

Abstract
We describe the application of 59Co NMR to the study of naturally occurring cobalamins. Targets of these investigations included vitamin B12, the B12 coenzyme, methylcobalamin, and dicyanocobyrinic acid heptamethylester. These measurements were carried out on solutions and powders of different origins, and repeated at a variety of magnetic field strengths. Particularly informative were the solid-state central transition NMR spectra, which when combined with numerical line shape analyses provided a clear description of the cobalt coupling parameters. These parameters showed a high sensitivity to the type of ligands attached to the metal and to the crystallization history of the sample. 59Co NMR determinations also were carried out on synthetic cobaloximes possessing alkyl, cyanide, aquo, and nitrogenated axial groups, substituents that paralleled the coordination of the natural compounds. These analogs displayed coupling anisotropies comparable to those of the cobalamins, as well as systematic up-field shifts that can be rationalized in terms of their stronger binding affinity to the cobalt atom. Cobaloximes also displayed a higher regularity in the relative orientations of their quadrupole and shielding coupling tensors, reflecting a higher symmetry in their in-plane coordination. For the cobalamines, poor correlations were observed between the values measured for the quadrupole couplings in the solid and the line widths observed in the corresponding solution 59Co NMR resonances.
Keywords: vitamin B12, cobaloximes, solid-state NMR
Despite a very low biological abundance, cobalt plays a unique role in the metabolism of several living organisms. Humans, for instance, possess in the order of 1 mg of this metal distributed throughout all body cells, and yet supporting this minute quantity by the average ingestion of ≈100 ng of cobalt per day is essential for maintaining a normal physiology (1). The importance of cobalt comes from its participation in the B12 family of compounds, whose active forms are responsible for catalyzing a wide variety of processes related to nucleic acid, protein, and lipid syntheses, as well as for maintaining the normal function of epithelial and nervous cells (2, 3). Cobalamins also stand out as nature’s most complex nonpolymeric structures, one of its most stable organometallic complexes, and the only biomolecules exhibiting covalent metal-carbon bonds. In view of all of these factors it is not surprising that vitamin B12 and its derivatives have evolved into some of the most intensively studied systems in modern chemistry, with analyses related to their structures and functions leading to record-breaking achievements in several areas, including x-ray diffraction (4, 5), 1H NMR (6), 13C pulsed NMR (7), and chemical synthesis (8, 9). Given their low concentration and high complexity, however, important questions remain regarding the relation between the biological functions of B12 derivatives and their chemical structures (10, 11). These issues could be further clarified with the aid of tools capable of monitoring the electronic environment of the cobalt atom as well as the coordination changes that the metal undergoes when participating in biochemical transformations. The present study demonstrates the feasibility of carrying out detailed characterizations of this kind with the aid of a hitherto untested spectroscopic probe: solid-state cobalt NMR.
59Co, cobalt’s main magnetically active isotope, is a nucleus whose NMR observation should in principle be facile. It is 100% naturally abundant, possesses a relatively high magnetogyric ratio, and by virtue of the magnetic mixing of its occupied and excited d orbitals it may experience substantial paramagnetic deshieldings (>15,000 ppm) that will reveal even subtle changes in chemical environments (12). Complicating these measurements is the S = 7/2 character of the isotope, associated with a quadrupole moment that provides a very efficient relaxation mechanism and consequently leads to broad solution phase resonances of difficult observation. Though free from this tumbling-induced relaxation solid NMR observations also can be expected problematic, as the anisotropy of the quadrupole and shielding interactions will endow nuclei in different crystallites with widely different resonance frequencies. Yet as is demonstrated in this study, a number of simple experimental procedures allow one to collect quantitative solid-state 59Co NMR line shapes from cobalamin powders. These data can be obtained with good signal-to-noise ratios, and their interpretation provides a wealth of local coupling information pertaining the cobalt sites. In an effort to further analyze these data investigations were extended to synthetic cobaloximes, model compounds that like cobalamins possess an in-plane nitrogenated coordination and also can establish covalent cobalt-carbon axial bonds (13). These data, as well as the overall trends revealed by the measurements, are discussed in the following sections.
MATERIALS AND METHODS
Materials.
The B12 derivatives analyzed in this study are presented in Fig. 1. Although solid spectra of good quality could be recorded from these compounds as purchased (Sigma and Fluka), the NMR features changed on crystallizing them by slow evaporation from adequate solvents. These changes were explored by using H2O for the crystallization of vitamin B12, the B12 coenzyme, and methylcobalamin, and CHCl3 for the crystallization of the dicyanocobyrinic acid heptamethylester. Nearly saturated solutions using these solute/solvent combinations (≈5 mg/ml in H2O, 10 mg/ml in CHCl3) also were used in the acquisition of the 59Co liquid NMR spectra.
Figure 1.

Natural corrinoids analyzed in this study.
To obtain further insight into the cobalamin data, a series of analogs involving cobalt and the dimethylglyoximate anion (DH−) were prepared and analyzed by 59Co NMR. Axial ligands in these synthetic cobaloximes (Fig. 2) were chosen to reflect the alkyl, cyano, and benzimidazole substitutions in the natural B12 derivatives; a methyl/aquo form also was included because of its resemblance to aquocobalamin, as was a diamino form that can be considered as the parent compound of the series. This last complex was prepared following an early literature procedure (14); the methyl/base, methyl/aquo, and cyano/base cobaloximes were prepared based on procedures developed by Schrauzer (15); K[(CN)2Co(DH)2] was obtained by heating a solution of [(NH3)2Co(DH)2]Cl with KCN (16). After their preparation, all compounds were suitably purified, crystallized, and identified by 400 MHz 1H NMR and elemental analysis (Midwest Microlab, Indianapolis IN).
Figure 2.

Structures of the cobaloximes analyzed in this study, which include all the chemical substitution patterns of the investigated B12 derivatives.
NMR Measurements.
Solid-state 59Co NMR measurements were carried out at 25°C and at magnetic field strengths of 4.7, 7.1, and 11.8 Tesla, corresponding to Larmor frequencies of 48.0, 72.5, and 119.7 MHz. Room temperature cobalamin solution signals could be detected only at 11.8 Tesla, and therefore all liquid-state 59Co measurements are reported at this field. Acquisitions were carried out by using laboratory-built NMR instrumentation of similar design, based on broad-band homodyne spectrometers controlled by Tecmag pulse programmers and equipped with high-power amplifiers and probeheads. Given the short spin-lattice relaxation times observed in both the liquid and solid phases, a large number of scans (2–4⋅106) could be co-added into the final time-domain signals. All 59Co spectra were externally referenced to 1 M aqueous [Co(NH3)6]Cl3, and subsequently converted to ppm down-field from 1 M aqueous K3[Co(CN)6] for the sake of literature consistency. Single-pulse Bloch decays were used in the acquisition of solution data (π/2 pulses ≈ 2.5 μs), but a more sophisticated approach was used in the solid measurements. Because the most revealing cobalamin features arose from these data, we briefly dwell on their nature, mode of collection, and interpretation.
Solid-State 59Co NMR.
The dominant local interaction affecting solid-state 59Co NMR spectra is usually the quadrupolar coupling. To first-order this interaction endows all allowed Δm = ±1 NMR transitions except the −1/2 ↔ +1/2 one with a broadening proportional to the full strength of the quadrupole coupling constant (several MHz for cobalamins and cobaloximes), thus making them unobservable in powdered samples (17). The remaining central transition is much sharper than the rest, but is still affected by the quadrupole interaction via second-order effects (18) as well as by conventional shielding and dipolar anisotropies. Even in the absence of decoupling these dipolar effects are orders of magnitude smaller than the local couplings and can be considered merely as a residual broadening mechanism. This consideration allows one to express the observable frequencies of the central transition spectra as (19)
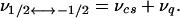 |
1 |
The first term in this equation represents the chemical shift and can be written (in Hz) as (20)
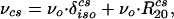 |
2 |
where νo is the site’s Larmor frequency, δisocs its isotropic shielding (in ppm), and R20cs is the irreducible spherical tensor component describing its shift anisotropy. This anisotropy usually is expressed in terms of second-rank Wigner matrices 𝒟ij(2)(Ωcs)
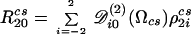 |
3 |
where the {ρ2ics}i=−2,2 are principal components defined by the chemical shift anisotropy parameter δanisocs and the shift asymmetry ηcs, and the Euler angles Ωcs describe the orientation between the external magnetic field and the principal axis system of the shielding tensor. The second term in Eq. 1 also can be expanded in spherical components as
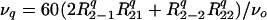 |
4 |
where the {R2jq}j=−2,2 elements depend again on a quadrupole coupling constant (e2qQ/h), a quadrupole asymmetry parameter ηq, and Euler angles Ωq. Rather than using two independent sets of powder angles (Ωcs and Ωq) it is convenient to express both quadrupole and shielding frequencies in terms of a single set; this simplification can be achieved by rewriting Eq. 3 as
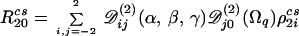 |
5 |
where (α, β,γ) denotes a structure-dependent transformation from the shielding to the quadrupole principal axes system. It follows from these arguments that the powder line shape arising from a single 59Co site will depend on eight a priori unknown parameters (δisocs, δanisocs, ηcs, e2qQ/h, ηq, α, β, γ), which can be determined by numerically fitting the experimental patterns while assuming an equiprobable Ωq distribution. An important aid in these determinations is variable magnetic field operation, as then the different νo dependences of νcs and νq (Eqs. 2 and 4) help remove ambiguities that otherwise may remain in the parameter fit. Another tempting alternative consists of collecting data while rapidly spinning the sample with respect to the external magnetic field (20, 21). When applied to most cobalamins, however, this approach resulted in little or no observable signals even when using fast spinning rates and week-long averaging times, probably as a result of a fast decay of the signals, which also conspired against the effective use of rotor-synchronized refocusing pulses. Consequently, we centered our attention on retrieving reliable static 59Co line shapes amenable to quantitative simulation. Although such powder spectra became visible for many of the samples in short acquisition times by using single, strong pulses, these line shapes were distorted and could not be fitted by using the idealized formalism described above. Part of these problems arose from the spectrometer’s dead-time (≈10 μs), a complication that was removed by using conventional spin-echo sequences (τecho = 30–50 μs) with an appropriate phase cycling (22). Distortions also arose from quadrupole-induced nutation effects that endowed different crystallites with unequal excitation angles (23); this complication was dealt with by using weak radio frequency fields (π/2 pulses ≈ 25 μs) so as to ensure that all spins fulfill the “strong coupling” regime and thus nutate at identical rates. A final complication originated in the large widths of the central transition resonances, which could only be partially excited by using a single on-resonance excitation pulse. This source of distortion was compensated by suitably co-adding into the final line shapes several subspectra recorded at different offsets, approximately 100 kHz from one another. Extensive tests on model 59Co compounds with known coupling parameters revealed that these precautions lead to highly reliable experimental line shapes; a complete description of this analytical procedure will be reported elsewhere.
RESULTS AND DISCUSSION
B12 Derivatives.
Fig. 3 illustrates representative 59Co solution NMR spectra recorded on the B12 derivatives introduced in Fig. 1, together with superimposed best fits of these line shapes to Lorentzian resonances, revealing their widths and isotropic shifts. Though recorded in different solvents, the basic trends displayed by these shifts can be understood in terms of classical “atom in a molecule” formalisms of 59Co NMR shielding. According to these crystal field models, peak positions are dictated by paramagnetic shielding contributions, which to a first approximation are inversely proportional to the ΔE separation between occupied and vacant 3d metal orbitals (24, 25). Therefore, as ligands increase their binding affinity and this crystal field splitting grows, up-field shifts for the 59Co resonances result. This shielding trend can be clearly followed in the benzimidazole-substituted series, where compounds with stronger alkyl ligands (methylcobalamin and adenosylcobalamin) exhibit their resonances up-field from the cyano-bound vitamin B12. Even the 250-ppm difference between the alkylated coenzymes is understandable in terms of crystal field splittings, as the smaller methyl group can be expected to act as a stronger ligand than the sterically hindered adenosyl moiety. As the benzimidazole group in vitamin B12 is replaced by a cyano in the dicyanocobyrinic derivative an even larger up-field shift occurs, as expected from the O < N < C ligand field strength that is predicted by the spectrochemical series. Notably, this detailed description of the changes undergone by the metal orbitals of B12 on axial substitution is not available from electronic spectra, as d-d metal transitions in cobalamins are masked by the much more intense π-π absorptions of the corrinoid rings.
Figure 3.

Solution 59Co NMR spectra of natural B12 derivatives. Superimposed on the experimental traces are best Lorentzian fits characterized by the isotropic shift δisocs and full-width-at-half-height (FWHH) values indicated on the right.
In addition to isotropic shifts, solution resonances can, in principle, provide information about the electronic field gradients surrounding the metal by analysis of their line widths (26). As mentioned earlier, however, a more direct alternative to measuring quadrupole interactions consists of analyzing the 59Co NMR line shapes observed in the solid. Fig. 4 presents these powder patterns as recorded at different magnetic fields, both for the B12 derivatives as originally purchased as well as after their crystallization from suitable solvents. Superimposed on these powder patterns are best-fit simulations obtained by a numerical iterative fit based on Eq. 1. Error margins in these simulations were estimated by making them depart from their optimized values until obtaining clearly inaccurate reproductions of the spectral line shapes; a summary of the resulting values is provided in Table 1.
Figure 4.

Superimposed experimental (thick lines) and simulated (thin lines) 59Co solid-state NMR spectra corresponding to the indicated B12 derivatives. (Left) Spectra recorded on the samples as purchased. (Right) Spectra recorded on samples crystallized by slow evaporation. Line shapes were fitted at all magnetic fields simultaneously as described in the text, by using the coupling parameters summarized in Table 1.
Table 1.
Solid-state 59Co NMR parameters measured for the B12 derivatives
| Compound | (e2qQ/h) (MHz) | ηq | δisocs* (ppm) | δanisocs (ppm) | ηcs | (α, β, γ) (degrees) |
|---|---|---|---|---|---|---|
| Vitamin B12† | 17.7 ± 0.6 | 0.2 ± 0.2 | 4,800 ± 100 | −800 ± 60 | 0.2 ± 0.2 | (135 ± 40, 45 ± 25, 45 ± 40) |
| B12 coenzyme† | 23.5 ± 0.6 | 1.0 ± 0.2 | 4,500 ± 100 | 850 ± 60 | 0.6 ± 0.2 | (45 ± 30, 160 ± 20, 45 ± 30) |
| Methylcobalamin† | 19.0 ± 0.6 | 0.4 ± 0.2 | 4,340 ± 120 | 1,690 ± 80 | 0.3 ± 0.2 | (45 ± 40, 150 ± 25, 50 ± 40) |
| Dicyanocobyrinic acid† heptamethylester | 26.7 ± 0.8 | 0.9 ± 0.4 | 4,300 ± 200 | −880 ± 100 | 0.7 ± 0.2 | (0 ± 35, 95 ± 35, 0 ± 35) |
| Vitamin B12‡ | 26.1 ± 0.4 | 0.1 ± 0.2 | 4,650 ± 100 | −650 ± 100 | 0.2 ± 0.2 | (45 ± 25, 40 ± 20, 20 ± 20) |
| B12 coenzyme‡ | 22.0 ± 2.0 | 0.7 ± 0.4 | 4,700 ± 300 | −1,000 ± 200 | 1.0 ± 0.4 | (45 ± 35, 75 ± 35, 0 ± 35) |
| Methylcobalamin‡ | 12.1 ± 0.4 | 0.3 ± 0.2 | 4,250 ± 100 | 1,760 ± 80 | 0.4 ± 0.2 | (25 ± 40, 60 ± 25, 0 ± 40) |
| Dicyanocobyrinic acid‡ heptamethylester | 26.7 ± 0.8 | 0.9 ± 0.4 | 4,300 ± 200 | −880 ± 100 | 0.7 ± 0.2 | (0 ± 35, 95 ± 35, 0 ± 35) |
Externally referenced to δK3[Co(CN)6] = 0 ppm.
As purchased samples.
Slowly crystallized samples.
According to the spectral fits all compounds exhibit similar isotropic chemical shifts in their liquid and solid phases, suggesting that no major chemical transformations affect them on dissolution. This finding is natural in view of the known stability of most diamagnetic B12 derivatives. Also evident from the data, however, are considerable changes in the solid-state NMR line shapes depending on the crystallization conditions of the compounds. Of all of the analyzed samples it is the vitamin B12 that exhibits the largest variations, including an increase in its quadrupole coupling and a sharpening of its powder singularities. Changes in molecular architecture for these compounds have in fact been characterized by x-ray diffraction, but these changes have involved conformational rearrangements of the macrocyclic substituents and changes in the water molecules of crystallization (4, 27). It is unlikely that structural variations taking place so far removed from the central metal positions will be capable of introducing the large line shape changes that are displayed by the solid 59Co NMR spectra. We consequently are inclined to ascribe these spectral variations to hitherto uncharacterized rearrangements of the axial ligands, or to conformational changes involving the cores of the corrinoid macrocycles. If further confirmed, this type of flexibility would provide additional support for mechanistic models that ascribe an important role in the dissociation of cobalt-carbon bonds to motions in the corrinoid rings (10). Furthermore, such structural variations would explain why nonmonotonic correlations emerge when comparing the 59Co solid-state quadrupole coupling constants with the line widths observed for the resonances of the various compounds in solution, where the cobalt ligands may be undergoing substantially different conformational dynamics.
In contrast to what happens with the quadrupole coupling tensors, the 59Co shielding tensors remain fairly constant before and after the crystallization of the various compounds. This behavior can be understood in terms of a weaker dependence of these tensors on the geometries of the chelates, coupled to a stronger dependence on their chemical constitution. From analyzing the changes undergone by the various principal elements of these shielding tensors we had expected to identify “in-plane” components that would be dictated by the corrinoid macrocycles, and “out-of-plane” components depending mainly on the axial substituents. The absence of such trend suggests that the principal axes of these shielding tensors bear no simple geometrical relations to the structures of the chelates. Additional evidence regarding this inability of the macrocycles to provide fixed reference frames for the coupling tensors is provided by the substantial variations undergone by the Euler angles describing their relative orientations, and by the large tensor asymmetry parameters observed for most derivatives. These characteristics are considerably different from the ones we recently have observed in the 59Co NMR of solid cobaltoporphyrins, where small shielding asymmetries always reflected a strong pseudo-C4 in-plane symmetry and where the two coupling tensors nearly coincided (28). Also worth noting are the much larger deshieldings, larger chemical shift anisotropies and smaller quadrupole couplings that characterized those aromatic compounds when compared with the B12 derivatives, indicating their overall unsuitability as models for the cobalamins. This behavior is quite different from the one exhibited by the glyoximate-based chelates, as further discussed in the next paragraph.
Cobaloximes.
Additional information regarding how the 59Co NMR data in Table 1 relate to B12’s electronic and molecular structures should become available from sufficiently accurate quantum chemical calculations. Although such efforts are under way, their reliable completion in systems as complex as cobalamins currently is not granted. Therefore we decided to complement the experimental B12 observations with comparisons to 59Co quadrupole and shielding parameters observed in cobaloximes (Fig. 2), compounds that have been extensively used as structural and chemical analogs of the natural corrinoids (10, 13). Representative solid-state 59Co NMR spectra acquired on these dimethylglyoximates are presented in Fig. 5, together with their best-fit simulations to Eq. 1. The quadrupole and shielding parameters arising from these simulations are summarized in Table 2, as are the solution 59Co NMR features observed for these derivatives (spectra not shown). It is worth noting that one of the compounds in this series, (CH3)(Py)Co(DH)2, previously had been analyzed by using pure quadrupole resonance techniques that afforded almost identical (e2qQ/h) and ηq values as our high-field NMR procedure (29).
Figure 5.

Superimposed experimental (thick lines) and simulated (thin lines) 59Co NMR powder line shapes obtained for the indicated cobaloximes as a function of external magnetic field. The spectral parameters arising from these line shape fits are summarized in Table 2.
Table 2.
Solid and solution phase 59Co NMR parameters measured for synthetic cobaloximes*
| Compound | (e2qQ/h) (MHz) | ηq | δiso,solidcs (ppm) | δaniso,solidcs (ppm) | ηcs | (α, β, γ)† (degrees) | δiso,solutioncs (ppm) | Solution width (FWHH, kHz) |
|---|---|---|---|---|---|---|---|---|
| (CH3)(Py)Co(DH)2 | 29.6 ± 0.4 | 0.1 ± 0.2 | 3,640 ± 100 | 700 ± 50 | 0.3 ± 0.2 | (30, 90, −10) | 3,645 ± 5 | 2.2 ± 0.2‡ |
| (CH3)(Im)Co(DH)2 | 32.1 ± 0.4 | 0.1 ± 0.2 | 3,620 ± 100 | 680 ± 50 | 0.4 ± 0.2 | (10, 90, 45) | 3,620 ± 5 | 3.7 ± 0.2‡ |
| (CN)(Py)Co(DH)2 | 27.7 ± 0.4 | 0.0 ± 0.2 | 4,100 ± 100 | −630 ± 50 | 0.5 ± 0.2 | (15, 90, 50) | 4,150 ± 10 | 11 ± 0.5§ |
| K[(CN)2Co(DH)2] | 40.2 ± 0.4 | 0.5 ± 0.2 | 3,200 ± 100 | −500 ± 50 | 0.5 ± 0.2 | (10, 0, 90) | 3,270 ± 20 | 31 ± 1.0§ |
| (CH3)(H2O)Co(DH)2 | 18.4 ± 0.4 | 1.0 ± 0.2 | 4,220 ± 100 | 430 ± 50 | 0.6 ± 0.2 | (20, 80, −10) | 4,220 ± 5 | 4.8 ± 0.2¶ |
| [(NH3)2Co(DH)2]Cl | 30.7 ± 0.4 | 0.6 ± 0.2 | 5,320 ± 100 | −1,570 ± 50 | 0.2 ± 0.2 | (0, 90, 90) | 5,371 ± 5 | 7.6 ± 0.3¶ |
All isotropic shifts externally referenced to δK3[Co(CN)6] = 0 ppm.
Average angular errors estimated at ± 25°.
Measured in acetone using ≈15 mg/ml solutions.
Measured in dimethylsulfoxide using ≈10 mg/ml solutions.
Measured in water using ≈7 mg/ml solutions.
The changes observed in the solution 59Co chemical shifts within the cobaloxime series conform to the trends expected from the spectrochemical series, as well as to the behavior exhibited by the corrinoids. Thus replacement of an axial methyl by a cyano group brings about a moderate down-field displacement of the resonance, replacement of axial cyano groups with nitrogenated bases introduces an additional 1,000 ppm down-field shift per substitution, and the even weaker H2O ligand shifts the 59Co peak by another 500 ppm with respect to a base. When comparing the isotropic chemical shifts of B12 and of cobaloxime chelates with similar axial substituents (cyano/base, alkyl/base, cyano/cyano) the latter consistently appear between 500 and 800 ppm further up-field than the former, indicating that the ligating ability of corrinoid macrocycles is on average weaker than that of the dimethylglyoximates. The widths of the cobaloxime solution resonances are also generally smaller than those of the B12 derivatives, probably reflecting differences in the overall correlation times of these molecules.
The features displayed by the solid-state cobaloxime spectra resemble those observed for the natural B12 derivatives, even if sharper powder singularities point toward a higher degree of crystallinity throughout the former samples. Although shielding anisotropies in both cobaloximes and cobalamins are also very similar, quadrupole couplings are consistently smaller in the latter, probably as a result of cobalt’s weaker binding to the corrinoid rings (27, 30). Another spectral difference between the two series of compounds concerns the Euler angles between their shielding and quadrupolar 59Co tensors, whose values showed no definite trends in the cobalamins but possess z-axes that are nearly orthogonal or parallel to one another in the cobaloximes. This difference is likely a reflection of the higher symmetry characterizing the in-plane cobaloxime ligands, which when combined to a stronger metal binding affinity leads to more regular patterns in the orientations of the principal axes systems.
CONCLUSIONS
The results presented in the preceding section demonstrate the potential usefulness of 59Co NMR for studying vitamin B12, its natural derivatives, and chemical analogs. In spite of the structural complexities of some of these compounds and of the presence of large quadrupole and shielding couplings, powder spectra with good signal-to-noise ratios could be recorded even at low magnetic field strengths. In fact, a number of circumstances combined to endow these solid-state spectra with a much higher quality than their solution counterparts, so that their acquisition on larger B12 holoenzymatic complexes should be feasible. The quantitative interpretation of these spectra was made possible by the combined use of suitable NMR protocols and numerical simulations, which provided insight into the structures of the complexes and binding affinities of the various ligands. These results have been analyzed so far only qualitatively in terms of classical 59Co crystal field models, but further information regarding ground and excited state wave functions should become available from more detailed quantum chemical calculations.
The 59Co spectral changes observed throughout the cobalamin samples suggest the presence of a hitherto uncharacterized conformational variability in the solid phases of these derivatives. The elucidation of these structures is worth pursuing by both traditional crystallization techniques, as well as by using sample spinning NMR methods capable of enhancing the spectral resolution (31, 32) and of yielding internuclear distance information (33). Also worth exploring is the potential presence of molecular dynamics in these solids, as well as the apparent discrepancies arising when comparing the solution resonance line widths with the magnitudes of the solid state quadrupole coupling tensors. We currently are attempting to address these issues by using a combination of solution and solid-phase variable-temperature NMR.
Acknowledgments
This work was supported by the National Science Foundation through Grants DMR-9420458 and CHE-9502644 (CAREER Award). A.M. acknowledges the University of Illinois at Chicago for a Dean Fellowship. L.F. is a Camille Dreyfus Teacher-Scholar Awardee (1996–2001), Beckman Young Investigator (1996–1998), University of Illinois Junior Scholar (1996–1999), and Alfred P. Sloan Fellow (1997–2000).
Note Added in Proof
After this manuscript was submitted, Prof. W. P. Power and coworkers (University of Waterloo) reported a single crystal 59Co NMR analysis of vitamin B12, which included coupling parameters that are remarkably similar to the ones we observe for the recrystallized powdered sample. We are grateful to Prof. Power for sharing these data with us.
References
- 1.Young R S. Cobalt in Biology and Biochemistry. New York: Academic; 1979. [Google Scholar]
- 2.Lippard S J, Berg J M. Principles in Bioinorganic Chemistry. Mill Valley, CA: Univ. Sci. Books; 1994. [Google Scholar]
- 3.Dolphin D, editor. B12. New York: Wiley; 1982. [Google Scholar]
- 4.Hodgkin D C, Kamper J, Mackay M, Pickworth J, Trueblood K N, White J G. Nature (London) 1956;178:64–66. doi: 10.1038/178064a0. [DOI] [PubMed] [Google Scholar]
- 5.Hodgkin D C. In: Vitamin B12. Zagalak B, Friedrich W, editors. Berlin: de Gruyter; 1979. pp. 19–36. [Google Scholar]
- 6.Hill, H. A. O., Pratt, J. M. & Williams, R. J. P. (1965) J. Chem. Soc. 2859–2865. [DOI] [PubMed]
- 7.Doddrell D, Allerhand A. Proc Natl Acad Sci USA. 1971;68:1083–1088. doi: 10.1073/pnas.68.5.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woodward R B. Pure Appl Chem. 1973;33:145–177. doi: 10.1351/pac197333010145. [DOI] [PubMed] [Google Scholar]
- 9.Eschenmoser A. Naturwissenschaften. 1974;61:513–525. doi: 10.1007/BF00606511. [DOI] [PubMed] [Google Scholar]
- 10.Halpern J. Science. 1985;227:869–875. doi: 10.1126/science.2857503. [DOI] [PubMed] [Google Scholar]
- 11.Drennan C L, Huang S, Drummond J T, Matthews R G, Ludwig M L. Science. 1994;266:1669–1674. doi: 10.1126/science.7992050. [DOI] [PubMed] [Google Scholar]
- 12.Harris R K, Mann B E. NMR and the Periodic Table. New York: Academic; 1970. [Google Scholar]
- 13.Schrauzer G N. Acc Chem Res. 1968;1:97–103. [Google Scholar]
- 14.Nakatsuka Y, Iinuma H. Bull Chem Soc Japan. 1936;11:48–54. [Google Scholar]
- 15.Schrauzer G N. Inorg Syn. 1968;11:61–70. [Google Scholar]
- 16.Maki N. Bull Chem Soc Japan. 1965;38:2013–2015. [Google Scholar]
- 17.Abragam A. The Principles of Nuclear Magnetism. Oxford: Oxford Univ. Press; 1962. [Google Scholar]
- 18.Cohen M H, Reif F. Solid State Phys. 1957;5:321–438. [Google Scholar]
- 19.Silver A H, Bray P J. J Chem Phys. 1958;29:984–990. [Google Scholar]
- 20.Haeberlen U. High Resolution NMR in Solids. New York: Academic; 1976. [Google Scholar]
- 21.Wooten E W, Muller K T, Pines A. Acc Chem Res. 1992;25:209–215. [Google Scholar]
- 22.Kunwar A C, Turner G L, Oldfield E. J Magn Reson. 1986;69:124–127. [Google Scholar]
- 23.Samoson A, Lippmaa E. Chem Phys Lett. 1983;100:205–208. [Google Scholar]
- 24.Freeman R, Murray G R, Richards R E. Proc R Soc London. 1957;A242:455–467. [Google Scholar]
- 25.Mason J. Chem Rev. 1987;87:1299–1312. [Google Scholar]
- 26.Spiess H W. Rotation of Molecules and Nuclear Spin Relaxation. New York: Springer; 1978. [Google Scholar]
- 27.Glusker J P. In: B12. Dolphin D, editor. New York: Wiley; 1982. pp. 23–106. [Google Scholar]
- 28.Medek A, Frydman V, Frydman L. J Phys Chem B. 1997;101:8959–8966. [Google Scholar]
- 29.LaRossa R A, Brown T L. J Am Chem Soc. 1974;96:2072–2081. [Google Scholar]
- 30.Rossi M, Glusker J P, Randaccio L, Summers M F, Toscano P J, Marzilli L G. J Am Chem Soc. 1985;107:1729–1738. [Google Scholar]
- 31.Schaefer J, Stejskal E O. J Am Chem Soc. 1976;98:1031–1032. [Google Scholar]
- 32.Frydman L, Harwood J S. J Am Chem Soc. 1995;117:5367–5368. [Google Scholar]
- 33.Guillon T, Schaefer J. Adv Magn Reson. 1989;13:57–83. [Google Scholar]