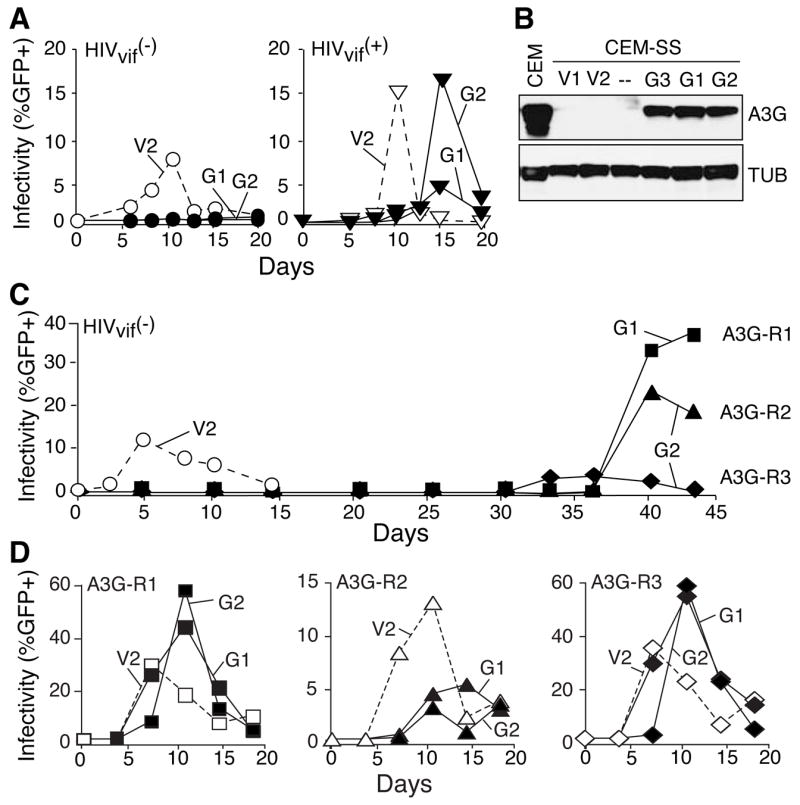
(A) Short-term spreading infection data for Vif-deficient (circles) or Vif-proficient (inverted triangles) viruses. Throughout the manuscript, a unique symbol represents each virus, open symbols and dashed lines represent virus growth on vector control cells (e.g., V2) and filled symbols and solid lines represent virus growth on APOBEC3-expressing cells (e.g., G1, G2).
(B) An immunoblot showing APOBEC3G levels in CEM, vector control CEM-SS clones (V1 and V2), CEM-SS (--) and three independently derived APOBE3G-expressing CEM-SS clones (G1, G2 and G3). The same membrane was stripped and probed for Tubulin (TUB). Complementary immunoblot data comparing model cell lines to primary cells are shown in Figure S1.
(C) Highlights of the long-term spreading infection experiments for Vif-deficient viruses on vector control or APOBEC3G-expressing CEM-SS cell lines. 45/48 cultures showed no virus replication on APOBEC3G-expressing cells (flat lines not graphed). The 3 APOBEC3G-resistant isolates that eventually emerged were A3G-R1 (squares), A3G-R2 (triangles) and A3G-R3 (diamonds).
(D) Spreading infections of A3G-R1, -R2 or -R3 on CEM-SS clones expressing APOBEC3G (G1, G2) or a vector control (V2). Parental viruses were also analyzed in parallel and these data resembled those in Figure 1A (not shown). See the APOBEC3G panels in Figure S2 for complementary data and Table S1 additional information.
Two independent APOBEC3G-expressing CEM-SS clones were each split into 24 wells, infected with Vif-deficient HIV-1IIIB and maintained continuously for 6 weeks. Cell-free supernatants were used to periodically assay for the presence of viable virus using reporter cells in which a Tat-activated HIV-1 LTR promoter drives GFP expression and therefore provides a quantitative measure of virus infectivity (Experimental Procedures). Although most cultures showed no signs of replicating virus, one culture from experiment 1 and two cultures from experiment 2 began to produce virus around the 5-week time point (Figure 1C). These virus isolates were called A3G-R1, -R2 and -R3.
To eliminate cell-based explanations for the apparent resistance phenotypes, such as loss of APOBEC3G expression, supernatants containing A3G-R1, -R2 and -R3 were purified and used to infect fresh APOBEC3G-expressing cells. All three of the virus isolates were now clearly capable of growth on APOBEC3G-expressing cells, indicating that they had evolved a strong, heritable resistance to APOBEC3G (Figure 1D). These virus replication kinetics were similar to those of the Vif-proficient virus on APOBEC3G-expressing cells, and they were stable over a minimum of 5 passages, each using fresh APOBEC3G-expressing cells (Table S1). These data demonstrated that APOBEC3G is capable of providing a long-term block to Vif-deficient HIV-1 replication, which is only rarely alleviated by resistance mutation(s).
To ask whether the APOBEC3G-resistant viral isolates had restored Vif function, we assessed whether the resistant isolates could replicate in two human T cell lines, CEM and H9, where Vif is required to overcome APOBEC3G, the related restriction factor APOBEC3F and potentially other cellular proteins (e.g., Figures 1, 2, S1 & [3, 9]). Curiously, none of the APOBEC3G-resistant isolates replicated in CEM or H9 cells indicating that Vif was still non-functional and that something other than APOBEC3G was still inhibiting virus replication (Figures 2A & S1). Proviral DNA sequencing confirmed that the resistant isolates retained the original vif nonsense mutations.
Figure 2. APOBEC3G-Resistant Isolates Are Still Susceptible to APOBEC3F.

(A & B) Replication kinetics of the parental viruses and A3G-R1, -R2 and -R3 on CEM cells or APOBEC3F-expressing CEM-SS cells (F1 or F2). These experiments were done in parallel, so the vector control data (V2) shown in the CEM panels are also applicable to the APOBEC3F panels. The A3G-R2 and -R3 data on APOBEC3F lines are presented in one panel to make space for Figure 2C. A3G-R1, -R2 and -R3 also spread with rapid kinetics on APOBEC3G-expressing cells (G2; not shown). The higher overall titers of the APOBEC3G resistant viruses warranted expanded Y-axes. Symbol and line designations are identical those in Figure 1. See Figures S1 & S2 for complementary data.
(C) An immunoblot of APOBEC3F levels in two APOBEC3F-expressing CEM-SS clones (F1 and F2), parental CEM-SS cells (--) and two non-permissive cell lines (H9 and CEM). The same membrane was stripped and probed for Tubulin (TUB).
APOBEC3F and APOBEC3G are thought to function similarly, by gaining access to assembling HIV-1 particles and deaminating nascent cDNA (e.g., [6, 9–11]). APOBEC3B and APOBEC3DE have also been implicated in HIV-1 restriction (e.g., [10, 12, 13]). To test whether any of these APOBEC3 proteins could mediate restriction of the APOBEC3G-resistant isolates, viral spreading infection experiments were done with CEM-SS clones that expressed CEM-like levels of APOBEC3F and independently with clones that expressed HA-tagged APOBEC3G, APOBEC3F, APOBEC3B or APOBEC3DE (Figures 2 & S2).
APOBEC3B or APOBEC3DE did not inhibit the replication of the Vif-deficient parental virus, let alone the APOBEC3G-resistant derivatives (Figure S2). These data suggested that neither of these is relevant to HIV-1 restriction in the CEM-SS system, but additional experiments will be necessary to determine whether these proteins influence HIV-1 in other cell types. In contrast, APOBEC3F inhibited the replication of both the Vif-deficient virus and the APOBEC3G-resistant isolates, but not the Vif-proficient parent (Figures 2 & S2). Inhibition was not attributable to differences in protein levels (Figure S2). These data therefore (i) indicated that resistance is specific to APOBEC3G and that the mechanisms of restriction by APOBEC3F and APOBEC3G differ at some fundamental level, (ii) supported the hypothesis that APOBEC3F is at least in part responsible for the inability of Vif-deficient viruses to replicate on CEM cells, and (iii) discouraged most trivial explanations for APOBEC3G resistance such as improved fitness or growth rate (as cross-resistance to APOBEC3F would likely have been observed in such scenarios).
The APOBEC3G-resistance mutations were identified by sequencing proviral DNA. Remarkably, the APOBEC3G-resistant isolates only had two types of mutation in common ---> an A200T-or-C non-coding transversion and a Vpr truncation mutation (Figure 2 & Table S2). It is likely that these two mutations arose independently, as the nature of these and several associated mutations suggested that both APOBEC3G-dependent and -independent mechanisms contributed (Table S2). To ask whether a Vpr-truncation and an A200 transversion combined to confer resistance, these mutations were incorporated into Vif-deficient molecular clones and used for spreading infection experiments (Figures 3C & S3; an ‘M’ suffix label distinguishes molecular clone-derived viruses from original isolates). All of the molecular clones with both mutations, A3G-R1M, -R2M or -R3M, spread efficiently on APOBEC3G-expressing cells, whereas viruses with either mutation alone were unable to replicate. Thus, both mutations were required for Vif-deficient viruses to overcome APOBEC3G-dependent restriction.
Figure 3. HIV-1 Mutations that Confer Vif-independent Resistance to APOBEC3G.

(A) A schematic of HIV-1IIIB indicating the coding regions and relevant restriction sites (GenBank Accession EU541617). Asterisks denote the tandem stop mutations in vif.
(B) Summaries of the types of base substitutions found in A3G-R1, -R2 and -R3 proviral DNA. Each horizontal line is a composite of 4 independent proviral DNA sequences from large or small PCR amplicons (solid and dashed lines, respectively). G-to-A substitutions are depicted by vertical bars and other substitutions by closed circles. Vertical arrows highlight the common A-to-T/C and vpr-inactivating mutations. See Figure S4 for an overview of 800 kb of sequencing data.
(C) Replication kinetics of molecular clone-derived viruses with the indicated genotypes. Symbol and line designations are identical those in Figure 1, except the double mutants share the same symbols as the APOBEC3G-resistant triple mutant (diamonds for A3G-R3M). See Figure S3 for supporting data, including experiments with A3G-R1M, A3G-R2M and their double mutant derivatives.
Mammalian retroviruses use multiple strategies to escape the APOBEC3 proteins of their hosts. HIV-1 and SIV use Vif to degrade APOBEC3s, foamy viruses use Bet to inhibit APOBEC3s and others such as HTLV, MuLV and MPMV simply appear to exclude APOBEC3s from encapsidation (e.g., [5, 14–19]). However, the frequent G-to-A hypermutations observed while identifying the APOBEC3G resistance mutations strongly suggested that the resistance mechanism described here would be unique. Indeed, extensive proviral DNA sequence analyses showed that the resistant viruses were being hypermutated at a significant frequency, 2-fold lower than that of the Vif-deficient parental virus (1.5 G-to-A versus 3.3 G-to-A per kb, respectively), but still much higher than the Vif-proficient virus (0.03 G-to-A per kb) (Figure S4).
These hypermutation data implied that significant but perhaps lower levels of APOBEC3G were being encapsidated. To address this directly, APOBEC3G encapsidation was quantified in spreading infection experiments using CEM-SS cells expressing a catalytically defective but otherwise fully intact APOBEC3G protein that does not inhibit Vif-deficient HIV-1 (A3G-E259Q; e.g., [2, 8, 20]). Immunoblots of virion-associated proteins from the spreading infection peaks demonstrated that the resistant molecular clones packaged 2- to 3-fold less APOBEC3G than the Vif-deficient parent (Figures 4A & S5). Moreover, the APOBEC3G resistance phenotype and the partial encapsidation defect were apparent in a single round of virus replication in APOBEC3G-expressing CEM-SS cells (Figure S6 & Online Discussion). Taken together, the hypermutation and packaging data demonstrated that the resistant viruses had evolved a novel tolerance mechanism for escaping APOBEC3G-dependent restriction. This mechanism may be particularly applicable to a population of viruses, as the population clearly expands while many individual viruses are undoubtedly being restricted.
Figure 4. A200T-or-C Contributes to APOBEC3G Resistance by Enhancing HIV-1 Transcription, Increasing Virus Titers and Diminishing APOBEC3G Encapsidation.
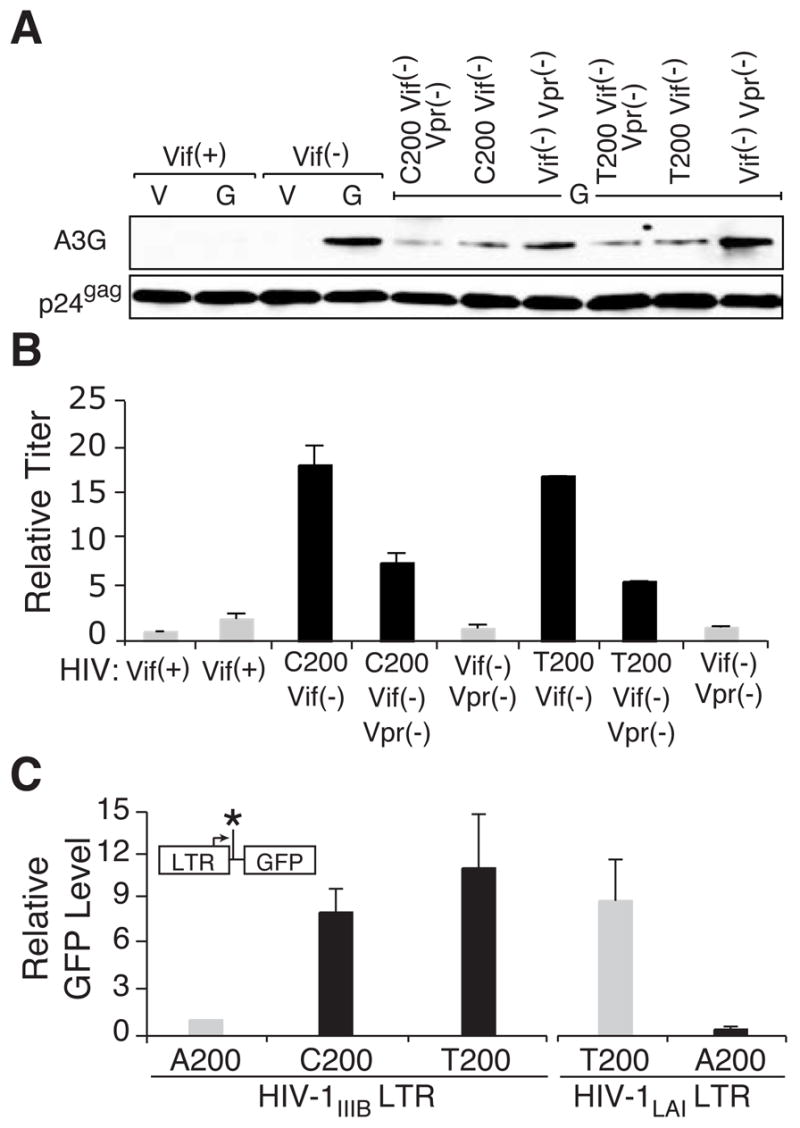
(A) Immunoblots showing the level of virus particle-associated APOBEC3G-E259Q or p24Gag from a spreading infection experiment with the indicated viruses. Relative to the Vif(−) virus with an APOBEC3G/p24Gag ratio normalized to 1 (n=3), the Vif(−)/Vpr(−), A200T-orC/Vif(−) and A200T-orC/Vif(−)/Vpr(−) viruses had mean ratios of 1.1 ± 0.36 (n=9), 0.25 ± 0.10 (n=9) and 0.32 ± 0.17 (n=8), respectively. See Figure S5 for additional quantification and Figure S6 for complementary single-cycle data.
(B) Relative titers of the indicated viruses produced after a single-round of replication on CEM-SS cells expressing APOBEC3G-E259Q. Each histogram bar shows the mean and s.d. of 3 independently derived supernatants (some error bars were too small to graph). The titer for the Vif-proficient virus was normalized to 1 to facilitate comparisons.
(C) Expression level of the indicated LTR-GFP reporter constructs in HEK-293 cells. Each histogram bar shows the mean and s.d. of 3 independent replicas. The GFP expression level of the A200 HIV-1IIIB construct was normalized to 1 to facilitate comparisons. The inset depicts the LTR-GFP expression construct, with position of nucleotide 200 marked by an asterisk.
Interestingly, the APOBEC3G packaging defect was also apparent in Vif-deficient A200T-or-C viruses (Figure 4A). This correlated inversely with data showing that the resistant isolates and A200T-or-C mutants yielded higher virus titers (Figure 4B). One way that these two results may be linked is for the A200T-or-C mutation to upregulate HIV-1 transcription, which would produce more substrate for particle production and in turn cause less APOBEC3G to be packaged (i.e., more particle assembly effectively dilutes available cellular APOBEC3G). In support of such a mechanism, the A200T-or-C mutation is located on the promoter proximal end of a demonstrated transcriptional activator-binding site (DBF; [21]). Therefore, to test whether the A200T-or-C mutation triggers higher levels of transcription, we made a series of LTR-promoter-driven GFP expression constructs containing A200, T200 or C200, transfected HEK-293 cells and monitored GFP expression by flow cytometry. Each of the A200 transversion constructs caused an 8- to 10-fold increase in GFP expression (Figure 4C). It is notable that most HIV-1 isolates already have a pyrimidine at position 200 (http://www.hiv.lanl.gov/content/hiv-db). We therefore tested whether the reciprocal mutation would diminish promoter activity and found that a T200A compromised the activity of the HIV-1LAI promoter (Figure 4C). Taken together, these data indicated that transcriptional up-regulation underlies both the observed titer increases and the APOBEC3G encapsidation defect, thereby constituting an integral part of the APOBEC3G-resistance mechanism.
Discussion
Our studies demonstrate that APOBEC3G is capable of blocking the replication of Vif-deficient HIV-1 over an extended duration. Virus replication was only detectable after two mutations appeared ---> a non-coding A200T-or-C transversion and a Vpr null mutation. The fact that significant virus evolution was required for Vif-deficient viruses to bypass restriction by APOBEC3G demonstrates that this arm of the innate immune response is capable of imposing a lethal block to virus replication. Our studies therefore provide an important proof-of-principle that can be appreciated by drawing analogies to HIV-1 drug resistance. The potency of APOBEC3G can now be compared to that of bona fide anti-retroviral drugs such as reverse transcriptase inhibitors, as both impose blocks to virus replication such that resistance mutations provide the only means of escape. The combination of APOBEC3G and APOBEC3F may be further likened to two anti-retroviral drugs that block replication through distinct paths (e.g., nucleoside and non-nucleodside reverse transcriptase inhibitors), as resistance to APOBEC3G did not confer cross-resistance to APOBEC3F. Our studies therefore provide strong additional justifications for efforts to develop Vif-neutralizing therapeutics.
The novel combination of resistance mutations reported here also provides a unique opportunity to better understand the mechanism of APOBEC3G-dependent HIV-1 restriction. It is remarkable that APOBEC3G did not select for a restoration of Vif function. This strongly indicates that Vif has other potentially more important roles in the biology of HIV-1 and related lentiviruses. Together with prior work [6, 9–11], our studies suggest that at least one of those roles will be APOBEC3F neutralization.
Our data indicate that the A200T-or-C mutation contributes to APOBEC3G resistance by elevating LTR transcription, increasing virus production and ultimately causing less APOBEC3G encapsidation (Figure S7). Diminished APOBEC3G levels thereby contribute to an overall shift from lethal to sub-lethal levels of restriction (i.e., a shift to tolerable APOBEC3G levels). This suggests that anything the virus does to minimize APOBEC3G packaging is likely to prove beneficial. Some viruses like HTLV, MuLV and MTMV appear to have taken this to the extreme by simply excluding APOBEC3G from particles [14–17].
A Vpr-inactivating mutation was also required for Vif-deficient HIV-1 to evolve resistance to APOBEC3G. Vpr-inactivation was not observed in Vif-deficient viruses that replicated on non-APOBEC3G-expressing CEM-SS cells (not shown). These unexpected results suggested that Vpr might actually facilitate APOBEC3G-dependent restriction by, for instance, packaging a cellular factor (Figure S7). Such a model is appealing because Vpr is abundant in virions and it has been shown to interact with many proteins (e.g., uracil DNA glycosylase UNG2, Vpr binding protein 1 and others; reviewed in [22]). However, the most obvious Vpr-binding candidate, UNG2, is not involved in APOBEC3G-dependent restriction [7, 8, 23, 24]. Much additional work will therefore be necessary to identify this putative restriction co-factor and test other tenable models.
In addition to the clear relevance to HIV-1, the novel tolerance mechanism described here has several broad implications. It highlights the distinct possibility that some retroviruses may have evolved to co-exist and possibly even benefit from the mutagenic activity of the APOBEC3 proteins. For example, the majority of reverse-transcribing viruses and transposons lack obvious APOBEC3 neutralizing factors such as Vif or Bet (e.g., [5, 18, 19]). Moreover, it is unlikely that all remaining retroelements escape restriction by HTLV/MuLV/MPMV-like avoidance mechanisms (e.g., [14–17]). Thus, based on current data, the APOBEC3G ‘resistance by tolerance’ mechanism described here may well apply to many viruses. Indeed, G-to-A hypermutations are evident in multiple viruses such as other lentiviruses, the hepadnavirus HBV, the gammaretrovirus SNV, the endogenous murine retrovirus Pmv and the DNA tumor virus HPV (e.g., [25–29]). Therefore, the ability to tolerate and perhaps even regulate APOBEC3-dependent restriction has clear implications for virus evolution, immune escape and drug resistance.
Supplementary Material
Supplemental Data
Experimental Procedures, additional Discussion, Tables S1 & S2 and Figures S1 to S7 are included as Supplemental Information for Online Publication.
Acknowledgments
We thank P. Bieniasz, J. Coffin, S. Goff, A. Haase, P. Hackett, L. Mansky, V. Planelles and P. Southern for valuable feedback, M. Malim, J. Lingappa and M. Stevenson for key reagents and M. Hertzberg and L. Mansky for the generous provision of facilities. These studies were supported by grants from the NIH (AI064046), Campbell Foundation and University of Minnesota (GIA and AHC-FRD). G.H., J.S.A. and R.S.H., respectively, were supported in part by a CIHR pre-doctoral studentship, a Medical Scientist Training Program grant (T32 GM008244 with supplemental support from the Mayo Foundation) and a Searle Scholarship. The University of Minnesota AGAC assisted with DNA sequencing and the SCI provided computational support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 2.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 3.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 4.Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424:94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 6.Liddament MT, Brown WL, Schumacher AJ, Harris RS. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr Biol. 2004;14:1385–1391. doi: 10.1016/j.cub.2004.06.050. [DOI] [PubMed] [Google Scholar]
- 7.Mbisa JL, Barr R, Thomas JA, Vandegraaff N, Dorweiler IJ, Svarovskaia ES, Brown WL, Mansky LM, Gorelick RJ, Harris RS, et al. Human Immunodeficiency Virus Type 1 cDNAs Produced in the Presence of APOBEC3G Exhibit Defects in Plus-Strand DNA Transfer and Integration. J Virol. 2007;81:7099–7110. doi: 10.1128/JVI.00272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schumacher AJ, Haché G, MacDuff DA, Brown WL, Harris RS. The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J Virol. 2008;82:2652–2660. doi: 10.1128/JVI.02391-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 2004;23:2451–2458. doi: 10.1038/sj.emboj.7600246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 2004;14:1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 11.Zheng YH, Irwin D, Kurosu T, Tokunaga K, Sata T, Peterlin BM. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J Virol. 2004;78:6073–6076. doi: 10.1128/JVI.78.11.6073-6076.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dang Y, Wang X, Esselman WJ, Zheng YH. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J Virol. 2006 doi: 10.1128/JVI.01123-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doehle BP, Schafer A, Cullen BR. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology. 2005;339:281–288. doi: 10.1016/j.virol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 14.Abudu A, Takaori-Kondo A, Izumi T, Shirakawa K, Kobayashi M, Sasada A, Fukunaga K, Uchiyama T. Murine retrovirus escapes from murine APOBEC3 via two distinct novel mechanisms. Curr Biol. 2006;16:1565–1570. doi: 10.1016/j.cub.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 15.Derse D, Hill SA, Princler G, Lloyd P, Heidecker G. Resistance of human T cell leukemia virus type 1 to APOBEC3G restriction is mediated by elements in nucleocapsid. Proc Natl Acad Sci U S A. 2007;104:2915–2920. doi: 10.1073/pnas.0609444104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doehle BP, Schafer A, Wiegand HL, Bogerd HP, Cullen BR. Differential sensitivity of murine leukemia virus to APOBEC3-mediated inhibition is governed by virion exclusion. J Virol. 2005;79:8201–8207. doi: 10.1128/JVI.79.13.8201-8207.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doehle BP, Bogerd HP, Wiegand HL, Jouvenet N, Bieniasz PD, Hunter E, Cullen BR. The betaretrovirus Mason-Pfizer monkey virus selectively excludes simian APOBEC3G from virion particles. J Virol. 2006;80:12102–12108. doi: 10.1128/JVI.01600-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lochelt M, Romen F, Bastone P, Muckenfuss H, Kirchner N, Kim YB, Truyen U, Rosler U, Battenberg M, Saib A, et al. The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc Natl Acad Sci U S A. 2005;102:7982–7987. doi: 10.1073/pnas.0501445102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russell RA, Wiegand HL, Moore MD, Schafer A, McClure MO, Cullen BR. Foamy virus Bet proteins function as novel inhibitors of the APOBEC3 family of innate antiretroviral defense factors. J Virol. 2005;79:8724–8731. doi: 10.1128/JVI.79.14.8724-8731.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holmes RK, Koning FA, Bishop KN, Malim MH. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J Biol Chem. 2007;282:2587–2595. doi: 10.1074/jbc.M607298200. [DOI] [PubMed] [Google Scholar]
- 21.el Kharroubi A, Verdin E. Protein-DNA interactions within DNase I-hypersensitive sites located downstream of the HIV-1 promoter. J Biol Chem. 1994;269:19916–19924. [PubMed] [Google Scholar]
- 22.Andersen JL, Planelles V. The role of Vpr in HIV-1 pathogenesis. Curr HIV Res. 2005;3:43–51. doi: 10.2174/1570162052772988. [DOI] [PubMed] [Google Scholar]
- 23.Kaiser SM, Emerman M. Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase APOBEC3G. J Virol. 2006;80:875–882. doi: 10.1128/JVI.80.2.875-882.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langlois MA, Neuberger MS. Human APOBEC3G can restrict retroviral infection in avian cells and acts independently of both UNG and SMUG1. J Virol. 2008;82:4660–4664. doi: 10.1128/JVI.02469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jern P, Stoye JP, Coffin JM. Role of APOBEC3 in genetic diversity among endogenous murine leukemia viruses. PLoS Genet. 2007;3:2014–2022. doi: 10.1371/journal.pgen.0030183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kristbjornsdottir HB, Andresdottir V, Svansson V, Torsteinsdottir S, Matthiasdottir S, Andresson OS. The vif gene of maedi-visna virus is essential for infectivity in vivo and in vitro. Virology. 2004;318:350–359. doi: 10.1016/j.virol.2003.09.044. [DOI] [PubMed] [Google Scholar]
- 27.Pathak VK, Temin HM. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: substitutions, frameshifts, and hypermutations. Proc Natl Acad Sci U S A. 1990;87:6019–6023. doi: 10.1073/pnas.87.16.6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suspene R, Guetard D, Henry M, Sommer P, Wain-Hobson S, Vartanian JP. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc Natl Acad Sci U S A. 2005;102:8321–8326. doi: 10.1073/pnas.0408223102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vartanian JP, Guetard D, Henry M, Wain-Hobson S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science. 2008;320:230–233. doi: 10.1126/science.1153201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data
Experimental Procedures, additional Discussion, Tables S1 & S2 and Figures S1 to S7 are included as Supplemental Information for Online Publication.