

Abstract
Purpose
Adoptive cellular immunotherapy has promise as an approach to eradicate established tumors. However, a significant hurdle in the success of cellular immunotherapy involves recently identified mechanisms of immune suppression on cytotoxic T-cells at the effector phase.
Transforming growth factor-β (TGF-β) is one of the most important of these immunosuppressive factors because it affects both T-cell and macrophage functions. We thus hypothesized that systemic blockade of TGF-β signaling combined with adoptive T-cell transfer would enhance the effectiveness of the therapy.
Experimental Design
Flank tumors were generated in mice using the OVA-albumin (OA) expressing thymoma cell line, EG7. Splenocytes from transgenic OT-1 mice (whose CD8 T-cells recognize an immunodominant peptide in OA) were activated in vitro and adoptively transferred into mice bearing large tumors in the presence or absence of an orally available TGF-β receptor-I kinase blocker (SM16).
Results
We observed markedly smaller tumors in the group receiving the combination of SM16 chow and adoptive transfer. Additional investigation revealed that TGF-β receptor blockade increased the persistence of adoptively transferred T-cells in the spleen and lymph nodes, increased numbers of adoptively transferred T-cells within tumors, increased activation of these infiltrating T-cells, and altered the tumor microenvironment with a significant increase in TNF-α and decrease in arginase mRNA expression
Conclusions
We found that systemic blockade of TGF-β receptor activity augmented the anti-tumor activity of adoptively transferred T-cells and may thus be a useful adjunct in future clinical trials.
Keywords: tumor immunology, immunosuppression, TGFβ, Cytotoxic T-cells, cytokines, adoptive transfer
INTRODUCTION
Adoptive T cell therapy of solid tumors has been a goal for decades (1–3). Although significant progress has been made in animal models and in early clinical trials, this approach remains challenging, with clinical successes primarily restricted to small numbers of patients with melanomas (4–6). Steady progress has been made over the past twenty years in developing more successful strategies for adoptive transfer of T-cells to treat cancer (1,6). There have been advancements in the generation of more effective T-cells for transfer by developing better ways to expand T-cells, by using transgenic T-cell receptors, T-cell clones, or chimeric T-cell receptors (6–8) and progress in developing ways to increase the proliferation and persistence of transferred T-cells (i.e. lymphodepletion, cyclophosphamide, total body irradiation, and concurrent vaccination with antigen (4, 9–13).
Generating antigen-specific cytotoxic T cells is not enough, however. For example, despite the presence of large numbers of antigen-specific T-cells in the blood, spleen, and lymph nodes in vaccine-treated patients, tumor progression can still occur (14,15). This suggests that additional manipulations aimed at increasing trafficking of effector T-cells into tumors and/or preventing these T-cells from either undergoing apoptosis or loss of effector function within the lymphoid system, the blood, and/or the tumor microenvironment could be very valuable (7).
It has been increasingly realized that the tumor microenvironment is extremely immunosuppresive due to the effects of inhibitory leukocytes (such as CD4+/CD25+ T-regulatory cells, Type 1 regulatory T-cells, B-cells, and immature myeloid and dendritic cells), down regulation of activating chemokines and cytokines, and the presence of immunosuppressive agents such as vascular endothelial cell growth factor, interleukin-10 (IL-10), prostaglandin E2, arginase, reactive oxygen species, indoleamine 2,3-dioxygenase, soluble Fas, soluble Fas Ligand, etc. (3, 16–18) One of the most important of these T-cell immunosuppressive factors is TGF-β, a cytokine produced by tumor cells and nearly every immunologic cell type, including T and B cells, macrophages, dendritic cells (DC) and platelets (19–22).
TGF-β affects both the tumor microenvironment and T-cells directly. TGF-β has been shown to modulate tumor-associated macrophages and shift them from a more cytotoxic M1 phenotype to a more tumor-supportive M2 phenotype (21). An especially important consequence of this change may be the ability of TGF-β to stimulate the production of arginase by M2 macrophages (23, 24). Arginase production in the tumor microenvironment by myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses (25). TGF-β also serves to inhibit leukocyte migration into tumors (26).
In addition, there are a number of direct effects on T-cells. TGF-β affects the expression of key transcription factors and cytokines involved in T cell development, differentiation, and activation (reviewed in 21). TGF-β inhibits antigen presentation (27), shifts the T cell repertoire to a primarily TH2 phenotype, and exerts inhibitory effects on T cell proliferation by modulating the expression and signaling function of IL-2 and IL-2R (21). Suppression of Tbet and GATA-3 mRNA expression levels by TGF-β hinders CD4 and CD8 T cell differentiation (20). Additionally, TGF-β supports the maintenance of FOXP3 expression, regulatory function and homeostasis in peripheral CD4+CD25+ regulatory T cells, including those found in the tumor microenvironment (28, 29). Perhaps most importantly for adoptive transfer, TGF-β induces apoptosis in activated T-cells, attenuates the acquisition and expression of T-cell effector function, and directly acts on CTLs to inhibit expression of cytolytic gene targets (such as perforin granzymes, and interferon-γ) (30–32).
Accordingly, we hypothesized that globally blocking TGF-β function would augment the anti-tumor responses produced by adoptive transfer of T-cells. T-cells with specificity for the immunodominant peptide of chicken ovalbumin were generated from OT-1 transgenic mice and adoptively transferred into mice bearing large EG7 thymomas expressing the cognate ligand (chicken ovalbumin) in the presence or absence of the small, orally available TGF-β type-1 receptor kinase (ALK5) inhibitor, SM16 (33). We have recently shown that it is highly effective in blocking the phosphorylation of SMAD2/3 within tumors (34).
Our results show that TGF-βR blockade markedly augmented the efficacy of adoptive transfer, resulting in cures of most animals. Potential mechanisms included increased persistence of adoptively transferred T-cells in the spleen and lymph nodes, increased numbers of adoptively transferred T-cells within tumors, increased activation of these infiltrating T-cells, and altered the tumor microenvironment with significant increases in TNF-α decreases in arginase mRNA expression
MATERIALS AND METHODS
Cell lines
EG7 is a derivative of a parental murine thymoma cell line (EL4) that was transfected with an OVA cDNA construct. EG7 cells were cultured and maintained in media consisting of RPMI media (Gibco) supplemented with 100mg/ml G418, 10% fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 100mM sodium pyruvate and 2.5% β-mercaptoethanol. EL4 cells were cultured in similar media without G418. The cell lines were regularly tested and maintained negative for Mycoplasma contamination. The cells grow in mice with a C57B6 background (35).
Mice
Pathogen free female C57/B6 mice (6–8 weeks old) were purchased from Taconic laboratories (Germantown, NY). OT-1 mice express a transgenic MHC-1 restricted TCR specific for the ovalbumin peptide (SIINFEKL) (36). For adoptive transfer/T cell tracking studies, a double transgenic mouse strain in which GFP is expressed by all T-cells (DPEGFP) was crossed to the OT-1 strain (OT-1×gfp mice) (37). The mice are on the C57/B6 background, backcrossed for more than seven generations.
Animals were housed in the animal facility at the Wistar Institute (Philadelphia, PA). The Animal Use Committees of the Wistar Institute approved all protocols in compliance with the care and use of animals.
In Vitro T Cell Stimulation
OT-1 or OT-1× gfp mice were anesthetized and spleens were harvested and strained through a 70-um filter with PBS with 10% FBS. Red blood cells were removed with lysing buffer (BD Pharmingen) for 15 minutes at room temperature. After washing, cells were counted and placed in tissue culture at a final concentration of 1× 106 cells/ml. OT-1 splenocytes were activated with media containing the SIINFEKL peptide (2 μg/ml) (Bachem, King of Prussia, PA) on Day 0. After two hours, cells were spun down and placed in media without peptide. Cells were stimulated with mIL-2 (20ng/ml) (R&D Systems, Minneapolis, MN) on Day 2, 4 and 6, harvested, and analyzed with flow cytometry or used for adoptive transfer and T cell tracking studies on Day 7.
Monoclonal Antibodies
The following antibodies were obtained from BD Biosciences PharMingen (San Diego, CA) and used for flow cytometry: anti-CD8-PE, CD8-APC, CD8-FITC, CD69-PE, CD44-FITC, CD25-PE, CD62L-APC, V-alpha2-PE and CD16/32.
Flow Cytometric Analysis
Phenotypic Profile of T cells generated ex-vivo
Activated cells were harvested and one million cells per sample were Fc blocked with anti-mouse unconjugated CD16/32 antibody for 15 minutes at 4°C, washed and then incubated for 50 minutes at 4°C with antibodies. Data acquisition was performed on a FACSCalibur (BD Biosciences) and data analysis was completed using FloJo software (Tree Star, Inc, San Carlos, CA).
In Vitro Cytotoxicity Assay
Chromium-51 release cytotoxicity assays were used to determine the lytic activity of OT-1 effector cells in vitro as previously described (38).
TGF-β Kinase Inhibitor, SM16
SM16, a 430MW ALK4/ALK5 kinase inhibitor produced by BiogenIdec has recently been described in detail (including its chemical structure) (33). This small molecule can be administered i.p. or formulated in mouse chow which allows for daily oral administration (34). We used the chow formulation for all studies. We have previously shown that SM16 chow at a dose of 0.45 g/kg of chow is well tolerated by the animals, results in therapeutic drug levels, and effectively blocks SMAD2 phosphorylation within tumor cells (34).
Animal tumor models
Tumors were established with subcutaneous flank injections of 1× 106 cells suspended in 100μl PBS. Tumors were measured twice weekly and volumes were estimated using the formula 3.14 × [largest diameter × (perpendicular diameter) 2]/6. Treatment was administered when tumors were ~200 mm3 in size and mice followed for tumor growth. Mice were sacrificed when the tumors became >10% body weight or the mice demonstrated signs of distress.
TGF-β blockade and adoptive transfer
When the tumors reached a minimal volume of 200mm3, one group of mice was started on SM16 chow at a dose of 0.45 g/kg chow. Three days later, 10 × 106 OT-1 cells that had been stimulated for seven days in vitro, were adoptively transferred via tail vein injection. The average tumor volumes of the combination treatment group over time was plotted and compared to untreated control, SM16 chow only and adoptive transfer only groups. Each experiment had 5–8 mice per group and the study was repeated four times.
Adoptive Transfer, T Cell Tracking Studies
EG7 tumors were established and mice were started on control or SM16 chow. Three days after starting chow, 20 × 106 OT-1×gfp T cells activated for 7 days, were adoptively transferred via tail vein injection in control or SM16 chow-fed mice. Spleens, lymph nodes and tumors were harvested three days after adoptive transfer. Tissues were harvested from three groups: (1) control chow, no treatment (2) control chow + adoptive transfer) and (3) SM16 chow + adoptive transfer. Spleens and lymph nodes were processed as described above. Tumors were chopped and single cell suspensions obtained with collagenase/DNase digestion at 50°C for 1 hour. Cells were run on a ficoll gradient, filtered, washed and counted. All cells were then subjected to FACS using anti-CD8 antibody and/or gated appropriately to determine the percentage of GFP+ adoptively transferred cells.
Intracellular Cytokine Staining
Intracellular interferon-γ staining was performed with a fixation/permeabilization solution kit (BD Biosciences Pharmingen). Briefly, cells were isolated as detailed above and 1.5 × 106 cells were stimulated in a 96 well plate for 5 hrs at 37°C with either 1) 1 μg/ml of SIINFEKL peptide plus 50 units/ml of IL-2 (Roche), 2) 50 units/ml of IL-2, or 3) 50 ng/ml phorbol-12-myristate-13-acetate (PMA) plus 1 μg/ml of ionomycin. One μl/ml of GolgiPlug was present for the entire incubation. Following several washes, cells were stained with the desired cell surface markers as detailed above, washed, and fixed and permeabilized with Cytofix/Cytoperm solution for 20 min at 4°C. Following 2 washes with PermWash, cells were stained with a PE-labeled interferon-γ antibody for 30 min at 4°C, washed, and analyzed on the Becton-Dickinson FACSCalibur flow cytometer. In one set of controls, cells were first treated with an unlabeled anti-IFN-γ antibody before the labeled anti-IFN antibody to quantify non-specific binding and set highly stringent gates.
RNA isolation and real time, reverse transcription-PCR
Quantitation of EG7 tumor mRNA levels was performed as previously described (39). EG7 tumor-bearing mice were started on control or SM16 chow. Three days later, tumors from these mice were harvested and total RNA isolated using Triszol reagent (Invitrogen, Carlsbad, CA). Semi-quantitative analysis of gene expression was done using a Cepheid Smart Cycler following the manufacturer’s protocol for SYBR Green kit supplied by Roche (Sunnyvale, CA). cDNA concentrations from each gene pool were normalized using β-actin as a control gene. Relative levels of expression of each of the selected gene (fold change versus saline control) were determined. Each sample was run in triplicate or quadruplicate.
Statistical Analyses
Data are presented as mean+/− SEM. For tumor size studies, data from multiple experiments (4 studies) were normalized (by dividing each experimental value by the average tumor volume at the time of adoptive transfer) and combined. Data comparing difference between two groups were assessed using unpaired Student t-tests. Comparisons with more than two groups were done using ANOVA and confirmed with the Kruskal-Wallis test. ANOVA analyses were adjusted for multiplicity of analyses by the Bonferroni method, correcting for the number of time points where an analysis was done. Pairwise comparisons of the means of treatment groups within time points were analyzed using the Tukey method. Difference were considered significant when P<0.05.
RESULTS
Generation of OT-1 Effector CD8+ T-cells for Adoptive Transfer
To evaluate the combination therapy of adoptive transfer and TGF-β blockade, we used a well-established tumor model with a known tumor antigen: EG7 thymoma cells expressing the neo-antigen chicken ovalbumin (35). T-cells for transfer were derived from OT-1 transgenic mice in which the T-cells express an MHC Class-I-restricted T-cell receptor (TCR) that recognizes the ovalbumin (OVA) peptide SIINFEKL (36). As previously reported (38, 40), after splenocytes were activated by peptide and IL-2, the cells were predominately CD8+ (>95%). Their phenotype was CD44HI, CD69HI, CD25HI and CD62LLO (Supplemental Figure 1A), consistent with activated effector T cells, and they were very active in an in vitro chromium cytotoxicity assay (Supplemental Figure 1B). EG7 cells were killed by activated OT-1 CD8+ T cells (>50% specific lysis) even at low E:T ratios (3:1). EL4 cells, the parental line of EG7 lacking ovalbumin expression, were not killed by activated OT-1 cells (Supplemental Figure 1B).
TGF-βR1 blockade enhances the efficacy of adoptive transfer
To test the system under stringent conditions, we transferred activated T-cells to mice with large (>200 mm3) flank tumors. An example of one such study is shown in Figure 1A (n= 8 mice in each group). Injection of EG7 cells into the flanks of C57/B6 mice led to rapid tumor growth (Fig. 2A, diamonds). Intravenous injection of 10 million activated OT-1 cells on Day 12 (200 mm3 sized tumors) led to a slowing of tumor growth, but no tumor regression (Fig. 1A, squares). This dose of cells is similar to that used by other investigators (38). We also treated established EG7 tumors on Day 9 (>150 mm3 sized tumors) with the TGF-β inhibitor, SM16, formulated in chow at a dose of 0.45 g/kg chow. This dose was previously demonstrated to have minimal toxicity and effectively block SMAD2/3 within tumors (34). As shown in Figure 1A (triangles), SM16 induced a slowing of tumor growth, but no tumor regression. In contrast, animals treated with SM16 Chow on Day 9, followed by adoptive transfer on Day 12, showed complete tumor regression (Figure 1A, crosses).
Figure 1. TGF-β Receptor Blockade and adoptive transfer of activated OT-1 splenocytes causes tumor regression.
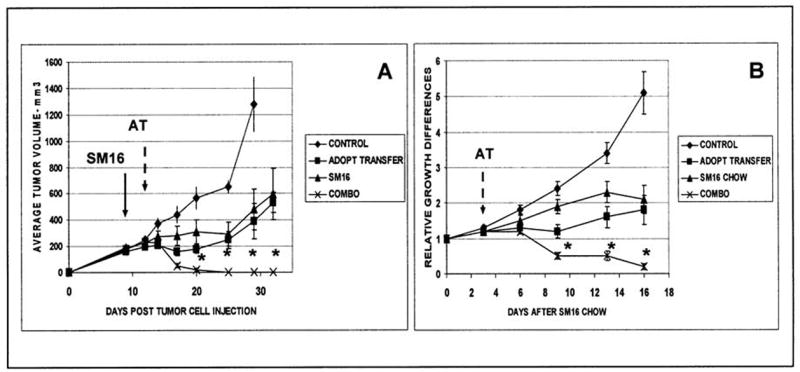
A. Representative Experiment. EG7 tumor cells were injected on the flanks of C57/B6 mice on Day 0 (n=8 in each group). On Day 9, established EG7 tumors were left on control chow (control and adoptive transfer groups) or begun on SM16 chow (solid arrow: SM16 and combo group). On Day 12, the adoptive transfer group and the combination group (combo) received an intravenous injection of 10 million activated OT-1 cells (AT- dashed arrow). Both SM16 and activated OT-1 cells caused a slowing of tumor growth (tumors were significantly smaller than control from Day 20 onwards). However, all animals treated with the combination had complete tumor regression up till Day 32. Tumor size in the combination group was significantly smaller (*p<0.05 after Bonferroni correction for multiplicity) than all of the other groups from Day 20 onwards.
B. Combined Data. This experiment was repeated 3 more times with similar results. This panel shows the data combined from the four different experiments (N=26 in each group). To control for minor differences in tumor size at the beginning of each experiment, the average tumor volume at each time point was normalized to the volume at the start of treatment. In this panel, Day 0 represents the day SM16 chow was started and adoptive transfer was performed three days later (arrow). These data are similar to the individual study shown above, with significant regression in the combination group compared to all others (* = p<0.05 after Tukey correction).
Figure 2. TGF-β Receptor Blockade Increases the Number of Transferred T-cells in spleen and lymph node.

Twenty million activated GFP-expressing OT-1 splenocytes were injected into tumor-bearing animals on control diets or SM16 diets. Three days after transfer, spleen and lymph nodes were isolated and subjected to FACS analysis to identify transferred CD8+/GFP+ T-cells.
Panel A shows results from Spleen and Panel B from lymph nodes. The upper panels show representative FACS tracings (CD8 on the X-axis and GFP on the Y axis) from control animals (no adoptive transfer), animals on control chow who received adoptive transfer of T cells, and animals on SM16 chow who received adoptive transfer of T cells. The transferred cells (CD8+/GFP+) are visualized in the right upper quadrant of each tracing. The lower graph shows the average values of the CD8+/GFP+ populations in each group for three independent experiments. *= p<0.05 from control and SM16 + AT; ** = p<0.05 from control and Control chow plus adoptive transfer.
Figure 1B shows data combined from the four different independent experiments (N=26 in each group). To control for minor differences in tumor size at the beginning of each experiment, the average tumor volume at each time point was normalized to the volume at the start of treatment. Day 0 represents the day SM16 chow was started and adoptive transfer was performed three days later (arrow). Again, these data show that: 1) untreated tumors grew rapidly, 2) tumors treated with either SM16 chow or adoptive transfer alone experienced a delay in tumor growth, and 3) tumors treated with combined therapy had striking decreases in size. ANOVA analyses with a Bonferroni correction showed significant overall differences between treatment groups beginning on day 9 (9 days after beginning chow, 6 days after adoptive transfer). From day 9 to day 16, tumors treated with the combination regimen had significantly more (p<0.05) tumor regression than either treatment alone, an average of 80% decrease in tumor volumes, with complete regression in the majority of the tumors (Fig. 1B).
These data show that the addition of TGF-β receptor blockade markedly augmented adoptive transfer therapy.
Mechanisms
Given this augmented effect of combination treatment, we explored a number of possible mechanisms. We took advantage of the availability of a double transgenic mouse (OT-1.gfp) in which the OT-1 T-cells are expressing green fluorescent protein (GFP). Splenocytes from OT-1×gfp mice were activated for seven days as described above. On Day 7, cells underwent flow cytometric analysis to confirm the presence of both the green fluorescence protein (96.9%) and the transgenic TCR indicated by the V-alpha2 chain (93.5%) on CD8+ T cells at the time of transfer (Supplemental Fig. 1A).
TGF–βR1 blockade increases the number of antigen specific T-cells in the spleens and lymph nodes
One mechanism by which TGF–βR1 blockade might augment adoptive transfer could be to increase the proliferation or persistence of the transferred T-cells. To evaluate this possibility, we injected activated GFP-expressing OT-1 cells into tumor-bearing animals on control or SM16 chow. Three days after transfer, spleen, lymph nodes (and tumors- see below) from treated mice were isolated and subjected to FACS analysis to identify transferred CD8+/GFP+ T-cells.
FACS analysis of isolated spleens and lymph nodes from EG7 tumor-bearing mice showed a clearly detectable subset of CD8+/GFP+ adoptively transferred cells in the spleen (8–14% of the CD8+ cells) (Fig. 2A) and lymph nodes (1.5–2.5% of cells) (Fig. 2B). In both spleen and lymph node there were significantly more GFP+ (p<0.05) cells in SM16-treated animals vs. control (Fig. 2A & 2B).
TGF-βR1 blockade increases the number of Tumor-associated T-cells
Another way by which TGF–β blockade might augment adoptive transfer could be by increasing the number of adoptively transferred cells within tumors. To assess this, FACS analysis of tumors in mice adoptively transferred with or without SM16 chow was performed, three days after cell transfer. After adoptive transfer, a small, but easily detectable population (estimated to be approximately 110,000 cells) was identified (Fig. 3A vs 3B). In animals treated with SM16 chow and given OT-1 cells, however, there was a significant (P<0.01) 3–4 fold increase in the number of transferred cells in tumors (430,000 cells) (Fig. 3C). The average values for three independent experiments are shown in the lower panel of Figure 3.
Figure 3. TGF-β Receptor Blockade Increases the Number of Transferred T-cells in Tumors.
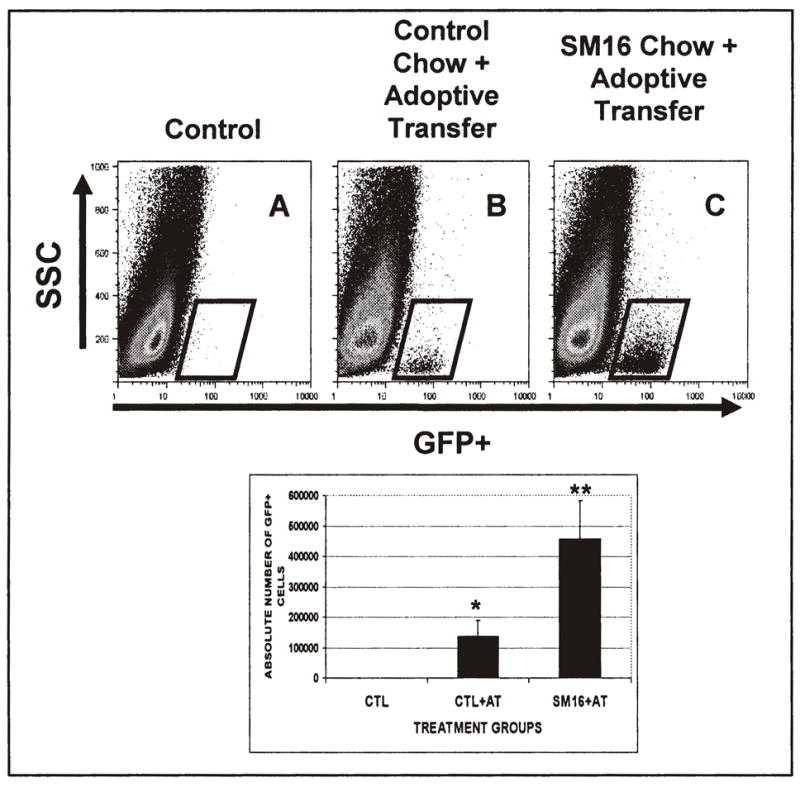
Twenty million activated GFP-expressing OT-1 cells were injected into tumor-bearing animals on control diets or SM16 diets. Three days after transfer, the tumors were isolated, digested, subjected Ficoll purification, and analyzed by FACS to identify transferred CD8+/GFP+ T-cells. The upper panels show representative FACS tracings (GFP on the X-axis and Side Scatter on the Y axis) from control animals (no adoptive transfer), animals on control chow who received adoptive transfer of T cells, and animals on SM16 chow who received adoptive transfer of T cells. Transferred cells (GFP+) are visualized in the boxes. The lower graph shows the average number of GFP+ cells in the tumors in each group for three independent experiments. *= p<0.05 from control and control chow plus AT; ** = p<0.05 from control and Control chow plus adoptive transfer.
TGF-βR1 blockade augments the effector function of transferred CD8+ T-cells
TGF-β can inhibit the activation of effector T-cells (19, 21, 31, 32). We therefore performed FACS on T-cells isolated from either spleens or tumors of control and treated animals and assessed the ability of these cells to produce INF-γ in response to non-specific activating signals (IL-2 and PMA/ionomycin) and to the specific activation signal of the OT-1 peptide (SIINFEKL) plus IL-2.
The effect of systemic TGF-βR1 blockade were determined in isolated splenocytes from adoptively transferred tumor-bearing animals. As expected, the percentages of GFP+ cells that expressed INF-γ increased after stimulation with IL-2 alone (19–22%), PMA (34–42%) and peptide plus IL-2 (59–78%). However, there were no significant differences between control and SM16-treated animals in their ability to induce INF-γ following stimulation. These results suggest that TGF-β inhibition does not increase the effector function of the transferred T cells present in spleen (Supplemental Figure 2).
The effector activity of the tumor-associated GFP+ (transferred T) cells were also investigated. Due to the very small numbers of T-cells within tumors, we pooled cells from four tumors, and following Ficoll enrichment, stimulated the cells with IL-2, PMA/ionomycin, or IL-2 plus SIINFEKL peptide, and then performed FACS to detect intracellular IFN-γ staining. Figure 4 shows two independent experiments. As expected, the amount of INF-γ produced by TILs was much lower than was seen in splenocytes (41, 42). However, CD8+ T-cells from the SM16-treated animals showed enhanced IFN-γ staining (2–3 fold increase) after non-specific (IL-2 or PMA) or specific (IL-2 plus SIINFEKL peptide) stimulation compared with TILs from animals treated with control chow. Thus, blockade of TGF-βR signaling appears to enhance responsiveness of intratumoral T-cells, but not splenic T-cells following adoptive transfer.
Figure 4. TGF-β Receptor blockade augments the effector function of adoptively transferred T-cells in tumor.
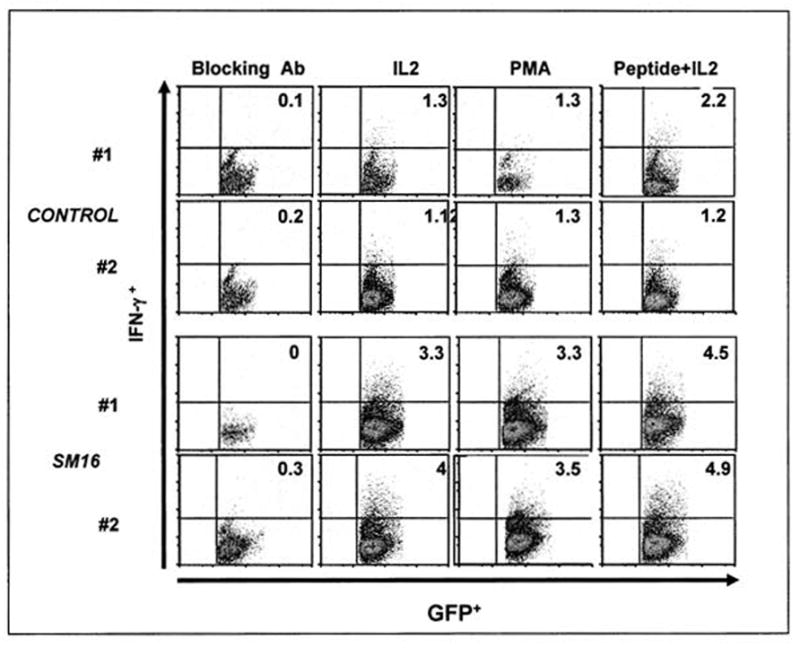
Twenty million activated GFP-expressing OT-1 cells were injected into tumor-bearing animals on control diets or SM16 diets. Three days after transfer, tumors were isolated and digested. Due to the very small numbers of T-cells within tumors, we pooled cells from 4 tumors, did a Ficoll purification, stimulated the cells with IL-2, PMA/ionomycin, or IL-2 plus SIINFEKL peptide, and then performed FACS to detect intracellular IFN-γ staining using PE-labeled anti-IFN-γ antibody (X-axis is GFP, Y-axis is IFN-γ). The two left hand panels were treated with an unlabeled anti-IFN–γ antibody (blocking antibody) before addition of PE-labeled antibody and were used to identify non-specific binding and define our stringent gates. Each row shows an independent experiment. The percent of activated cells (GFP+/IFN-γ+) of the total GFP+ (CD8+) cells are shown in the upper right hand quadrant of each FACS panel.
TGF-β blockade alters the tumor microenvironment
Although it is likely that many of these effects on T-cells were direct (i.e. due to blockade of TGF-β signaling on T-cells themselves), TGF-β receptor blockade could also have effects on the tumor microenvironment, generating conditions that might favor enhanced T-cell trafficking or cytotoxic T-cell activity. To evaluate the effect of TGF-β inhibition on the tumor microenvironment in this model, EG7 tumor-bearing mice were treated with SM16 chow for three days, tumors were harvested and real time RT-PCR performed to determine the expression levels of key cytokines, chemokines and immunomodulatory enzymes. The results in Table 1 show that tumors from animals in which the TGF-β receptor was blocked with SM16 displayed significant (p<0.01) increases in the mRNA for IP-10 (CXCL10) (2 fold), MIG (CXCL9) (2.5 fold), TNF-α (6.4 fold), and IL-12 (2.7 fold). Upregulation of T-cell chemoattractants and cytokine expression would be consistent with increased trafficking of CD8+ T-cells into the tumors. In addition, the message for arginase was significantly decreased (p < 0.001) by 60%. There were no significant changes in the expression of RANTES (CCL5) or TGF-β. These results suggest that inhibition of TGF-β signaling alters the tumor microenvironment by significantly changing intratumoral expression of key immunomodulatory cytokines, chemokines and enzymes.
Table 1.
TGF-β inhibition upregulates mRNA (as measured by real-time RT-PCR for some pro-inflammatory cytokines, chemokines and chemo-attractants)
| Primer | Fold change | p-value |
|---|---|---|
| IP-10 (CXCL10) | 2.0 | 0.001 |
| MIG (CXCL9) | 2.5 | 0.01 |
| Rantes (CCL5) | 1.0 | NS |
| TNF-α | 6.4 | <0.001 |
| iNOS | 1.4 | 0.03 |
| IL-12 | 2.7 | 0.001 |
| Arginase | 0.4 | <0.001 |
| TGF-β | 1.0 | NS |
DISCUSSION
As discussed in detail above (and recently reviewed in Ref. 22), there are many potential mechanisms by which TGFβ in the tumor microenvironment may function to directly and indirectly inhibit the function of anti-tumor T-cells. We thus hypothesized that blockade of TGF-β function might augment cancer immunotherapy induced by adoptive transfer of T-cells.
To inhibit TGF-β-mediated effects, we used SM16, a small, highly specific, orally available ALK4/5 kinase inhibitor that effectively blocks TGF-β or activin-induced SMAD2/3 phosphorylation in vitro and in tumors (33, 34). It should be noted that any inhibitor may have off-target effects, however, SM16 appears to be quite specific. In HepG2 cells, SM16 inhibits TGFβ-induced PAI-luciferase activity (IC50 64 nM) and TGFβ-or Activin-induced Smad2 phosphorylation at concentrations between 100 and 620 nM. SM16 was tested against >60 related and unrelated kinases and showed moderate off-target activity only against, Raf (IC50 1uM) and p38/SAPKa (IC50, 0.8 uM). SM16 exhibited no inhibitory activity against ALK family members, ALK1 and ALK6 (33).
To test our hypothesis, we used a well-established murine system of adoptive transfer, the EG7/OT1 model (35–37). There are advantages and limitations to this approach. The target antigen is not a natural tumor antigen and was highly expressed. However, although this system does not mimic most human tumors in these respects, it does allow for the generation of large numbers of uniformly activated T-cells (Supplemental Fig. 1A) that are highly cytotoxic and directed against a defined antigen (Supplemental Fig. 1B). These T-cells can be tracked in vivo using an antibody against the V-α2 chain of the TCR, tetramer staining, or by GFP staining (in double transgenic mice) (37). In order to give our system the most stringent test possible, we used adoptive transfer in large (200 mm3) established flank tumors and did not supplement treatment with total body irradiation, lymphodepletion, or vaccination. Another limitation is that we only evaluated the effect of TGF β blockade in one system. It will be important to validate this approach in other adoptive transfer models and these studies are ongoing. However, despite these limitations, our model is very similar to animal and clinical studies using T-cells with transgenic T-cell receptors or chimeric T-bodies (6) and thus provides a valuable “proof of principal” for this approach.
Consistent with most published studies (see Ref. 38), transfer of 10–20 million activated T-cells under these conditions only slowed tumor growth, but did not induce remission (Fig. 1). As we have seen in other models, systemic blockade of the TGF-βRI had some anti-tumor activity that resulted in a slowing of tumor growth (34). However, when we combined adoptive transfer with blockade of the TGF-βRI, we saw markedly augmented effectiveness, with consistent tumor regressions, most often leading to complete disappearance of the tumors (Fig. 1). Given the size of the tumors at treatment, the rapid growth rate of these tumors, and the lack of any conditioning or radiation, we feel these results reveal a very potent anti-tumor effect.
This augmentation of efficacy appears to be the result of multiple mechanisms, as might be expected from an approach that targets a cytokine that has effects in both the T-cells and the host. Similar to the studies using T-cells expressing a dominant negative TGF-βRII (43–48), we observed increased numbers of the transferred T-cells in the spleens and lymph nodes of the SM16-treated animals (Fig. 2A and B). We did not determine whether this was due to increased proliferation and/or decreased cell death, but this finding is consistent with the observation that TGF-β is both an inhibitor of CD8+ T-cell growth and an inducer of T-cell apoptosis (21, 30, 45). We postulate increased persistence of adoptively transferred cells was one factor that contributed to enhanced efficacy, however this conclusion is tentative, since measuring proliferation in this short term model is not feasible due to fact that the vigorous in vitro T-cells stimulation prior to transfer commits the cells to multiple rounds of replication (49).
Accompanying the increased numbers of adoptively transferred cells observed in the lymphoid tissue, we found increased numbers of transferred T cells within the tumor (Fig. 3). Although the actual number of labeled T-cells within the tumors is relatively low, similar to observations in other studies (50, 51), treatment with SM16 increased this population by approximately four-fold. Our experimental design did not allow us to determine whether this was due to increased trafficking of cells, enhanced proliferation of T-cells within the tumors, and/or decreased cell death within the tumor, but current studies are planned to address this issue.
Although we observed increased numbers of T-cells within the tumors, it is well established that the tumor microenvironment can inactivate the functional capacity of these CTLs to kill tumors (16, 41, 42). An important mechanism by which this inactivation occurs is the presence of TGF-β (22, 41, 42). We thus also evaluated the “quality” of the intratumoral T-cells by assessing their ability to undergo activation. As has been reported, there was an overall reduced ability of the intratumoral CD8+ T cells to produce INF-γ compared to the splenic CD8+ T cells, consistent with local tumor microenvironmental inactivation of these cells (41, 42). However, our results showed that intra-tumoral T-cells from the SM16-treated mice produced more intracellular IFN-γ after non-specific (Il-2 or PMA/ionomycin) stimulation and antigen-specific (antigenic peptide) stimulation than did T-cells from control-treated animals (Fig. 4). Thus, systemic TGF-βRI blockade with SM16 resulted in increased numbers of intra-tumoral T-cells that have higher levels of effector function. We postulate that these changes account for much of the enhanced therapeutic effects of the adoptively transferred cells.
Given that SM16 is a systemic agent that affects other cell types (i.e. tumor cells, macrophages, etc), in addition to direct effect on T-cells, we also evaluated the effect of this agent on the tumor microenvironment using real-time RT-PCR of tumor samples (Table 1). Blockade of TGF-βRI signaling resulted in increased mRNA levels of a number of pro-inflammatory cytokines and chemokines (such as MIG (CXCL-9), IP-10 (CXCL-10, IL-12, and TNF-α) that would be expected to augment trafficking of activated T-cells (52). The increased levels of iNOS, IL-12, and TNF-α suggest that TGF-β blockade produces a more TH-1 like environment and likely affects the tumor-associated macrophage phenotype favoring a more M1 (cytotoxic) versus M2 (alternatively activated) phenotype (23, 24, 53). This observation is consistent with an earlier report showing improvement of macrophage dysfunction by administration of anti-transforming growth factor-beta antibody in EL4-bearing hosts 54). Furthermore, a decrease in tumor arginase levels would also be expected to augment T-cell persistence and function (25). Thus, SM16 appears to induce a less immunosuppressive tumor microenvironment that would encourage the migration of T-cells into the tumor and further enhance the ability of CD8+ T-cells to kill tumor cells directly and indirectly (i.e. through cytokine-mediated activation of tumor associated macrophages).
Our data are consistent with the direct effects of TGF-β inhibition on T-cells as studided by introducing a dominant negative TGF-β receptor into mouse and human T-cells using transgenic approaches or by transfection (43–48). This strategy has consistently led to enhanced T-cell proliferation, higher numbers of T-cells within tumors, and better anti-tumor effects. However, there may be some limitations to this approach if applied to patients. T-cell manipulation and re-infusion in the clinical setting is technically challenging and expensive. There are concerns that malignancy could be induced by insertional mutagenesis or by the introduction of genes (like TGF–β) that could inhibit apoptosis or function as a potential tumor-suppressor gene. In support of this possibility, Lucas et al. reported that the reduced TGF-β signaling associated with the transgenic introduction of a dominant-negative TGF-β receptor in mice led to rapid expansion of a CD8+ memory T-cell population and subsequent transformation to leukemia/lymphoma (45). This side effect was not observed, however, by Lacuesta et al (47) using multiple infusions of adoptively transferred, retrovirally-transduced mouse T-cells encoding a dominant-negative TGF-β receptor.
In summary, our data show that systemic blockade of TGF-β function using the TGF-βRI blocker SM16 markedly augmented the efficacy of adoptively transferred T-cells. This augmentation is likely due to multiple mechanisms that include an altered tumor microenvironment, increased numbers of CTLs in the spleen and lymph nodes, increased numbers of CTLs within the tumors, and increased CTL effector function (as measured by IFN-γ release after stimulation) in intra-tumoral T-cells. Although we detected only small numbers of CD4+/CD25+ T-regulatory cells in this model (data not shown), inhibition of this pathway by a TGF-β function blocker may be important in other models (28, 29).
A number of TGF-β blocking agents are in active clinical development (55, 56). If these drugs show acceptable toxicity, our data indicate that systemic blockade of TGF-β function in conjunction with adoptive transfer may be useful. Even if long-term administration of these agents proves to be limited by side effects, our study suggests that one important use for these agents could be short term administration in combination with adoptive T-cell strategies.
Supplementary Material
Supplemental Figure 1. Characterization of Activated OT-1 Splenocytes
A. OT-1 splenocytes stimulated for seven days have the phenotypic profile of activated effector T cells.
Splenocytes were harvested from OT-1xgfp mice and activated with media containing the SIINFEKL peptide for two hours. Cells were stimulated with mIL-2 (20ng/ml) on Day 2, 4 and 6, harvested on Day 7 and analyzed with flow cytometry. Plots show expression of CD8 (X-axis) versus CD44, CD69, CD25, CD62L, V-α-2 TCR chain, and GFP (Y-axis). The number in the right upper quadrant represents the percentage positive of total cells. Cells were almost exclusively CD8+/V-α-2+/GFP+ and had an activated phenotype (CD44HI, CD69 HI, CD25 HI, CD62LLow)
B. Activated OT-1 effector cells demonstrate cytotoxic activity in vitro.
Splenocytes were harvested from OT-1 mice as above, activated for 7 days and reacted with chromium51-labeled tumor cells at varying effector to target (E:T) ratios. EG-7 cells express the target ova antigen, EL-4 cells do not. The activated OT-1 cells were highly efficient in lysing EG-7 cells, even at very low E:T ratios, while having no effect on the EL-4 cells.
Supplemental Figure 2. Splenocytes from adoptively transferred tumor-bearing animals expressed increased IFN-γ after stimulation with IL-2 alone, PMA and peptide plus IL-2.
Three days after transfer of 20 million activated, GFP-labeled OT-1 cells, spleens were isolated and digested. Cells were stimulated with IL-2, PMA/ionomycin, or IL-2 plus SIINFEKL peptide, and subjected to FACS to detect intracellular IFN-γ staining (X-axis is GFP, Y-axis is IFN-γ). The two left hand panels were treated with an unlabeled anti-IFN–γ antibody (blocking antibody) before addition of labeled antibody and used to identify non-specific binding and define gates. The percent of activated cells (GFP+/IFN-γ+) of the total GFP+ cells are shown in the upper right hand quadrant.
Acknowledgments
Grant support:
NCI PO1 CA 66726
T32 CA 09140
References
- 1.Rosenberg SA, Yang JC, Restifo NP. Cancer Immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hinrichs CS, Gattinoni L, Restifo NP. Programming CD8+ T cells for effective immunotherapy. Curr Opin Immunol. 2006;18:363–70. doi: 10.1016/j.coi.2006.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knutson KL, Wagner W, Disis ML. Adoptive T cell therapy of solid cancers. Cancer Immunol Immunother. 2006;55:96–103. doi: 10.1007/s00262-005-0706-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–57. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan RA, Dudley ME, Wuderlich JR, et al. Cancer Regression in Patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.June C. Adoptive T cell therapy for cancer in the clinic. J Clin Invest CI. 2007;117:1466–76. doi: 10.1172/JCI32446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leen AM, Rooney CM, Foster AE. Improving T cell therapy for cancer. Annu Rev Immunol. 2007;25:243–65. doi: 10.1146/annurev.immunol.25.022106.141527. [DOI] [PubMed] [Google Scholar]
- 8.Yee C, Thompson JA, Byrd D, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: In vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci NAS. 2002;99:16168–73. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palmer DC, Balasubramaniam S, Hanada K, et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction. J Immunol. 2004;173:7209–16. doi: 10.4049/jimmunol.173.12.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gattinoni L, Finkelstein SE, Klebanoff CA, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–12. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang LX, Shu S, Plautz GE. Host lymphodepletion augments T cell adoptive immunotherapy through enhanced intratumoral proliferation of effector cells. Cancer Res. 2005;65:9547–54. doi: 10.1158/0008-5472.CAN-05-1175. [DOI] [PubMed] [Google Scholar]
- 12.Paulos CM, Wrzesinski C, Kaiser A, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin InvestCI. 2007;117:2197–204. doi: 10.1172/JCI32205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bracci L, Moschella F, Sestili P, et al. Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation, and specific tumor infiltration. Clin Cancer Res. 2007;13:644–53. doi: 10.1158/1078-0432.CCR-06-1209. [DOI] [PubMed] [Google Scholar]
- 14.Lee PP, Yee C, Savage PA, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. 1999;5:677–85. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- 15.Rosenberg SA, Sherry RM, Morton KE, et al. Tumor Progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Clin InvestCI. 2005;175:6169–76. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- 16.Gajewski TF, Meng Y, Harlin H. Immune suppression in the tumor microenvironment. J Immunother. 2006;29:233–40. doi: 10.1097/01.cji.0000199193.29048.56. [DOI] [PubMed] [Google Scholar]
- 17.Lizee G, Radvanyi LG, Overwijk WW, Hwu P. Improving antitumor immune responses by circumventing immunoregulatory cells and mechanisms. Clin Cancer Res. 2006;12:4794–803. doi: 10.1158/1078-0432.CCR-06-0944. [DOI] [PubMed] [Google Scholar]
- 18.Kim R, Emi M, Tanabe K, Arihiro K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res. 2006;66:5527–36. doi: 10.1158/0008-5472.CAN-05-4128. [DOI] [PubMed] [Google Scholar]
- 19.de Visser KE, Kast WM. Effects of TGF-beta on the immune system: implications for cancer immunotherapy. Leukemia. 1999;13:1188–99. doi: 10.1038/sj.leu.2401477. [DOI] [PubMed] [Google Scholar]
- 20.Gorelik L, Flavell RA. Transforming growth factor-beta in T-cell biology. Nat Rev Immunol. 2002;2:46–53. doi: 10.1038/nri704. [DOI] [PubMed] [Google Scholar]
- 21.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 22.Teicher BA. Transforming growth factor-beta and the immune response to malignant disease. Clin Cancer Res. 2007;13:6247–51. doi: 10.1158/1078-0432.CCR-07-1654. [DOI] [PubMed] [Google Scholar]
- 23.Munder M, Eichmann K, Modolell M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol. 1998;160:5347–54. [PubMed] [Google Scholar]
- 24.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166–73. doi: 10.4049/jimmunol.164.12.6166. [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez PC, Quiceno DG, Zabaleta J, et al. Arginase 1 production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 26.Mrass P, Weninger W. Immune cell migration as a means to control immune privledge: lesons from the CNS and tumors. Immunol Rev. 2006b;213:195–212. doi: 10.1111/j.1600-065X.2006.00433.x. [DOI] [PubMed] [Google Scholar]
- 27.Kobie JJ, Wu RS, Kurt RA, et al. Transforming growth factor beta inhibits the antigen-presenting functions and antitumor activity of Dendritic cell vaccines. Cancer Res. 2006;63:1860–4. [PubMed] [Google Scholar]
- 28.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp MedEM. 2005;7:1061–7. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu VC, Wong LY, Jang T, et al. Tumor evasion of the immune system by converting CD4+/CD25− T cells into CD4+CD25+ T regulatory cells: Role of tumor-derived TFG-β. J Immunol. 2007;178:2883–92. doi: 10.4049/jimmunol.178.5.2883. [DOI] [PubMed] [Google Scholar]
- 30.Chang CJ, Liao CH, Wang FH, Lin CM. Transforming growth factor-beta induces apoptosis in antigen-specific CD4+ T cells prepared for adoptive immunotherapy. Immunol Letters. 2003;86:37–43. doi: 10.1016/s0165-2478(02)00307-3. [DOI] [PubMed] [Google Scholar]
- 31.Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–80. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 32.Ahmadzadeh M, Rosenberg SA. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J Immunol. 2005;174:5215–23. doi: 10.4049/jimmunol.174.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fu K, Corbley MJ, Sun L, et al. An Orally Active Inhibitor of the TGF-β Type I Receptor, ALK5, Inhibits Vascular Fibrosis and Adventitial Myofibroblast Induction in the Rat Carotid Balloon Injury Model. Arteriosclerosis, Thrombosis, Vascular Biology. doi: 10.1161/ATVBAHA.107.158030. Epub ahead of print: Jan. 17, 2008. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki E, Kim S, Cheung HK, Corbley M, et al. A Novel Small Molecule Inhibitor of TGF-beta type I Receptor Kinase (SM16) Inhibits Murine Mesothelioma Tumor Growth in vivo and Prevents the Extent of Tumor Recurrence After Surgical Resection. Cancer Res. 2007;67:2351–9. doi: 10.1158/0008-5472.CAN-06-2389. [DOI] [PubMed] [Google Scholar]
- 35.Moore MW, Carbone FR, Bevan MJ. Introduction of soluble protein into the Class I pathway of antigen processing and presentation. Cell. 1988;54:777–85. doi: 10.1016/s0092-8674(88)91043-4. [DOI] [PubMed] [Google Scholar]
- 36.Carbone FR, Stery SJ, Butler J, Rodda S, Moore MW. T cell receptor a-chain pairing determines the specificity of residue 262 within the Kb-restricted, ovalbumin257–264 determinant. Intl Immunol. 1992;4:861–7. doi: 10.1093/intimm/4.8.861. [DOI] [PubMed] [Google Scholar]
- 37.Mrass P, Takano H, Ng LG, et al. Random migration precedes stable target cell interactions of tumor-infiltrating T cells. J Exp Med. 2006a;203:2749–61. doi: 10.1084/jem.20060710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang H, Xiang J. Synergistic effect of lymphotactin and interferon-γ-inducible protein-10 transgenr expression in T-cell localization and adoptive T-cell therapy of tumors. Int J Cancer. 2004;109:817–25. doi: 10.1002/ijc.20043. [DOI] [PubMed] [Google Scholar]
- 39.Jassar A, Suzuki E, Kapoor V, et al. Activated Tumor-Associated Macrophages and CD8+ T-cells are the Key Mediators of Anti-tumor Effects of the Vascular Disrupting Agent 5,6 Di-methylxanthenone-4-acetic Acid (DMXAA) in Murine Models of Lung Cancer and Mesothelioma. Cancer Res. 2005;65:11752–61. doi: 10.1158/0008-5472.CAN-05-1658. [DOI] [PubMed] [Google Scholar]
- 40.Gattinoni L, Klebanoff CA, Palmer DC, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1616–26. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Radoja S, Sai M, Frey AB. CD8+ tumor-infiltrating lymphocytes are primed for Fas-mediated activation-induced cell death but are not apoptotic in situ. J Immunol. 2001;166:6074–83. doi: 10.4049/jimmunol.166.10.6074. [DOI] [PubMed] [Google Scholar]
- 42.Radoja S, Saio M, Schaer D, et al. CD8+ Tumor-inflating T cells are deficient in perforin-mediated cytolytic activity due to defective microtubule-organizing center mobilization and lytic granule exocytosis. J Immunol. 2001;167:5042–51. doi: 10.4049/jimmunol.167.9.5042. [DOI] [PubMed] [Google Scholar]
- 43.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nature. 2001;7:1118–22. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 44.Bollard CM, Rossig C, Calonge MJ, et al. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99:3179–87. doi: 10.1182/blood.v99.9.3179. [DOI] [PubMed] [Google Scholar]
- 45.Lucas PJ, McNeil N, Hilgenfeld, et al. Transforming growth factor-beta pathway serves as a primary tumor suppressor in CD8+ T cell tumorgenesis. Cancer Res. 2004;64:6524–29. doi: 10.1158/0008-5472.CAN-04-0896. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Q, Yang X, Pins M, et al. Adoptive transfer of tumor-reactive transforming growth factor-beta-insensitive CD8+ T cells: Eradiation of autologous mouse prostate cancer. Cancer Res. 2005;65:1761–9. doi: 10.1158/0008-5472.CAN-04-3169. [DOI] [PubMed] [Google Scholar]
- 47.Lacuesta K, Buza E, Hauser H, et al. Assessing the safety of cytotoxic T lymphocytes transduced with a dominant negative transforming growth factor-beta receptor. J Immunother. 2006;29:250–60. doi: 10.1097/01.cji.0000192104.24583.ca. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Q, Yang X, Kundu SD, et al. Blockade of transforming growth factor-beta signaling in tumor-reactive CD8+ T cells activates the antitumor immune response cycle. Mol Cancer Ther. 2006;5:1733–43. doi: 10.1158/1535-7163.MCT-06-0109. [DOI] [PubMed] [Google Scholar]
- 49.Petersen CC, Petersen MS, Agger R, Hokland ME. Accumulation in tumor tissue of adoptively transferred T cells: a comparison between intravenous and intraperitoneal injection. J Immunother. 2007;29:241–9. doi: 10.1097/01.cji.0000203078.97493.c3. [DOI] [PubMed] [Google Scholar]
- 50.Kircher MR, Allport JR, Grages EE, et al. In vivo high resolution three-dimensional imaging of antigen-specific cytotoxic T-lymphocyte trafficking to tumors. Cancer Res. 2003;63:6838–46. [PubMed] [Google Scholar]
- 51.Pittet MJ, Grimm J, Berger CR, et al. In vivo imaging of t cell delivery to tumors after adoptive transfer therapy. Proc Nat Acad Sci. 2007;104:12457–61. doi: 10.1073/pnas.0704460104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vicari AP, Caux C. Chemokines in cancer. Cytokine Growth Factor Rev. 2002;13:143–54. doi: 10.1016/s1359-6101(01)00033-8. [DOI] [PubMed] [Google Scholar]
- 53.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 54.Maeda H, Tsuru S, Shiraishi A. Improvement of macrophage dysfunction by administration of anti-transforming growth factor-beta antibody in EL4-bearing hosts. Jpn J Cancer Res. 1994;85:1137–43. doi: 10.1111/j.1349-7006.1994.tb02919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-β signaling inhibitors for cancer therapy. Nat Rev Drug Discov. 2004;3:1011–22. doi: 10.1038/nrd1580. [DOI] [PubMed] [Google Scholar]
- 56.Arteaga CL. Inhibition of TGFbeta signaling in cancer therapy. Curr Opin Genet Dev. 2006;16:30–7. doi: 10.1016/j.gde.2005.12.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Characterization of Activated OT-1 Splenocytes
A. OT-1 splenocytes stimulated for seven days have the phenotypic profile of activated effector T cells.
Splenocytes were harvested from OT-1xgfp mice and activated with media containing the SIINFEKL peptide for two hours. Cells were stimulated with mIL-2 (20ng/ml) on Day 2, 4 and 6, harvested on Day 7 and analyzed with flow cytometry. Plots show expression of CD8 (X-axis) versus CD44, CD69, CD25, CD62L, V-α-2 TCR chain, and GFP (Y-axis). The number in the right upper quadrant represents the percentage positive of total cells. Cells were almost exclusively CD8+/V-α-2+/GFP+ and had an activated phenotype (CD44HI, CD69 HI, CD25 HI, CD62LLow)
B. Activated OT-1 effector cells demonstrate cytotoxic activity in vitro.
Splenocytes were harvested from OT-1 mice as above, activated for 7 days and reacted with chromium51-labeled tumor cells at varying effector to target (E:T) ratios. EG-7 cells express the target ova antigen, EL-4 cells do not. The activated OT-1 cells were highly efficient in lysing EG-7 cells, even at very low E:T ratios, while having no effect on the EL-4 cells.
Supplemental Figure 2. Splenocytes from adoptively transferred tumor-bearing animals expressed increased IFN-γ after stimulation with IL-2 alone, PMA and peptide plus IL-2.
Three days after transfer of 20 million activated, GFP-labeled OT-1 cells, spleens were isolated and digested. Cells were stimulated with IL-2, PMA/ionomycin, or IL-2 plus SIINFEKL peptide, and subjected to FACS to detect intracellular IFN-γ staining (X-axis is GFP, Y-axis is IFN-γ). The two left hand panels were treated with an unlabeled anti-IFN–γ antibody (blocking antibody) before addition of labeled antibody and used to identify non-specific binding and define gates. The percent of activated cells (GFP+/IFN-γ+) of the total GFP+ cells are shown in the upper right hand quadrant.