

Abstract
Analysis of genetic interactions has been extensively exploited to study gene functions and to dissect pathway structures. One such genetic interaction is synthetic lethality, in which the combination of two non-lethal mutations leads to loss of organism viability. We have developed a dSLAM (heterozygote diploid-based synthetic lethality analysis with microarrays) technology that effectively studies synthetic lethality interactions on a genome-wide scale in the budding yeast Saccharomyces cerevisiae. Typically, a query mutation is introduced en masse into a population of ~6,000 haploid-convertible heterozygote diploid Yeast Knockout (YKO) mutants via integrative transformation. Haploid pools of single and double mutants are freshly generated from the resultant heterozygote diploid double mutant pool after meiosis and haploid selection and studied for potential growth defects of each double mutant combination by microarray analysis of the “molecular barcodes” representing each YKO. This technology has been effectively adapted to study other types of genome-wide genetic interactions including gene-compound synthetic lethality, secondary mutation suppression, dosage-dependent synthetic lethality and suppression.
1. Introduction
Genome sequencing has allowed scientists to identify the majority of the parts list of an organism. Now a daunting task is to understand the function(s) of each gene and how genes functionally interconnect to form a cellular network that defines life. Studies of genetic interactions have been extremely useful for both characterizing gene function and dissecting pathway structures in model organisms. One such genetic interaction is Synthetic Lethality (SL) in which two mutations are separately non-lethal but their combination causes lethality (1). This interaction normally reflects a compensatory relationship between two genes, which operate either in the same pathway or in two distinct but highly related pathways (2).
The baker’s yeast Saccharomyces cerevisiae has long been an excellent model organism for studying eukaryotic molecular and cellular biology thanks to its relative simplicity, the availability of sophisticated genetic and biochemical tools, and its conserved basic biological processes with other systems. In the wake of the sequencing of the S. cerevisiae genome, genome-wide “bar-coded” yeast knockout (YKO) mutants (Figure 1A) representing defined null mutations for nearly every gene in the yeast genome have been constructed and have greatly facilitated functional characterization of the yeast genome (3, 4). In particular, systematic analysis of genome-wide gene-gene synthetic lethality (GGSL) interactions and other types of genetic interactions provided a very effective way to study gene functions and to dissect pathway topologies in yeast (5–7).
Figure 1. Simplified structural diagrams for the YKO construct, pXP346, and pSO142-4.
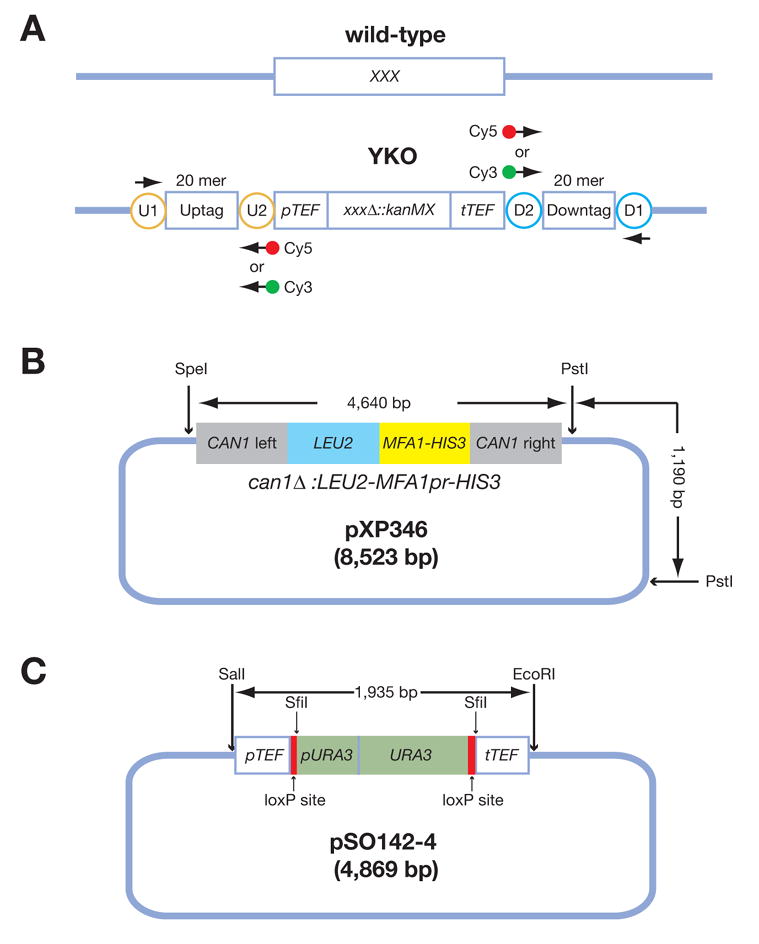
(A) A diagram for the yeast knockout construct. Each YKO consists of a kanMX module that confers resistance to the antibiotic G418 flanked by unique 20 mer “molecular barcodes” or “Tags” called the “Uptag” and “Downtag.” All “Uptags” are themselves flanked by a common set of priming sites (U1 and U2 within the orange circles) and all “Downtags” are flanked by another set of common priming sites (D1 and D2 within the cyan circles). These common priming sites allow for PCR amplification and microarray analysis of all Uptags or all Downtags in a population. “XXX” stands for any yeast gene. (B) A simplified diagram of pXP346, which contains the can1Δ::LEU2-MFA1pr-HIS3 reporter. This plasmid needs to be digested with SpeI and PstI to release the reporter for integration into the CAN1 locus of the yeast genome via homologous recombination. (C) A diagram of pSO142-4, which contains the URA3-loxP cassette. URA3 targeting construct pSO142 can be integrated into the existing kanMX4-marked YKO strains based on its sequence homology to the promoter and terminator sequences, derived from the Ashbya gossypii TEF gene. URA3, driven by its own promoter, is flanked by loxP sites allowing subsequent excision/marker swaps upon expression of Cre recombinase, increasing the flexibility of the marker for future manipulations. The SwaI and SfiI restriction sites were introduced for diagnostic purposes. In the first step, a loxP-URA3 cassette containing SwaI and SfiI sites was constructed using PCR. The loxP-URA3 cassette was amplified as two separate fragments, loxP-partial URA3 (loxP-URA3’) and partial URA3-loxP (‘URA3-loxP). The loxP, SwaI and SfiI sequences (introduced on primers) were fused to the 5′ and 3′ ends of the URA3 fragment via three sequential rounds of PCR. The URA3 fragment was amplified from pRS406. The loxP sequence was obtained from (25). Both PCR products were then co-transformed into a nej1 ::kanMX4 strain. The goal was to have the two PCR products homologously replace the kan ORF sequence. Integrative transformation was made possible by the following features on the two PCR products: loxP-URA3 contains 45 bp sequence homology to the TEF promoter, while URA3-loxP contains 48 bp sequence homology to the TEF terminator. The 3′ end of loxP-URA3’ and the 5′ end of ‘URA3-loxP overlap by 111 bp. nej1_ was used to decrease the efficiency of non-homologous end-joining. We first selected for Ura+ transformants. Next, we assayed and identified Ura+ transformants that are G418s, indicating that URA3 gene was integrated into kanMX4; one such strain was YSO205. In the second step, genomic DNA isolated from YSO205 was used as template to amplify the pTEF-loxP-URA3-loxP-tTEF fragment (1965 bp) using primers U2 and D2. The PCR products were then digested with SalI and EcoRI and cloned into pBSIIKS(−). Primer U2 contains a SalI site, while D2 contains EcoRI site.
Note: The plasmid backbones in both panels B and C are not drawn to scale.
We have recently developed a technology called dSLAM (heterozygous diploid-based Synthetic Lethality Analysis on Microarrays) that exploits heterozygous diploid YKOs to detect genome-wide synthetic lethality (8). This methodology combines the excellent genetic quality of the heterozygote diploid YKOs, the convenience of handling them in pooled form, and the efficiency of a microarray analysis of abundance of YKOs in the population. The ‘d’ in ‘dSLAM’ also highlights the fact that both the control (single mutant) and experimental (double mutant) pools are derived from the same molecularly manipulated heterozygote diploid pool, which alleviates experimental noises introduced by two separate transformations necessary for a haploid SLAM experiment (9).
The principal stages of dSLAM are outlined in Figure 2. First, a specialized haploid selective marker called the “SGA (synthetic genetic array) reporter” (8, 10) is incorporated into the heterozygote diploid YKO strains originally constructed by the Saccharomyces Genome Deletion Project (http://www-sequence.stanford.edu/group/yeast_deletion_project/deletions3.html). Next, a query mutation is introduced into a pool of such haploid-convertible heterozygote YKOs to make a heterozygous double mutant pool of the query mutation and genome-wide YKOs. This double mutant pool is subsequently converted into a pool of haploid single mutants and a pool of haploid double mutants by exploiting the SGA reporter after meiosis. For each YKO, its relative growth rate as a single mutant and as a double mutant (in combination with the query mutation) are indirectly compared by microarray analysis of the abundance of the “molecular barcodes” or “Tags” in both the haploid single (Control, C) and double (Experiment, E) mutant pools. An SL interaction is revealed by a high Control/Experiment (or C/E) ratio of the Tag hybridization signal intensity (8).
Figure 2. A flowchart for a dSLAM screen.
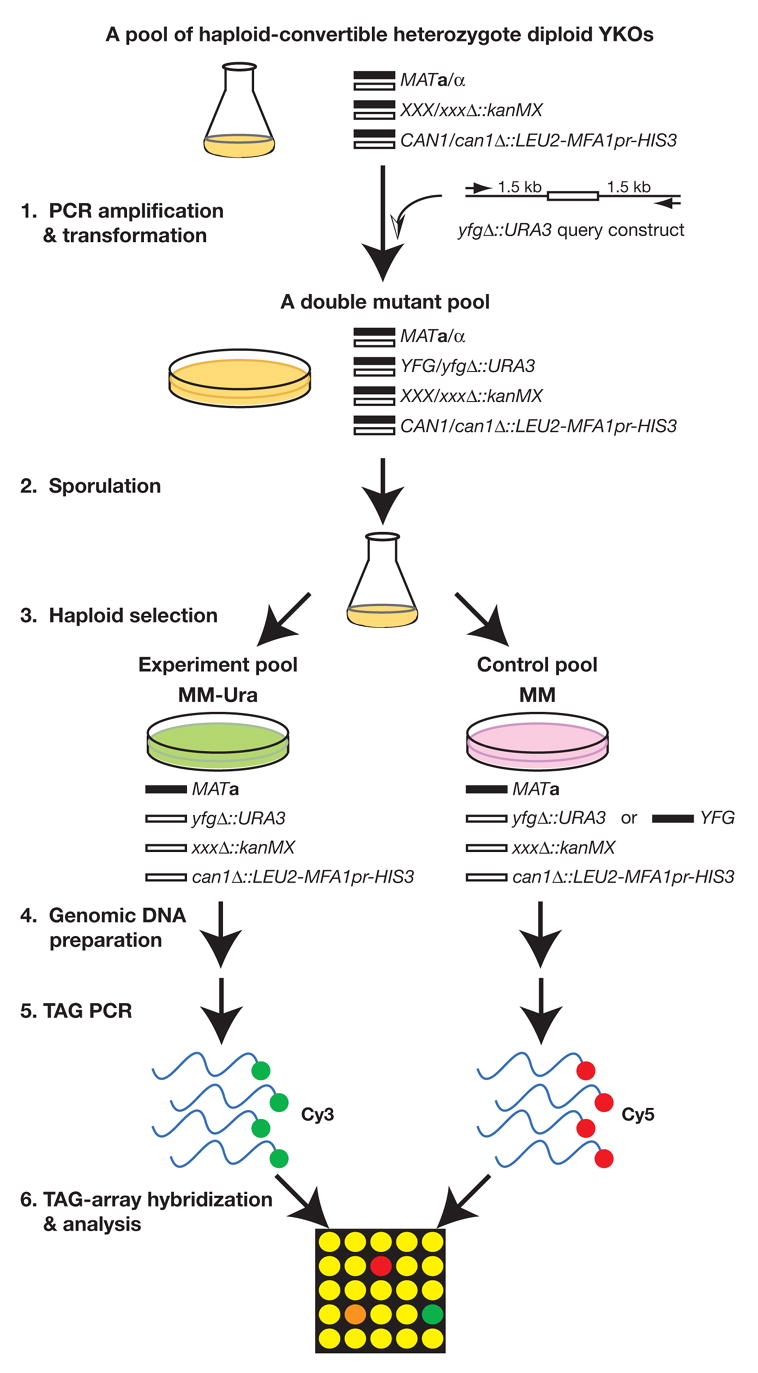
Step 1. A query construct yfgΔ::URA3 is PCR-amplified and transformed en masse into a pool of haploid convertible heterozygote diploid YKOs. Step 2. The resultant double mutant pool is sporulated. Step 3. Haploid experiment (double mutants) and control (single and double mutants) pools are freshly and independently generated from the same sporulation culture by plating on the haploid selection media MM-Ura and MM, respectively. Step 4. Genomic DNA samples are prepared from these two haploid pools. Step 5. The TAGs from both pools are PCR-amplified with differentially labeled primers (Cy3 and Cy5). Step 6. The PCR-amplified dye-labeled TAGs are hybridized to a TAG-array and analyzed for the relative abundance of each TAG in both control and experiment pools. “YFG” stands for “Your Favorite Gene;” “XXX” stands for any gene in the yeast genome; “MM” stands for the haploid selection “Magic Medium” (see Table 3).
In addition to discussing dSLAM itself, we also describe confirmatory assays that are useful in weeding out false positive findings arising from the extreme sensitivity of a TAG-array based assay, which includes a PCR-based amplification step. We also illustrate the ease with which dSLAM can be adapted to characterize various other kinds of cell growth-based genetic interactions genome-wide, including SL interactions between a non-knockout allele and genome-wide YKOs, gene-compound synthetic lethality (GCSL), genetic suppression by a second mutation, dosage-dependent synthetic lethality and suppression, and synthetic haplo-insufficiency.
2. dSLAM analysis of gene-gene synthetic lethality
2.1 The dSLAM procedure
2.1.1 Constructing a haploid-convertible heterozygote YKO pool
One preparative step required for performing a dSLAM screen is the construction of a pool of haploid-convertible heterozygote diploid YKOs. These can be made in one of two ways. 1) We have individually incorporated the modified SGA reporter can1Δ::LEU2-MFA1pr-HIS3 into the full set of heterozygote diploid YKOs originally constructed by the Saccharomyces Genome Deletion Project (Boeke et al., unpublished data). This entire set of haploid-convertible heterozygous diploid YKO strains, available from Open Biosystems and ATCC, can be spotted on Nunc OmniTrays containing solid YPD plus 200 μg/ml G418 (GIBCO, Cat # 11811-031) in a 96-well format. The cells are deposited onto the plates from liquid culture with a 96-pin replicator (BOEKEL, Model 140500), generating patches about 5 mm in diameter after growth. It is wise to make two or more copies of each plate in case of sporadic contamination. All strains are incubated at 30°C for 2 days, scraped with filter-sterilized 15% glycerol (2 x 5 ml/plate), and mixed (with a stir bar and stir plate) to make a pool at a concentration of approximately 60 OD600nm/ml and stored as 1 or 2 ml aliquots at -80°C. Because all strains have similar growth rates, all patches should be highly uniform in cell number and state. 2) If the “original” set of heterozygote YKOs (4) (distributed by Open Biosystems, ATCC, Invitrogen, Euroscarf and previously, Research Genetics) that does not contain the SGA reporter is available, a researcher can make his/her own haploid-convertible YKO pool using a brief procedure that follows. First, a pool of the original heterozygote YKOs should be constructed exactly as described above. 10–20 μg of plasmid pXP346 (8) that carries the SGA reporter, can1Δ::LEU2-MFA1pr-HIS3, is digested with SpeI and PstI (Figure 1B) in a 500 μl reaction volume and concentrated by ethanol precipitation. This digested plasmid can then be transformed en masse into the pool of original heterozygote diploid YKOs using a high efficiency integrative transformation protocol described in the “transformation of pool aliquots” section (see below). All cells from one transformation are suspended in 4 ml of 5 mM CaCl2 for 5–15 minutes and plated (400 μl/plate) onto ten 150 x 25 mm Petri dishes containing SC-Leu solid medium for LEU2 selection. Before plating, dilutions of 100-fold should be plated on a 100 X 25 mm SC-Leu Petri dish to carefully titer the yield of transformants. > 5.0 x 105 independent Leu+ transformants are preferred to avoid stochastic underrepresentation of any particular YKO mutant from this population. The Leu+ cells on each of the ten 150 x 25 mm Petri dishes are harvested with filter-sterilized 15% glycerol (2 x 5 ml/plate), pooled, mixed well and stored as 1 or 2 ml aliquots at -80°C.
For reasons we do not understand, a small number of our individually constructed haploid-convertible heterozygote YKOs of non-essential genes do not give rise to a sufficient number of viable haploid mutant cells when plated on the synthetic “magic medium” (MM; SC-Leu-His-Arg +Canavanine +G418; Table 3), which selects for haploid YKO mutants after sporulating the diploids. For example, we have noticed that the mutants of DIA2, HTZ1, SGS1, and TSA1 behaved in this manner. This relative growth defect of certain derived haploids compromises the quality of the pool for these mutants. Interestingly, most of these mutants actually behaved fine in our en masse created haploid-convertible pool (8). However, an en masse created haploid-convertible pool has its own shortcomings in that mutants exhibiting haplo-insufficiency for growth (mainly ribosome protein gene mutants) will likely be underrepresented after integrating the can1Δ::LEU2-MFA1pr-HIS3 reporter and selection for Leu+ transformants. With these considerations, we have recently started mixing aliquots of the haploid-convertible heterozygote YKO pools made by the two different methods to make pools for dSLAM screens, an approach that has worked very well.
Table 3.
A recipe for the haploid-selection “magic medium.”
| Ingredient | Quantity |
|---|---|
| Dextrose | 20 g |
| Difco yeast nitrogen base without amino acids and ammonium sulfate | 1.7 g |
| SC-Leu-His-Arg drop out mix | 2 g |
| Sodium (or potassium) glutamate | 1 (or 1.2) g |
| G418 | 0.2 g |
| L-Canavanine | 60 mg |
| dH2O | to 500 ml |
Note: This recipe makes 500 ml of 2 x magic medium liquid, which is subsequently filter-sterilized and mixed with an equal volume of 4% agar to make enough medium for ten 150 x 25 mm Petri dishes with 100 ml per plate. G418 is purchased from GIBCO (Cat # 11811-031) and L-canavanine from Sigma (Cat # C1625). An SC drop out mix contains 6 grams of each amino acid except for L-leucine (12 g). It also contains uracil (6 g) and adenine hemisulfate (1.5 g). Any combination of these components can be “dropped out”; thus SC-Leu-His-Arg refers to SC drop out mix that lacks L-leucine, L-histidine, and L-arginine. To make this drop out mix, each component is weighed, with clumps broken up with a mortar and pestle, and transferred into a large bottle. This bottle is shaken vigorously after each component is added. One can also buy from a “Hopkins mix” QBiogene, which is prepared according to this formula.
2.1.2 Making a query construct for integrative transformation
For each dSLAM screen, a query construct intended for high efficiency gene disruption is needed. We typically use a yfgΔ::URA3 or a yfgΔ::natMX cassette as the query construct (yfgΔ stands for disruption of your favorite gene). First, a URA3 cassette (e.g. plasmid pSO142-4 cut with SalI and EcoRI, Figure 1C) or a natMX cassette (e.g. pAG25 cut with NotI (11)) is transformed into the existing haploid or diploid YKO mutant (kanMX cassette) of the corresponding gene intended as the query. In all three cassettes, the selectable markers are flanked by a common set of promoter and terminator sequences (pTEF/tTEF) that facilitate swapping the kanMX with either URA3 or natMX via homologous recombination. Ura+ G418s or CloNatR G418s colonies are selected as the query construct template strains. In fact, we have made a set of genome-wide haploid-convertible heterozygote diploid YKOs in which URA3 replaces kanMX as the selectable marker as described above and have converted those diploids of the non-essential genes into haploids (Boeke et al, unpublished data). However for those genes that lack an existing YKO mutant, query construct template strains can be constructed via PCR-mediated gene disruption (12, 13). Genomic DNA from a haploid yfgΔ::URA3 strain is isolated (see below) and used as template for PCR amplification of the query construct using conditions described in Table 1. The PCR primers should be designed so that the PCR product contains ~1.5 kb sequences flanking the target gene, with the long homology necessary for high efficiency integrative transformation of the pool (9). This design should also consider excluding known autonomous replication element sequences (ARSs), which allows for low-frequency ectopic propagation of circularized DNA, presumably following non-homologous end joining, from the query construct.
Table 1.
PCR conditions for a dSLAM query construct.
| Ingredient | Volume |
|---|---|
| 10 x ExTaq (or LA Taq) Buffer | 10 μl |
| dNTP (2.5 μM each) | 8 μl |
| Forward primer (20 μM) | 5 μl |
| Reverse primer (20 μM) | 5 μl |
| Template DNA (200 ng/μl) | 2 μl |
| ExTaq or LA Taq DNA polymerase (5 units/μl) | 0.5 μl |
| dH2O | 69.5 μl |
| Total | 100 μl |
| 94°C 5′; 35 x (94°C 20″, 55°C 20″, 72°C 3′30″); 72°C 5′; 4°C hold | |
Note: The PCR annealing temperature should be adjusted according to the melting temperatures (Tm) of the primers. We typically use primers with Tm values ranging from 52°C to 60°C. 5 μl of DMSO should be added to the PCR mixture if the template, for example the natMX cassette, is G/C-rich.
To get sufficient DNA for high yield transformation, for each query construct we typically do 3 x 100 μl PCR reactions, which are pooled and ethanol-precipitated with 0.025 volumes of 4 M ammonium acetate (pH7.0) and 2.5 volumes of 100% ethanol. The DNA pellet is washed once with 300 μl of 70% ethanol, dried in a speed vacuum (Thermo Electron Corporation, Model: DNA 120 Speedvac system), and dissolved in 28 μl of distilled water or 1 x TE. ExTaq (Takara, RR001) or LA Taq (Takara, RR002) DNA polymerase is critical to the success of this long range PCR in our hands; the latter gives a better yield of PCR products but is more expensive. We have also noticed dramatically different performance of different PCR machines in amplifying both the query constructs and fluorescent dye-labeled TAGs for microarray hybridization (see below). For example, we have noted that when 96 large volume (100 μl/reaction) PCR reactions are run simultaneously, amplifications can fail in certain Peltier instruments, whereas we see very reproducible success with Perkin Elmer 9600 PCR machines. It is thus important to find a good PCR machine and stick with it.
2.1.2 Transformation of pool aliquots
For each transformation, 6.25 OD600nm (1 OD600nm = ~ 2 x 107 yeast cells) of the haploid-convertible heterozygous diploid YKO pool is inoculated into 50 ml YPD liquid (starting at 0.125 OD600nm/ml in a 250 ml Erlenmeyer flask) and shaken at 200 rpm at 30°C for 5.5 hours to a cell density of ~0.5 OD600nm/ml. The culture is harvested by spinning at 5000 rpm in a Sorvall RT6000B centrifuge for 2 min at 4°C or room temperature, washed once in 10 ml of water and subsequently in 10 ml of 0.1 M lithium acetate (LiOAc), and finally resuspended in residual 0.1 M LiOAc in a total volume of 100 μl in a 1.5 ml Eppendorf tube. A transformation mixture is freshly made according to Table 2 and added to the 100 μl of yeast competent cells. The transformation reaction is immediately mixed well by pipetting with a P1000 pipetter followed by vortexing (VMR, Vortexer 2) at top speed for 5–10 seconds, then incubated at 30°C for 30 minutes. 72 μl of dimethyl sulfoxide (DMSO; Qbiogene DMSO0001, Molecular Biology Grade) is subsequently added to the transformation reaction and immediately mixed thoroughly by vortexing at top speed for 5–10 seconds. DMSO is intrinsically sterile and no further sterilization is needed. It is also fairly stable when stored at room temperature and can be repeatedly opened and used for years. The mixture is then incubated in a 42°C water bath for 13 minutes. After the heat-shock, cells are spun down at 3600 rpm in an Eppendorf centrifuge (Model 5417 C) for 30 seconds and after aspirating the supernatant, the cells are resuspended and incubated in 400 μl of 5 mM CaCl2 for 5–15 minutes at room temperature. Incubation of cells in 5 mM CaCl2 for a short period of time improves transformation efficiency by 2 to 3-fold; however, prolonged incubation (>30 minutes) of cells in CaCl2 will reduce transformation efficiency. For transformation with a yfgΔ::URA3 cassette, 1/2000 of the transformation is spread on a regular (100 x 10 mm) SC-Ura plate for calculating transformation yields. The remaining cells from the transformation are plated on a single 150 x 25 mm Petri dish containing SC-Ura for URA3 selection. ~40 autoclaved glass beads (Fisher Scientific, Cat # 11-312A, Diameter 3 mm) are used to evenly spread the cells; these should be left inside the plate, which is inverted and incubated at 30°C for 2 days. The spreading procedure should be gentle in the beginning to avoid splashing of cell suspension. A good transformation will give rise to 5–20 x 105 transformants, which form a confluent lawn of cells.
Table 2.
A recipe for the high efficiency yeast transformation mixture.
| Ingredient | Volume |
|---|---|
| 50% polyethylene glycol (PEG-3350, Sigma) | 480 μl |
| 1 M LiOAc | 72 μl |
| Sheared, heat-denatured herring sperm DNA (10 mg/ml) | 40 μl |
| Query construct DNA (~0.5–1 μg/μl) | 28 μl |
| Total | 620 μl |
Note: To prepare the herring sperm DNA stock solution, 1 gram of dry sample (Sigma, D4489) is first dissolved in 100 ml of 1 x TE buffer by stirring overnight in a cold room. The DNA is then sheared by sonication (3 x 10 seconds with 1 minute interval sitting in ice), aliquoted (0.5–1 ml/aliquot), and stored at −20°C. For best transformation efficiency, an aliquot of herring sperm DNA should not be repeatedly heat-denatured and frozen; always use herring sperm DNA from a new tube.
For transformation with an yfgΔ::natMX cassette, the 400 μl CaCl2 suspended cells should be transferred to a 250 ml Erlenmeyer flask containing 50 ml fresh YPD and shaken at 30°C for 2–3 hours to allow expression of the CloNat-resistance gene. Cells are then spun down and resuspended in residual YPD in a total volume of 400 μl and plated on a single 150 x 25 mm plate containing YPD plus 50 μg/ml CloNat. This plate is incubated at 30°C for 2 days and replica-plated to another YPD plus CloNat plate to reduce the background of untransformed cells. The new plate is incubated at 30°C for another day. Transformation yield can also be monitored by plating a small aliquot on a regular Petri dish containing CloNat as outlined above.
If multiple dSLAM screens are performed simultaneously, one should grow a single larger culture of the haploid-convertible heterozygote pool (e.g. 500 ml in a 2-liter flask for 10 screens) to prepare competent cells. A scaled-up master transformation mixture (omitting query construct DNA) for multiple transformations is also preferred.
Each transformation should aim for >5 x 105 independent stable transformants to prevent random drop out of YKO mutants from the pool. The yield of a transformation is determined by a number of factors but is largely proportional to the amount of query DNA put into the transformation reaction. The number (~5 x 108 or ~25 OD600nm cells) and the growth stage (after 2 doublings) of the competent cells also greatly affect transformation yield. In addition, bigger ORFs are typically more difficult to disrupt than smaller ORFs (9). The exact reagents and experimental procedures used can also affect transformant yield but we recommend using those described here.
One potential problem for dSLAM screens are the autonomous replication sequences (ARSs) harbored by a small number of query constructs. Some of these ARSs are well characterized and easy to avoid but others are not so clear. An ARS allows circularized query construct DNA to propagate ectopically without integrating and disrupting the target gene after transformation into yeast cells. A dSLAM screen with such a query construct can generate useless or even misleading data. The potential ARS activity of a yfgΔ::URA3 query construct can and should be monitored experimentally. Briefly, a few hundred independent Ura+ transformants on a regular Petri dish (100 x 10 mm) are sequentially replica-plated onto a second SC-Ura plate (to minimize untransformed Ura− background cells), a YPD plate (allowing mitotic loss of the URA3 marker as would frequently occur with unstable ARS plasmids), and a plate of synthetic medium contain 0.1% of 5-FOA (5-Fluoroorotic Acid; for selecting cells without the URA3 marker). For a query construct with strong ARS activity, the ectopically propagating URA3-containing DNA is rapidly lost and a significant portion of the original Ura+ colonies will become resistant to 5-FOA. When this occurs the investigator may want to consider redesigning the query construct, depending on the degree of the problem and whether the extrachromosomal elements persist through meiosis.
2.1.3 Sporulation and haploid selection
The transformants (or heterozygous diploid double knockouts) are harvested by adding 5 ml of sterile H2O to the cells and swirling gently. The motion of the glass beads helps to dislodge and resuspend the cells. Cell suspensions are transferred to 15 ml conical tubes and another 5 ml of sterilized water is used to rinse residual cells off the plate. The two cell suspensions are combined, giving rise to a transformed pool of ~8–9 ml cells at a typical density of ~80 OD600nm/ml. Aliquots are saved for future use by freezing in 15% glycerol at −80°C. In the mean time, ~25 OD600nm of the pool can be immediately inoculated into 50 ml fresh YPD liquid and shaken in a 250 ml Erlenmeyer flask at 30°C for 3 hours. Cells are harvested by centrifugation, washed with 25 ml sterile water, and incubated in 50 ml of liquid sporulation medium (1% w/v potassium acetate, 0.005% w/v zinc acetate, 0.3 mM histidine) at 25–30°C with shaking at 200 rpm for 5 days. Incubation at >30°C dramatically decreases sporulation efficiency, which can be monitored with a normal phase contrast microscope. A ≥25% sporulation efficiency is considered sufficient.
Sporulation cultures are harvested by centrifugation and washed once in 20 ml of sterilized water. For each type of haploid selection medium (see below), 2–4 OD600nm of sporulated cells are evenly spread on one 150 X 25 mm plate using glass beads and incubated at 30°C for 2 days. Again, the glass beads used for spreading are left inside the plates for harvesting freshly converted haploid cells later. The rest of the sporulation culture can be stored in 15% glycerol at −80°C for future use.
For simplicity, we will discuss below only the medium used in a dSLAM screen using a URA3 query construct. In such a screen, a basic synthetic haploid selection “magic medium” (MM; SC-Leu-His-Arg +Canavanine +G418; Table 3) is used to select a control pool of haploid single and double mutant MATa YKOs whereas MM-Ura medium (the same as MM except lacking uracil) is used to select the experimental pool of haploid double mutants. Importantly, both pools derive from a common sporulated culture by exploiting the SGA reporter. We note that as the control, we have previously used an MM +5-FOA (0.1%) medium, which selects for G418R Ura− single mutants only, rather than MM, on which both single and double mutants can grow (8). These two different controls perform equally well in identifying strong synthetic interactions but the MM + 5-FOA control in general gives a slightly higher yield of weak synthetic interactions. However, we have also found that some YKO single mutants are intrinsically sensitive to 5-FOA and thus have discontinued using 5-FOA containing control medium. However, it is better to use both types of controls if multiple replicates of the same dSLAM screen are planned. We have previously shown that a dSLAM experiment can be exploited to identify YKOs whose growth is improved by the query mutation (genetic suppression) by looking for TAGs with low C/E ratios (8). This works best when using MM + 5-FOA to select for the single mutant pool as the control.
2.1.4. Genomic DNA isolation
Cells grown on the haploid selection medium are harvested as described above. A normal yield is ~600 OD600nm cells per freshly generated haploid pool. From this we use an aliquot of 25 OD600nm cells for isolating genomic DNA and freeze down (at −20°C or −80°C) another aliquot as a backup sample. The following is a protocol for isolating high quality genomic DNA as the template for PCR amplification of both the query constructs described above and the fluorescent-labeled TAGs described below.
Step 1. Eppendorf tubes each containing 25 OD600nm cells are spun at 3600 rpm for 30 seconds and the liquids are discarded. 200 μl of glass beads (Biospec products, Inc., Cat # 11079105, ~0.5 mm in diameter), 200 μl of Winston-Hoffman (14) lysis buffer (2% Triton X-100, 1% SDS, 100 mM NaCl, 10 mM Tris.HCl pH 8.0, 1 mM EDTA pH 8.0) (14), and 200 μl of phenol/chloroform/isoamyl alcohol (PCI, 25/24/1; Sigma, P2069) are added to each tube.
Step 2. These tubes are spun at 5000 rpm for 30 seconds. The cells will collect between the organic and aqueous phases.
Step 3. The tubes are then vortexed with an Eppendorf tube shaker (4132 or similar model) at top speed for 10 minutes at room temperature to lyse the yeast cells.
Step 4. The glass beads and liquid are briefly spun off the wall and cap of each tube in a microcentrifuge.
Step 5. 450 μl of 1x TE is then added into each tube and mixed well with the cell extracts by inverting.
Step 6. The eppendorf tubes are spun at >12,000 rpm in a microcentrifuge for 3 minutes to separate the DNA-containing aqueous phase from the cell debris, beads, and the organic phase.
Step 7. For each sample, 300 μl of PCI is added to a new tube, into which as much as possible of the DNA-containing aqueous phase (~650 μl) generated from the previous step is also transferred.
Step 8. The aqueous and organic phases are mixed by vortexing the tube with the Eppendorf tube shaker for 5 minutes and subsequently separated by centrifugation at >12,000 rpm for 3 minutes.
Step 9. ~550 μl of aqueous liquid is carefully transferred to a new tube containing 300 μl chloroform followed by a repeat of step 8.
Step 10. ~ 500 of the aqueous phase is carefully transferred to a new Eppendorf tube containing 10 μl of RNase A (Sigma, R5500; 2 mg/ml) and incubated at 37°C for 30 minutes to eliminate RNA molecules. Afterwards, 300 μl of PCI is added and step 8 is repeated.
Step 11. ~400 μl of aqueous liquid is carefully transferred to a new tube that contains 300 μl chloroform. This again is followed by a repeat of step 8.
Step 12. 300 μl of the DNA-containing aqueous liquid is carefully transferred to a new tube. 750 μl of 100% ethanol and 7.5 μl of 4 M ammonium acetate (pH 7.0) are subsequently added and mixed well. Genomic DNA is precipitated by spinning the tube at >12,000 rpm for 7 minutes. At this point, one should see a tiny whitish DNA pellet at the bottom of each tube.
Step 13. The DNA pellet is washed once with 300 μl of 70% ethanol, dried in a Speed-Vac for 10–20 minutes (medium heat), and dissolved in 100 μl of 1x TE, yielding a concentration of about 200–400 ng/μl.
For genomic DNA preparation from yeast cells, we have also used a Master PureTM yeast DNA purification kit (Epicenter, MPY80200). This works faster and is more amenable to processing many samples simultaneously. However, genomic DNA samples so isolated usually contain a large amount of RNA that can interfere with PCR amplification of the TAGs. Therefore, Master Pure™ DNA samples must be cleared of RNA molecules by incubation with RNaseA and subsequent extraction with PCI and chloroform as described above (steps 10–13) or cleaned with a Qiagen DNA purification column.
2.1.5. TAG-array PCR
For TAG-array hybridization, we PCR-amplify the TAGs from the genomic DNA isolated as described above by using universal primers U1 and U2 or D1 and D2, one of which is labeled with fluorescent cyanine dyes (Cy5 and Cy3) (Figure 1A). While we do not necessarily speak for all yeast researchers, our experience suggests that the potential of TAG-array analysis of the YKO mutants has been greatly limited by low hybridization signal intensity. This is especially true for samples labeled with Cy5, which is prone to oxidative degradation by ozone in the air (15). We believe that this poor performance mainly results from low hybridization of the dye-labeled TAG sequence to the array. Existing protocols for TAG-array PCR (3, 4, 16–18) typically yield labeled products that are predominantly double-stranded. The two strands must be separated by denaturation before hybridization of the dye-labeled strand to the microarray can occur. However, TAG-array hybridization is mediated by TAG-specific 20 mer oligonucleotide sequences that lie in the middle of the TAGs, whereas the PCR products are 56 bp double-stranded DNA molecules, which tend to re-anneal after denaturation and decrease TAG-array hybridization intensity.
To overcome this problem, we use an asymmetric PCR protocol to generate Cy5- and Cy3-labeled products that are predominantly single-stranded (19) (Table 4). This is achieved by increasing the concentration of labeled primer by 10-fold in the PCR reactions (Table 5) and increasing the number of PCR cycles from the typical 30 to 50 (Table 4). With this protocol, the unlabeled primer is consumed within 26 cycles, after which the dye-labeled PCR product is preferentially amplified in a linear fashion (19). When checked by native agarose gel electrophoresis (2%), the PCR products should contain a band that migrates faster than the usual double-stranded product and co-migrates with a single-stranded 56 mer. These bands can be visualized either on a Typhoon phosphorimager set to directly image the appropriate Cy-dye or by ethidium bromide staining. Estimation of relative PCR yields by ethidium bromide staining should take into account the fact that single-stranded DNA is stained about 90% less efficiently than the same molar quantity of double-stranded DNA and that Cy5 might quench the orange fluorescence of ethidium bromide
Table 4.
A recipe for TAG-array PCR.
| Ingredient | Quantity |
|---|---|
| 2 x Extaq premix (Takara, RR003) | 30 μl |
| Primer mixture | 3 μl |
| Genomic DNA template (~200 ng/μl) | 1 μl |
| MilliQ water | 26 μl |
| Total | 60 μl |
| 94°C 5′; 50 x (94°C 10″; 50°C 10″; 72°C 20″); hold at 4°C | |
Note: If multiple samples are to be analyzed simultaneously, one should make a master mixture excluding the template genomic DNA for each of the four types of PCR reactions: Uptag Cy5, Uptag Cy3, Downtag Cy5, and Downtag Cy3. Aliquots of the master mixture are then added to PCR tubes containing the individual template DNA samples, from which the TAGs are amplified.
Table 5.
Primer mixtures for TAG-array PCR.
| Type of primer mixture | Template to be labeled | Dye-labeled primer (50 μM) | Unlabeled primer (5 μM) |
|---|---|---|---|
| Uptag Cy5 | Control | 5’(Cy5)GTCGACCTGCAGCGTACG3’ | 5’GATGTCCACGAGGTCTCT3’ |
| Uptag Cy3 | Experiment | 5’(Cy3)GTCGACCTGCAGCGTACG3’ | 5’GATGTCCACGAGGTCTCT3’ |
| Downtag Cy5 | Control | 5’(Cy5)CGAGCTCGAATTCATCGAT3’ | 5’CGGTGTCGGTCTCGTAG3’ |
| Downtag Cy3 | Experiment | 5’(Cy3)CGAGCTCGAATTCATCGAT3’ | 5’CGGTGTCGGTCTCGTAG3’ |
Because we use a two-color (Cy5 and Cy3) hybridization format and there are two TAGs (Uptag and Downtag) for each YKO (Figure 1A), a total of four different PCR reactions (Uptag Cy5, Uptag Cy3, Downtag Cy5, and Downtag Cy3) are needed for each TAG-array experiment (Table 5). These reactions differ by the primers and template DNA samples used; also each PCR contains a Cy-labeled primer and an unlabeled primer, ensuring that the labeled product will be complementary to the sequences on the array. Uptags and Downtags are amplified separately because different universal primers are used (Table 5). We typically label TAGs from the control pool with Cy5 and those from the experimental pool with Cy3 (Table 5) so that a high Cy5/Cy3 ratio (red spot in scanned microarray image) indicates a synthetic lethality or fitness defect interaction. Primer stock solutions are prepared by first dissolving lyophilized DNA from the manufacturer in 10 mM Tris (pH 6.8) buffer at a concentration of 100 μM (for dye-labeled oligonucleotides) or 10 μM (for unlabeled oligonucleotides). Equal volumes of these two are then mixed to make a 10 x primer mixture for an asymmetric PCR. This can be stored at −20°C or used right away. The final concentrations of the primers when so mixed become 50 μM (labeled) and 5 μM (un-labeled) (Table 5).
In our hands, TAG-array PCR has been the most technically challenging step of the whole dSLAM technology. Genomic DNA templates of poor quality used in the PCR reactions can affect amplification of the TAGs and give rise to low hybridization signals. However, this is normally not a concern if the above-described genomic DNA isolation procedure is followed closely. We have previously also emphasized the importance of carefulness in avoiding PCR contamination (19) and we still think this is very important. However, the biggest problem for TAG-array analysis has been false positive results that seem to be PCR primer batch-dependent. The good news is that these are usually consistent within a primer batch and thus can be easily identified and systematically filtered out. Normally for each batch of primers, we do a control TAG-array experiment, in which the same genomic DNA (typically from a pool of freshly converted haploid single mutant pool) is used as the template in all four PCR reactions – a so-called self-self hybridization. Any TAG with a high Cy5/Cy3 signal intensity ratio in this control experiment will likely give rise to a false positive result in a real dSLAM experiment. Normally such false positives affect either the Uptag or the Downtag of a YKO but not both. Thus the affected YKOs can still be studied even if data for one of the two TAGs must be systematically filtered out.
To identify potential sources of the contamination in the primer stock solutions, we enlisted a non-yeast laboratory to prepare the stock solutions for us, using their own supplies. When false positives persisted, we made arrangements with the oligonucleotide manufacturer (Qiagen/Operon) to forego the default HPLC purification of the labeled primers, postulating that traces of previous orders were lodged in valves or other parts of the HPLC instrument and leached into the new order. This greatly reduced the false positives without a noticeable decrease in data quality. Now we routinely purchase both dye-labeled and unlabeled primers in desalted form, resulting in substantial cost savings, both in purification fees and in the four-fold higher yield.
2.1.6. TAG-Array hybridization
The TAG-array PCR products need to be hybridized to a high density TAG-array containing single-stranded oligonucleotides that are complementary to the fluorescent dye-labeled TAGs. Both Rosetta arrays (9, 17) and Affymetrix Tag3 arrays (4) have been used for TAG hybridization, although the former are not commercially available. However, for the majority of our dSLAM experiments, we have used a custom-designed “Hopkins TAG Array” (19) that is produced by Agilent Technologies (AMADID 011443) and available through a consortium purchasing arrangement (see http://barcode.princeton.edu). We disclose that no one affiliated with our research group receives any financial compensation for purchases of these arrays. The following is a hybridization protocol we use with the Hopkins TAG-array.
2.1.6.1. Masking the common priming sites on dye-labeled TAGs
All Uptags and all Downtags are flanked by a pair of common priming sites necessary for their PCR amplification as a population (Figure 1A). However, these common priming sites will promote the formation of double-stranded duplexes and may also cross-hybridize to microarray features with related sequences. They are normally masked by pre-incubation with an ~48-fold excess of complementary oligonucleotides (“blocking oligonucleotides”) before hybridization to a TAG-array (Table 6). These blocking oligonucleotides are purchased from Qiagen/Operon in desalted form and are dissolved at 1 mM in a 10 mM Tris buffer (pH 6.8). Equal volumes of the two Uptag blocking oligonucleotides are then mixed (500 μM final concentration of each; Table 6) and used right away or stored at −20°C for future use. Likewise, the two Downtag blocking oligos are mixed. 12 μl of blocking oligonucleotide mixture is combined with 50 μl of each of the corresponding Cy labeled PCR products and mixed in a new 1.5 ml microfuge tube, heated at 100°C for 1 minute, and incubated on ice for at least 2 minutes before addition to the hybridization buffer (see below). From this point on in the procedure, the Uptag and Downtag samples can be treated in separate tubes or pooled.
Table 6.
A description of oligonucleotides used in TAG-array hybridization.
| Name of oligonucleotide | oligonucleotide DNA sequence | stock concentration |
|---|---|---|
| Uptag blocking oligo 1 | 5’GATGTCCACGAGGTCTCT3’ | 1 mM |
| Uptag blocking oligo 2 | 5’CGTACGCTGCAGGTCGAC3’ | 1 mM |
| Downtag blocking oligo 1 | 5’CGGTGTCGGTCTCGTAG3’ | 1 mM |
| Downtag blocking oligo 2 | 5’ATCGATGAATTCGAGCTCG3’ | 1 mM |
2.1.6.2. Hybridization
The pre-blocked Uptag and Downtag samples described above are hybridized to the same “Hopkins TAG Array” (19). They are mixed well with the hybridization buffer (7.5 ml of 1 x SSTE with freshly added Dithiothreitol (DTT, 1 mM)) and then added to a hybridization chamber that holds a TAG Array. Here DTT is used in an effort to curb the destruction of Cy5 fluorescence by atmospheric ozone (see above). We have previously used the old-fashioned “Seal-A-Meal” plastic hybridization bags (4cm x 12cm; Kapak Corporation) for TAG-array hybridization. Now we have found that six-compartment polypropylene boxes with lids (Alpha Rho Inc., www.alpharho.com, Cat # 776C-6-P) are extremely economical and easy to use, especially for experiments involving more than six hybridizations at a time. When using these boxes, care should be taken to ensure that the microarray faces up before adding the hybridization mixture. After all hybridizations are set up in a box, the lid is closed and the box is then carefully wrapped with aluminum foil to shield light and retard evaporation. TAG-array hybridization is carried out overnight at 42°C in a hybridization oven (Robbins Scientific MODEL 400) with gentle rocking (8 rpm).
2.1.6.3. TAG-array scanning
A hybridized TAG-array is carefully removed from the hybridization chamber by grabbing one end with Teflon coated forceps. The array is slowly dipped into and pulled out of a 50-ml conical tube containing 50 ml of 6 x SSPE with 0.05% Triton X-100 and 1 mM DDT for 5 times and then similarly washed in 50 ml of 0.06 SSPE containing 1 mM DDT (also in a 50-ml conical tube). After the final dip into the 0.06 x SSPE buffer, the slide is very slowly pulled out of the washing solution to avoid residual liquid stuck on the slide. In general, we process one microarray at a time, taking care that the slide does not dry until after the final wash. All washes are carried out at room temperature. When washing is complete, the wash solution should sheet cleanly off the slide to leave a dry surface.
Each slide is scanned with a Genepix 4000B instrument (or other scanners) immediately after washing to minimize exposure of the dyes, especially Cy5, to atmospheric ozone. Generally speaking, laser power settings and photomultiplier tube (detector) voltages should be set so that signal intensities are maximized with no more than a few saturated features. With the Genepix 400B, a good starting point is 33% laser power and 600 volts for the photomultiplier tube (PMT) in each channel. High PMT voltages (>800 volts) should be avoided because they will increase background noise and lower the dynamic range. Scanner resolution should be 10 microns per pixel; there is no advantage to scanning at higher resolution, which is slower and generates unnecessarily large files. For each scan, we recommend saving the result as a multi-color image file (‘.tif’). For the sake of future data analysis, each image file should be systematically named, documented in a database-compatible format, and archived in at least two secure locations.
2.1.7. Data acquisition
2.1.7.1. Image analysis
We use the GenePix Pro software package (formerly version 4.0 for our published results, now version 6.0) that is integrated with the scanner to generate data files (‘.gpr’) from the raw data image files (‘.tif’) for statistical analysis. Other software programs may be equally suitable. Other files that might be helpful for later review or editing are the settings file used to analyze the image (‘.gps’) and the ancillary image files used to view the data (‘.jp1’, ‘.jp2’, etc).
2.1.7.2. Data analysis
All information about Hopkins TAG array design is available at the GEO database at NCBI (‘http://www.ncbi.nlm.nih.gov/projects/geo/query/acc.cgi’) under accession number GPL1444. For convenience, we have also constructed a ‘.gal’ (“GenePix Annotation List”) file that annotates everything except the sequence information inherent to the microarray features; this makes image segmentation virtually automatic except for minor positioning. In particular, the “Index” column of the ‘.gal’ file provides quick access to the different components of the array (comprehensive list of tags, replicate controls, hybridization controls, etc) in one sorting operation. This file is available in the ‘etc’ subdirectory of an installed hoptagInfo R package (‘http://www.r-project.org’).
Simple analyses can be carried out with a spreadsheet program such as Microsoft Excel merely by using the pre-calculated ‘F532 Median - B532’ and ‘F635 Median - B635’ values. (Medians provide better statistical robustness to artifacts such as dust, which can be difficult to avoid experimentally even with meticulous technique.) For features with no expected signal, these values are often remarkably close to zero (< 10). This simple approach is often sufficient for experiments whose goal is hypothesis generation. More ambitious analyses can be performed with the aid of an object-oriented programming interface that we have customized for our arrays using the R statistical programming language (‘http://www.r-project.org’). The necessary R packages (hoptag and hoptagInfo) are freely available (19).
2.1.7.3. Generating a candidate hit list
A typical dSLAM experiment yields a list of YKO mutations ranked according to their TAG hybridization control-to-experiment (C/E) ratios. A high C/E ratio indicates a potential synthetic lethal or fitness defect interaction between the corresponding YKO and the query mutation. Not all high C/E ratios result from such genetic interactions. In addition to the primer batch-dependent false positives mentioned in a previous section, high C/E ratios are expected for YKOs that cause uracil biosynthesis deficiency because the mutant cannot grow on the medium for selecting haploid double mutants (Table 7). There are also a few YKOs that we call “frequent diers,” which appear to grow better in medium containing uracil (Table 7). A high C/E ratio may also be observed with YKOs located adjacent to the query gene in the genome because the corresponding TAGs can be deleted by the query construct during transformation (8). False negatives are also a concern because a substantial fraction of TAGs (roughly 35%) will have signal intensities near background levels. The reasons for this are both biological and technical. Many freshly converted haploid YKO mutants are lethal or grow poorly and therefore are severely underrepresented in the pool. In addition, direct sequencing of the TAGs and their associated primer sites have revealed many mutations that may hinder their PCR amplification or hybridization to the array (20). Here we describe a protocol we use to select from a dSLAM screen the candidates meriting followup confirmation.
Table 7.
A list of YKOs normally excluded from RSA confirmation.
| ORF Name | Gene Name | Comment on single mutant phenotype |
|---|---|---|
| YBL025W | RRN10 | Extreme slow growth |
| YBL040C | ERD2 | Hard to confirm due to low spore viability |
| YBL058W | SHP1 | Extreme slow growth |
| YBR112C | CYC8 | Low spore viability |
| YBR179C | FZO1 | Extreme slow growth |
| YCR084C | TUP1 | Low spore viability |
| YCR094W | CDC50 | Extreme slow growth |
| YDL151C | BUD30 | Extreme slow growth on synthetic medium |
| YDR138W | HPR1 | Extreme slow growth |
| YDR173C | ARG82 | Extreme slow growth on magic medium |
| YDR364C | CDC40 | Extreme slow growth |
| YER040W | GLN3 | Requires uracil for optimal growth |
| YER044C | ERG28 | Extreme slow growth |
| YER073W | ALD5 | |
| YFR009W | GCN20 | Extreme slow growth on magic medium |
| YGL038C | OCH1 | Extreme slow growth |
| YGL240W | DOC1 | hard to confirm due to low spore viability |
| YGR056W | RSC1 | Might need uracil for optimal growth |
| YIL036W | CST6 | Requires uracil for optimal growth |
| YJL071W | ARG2 | Lethal on magic medium |
| YJL130C | URA2 | Uracil biosynthesis |
| YJL148W | RPA34 | Extreme slow growth |
| YJL183W | MNN11 | Extreme slow growth |
| YJR063W | RPA12 | Extreme slow growth |
| YKL048C | ELM1 | Hard to confirm due to low spore viability |
| YKL216W | URA1 | Uracil biosynthesis |
| YKR026C | GCN3 | Extreme slow growth on magic medium |
| YLR014C | PPR1 | Uracil biosynthesis |
| YLR015W | BRE2 | Requires uracil for optimal growth |
| YLR027C | AAT2 | Requires uracil for optimal growth |
| YLR074C | BUD20 | Low spore viability |
| YLR204W | QRI5 | Requires uracil for optimal growth |
| YLR357W | RSC2 | Might need uracil for optimal growth |
| YLR410W | VIP1 | Extreme slow growth |
| YLR420W | URA4 | Uracil biosynthesis |
| YLR442C | SIR3 | Lethal on magic medium |
| YML043C | RRN11 | Extreme slow growth |
| YML064C | TEM1 | Extreme slow growth |
| YML065W | ORC1 | Extreme slow growth |
| YML106W | URA5 | Uracil biosynthesis |
| YML107C | Might affect URA5 expression | |
| YMR205C | PFK2 | |
| YNL084C | END3 | Extreme slow growth |
| YNL171C | Hard to confirm due to low spore viability | |
| YNL243W | SLA2 | |
| YNL248C | RPA49 | Extreme slow growth |
| YNL268W | LYP1 | |
| YNL297C | MON2 | Requires uracil for optimal growth |
| YOR295W | UAF30 | Extreme slow growth |
Step 1. All TAGs with signal intensity in the control Cy5 channel less than 400 are discarded regardless of the hybridization ratio, which tends to be unreliable.
Step 2. All TAGs with primer batch-dependent false positives are systematically filtered out. This usually only affects either the Uptag or Downtag of a small subset of YKOs.
Step 3. The YKOs listed in Table 7 also are filtered out. These include mutants defective in uracil biosynthesis, other mutants that for obscure reasons require uracil for optimal growth, and some mutants that grow extremely slowly. While these YKOs could be considered real candidates, their potential interactions with the query mutation must be confirmed by tetrad analysis.
Step 4. For YKOs with data for both TAGs, take those with a C/E ratio >=1.5 or close to 1.5 in both TAGs (concordant TAG behavior) for individual confirmation. For genes with data for both TAGs but where the behaviors of the two TAGs are dramatically different (e.g. one ratio >=2.0 and the other <=1.0), take into consideration that TAGs with low signal intensity (barely above 400 in the control channel) are less reliable and go with the TAG with higher signal intensity in the control channel. For YKOs with data for only one TAG, take those with a C/E ratio >=2.0 for individual confirmation. YKOs with only one TAG C/E ratio >=1.5 can also be listed as candidates at the user’s discretion (e.g. if the deleted genes are functionally related to the query gene).
2.1.8. Stripping and reusing TAG-arrays
A TAG-array can often (but not always) be stripped of the hybridized fluorescent molecules and re-used up to two times without substantial reduction in performance. For the best results, a slide should be stripped right after each hybridization is scanned or at least on the same day as scanning. Sometimes the ink of the commercial barcode on the slide comes off during stripping and this causes a very high green fluorescence background on the slide. We thus recommend peeling off the commercial barcode from the slide before stripping. The original serial number can be inscribed on the top of the slide with a diamond-pen for documentation; this inscription also serves as an indicator of the array orientation. Any visible dust (such as ink and glass debris from inscription) should be blown off the array with clean compressed air (such as a “Dust-off” canister). Stripping of the array is carried out by soaking in 50 ml 1% SDS in a 50 ml conical tube at 65°C for 2–3 hours. After that, the array is washed with 6 x SSPE and 0.06 x SSPE washing buffers as described above and scanned at low resolution with a GenePix 4000B scanner to check for persistent signal. Addition of 10 mM EDTA and temperatures up to 75°C may be helpful. A stripped array can be stored for several months at room temperature.
2. 2. dSLAM materials and reagents
Yeast strains and plasmids: Donor strains for query constructs yfgΔ::URA3 or yfgΔ::natMX; a pool of haploid-convertible heterozygous diploid YKOs or the original heterozygote diploid YKOs if making your own haploid-convertible pool; pXP346 (SpeI/PstI); pSO142-4 (SalI/EcoRI); pAG25 (NotI) (11).
Media: Liquid YPD; Liquid sporulation medium (1% w/v potassium acetate, 0.05% w/v zinc acetate, 0.3 mM L-histidine); YPD plus 200 μg/ml G418 in OmniTray plates (for growing the YKOs to construct the pool); YPD plus 50 μg/ml CloNat plate (100 ml/150 x 25 mm Petri dish, for transformation with a yfgΔ::natMX query construct); SC-Ura plates (100 ml/150 x 25 mm Petri dish and 25 ml/100 x 10 mm Petri dish, for transformation with a yfgΔ::URA3 query construct); SC-Leu plate (100 ml/150 x 25 mm Petri dish, for making your own haploid-convertible pool en masse); haploid selection magic medium plate (MM; 100 ml/150 x 25 mm Petri dish; Table 3) or MM plus 0.1% 5-FOA plate (100 ml/150 x 25 mm Petri dish); magic medium lacking uracil plate (100 ml/150 x 25 mm Petri dish).
Buffers, Solutions, and other reagents: Sterile dH2O; 1 X TE (10 mM Tris.HCl, pH8.0; 1 mM EDTA); DTT (1 M stock); LiOAc solutions (0.1 M and 1 M); herring sperm DNA (Sigma, D4889, 10 mg/ml); CaCl2 (5 mM); DMSO (Qbiogen, DMSO0001, Molecular Biology Grade); 1 X SSTE (1M NaCl; 10 mM Tris.HCl, pH7.5; 0.5% Triton X-100); 6 X SSPE (0.9M NaCl; 60 mM NaH2PO4; 6 mM EDTA; adjust to pH7.4) plus 0.5% Triton X-100; 0.06 X SSPE; phenol/chloroform/isoamyl alcohol (25/24/1; pH7.9); chloroform; ethanol (70% and 100%); 4 M ammonium acetate (pH7.0); RNase A (2 mg/ml); Master PureTM yeast DNA purification kit (Epicenter, MPY80200); 10 mM Tris.HCl (pH6.8); 2X ExTaq premix (Takara, R003; for TAG-array PCR); ExTaq (Takara) or LA Taq (Takara); primers for PCR amplification of the query constructs and TAGs (Table 5) and oligonucleotides for TAG-array hybridization (Table 6).
Equipment and supplies: PCR machine; centrifuges for pelleting cells; microcentrifuge; 30°C incubator; 42°C water bath; glass beads; Speed vacuum; hybridization oven with a rocker; hybridization boxes; aluminum foil; microarray scanner; TAG-array; forceps; diamond-pen; compressed air.
3. Confirmation of dSLAM hits
3.1 Confirmation procedure
In order to confirm the candidate hit list generated from a dSLAM screen, we typically make the corresponding individual heterozygote diploid double mutant YKOs by transforming into each haploid-convertible heterozygote YKO a query construct and follow it up with either random spore analysis (RSA) exploiting the SGA reporter, tetrad dissection, or both after meiosis. We consider tetrad dissection as the gold standard for confirming synthetic lethality interactions, though it is labor intensive. RSA confirmation is a reliable alternative and the procedure is much faster. Here we describe protocols for both transformation and RSA in 96-well format so that up to thousands of synthetic interactions can be individually tested within 2 weeks (Figure 3).
Figure 3. A flowchart for high throughput yeast transformation and random spore analysis of synthetic lethality interactions.
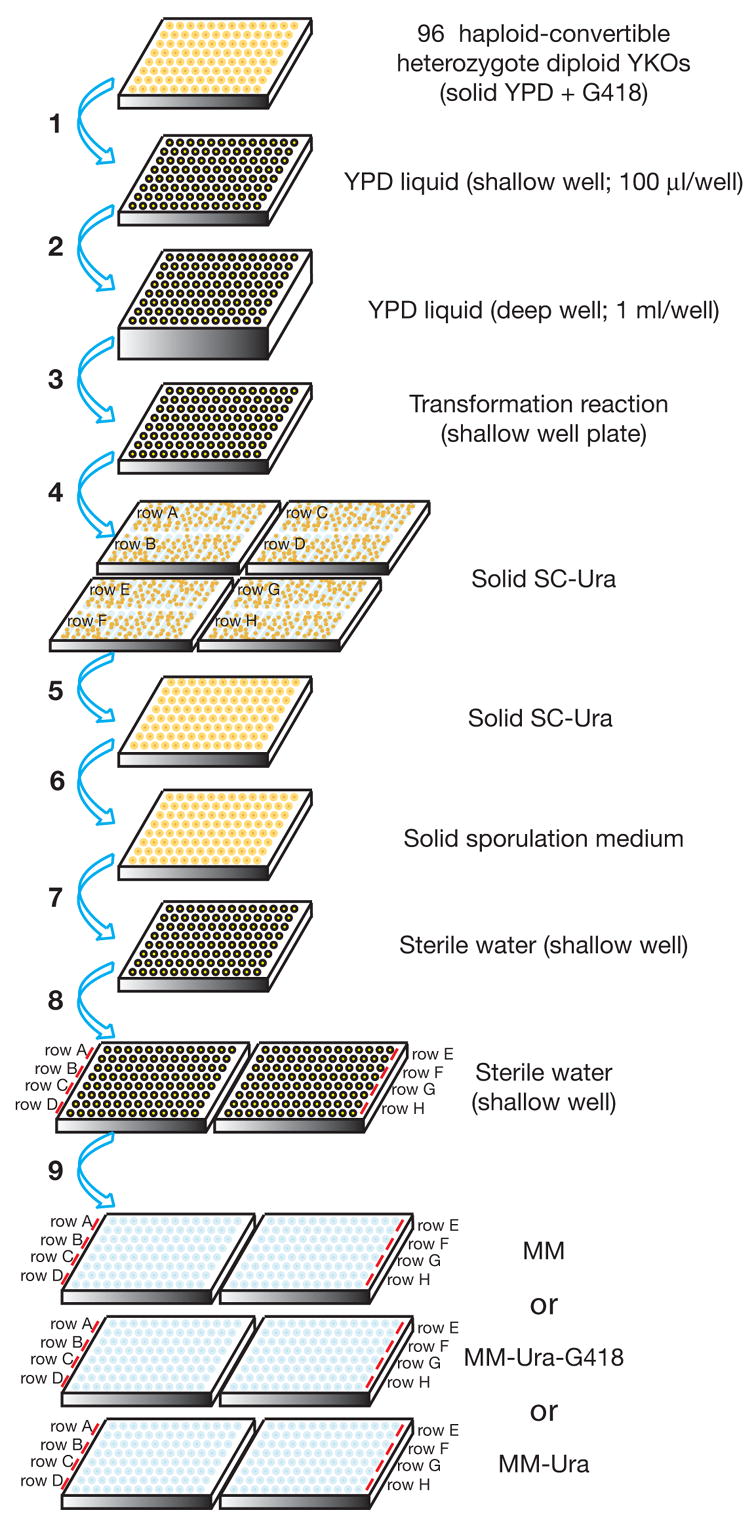
Step 1. Individual haploid-convertible heterozygote diploid YKOs are picked, inoculated onto OmniTrays containing solid YPD plus G418 (200 μg/ml) in a 96-well format, and incubated at 30°C for overnight. Step 2. These strains are inoculated into fresh liquid YPD (100 μl/well) in a shallow 96-well plate with a 96-pin replicator and incubated at 30°C overnight. Step 3. 25 μl of each of the overnight cultures is transferred to a deep 96-well plate containing fresh liquid YPD (1 ml/well). The plate is shaken at 200 rpm at 30°C for 4 hours. Step 3. Cells are pelleted, washed once in 0.1 M LiOAc (200 μl/well), and transferred to a shallow 96-well plate as a cell suspension in 0.1 M LiOAc (100 μl/well). Step 4. After incubation with the transformation mixture at 30°C for 30 minutes and subsequently at 42°C for 13 minutes, cells are pelleted, resuspended in 5 mM CaCl2 (30 μl/well) for 5–15 minutes, transferred row by row onto solid SC-Ura OmniTray plates, and incubated at 30°C for 2 days. Step 5. Two isolated colonies are picked for each strain and patched onto fresh SC-Ura plates back into the original 96-well format and incubated at 30°C overnight. Step 6. Cells are transferred to solid sporulation medium with a 96-pin replicator and incubated at room temperature (22 to 25°C) for 5 days. Step 7. The sporulated cultures are transferred to a shallow 96-well plate containing sterile water (100 μl/well) with a 96-pin replicator. Cells are resuspended in water by vigorous shaking. Step 8. Two sequential 10 x serial dilutions are made for each row of cell suspensions prepared at the previous step. Step 9. 4 μl of each cell suspension prepared at step 8 is spotted onto solid MM (for xxxΔ::kanMX single and xxxΔ::kanMX yfgΔ::URA3 double mutants), MM-Ura-G418 (for yfgΔ::URA3 single and xxxΔ::kanMX yfgΔ::URA3 double mutants), and MM-Ura (for xxxΔ::kanMX yfgΔ::URA3 double mutants) media and incubated at 30°C for 2–3 days.
Note: For simplicity, this diagram is an abbreviated version of the detailed procedure described in the text and the step numbers may not match those described in the text.
3.1.1 Constructing individual heterozygous diploid double mutants
Step 1. Based on a dSLAM candidate interaction list, individual haploid-convertible heterozygote diploid YKOs (Boeke et al, unpublished data) with corresponding gene deletions are picked, patched in a 96-well format on OmniTray plates containing solid YPD plus 200 μg/ml G418, and incubated at 30°C for overnight.
Step 2. These mutants are inoculated into shallow 96-well dishes that contain liquid YPD (100 μl/well) and incubated at 30°C overnight without agitation.
Step 3. The overnight cultures are resuspended by vigorous shaking on a Mini-Orbital shaker (Bellco Biotechnology) and transferred (25 μl/sample) with a multi-channel pipetter into deep 96-well dishes that contain fresh liquid YPD (1 ml/well) and glass beads (1 piece/well; 5 mm diameter). Each deep well dish is covered with a piece of Airpore filter (Qiagen; Cat # number 19571) after inoculation to prevent contamination during incubation. The cultures are shaken at 200 rpm at 30°C for 4 hours.
Step 4. Cells are harvested by spinning at 3600 rpm for 3 minutes. The Airpore filter is peered off each deep well dish and the medium damped. The deep-well dish is put upside down on a stack of autoclaved paper towels to drain the residual liquid.
Step 5. 200 μl of 0.1M LiOAc is added to each well and step 4 is repeated once. (The plates are left uncovered.)
Step 6. 100 μl of 0.1M LiOAc is added to each well. Cells are resuspended by vigorous shaking and transferred to a new shallow 96-well dish covered with a lid.
Step 7. Cells are pelleted as in step 4 and the LiOAc solution is removed from each well with a 12-channel pipetter (50–300 μl). Afterwards, 10 μl of 0.1M LiOAc is added to each well. (Note: If necessary, cells can sit in 0.1 M LiOAc at room temperature for several hours without obvious decline in transformation competency.)
Step 8. 500 μl herring sperm DNA (Sigma, D4889; 10 mg/ml) is heat-denatured by incubating at 100°C for 5 minutes and subsequently chilled on ice before use.
Step 9. For each 96-well plate, a transformation mixture is prepared in a 50 ml conical tube according to Table 8. A master mixture can also be made when transforming a query construct into multiple 96-well plates. This mixture is mixed well by votexing at top speed for 0.5–1 minute before being poured into a reservoir for 12-channel pipetting.
Table 8.
A master mixture for 96-strain transformation.
| Ingredient | Volume |
|---|---|
| 50% polyethylene glycol (PEG-3350, Sigma) | 6,240 μl |
| 1 M LiOAc | 822 μl |
| Heat-denatured herring sperm DNA (10 mg/ml) | 500 μl |
| Query construct (PCR product; 100 ng/μl) | 100 μl |
| DMSO | 936 μl |
| Total | 8,598 μl |
Step 10. Cells prepared in step 7 are resuspended by vigorous shaking on a mini-orbital shaker (Bellco Biotechnology), making it much easier to mix cells with transformation mixture in the next step.
Step 11. Immediately, 80 μl of transformation mixture is added to the competent cells using a manual 12-channel pipetter (50–300 μl) and mixed well immediately by pipetting 3 to 4 times. (Note: It is important that this mixing step is done row by row to avoid formation of cell clamps and reduction in transformation efficiency.) Cover each dish with a lid when transformations in all wells have been set up.
Step 12. The transformation reactions are incubated in a 30°C incubator for 30 minutes and subsequently in a 42°C water bath for 15 minutes. The dish bottom should make complete contact with water but the entire dish should not be submerged in water.
Step 13. Plates are spun at 3600 rpm for 3 minutes and the supernatants removed from each well with a multi-channel pipetter (50–300 μl).
Step 14. 30 μl of 5 mM CaCl2 is added to each well and cells are resuspended by vigorous shaking for 5 minutes on a mini-Orbital shaker (Bellco Biotechnology).
Step 15. Using a 12-channel pipetter, the contents of every two rows are transferred (row by row) to an OmniTray plate that contains solid SC-Ura (when using a yfgΔ::URA3 query construct). The OmniTray plate is tilted while pipetting to allow draining down of the cell suspension along the medium surface. Great care should be taken to avoid samples running into one another. Each of the transformation plates is labeled to reflect positions of the transformed strains (plate number and rows) in the original 96-well format. If selecting for drug-resistant markers, allow the cells to express the drug-resistance gene by growing in non-selective medium for 2 hours before transfer onto a drug-containing plate. For best results, the OmniTray plates with selection medium should be air-dried for 3–4 days to allow efficient drying of the cell suspension after draining.
Step 16. The transformation plates are incubated at 30°C for 2 days. Two independent transformants are picked and patched onto OmniTray plates containing the selection medium back in the original 96-well format. The plates are incubated overnight at 30°C. At least 95% of randomly picked transformants should be true double mutants.
3.1.2 Sporulation of double mutants
All 96 heterozygote diploid double mutants are transferred with a 96-pin replicator (BOEKEL, Model 140500) to an OmniTray containing solid sporulation medium (1% w/v potassium acetate, 0.05% w/v zinc acetate, 0.3 mM L-histidine, 2% agar) and incubated at room temperature (22–25°C) for 5 days.
3.1.3 Random spore analysis (RSA)
Cells from each sporulation plate are transferred, using a 96-pin replicator (BOEKEL, Model 140500), into a shallow 96-well dish containing sterile water (100 μl/well) and thoroughly resuspended by vigorous shaking. Two subsequent 10 x dilutions of the sporulated cells are carried out in 96-well dishes with sterile water. 4 μl of each dilution is spotted with a multi-channel pipetter onto OmniTray plates that contain solid MM, MM-Ura-G418, and MM-Ura media. The plates are dried, incubated at 30°C for 2–3 days, and the sizes of haploid colonies grown on the three different media are compared.
3.2. Reagents and materials for confirmation
Yeast strains: Donor strains for query constructs yfgΔ::URA3; a list of individual haploid-convertible heterozygous diploid YKOs.
Media: Liquid YPD; Solid YPD plus 200 μg/ml G418 (50 ml/OmniTray); SC-Ura plates (50 ml/OmniTray); Sporulation plates (50 ml/OmniTray; 1% potassium acetate, 0.05% zinc acetate, 0.3 mM L-histidine, 2% agar); haploid selection magic medium plate (MM; 50 ml/OmniTray); magic medium lacking uracil plate (MM-Ura; 50 ml/OmniTray); magic medium lacking both uracil and G418 (MM-Ura-G418; 50 ml/OmniTray).
Solutions and other reagents: Sterile dH2O; LiOAc (0.1 M and 1 M); Herring sperm DNA (Sigma, D4889; 10 mg/ml); CaCl2 (5 mM); DMSO (Qbiogen, DMSO0001, Molecular Biology Grade); ExTaq (Takara, RR001) or LA Taq (Takara, RR002).
Equipment: PCR machine; centrifuge with swinging buckets; 96-well plates (shallow and deep wells); 96-pin replicator (BOEKEL, Model 140500); mini-orbital shaker (Bellco Biotechnology); multi-channel pipetters; 30°C incubators with and without shaker; 42°C water bath.
4. Analysis of other types of genetic interactions
The dSLAM technology is quite versatile and can be modified easily to study other types of genetic interactions on a genome-wide scale. These include synthetic lethality interactions between a thermosensitive (Ts) allele of an essential gene and the genome-wide YKOs, gene-compound synthetic lethality (GCSL) interaction and drug-resistance, genetic suppression by a second mutation, dosage-dependent synthetic lethality and suppression, and synthetic haplo-insufficiency. Here we briefly survey the modifications needed for these purposes.
4.1. dSLAM with a Ts allele as the query
A typical dSLAM studies synthetic lethality between knockout mutations and needs to be modified in order to study similar genetic interactions involving essential genes. To systematically study the synthetic lethality interactions between an essential gene and genome-wide YKOs, we used a Ts allele of the essential gene as the query (8). First a yfgΔ::natMX cassette of the essential gene is transformed en masse into the haploid-convertible heterozygote diploids to make a double YKO pool using the protocol described in a previous section. Into this double mutant pool 5 μg of a URA3-marked, centromere-based plasmid that contains a Ts mutant allele of the same essential gene is then similarly transformed. The resultant plasmid-harboring double knockout pool is subsequently sporulated and haploid pools are selected on MM-Ura (to select for xxxΔ::kanMX single knockouts and xxxΔ::kanMX yfgΔ::natMX double knockouts as the control pool) and MM-Ura+CloNat (50 μg/ml; to select for xxxΔ::kanMX yfgΔ::natMX double knockouts as the experiment pool) at a temperature semi-permissive to the Ts mutant. The abundance of each YKO in these two pools is compared by microarray hybridization of the TAGs as described in previous sections. The haploid selection temperature is critical in such an experiment and should be optimized to give the best balance between growth of the double knockout cells and expression of the query mutant phenotype. We recommend selecting the haploid pools at multiple temperatures and comparing the relative growth rates of the corresponding single and double mutants at each temperature by TAG-array analysis.
In principle, this screen also provides an opportunity to screen for secondary mutations that suppress lethality caused by the Ts mutation at a restrictive temperature. For this purpose, haploid cells can be selected on MM-Ura+CloNat at the restrictive temperature. Cells grown under this condition might contain YKO mutations that allow the Ts mutant to survive and the identities of these YKOs can be easily revealed by TAG-array hybridization. However, some of these survivors might not be the results of secondary mutation suppression; instead, they may survive the restrictive temperature due to the presence of a wild-type copy of the essential gene on a chromosome mainly resulting from meiotic non-disjunction (8). Nevertheless, a list of candidate suppressors can be generated from this experiment and individually tested. In this case, multiple replicates of the experiment will likely be necessary in order to minimize false positive results caused by the random event of meiotic non-disjunction.
4.2. Gene-compound interaction analysis
dSLAM can be easily modified to study genome-wide YKOs for their sensitivity (or gene-compound synthetic lethality, GCSL) or resistance to a cytotoxic compound. For this purpose, the haploid-convertible heterozygote diploid pool is directly sporulated without transformation and haploid pools are freshly derived on MM (control pool) and MM plus the compound (experiment pool) at various concentrations. This allows identification of both compound-hypersensitive and -resistant haploid YKOs. Mutants killed by low concentrations of the compound are deemed hypersensitive and those surviving drug concentrations that kill a wild-type strain are considered resistant. By using this approach, we have found that mutants defective in microtubule biogenesis are hypersensitive to the microtubule depolymerization agent benomyl, mutants defective in DNA repair are hypersensitive to the DNA-damaging agent methyl methane sulfonate, and mutants affecting protein translation are resistant to arsenite (6, 8; Pan et al., in preparation).
When combined with a typical dSLAM screen, this also allows identification of pairs of mutations that modulate each other’s effects on the sensitivity of yeast cells to a compound. The procedure can be done essentially the same as a typical dSLAM screen except that both the haploid control and experimental pools are selected in the presence of the compound. With this approach, we have found that mutations in the DNA damage checkpoint pathway (rad17Δ, rad24Δ, and ddc1Δ) and those in the trans-lesion repair pathway (rev1Δ, rev3Δ, and rev7Δ) are synthetic hypersensitive to low concentrations of methyl methane sulfonate that are not inhibitory to single mutants of either pathway (Pan and Boeke, unpublished data).
4.3. Dosage-dependent synthetic lethality and suppression
We have previously shown that dSLAM could also be modified to study the genetic interaction between a gene overexpression and the genome-wide YKOs, including both dosage-dependent synthetic lethality and dosage suppression (8). Here a gene overexpression instead of mutation is used as the query. Briefly, a control plasmid (URA3) and a plasmid with the query gene expressed under control of a galactose inducible promoter (GAL1pr) are independently transformed en masse into the pool of haploid-convertible heterozygote diploids. From these, haploid pools containing the control plasmid and gene-overexpression plasmid are separately derived by growth on a magic medium lacking uracil, with galactose (2%) as the sole carbon source. The relative growth rates of each freshly generated YKO in these two populations are compared by TAG-array analysis. A dosage-dependent synthetic lethality interaction identifies a YKO whose growth is impaired when the query gene is overexpressed whereas a dosage suppression interaction identifies a mutant whose growth is improved by the query gene overexpression.
4.4. Synthetic haplo-insufficiency
Synthetic haplo-insufficiency is a less common genetic interaction is which two heterozygous mutations individually lack phenotypes but when combined produce phenotypes (21). This could be effectively exploited to study genetic interactions involving essential genes with the knockout mutations. For example, we have identified synthetic haplo-insufficiency interactions between the essential gene TUB1 and two nonessential genes TUB3 and RBL2, as well as with a number of essential genes required for the chromatin-remodeling activity RSC (8). For a synthetic haplo-insufficiency screen, both control (ura3Δ::URA3) and query (yfgΔ::URA3) constructs are independently transformed en masse into a pool of heterozygote diploid YKOs as described in a previous section. Because each transformation is started with 25 OD600nm cells, we typically plate each transformation onto at least 5 solid SC-Ura plates (100 ml/150 X 25 mm Petri dish) to increase the signal/noise ratio. The Ura+ transformants from each transformation are pooled with sterile water and studied by TAG-array analysis for relative growth rates of each heterozygote YKO in the two transformed pools.
5. Concluding remarks
Here we have given a detailed description of the dSLAM technology for studying genome-wide synthetic lethality interactions in yeast. This technology is robust and versatile and can be modified easily to study other types of genetic interactions on a genome-wide scale (6, 8). One major limitation of this technology is low hybridization of a subset of TAGs, presumably due to mutations in the TAG sequences or the common priming sites (20). Reconstruction of the corresponding heterozygote diploid YKOs with correct TAG sequences will likely be necessary to totally solve this problem and this will also further improve the performance of dSLAM. We also anticipate that the SLAM concept will be adapted to study synthetic lethality interactions in mammalian tissue culture systems by exploiting the genome-wide barcoded shRNA libraries available (22–24).
Acknowledgments
This work was supported in part by NHGRI grant HG02432 and by the Technology Center for Networks and Pathways (RR020839) to J.D.B. X.P. is a fellow of the Leukemia & Lymphoma Society. D.S.Y. was supported by fellowships from the Burroughs-Wellcome Center for Computational Biology at Johns Hopkins and by an NRSA fellowship from NHGRI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dobzhansky T. Genetics. 1946;31:269–90. doi: 10.1093/genetics/31.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartman JLt, Garvik B, Hartwell L. Science. 2001;291:1001–4. doi: 10.1126/science.291.5506.1001. [DOI] [PubMed] [Google Scholar]
- 3.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Davis RW, et al. Science. 1999;285:901–6. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 4.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M. Nature. 2002;418:387–91. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 5.Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, Punna T, Ihmels J, Andrews B, Boone C, Greenblatt JF, Weissman JS, Krogan NJ. Cell. 2005;123:507–19. doi: 10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 6.Pan X, Ye P, Yuan DS, Wang X, Bader JS, Boeke JD. Cell. 2006 doi: 10.1016/j.cell.2005.12.036. [DOI] [PubMed] [Google Scholar]
- 7.Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, Chen Y, Cheng X, Chua G, Friesen H, Goldberg DS, Haynes J, Humphries C, He G, Hussein S, Ke L, Krogan N, Li Z, Levinson JN, Lu H, Menard P, Munyana C, Parsons AB, Ryan O, Tonikian R, Roberts T, Sdicu AM, Shapiro J, Sheikh B, Suter B, Wong SL, Zhang LV, Zhu H, Burd CG, Munro S, Sander C, Rine J, Greenblatt J, Peter M, Bretscher A, Bell G, Roth FP, Brown GW, Andrews B, Bussey H, Boone C. Science. 2004;303:808–13. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- 8.Pan X, Yuan DS, Xiang D, Wang X, Sookhai-Mahadeo S, Bader JS, Hieter P, Spencer F, Boeke JD. Mol Cell. 2004;16:487–96. doi: 10.1016/j.molcel.2004.09.035. [DOI] [PubMed] [Google Scholar]
- 9.Ooi SL, Shoemaker DD, Boeke JD. Nat Genet. 2003 doi: 10.1038/ng1258. [DOI] [PubMed] [Google Scholar]
- 10.Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, Andrews B, Tyers M, Boone C. Science. 2001;294:2364–8. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 11.Goldstein AL, McCusker JH. Yeast. 1999;15:1541–53. doi: 10.1002/(SICI)1097-0061(199910)15:14<1541::AID-YEA476>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 12.Lorenz MC, Muir RS, Lim E, McElver J, Weber SC, Heitman J. Gene. 1995;158:113–7. doi: 10.1016/0378-1119(95)00144-u. [DOI] [PubMed] [Google Scholar]
- 13.Wach A, Brachat A, Pohlmann R, Philippsen P. Yeast. 1994;10:1793–808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- 14.Hoffman CS, Winston F. Gene. 1987;57:267–72. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- 15.Fare TL, Coffey EM, Dai H, He YD, Kessler DA, Kilian KA, Koch JE, LeProust E, Marton MJ, Meyer MR, Stoughton RB, Tokiwa GY, Wang Y. Anal Chem. 2003;75:4672–5. doi: 10.1021/ac034241b. [DOI] [PubMed] [Google Scholar]
- 16.Shoemaker DD, Lashkari DA, Morris D, Mittmann M, Davis RW. Nat Genet. 1996;14:450–6. doi: 10.1038/ng1296-450. [DOI] [PubMed] [Google Scholar]
- 17.Ooi SL, Shoemaker DD, Boeke JD. Science. 2001;294:2552–6. doi: 10.1126/science.1065672. [DOI] [PubMed] [Google Scholar]
- 18.Giaever G, Shoemaker DD, Jones TW, Liang H, Winzeler EA, Astromoff A, Davis RW. Nat Genet. 1999;21:278–83. doi: 10.1038/6791. [DOI] [PubMed] [Google Scholar]
- 19.Yuan DS, Pan X, Ooi SL, Peyser BD, Spencer FA, Irizarry RA, Boeke JD. Nucleic Acids Res. 2005;33:e103. doi: 10.1093/nar/gni105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eason RG, Pourmand N, Tongprasit W, Herman ZS, Anthony K, Jejelowo O, Davis RW, Stolc V. Proc Natl Acad Sci U S A. 2004;101:11046–51. doi: 10.1073/pnas.0403672101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stearns T, Botstein D. Genetics. 1988;119:249–60. doi: 10.1093/genetics/119.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paddison PJ, Silva JM, Conklin DS, Schlabach M, Li M, Aruleba S, Balija V, O’Shaughnessy A, Gnoj L, Scobie K, Chang K, Westbrook T, Cleary M, Sachidanandam R, McCombie WR, Elledge SJ, Hannon GJ. Nature. 2004;428:427–31. doi: 10.1038/nature02370. [DOI] [PubMed] [Google Scholar]
- 23.Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, Agami R, Ge W, Cavet G, Linsley PS, Beijersbergen RL, Bernards R. Nature. 2004;428:431–7. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- 24.Silva JM, Li MZ, Chang K, Ge W, Golding MC, Rickles RJ, Siolas D, Hu G, Paddison PJ, Schlabach MR, Sheth N, Bradshaw J, Burchard J, Kulkarni A, Cavet G, Sachidanandam R, McCombie WR, Cleary MA, Elledge SJ, Hannon GJ. Nat Genet. 2005;37:1281–8. doi: 10.1038/ng1650. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh K, Van Duyne GD. Methods. 2002;28:374–83. doi: 10.1016/s1046-2023(02)00244-x. [DOI] [PubMed] [Google Scholar]