

Abstract
ADP-ribosylation factor (ARF) proteins are involved in multiple intracellular vesicular transport pathways. Most studies have focused on the functions of ARF1 or ARF6 and little is known about the remaining ARF isoforms. Although the mammalian ARF proteins share a high degree of sequence identity recent evidence has indicated that they may control distinct trafficking steps within cells. A unanswered issue is the degree of specificity of ARF family members for different interacting proteins. To investigate potential functional differences between the human ARF proteins we have examined the localisation of all human ARF isoforms and their interactions with two ARF1 binding proteins, neuronal calcium sensor-1 (NCS-1) and phosphatidylinositol-4 kinase-IIIβ (PI4Kβ). Use of a fluorescent protein fragment complementation method showed direct interactions between ARFs 1, 3, 5 and 6 with NCS-1 but at different intracellular locations in live HeLa cells. Photobleaching experiments indicated that complementation did not detect dynamic changes in protein interactions over short time scales. A more specific interaction between ARFs 1/3 and PI4Kβ was observed. Consistent with these latter findings ARF1 but not ARF5 or 6 enhanced the stimulatory effect of PI4Kβ on regulated exocytosis suggesting a specific role for class I ARFs in the regulation of PI4Kβ.
Keywords: Golgi, exocytosis, secretion, GTPase, membrane traffic, lipid synthesis, fluorescence complementation
Introduction
ADP-ribosylation factor (ARF) proteins are small GTPases that are essential regulators of many intracellular vesicular trafficking pathways (1). The five human ARF isoforms (ARF2 is not present in the human genome) fall into three categories based on sequence similarities and comprise the class-I ARFs (ARFs 1 and 3), the class-II isoforms (ARFs 4 and 5) and a single class-III protein (ARF6). The most comprehensively studied ARF proteins are ARF1 and ARF6 (2). ARF1 is a key component in the formation of a number of different vesicular coat complexes and is essential to the dynamic assembly/disassembly of AP-1, AP-3, AP-4 and GGA1-3 based vesicle coats (3, 4). Through the dynamic control of AP-1 and AP-3 coat assembly at the trans-Golgi-network (TGN), ARF1 represents a critical regulator of cargo trafficking from this sorting hub to both the plasma-membrane and endosomes. ARF1 also exhibits a second Golgi localised trafficking function based on its ability to recruit soluble phosphatidylinositol-4 kinase IIIβ (PI4Kβ) (5). ARF6 exhibits a sub-cellular localisation pattern distinct to that of ARF1 and is distributed predominantly to the plasma-membrane and endosomes (6). This isoform recruits AP-2 (7) and is central to several aspects of cell surface receptor trafficking between the plasma-membrane and endosomal systems, a function that is dependent on its second well characterised activity in the regulation of actin based cytoskeleton (8). ARF1 and ARF6, in addition to possession of distinct effector proteins/pathways have also been found to display overlap with respect to effectors including phospholipase D (9) and phosphatidylinositol-4 phosphate 5-kinase (10) although isoform preference by phospholipase D has been observed (11). It is likely that the functional relevance of such common interactions in a physiological setting will depend on the relative expression levels of particular ARFs and the extent to which they co-distribute with a particular effector.
There is a paucity of data relating to the remaining ARF isoforms. A recent report analysing loss of function has indicated, however, that they are essential components in ARF mediated cellular activities and furthermore that specific ARF isoform pairs may act in tandem to regulate exclusive trafficking pathways (12). There are reports suggesting that the class-II isoforms, ARFs 4 and 5, do have unique effector targets including Arfophilin (13, 14), and rhodopsin (15) but the absolute selectivity of these interactions has not been fully investigated with the class-I and class-III proteins. Similarly, no studies thus far have attempted to ascertain if ARF1/ARF6 specific effectors also represent genuine targets for other ARF isoforms. Since all five human ARF proteins are highly homologous (>70% sequence identity between the most divergent isoforms ARF1 and ARF6) and taking into consideration the lack of experimental evidence concerning specificity of particular ARFs for effectors, we have attempted to address these issues. Firstly, we have compared the sub-cellular distributions of all five human ARFs expressed in live HeLa cells. Following on from this we have analysed the co-localisation and binding specificity of the human ARF isoforms for the documented ARF1 binding partners neuronal calcium sensor-1 (NCS-1) (16) and PI4Kβ (5, 17) through use of a fluorescence protein complementation methodology (18-20). Other techniques exist for examining protein-protein interactions in live cells such as fluorescence resonance energy transfer or FRET (21) but standard FRET protocols are open to numerous artefacts and variations of the method designed specifically to negate these issues such as acceptor bleaching (22) have also recently been exposed as open to potential artefact (23). We chose fluorescence fragment complementation for use in this study as it provides direct information concerning the presence of protein-protein interactions, can be used in live cells and requires no additional post-capture data manipulation to correct for potential artefacts. A previous study has shown an interaction between ARF1 and the exchange factor GBF1 using this approach (24).
The results presented indicate that human ARFs 1, 3, 4 and 5 all have unique yet somewhat overlapping sub-cellular distributions with ARF6 displaying the most divergent cellular localisation pattern by virtue of its strong plasma membrane association. All of the ARFs tested co-localised to a greater or lesser extent with NCS-1 and ARFs 1, 3, 5 and 6 were all found to directly bind to this protein based on fluorescence complementation analysis. The specificity of the ARFs was found to be different with respect to PI4Kβ interaction and only the class-I isoforms ARF1 and ARF3 were observed to bind this enzyme and reconstitute fluorescence. The functional significance of this observed specificity was confirmed in an assay of regulated exocytosis. These results provide evidence for a direct interaction between ARF family proteins and NCS-1 and PI4Kβ in live cells and also highlight a previously undetected promiscuity in binding to NCS-1 (17) but greater specificity in binding to PI4Kβ. These data are consistent with the idea that ARF proteins, although highly similar, are likely to control specific aspects of cellular function.
Results
Co-localisation of all human ARF isoforms with NCS-1
In order to characterise the ARF and NCS-1 constructs used in this study and to permit comparisons with split EYFP variants in later analyses we first examined their subcellular distributions in live HeLa cells when expressed alone. As both NCS-1 and ARF proteins are N-terminally myristoylated all constructs had tags added at the C-terminus. Figure 1 displays representative images of HeLa cells expressing ARFs 1, 3, 4, 5 and 6 tagged to full length EYFP and NCS-1 tagged with full length ECFP. We did not see any evidence for changes in cell morphology or changes in Golgi organisation (not shown) following over-expression of the proteins. NCS-1-ECFP exhibited a characteristic peri-nuclear as well as plasma membrane localisation consistent with previous reports (25, 26). ARF1-EYFP displayed a predominantly peri-nuclear distribution in addition to labelling of randomly distributed cytosolic puncta which may reflect the presence of Golgi derived coated transport vesicles. ARF-EYFPs 3 and 5 exhibited a similar more diffuse cytosolic/nuclear signal but with a concentration of both proteins observable in a peri-nuclear region. ARF4-EYFP appeared to concentrate largely to a Golgi like locale and there was little cytosolic/nuclear fluorescence with this protein. ARF6-EYFP distributed primarily to the plasma membrane as previously shown (27) and was also apparent on cytosolic punctate structures potentially consistent with its ability to associate with endosomal membranes (28). These localisation data are largely consistent with a previous study of ARF protein distributions in fixed vero cells (29). An important point is that over-expression of the tagged ARF constructs did not result in any case in detectable changes in cell morphology or cell behaviour.
Figure 1.
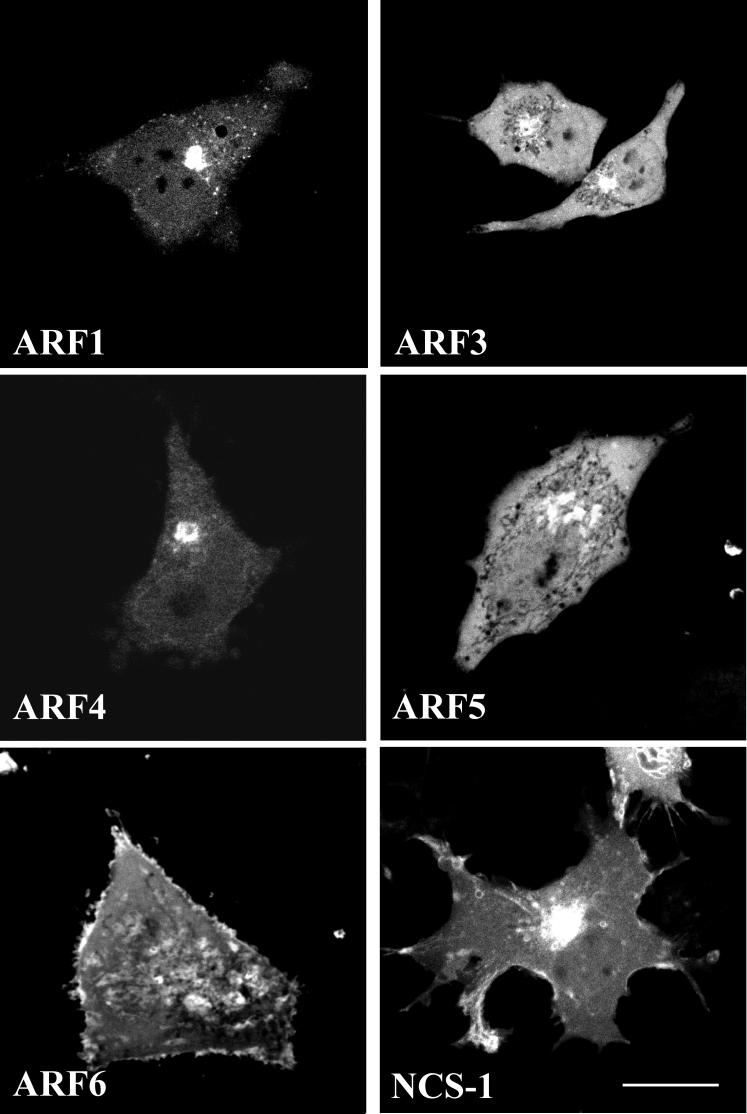
Sub-cellular localisation of human ARF proteins and NCS-1 in live HeLa cells.
Representative images of HeLa cells transfected with Human ARF 1, 3, 4, 5 and 6 proteins tagged with full length EYFP and NCS-1 with full length ECFP. Protein distributions were analysed 24 hrs post transfection by confocal microscopy. Scale bar, 10 μm.
When co-expressed with NCS-1, the distributions of the various ARF-EYFP proteins analysed remained unaltered compared to the ARF-EYFPs expressed alone (Figure 2). While some variability between cells was seen in the distribution of NCS-1 to the Golgi or plasma membrane, examination of a large number of cells showed that co-expression with any of the ARF-EYFP proteins tested did not consistently affect the characteristic Golgi/plasma membrane distribution of NCS-1-ECFP (Figure 2). NCS-1-ECFP exhibited some co-localisation with ARF1-EYFP at the Golgi complex although notably no co-localisation was observed between these proteins at the plasma membrane (labelled by NCS-1-ECFP) or cytosolic puncta (labelled by ARF1-EYFP) (Figure 2a). ARF-EYFPs 3, 4 and 5 co-localised extensively with NCS-1-ECFP in a peri-nuclear region, most likely the Golgi complex (Figure 2b, c and d) and ARF6-EYFP exhibited significant co-localisation with NCS-1 at the plasma membrane and to a lesser extent at an intra-cellular peri-nuclear compartment (Figure 2e). These data are consistent with the presence of NCS-1-ECFP and ARF-EYFP 1, 3, 4 and 5 at a Golgi locale and with ARF6-EYFP primarily localising to the plasma membrane and endosomal compartments.
Figure 2.
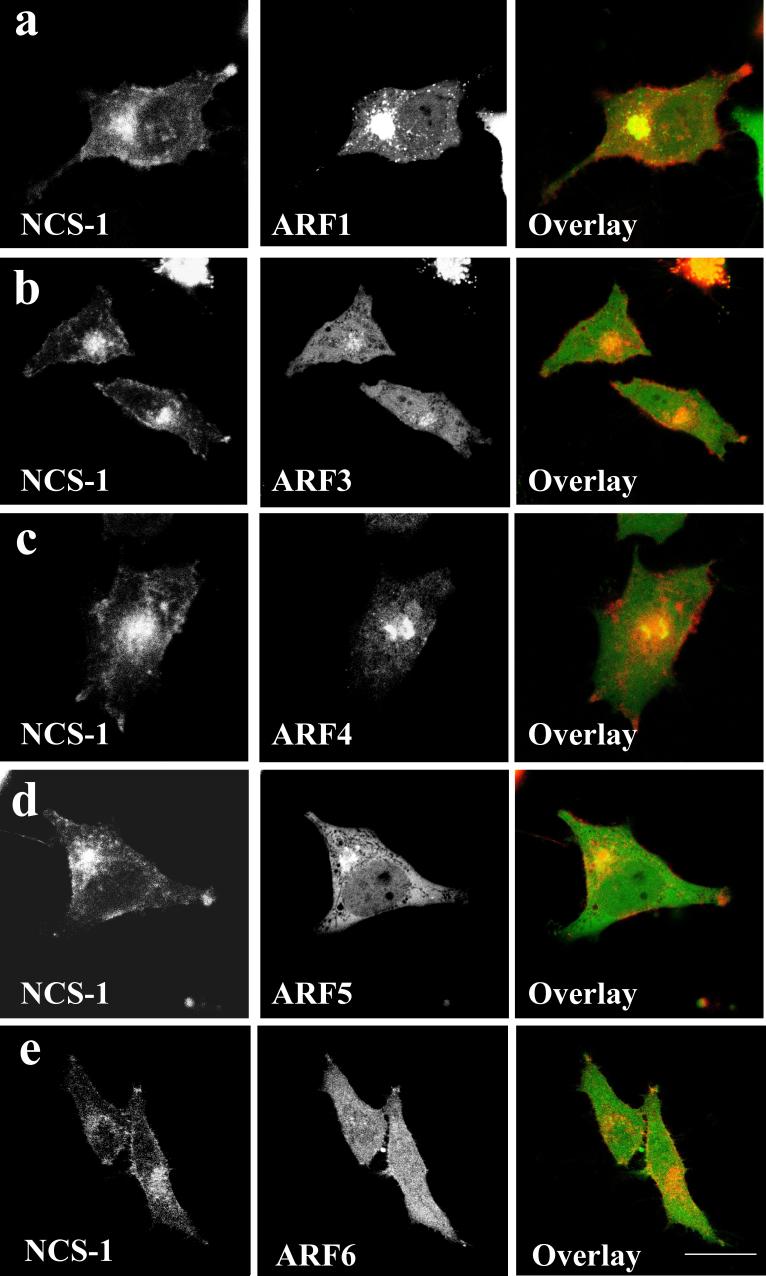
Co-localisation of NCS-1-ECFP with ARFs 1, 3, 4, 5 and 6-EYFP in live HeLa cells.
Representative images are shown of HeLa cells co-expressing NCS-1/ARF1 (a), NCS-1/ARF3 (b), NCS-1/ARF4 (c), NCS-1/ARF5 (d) and NCS-1/ARF6 (e). Protein distributions were determined using confocal microscopy 24 hrs post-transfection and extent of co-localisation shown by overlaying EYFP (Green) and ECFP (Red) channels from the same cell. Regions of co-localisation appear yellow in overlay images. Scale bar, 10 μm.
Use of fluorescence fragment complementation to examine direct interaction of NCS-1 with ARF1 in live cells
Initial data indicated a possible co-localisation between tagged NCS-1 and ARF1 (Figure 2) consistent with a previously observed direct interaction from in vitro and functional studies (17). We next employed a fluorescent protein fragment complementation approach in an attempt to identify direct protein binding in live cells. Vectors encoding an N-terminal fragment of EYFP (YFP-YN, residues 1-158) and a shorter C-terminal fragment (YFP-YC, residues 159-239) were generated as described in Materials and Methods. Cells were co-transfected with an ECFP-Golgi marker to verify transfection and to allow visualisation of the localisation of the Golgi. The Golgi marker contained the N-terminus of beta 1,4-galactosyltransferase which allows trans- and medial-Golgi targeting. When both empty parent vectors were co-transfected into HeLa cells along with the ECFP-Golgi marker to verify transfection, no detectable EYFP signal was observed (Figure 3a). ARF proteins were sub-cloned into the YFP-YC vector and, as a control, transfection of cells with the ARF1-YC construct alone failed to generate detectable EYFP fluorescence (Figure 3, b). All NCS-1 constructs were sub-cloned into YFP-YN and the reciprocal control of transfecting cells with NCS-1-YN alone again failed to generate a detectable EYFP signal (Figure 3c). In contrast, when NCS-1-YN and ARF1-YC were co-expressed an EYFP signal was reconstituted (Figure 3d) that showed co-localisation with the co-transfected CFP tagged Golgi marker. The fluorescence signal from the reconstituted EYFP was notably less intense than that seen with full length EYFP constructs but the efficiency of the fluorescence complementation was revealed by the fact that a signal was observed to a greater or lesser extent in essentially all transfected cells expressing the CFP-Golgi marker.
Figure 3.
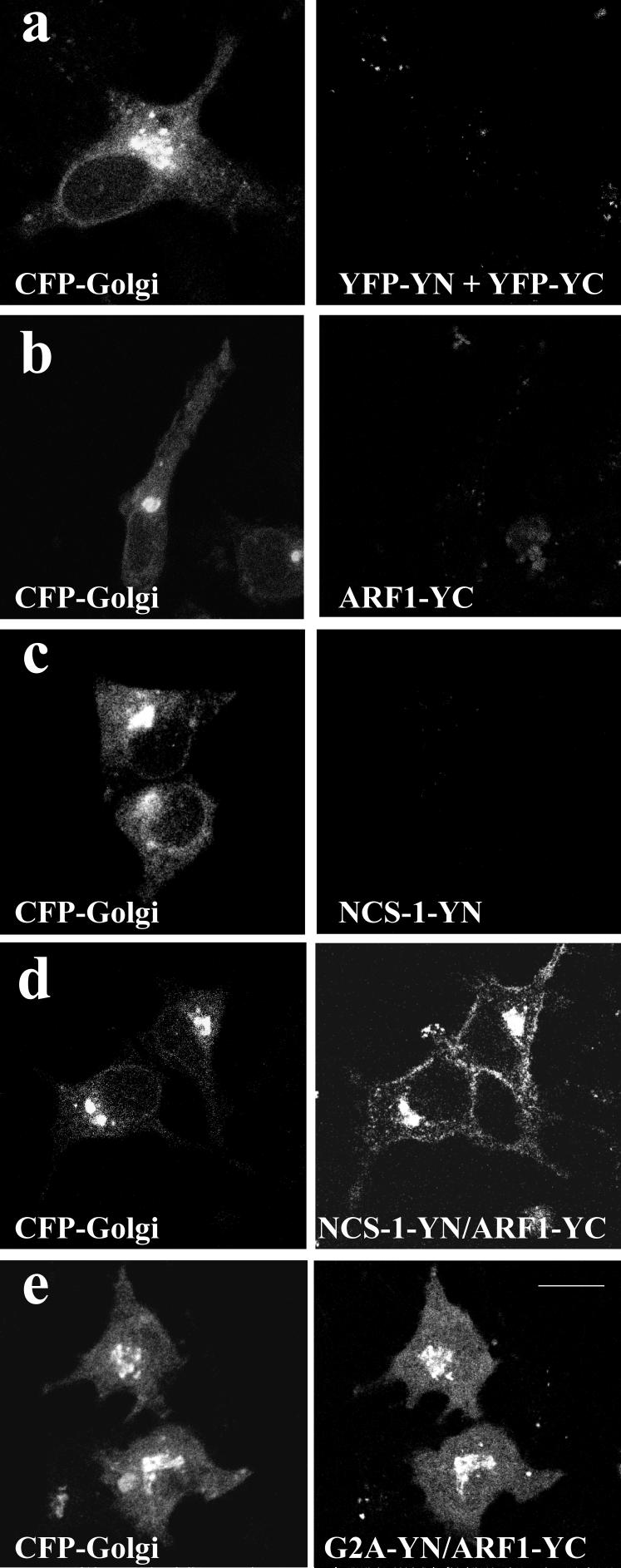
Fluorescent protein fragment reconstitution analysis to examine direct interaction between NCS-1 and ARF1 in live HeLa cells.
HeLa cells were transfected with a ECFP-tagged Golgi marker along with both empty split-EYFP cloning vectors (a), an ARF1-YC fragment construct alone (b), an NCS-1 fragment construct alone (c), NCS-1-YN and ARF1-YC in combination (d) and a construct encoding the myristoylation deficient G2A mutant of NCS-1 (e). ECFP (Red) and EYFP (Green) channels from individual cells were combined to show extent of co-localisation of reconstituted EYFP signal with the Golgi marker (yellow regions in overlay images). Scale bar, 10 μm.
Intriguingly, reconstituted EYFP fluorescence was also observed at the plasma membrane in cells co-expressing NCS-1-YN and ARF1-YC. We do not believe that this plasma-membrane localisation is simply an artefact of protein over-expression since it is not observed in cells co-expressing NCS-1 and ARF1 tagged with full length fluorescent proteins (Figure 2). We suggest, therefore, that this plasma membrane signal may represent a site of low affinity or transient interaction between NCS-1 and ARF1 that has previously evaded detection but which is efficiently visualised using this technique as the complex is trapped by interaction of refolded EYFP fragments and therefore is able to accumulate over time. We next decided to investigate the potential of a mutant form of the NCS-1 protein, NCS-1G2A, to generate reconstituted EYFP fluorescence when co-expressed with ARF1. The G2A mutation destroys the myristoylation site in NCS-1 which is essential for membrane association of the protein (25, 26) and as a result this mutant exhibits a diffuse cytosolic distribution (25). Cells expressing NCS-1G2A-YN and ARF1-YC were able to reconstitute EYFP fluorescence at the level of the Golgi complex however, in contrast to NCS-1-YN/ARF1-YC expressing cells, no fluorescence signal was detectable at the plasma membrane (Figure 3, e). These results would be consistent with ARF1-YC at the Golgi recruiting soluble NCS-1G2A-YN, with the trapped complex becoming apparent with accumulation over time. Unlike the situation with NCS-1-YN however, no plasma membrane signal was detected since neither ARF1-YC nor NCS-1G2A-YN are localised to this site when expressed alone.
Examination of high resolution images revealed that the reconstituted NCS-1-YN/ARF1-YC EYFP signal exhibited a discrete and highly localised sub-cellular distribution over the Golgi complex (Figure 4a). Interestingly, the reconstituted NCS-1-YN/ARF1-YC EYFP co-localised with the trans/medial Golgi marker on many but not all Golgi structures in most of the cells examined suggesting differential localisation of the reconstituted EYFP within the Golgi complex. It is unclear to what extent fluorescence fragment complementation is a slow irreversible process or whether it could be used to probe protein dynamics in living cells. We addressed this using by using fluorescence recovery after photobleaching (FRAP). Use of this technique has demonstrated that Golgi-localised fluorescently-tagged ARF1 rapidly recovers on the Golgi complex following photobleaching due to its recycling between the Golgi complex and the cytosol. To see if reconstituted NCS-1-YN/ ARF1-YC EYFP could rapidly form by recruitment from a cytosolic pool of the proteins, the characteristics of the signal were followed using a FRAP protocol. In control cells expressing ARF1-EYFP in the presence of NCS-1-ECFP, rapid recovery of ARF1-EYFP fluorescence was observed on the Golgi complex (Figure 4b) as previously described for ARF-1-GFP expressed alone (30). The main component of the recovery process occurred with a half-time of 10 seconds. In contrast, following photobleaching of the reconstituted NCS-1-YN/ARF1-YC EYFP fluorescence, only a very minor fast component of recovery was seen probably due to cytosolic diffusion followed by a slow decline attributable to photobleaching during imaging. No additional recovery on the Golgi complex was observed over the same time that ARF1-EYFP recovered. It is possible that the bleached reconstituted complexes are immobile on the Golgi complex, otherwise these data suggest that fluorescence fragment complementation may be relatively slow and not occur fast enough for it to be used in dynamic imaging over short time scales.
Figure 4.

Characterisation of the reconstituted NCS-1-YN/ ARF1-YC EYFP fluorescence on the Golgi complex.
(a) HeLa cells co-expressing NCS-1-YN/ ARF1-YC were examined and high resolution images of the reconstituted signal in cells co-transfected with Golgi-ECFP are shown. Expanded images of the boxed Golgi complex regions are shown in the inserts. The scale bars represent 5 μm or 1 μm in the insert. (b) HeLa cells were transfected with ARF1-EYFP and NCS-1-ECFP of NCS-1-YN and ARF1-YC EYFP. To determine the dynamics of ARF1-EYFP and the reconstituted NCS-1-YN/ ARF1-YC EYFP fluorescence, cells were imaged and the Golgi complex was photobleached using local high intensity laser light (at the time indicated by the arrow). Fluorescence intensity in the region of interest over the Golgi complex was monitored before and after photobleaching. The data were normalised to the initial fluorescence intensity for each cell and the recovery of fluorescence over the Golgi was monitored over time. The data are shown as mean ± SEM for 13 (ARF1-EYFP) or 14 (NCS-1-YN/ ARF1-YC) cells. The data for ARF1-EYFP was fitted to a double hyperbolic function (blue line) suggesting two components of recovery with half times of around 1 and 10s.
Direct interaction of NCS-1 with ARFs 3, 5 and 6 in live HeLa cells
Due to the potential co-localisation of the various ARF isoforms with NCS-1 (Figure 1), we tested the specificity of ARFs 3, 4, 5 and 6 for interaction with NCS-1. Co-expression of NCS-1-YN with ARFs 3, 5 and 6-YC in HeLa cells resulted in efficient reconstitution of EYFP fluorescence (Figure 5). ARF1-YC/NCS-1-YN expressing cells again show interaction at both the Golgi and plasma membrane (Figure 3a). ARF3 appeared to interact with NCS-1 predominantly at the Golgi complex however an interaction was also observed to a lesser extent on large cytosolic puncta which could represent Golgi membranes or distinct membrane bound organelles (Figure 5b). Co-expression of ARF4-YC and NCS-1-YN failed to generate a detectable EYFP fluorescence signal (Figure 5c) although it should be noted that ARF4-YC expression levels are significantly lower than for the other ARF proteins analysed in this study (Fig 5f) and hence we cannot formally rule out a direct interaction between the two proteins based on this data alone. NCS-1-YN and ARF5-YC co-expression also led to reconstitution of EYFP fluorescence although in this case the interaction appeared exclusively at the Golgi complex and was not observed on other cytosolic structures or the plasma membrane (Figure 5d). ARF6-YC also directly interacted with NCS-1-YN however in contrast to all other ARF isoforms tested that displayed a reconstitution of EYFP fluorescence, the site of interaction appeared predominantly at the plasma membrane. EYFP fluorescence could also be detected in ARF6-YC/NCS-1-YN cells on intracellular structures but the distribution of these sites did not overlap with the co-expressed Golgi marker (Figure 5e). These observations are consistent with findings showing that ARF1 and ARF6 do share extensive overlap in binding to other known effectors including phospholipase-D and phosphatidylinositol-4 phosphate 5-kinase (2).
Figure 5.

Specificity of human ARF proteins for binding to NCS-1 in live HeLa cells as determined by fluorescent protein fragment reconstitution and determination of protein expression levels by western blotting.
An NCS-1-YN fluorescent protein fragment construct was co-transfected into HeLa cells with ARF fragment constructs encoding ARF1 (a), ARF3 (b), ARF4 (c), ARF5 (d) or ARF6 (e). An ECFP-Golgi marker construct was simultaneously co-transfected. ECFP (Red) and EYFP (Green) channels from individual cells were combined to show extent of co-localisation of reconstituted EYFP signal with the Golgi marker (yellow regions in overlay images). Scale bar, 10 μm. Determination of fluorescent fragment protein expression levels by Western blotting (f).
Co-expression of split YFP constructs does not affect expression levels of either NCS-1 or ARF proteins
To check that co-expression of the proteins employed in this study did not affect their relative expression levels, which might provide an explanation for differences in observed sub-cellular localisation patterns, we analysed protein expression of all constructs by western blotting (Figure 5f). Levels of expressed NCS-1-YN remained essentially constant when co-expressed with either ARFs 1, 3, 4, 5 or 6 (Fig 5f). Similarly, the expression level of NCS-1G2A-YN was comparable to that of NCS-1-YN when both proteins were co-expressed with ARF1-YC (Figure 5f). The expression levels of all ARF-YC constructs when co-expressed with NCS-1-YN were found to be generally comparable with the exception of ARF4-YC which expressed at significantly lower levels than other ARFs (Figure 5f). The level of ARF1-YC expression with NCS-1G2A-YN and was found to be similar to that observed when expressed with NCS-1-YN.
Use of fluorescence fragment complementation to test a direct interaction between PI4Kβ and ARF1 in live HeLa cells
PI4Kβ is a common effector for both ARF1 and NCS-1 (5, 17, 31) and we therefore extended our study to attempt to detect interaction between PI4Kβ and ARF proteins in live cells. As an initial step, we characterised the cellular distribution of PI4Kβ tagged to ECFP and its possible co-localisation with ARF1-EYFP in HeLa cells (Figure 6). When expressed in isolation, PI4Kβ-ECFP exhibited a diffuse cytosolic signal in addition to a reproducible concentration of the protein in a peri-nuclear region (Figure 6a). Co-expression with ARF1-EYFP did not alter the cellular distribution of PI4Kβ-ECFP and a partial co-localisation was detectable only at a peri-nuclear locale (Figure 6b). The PI4Kβ coding sequence was sub-cloned into YFP-YN for co-expression with ARF1-YC. Co-expression of ARF1-YC with PI4Kβ-YN effectively reconstituted EYFP fluorescence at the Golgi as determined by co-localisation with the co-transfected ECFP Golgi marker (Figure 6c,d). A signal due to fluorescence complementation was observed in essentially all transfected cells expressing the co-transfected Golgi marker. Notably, the diffuse cytosolic signal observed in cells expressing PI4Kβ-ECFP was absent from ARF1-YC/PI4K-YN expressing cells and EYFP fluorescence was restricted exclusively to the Golgi.
Figure 6.

Use of fluorescence fragment complementation to examine interaction of PI4Kβ with ARF1 in live HeLa cells.
HeLa cells were transfected with PI4Kβ tagged to full length ECFP (a) and protein localisation determined by confocal microscopy. PI4Kβ-ECFP was co-expressed with ARF1-EYFP (b). The fluorescent fragment variant of PI4Kβ (PI4Kβ-YN) was used to co-transfect HeLa cells along with ARF1-YC (c) and ells were simultaneously co-transfected with an ECFP-tagged Golgi marker. Expanded images of the Golgi complex region of one cell are shown in (d). ECFP (Red) and EYFP (Green) channels from individual cells were combined to show extent of co-localisation (yellow regions in overlay images). Scale bar, 10 μm (a-c) or 5 μm (d).
Specificity of class-I ARF proteins for interaction with PI4Kβ
Little is known concerning the interactions/functions of ARFs 3, 4 and 5 with most studies focusing on ARF1 and ARF6 proteins. With this in mind we decided to examine whether ARF proteins other than ARF1 would be capable of interacting with PI4Kβ as so far only an interaction with ARF1 has been shown (5, 17). Analysis of reconstituted EYFP fluorescence in cells co-expressing PI4Kβ-YN with ARFs 1, 3, 4, 5 and 6-YC showed that in addition to ARF1 (Figure 7, a), the other class-I ARF used in this study, ARF3, was also capable of direct interaction with PI4Kβ (Figure 7, b). As was observed for ARF1/ PI4Kβ interaction, the reconstituted EYFP signal was highly localised and was only visible at discrete Golgi locations. Neither class-II ARFs tested, ARF4 and ARF5, nor the class-III ARF, ARF6 were able to support EYFP fluorescence reconstitution (Figure 7, c, d and e) suggesting a highly specific interaction between PI4Kβ and class-I ARF proteins. The differences in effectiveness of reconstitution were not due to differences in expression levels of the proteins apart from possibly ARF4 (Figure 7f). In order to use an independent approach to examine the specificity of ARF isoform interaction with PI4Kβ, use was made of RNA silencing using reagents developed and characterised previously for knock-down of ARF isoforms in HeLa cells (12). It had been shown that knock-down of single ARFs had no detectable effects whereas distinct phenotypes were seen with pair-wise knock-down. We, therefore, compared cells co-transfected with pSUPER plasmids targeted at ARF1 and ARF3 or ARF4 and ARF5 in combination and examined the effect on localisation of PI4Kβ-ECFP. Both combinations of pSUPER plasmid reduced cell survival. ARF4/5 pSUPER plasmids had no effect on the Golgi localisation of PI4Kβ-ECFP (Figure 7i) compared to control cells (Figure 7g). In contrast, in cells transfected with ARF1/3 p-SUPER plasmids, PI4Kβ-ECFP was entirely cytosolic (Figure 7h). These results are consistent with the above findings from fluorescence fragment complementation.
Figure 7.

Use of fluorescent fragment complementation and RNA interference to probe the specificity of human ARF isoform interactions with PI4Kβ in HeLa cells.
HeLa cells were co-transfected with PI4Kβ-YN and ARF1-YC (a), ARF3-YC (b), ARF4-YC (c), ARF5-YC (d) or ARF6-YC (e) along with an ECFP tagged Golgi marker. ECFP (Red) and EYFP (Green) channels from individual cells were combined to show extent of co-localisation (yellow regions in overlay images). Scale bar, 10 μm. Determination of protein expression levels of PI4Kβ and ARF isoforms fragment constructs was done by Western blotting (f). To examine the effect of pair-wise knock-down of ARF isoforms, HeLa cells were transfected to express PI4Kβ-ECFP alone as control (g) or alongside RNA interference constructs for ARF1 and 3 or ARF 4 and 5 as indicated.
Functional analysis of the specificity of ARF- PI4Kβ interactions in exocytosis
In order to test the functional significance of the specificity of ARF isoform interactions with PI4Kβ, and to investigate whether the observed specificity could be seen with another independent approach, we used a well characterised reporter assay for regulated exocytosis in transfected PC12 cells. This allows expression of proteins and analysis of exocytosis from the sub-population of transfected cells. PC12 cells were transfected to express growth hormone and additional constructs and growth hormone release measured under basal conditions and in response to stimulation with 300 μM ATP which increases intracellular calcium concentration through activation of purinergic receptors. It is known that over-expression of PI4Kβ in PC12 cells leads to an increase in evoked release of growth hormone (32). In our experiments we saw a significant increase in hormone release compared to control cells in response to ATP stimulation in cells transfected to express tagged PI4Kβ (Figure 8). In contrast , in this and in previous work (17) over-expression of ARF1 had no effect. We therefore examined the effect of combining ARF isoform over-expression with PI4Kβ over-expression and chose to examine ARF1 which interacts with PI4Kβ in the fluorescence reconstitution assay and ARF5 and 6 which do not. A further enhancement of growth hormone release over that due to PI4Kβ in these experiments was seen with ARF1 but not with ARF 5 or 6 (Figure 8). None of the expression constructs had any effects on total levels of growth hormone synthesised after transfection ruling out any detrimental effects of the constructs. These results are fully consistent with the specificity seen in the fluorescence reconstitution assay and suggest that there is a specific functional role for the Class I ARF isoforms in PI4Kβ activation and exocytosis.
Figure 8.

Analysis of the effects of PI4Kβ and ARF isoforms in PC12 cells. PC12 cells were co-transfected to express growth hormone along with the indicated constructs.
Cells co-transfected with YFP-YN were used as controls. Growth hormone released with measured after a 15 min incubation in the absence or presence of 300 μM ATP. The data for release growth hormone were calculated as a percentage of total cellular growth hormone and release elicited by ATP over basal release is shown. Growth hormone release was significantly increased by co-transfection with PI4Kβ and this was enhanced by ARF1 but not ARF5 or ARF6. (n= 20 for all conditions except ARF1-YC where n=6. All data are shown as mean ± SEM and statistical comparisons were carried out using an unpaired t-test.)
Discussion
In this study we have assessed whether the five highly homologous proteins of the human ARF family are capable of exhibiting selectivity in their associations with two documented ARF1 interacting proteins NCS-1 and PI4Kβ. NCS-1 and PI4Kβ also interact directly with each other (17, 31) and this has been confirmed in live cells (32) and genetically for the yeast orthologues (33). Our new data provide cellular evidence for the NCS-1/ARF and PI4Kβ/ARF interactions previously studied mainly as in vitro interactions (5, 17). The approach involved over-expression of the ARF proteins. Previous studies have shown that mutants of ARFs including ARF1 (34) and ARF6 (27) can disrupt cellular function including changing Golgi organisation following expression of constitutively active ARF mutants (17, 34). In contrast, wild type ARF1 and ARF6 over-expression were without effect (27, 30, 34). Similarly we did not observe any detectable deleterious effects of expression of any of the ARF constructs used in this study. Using fluorescence complementation we present evidence for both a promiscuous interaction between NCS-1 and all three classes of human ARF proteins and a second more specific interaction of PI4Kβ exclusively with class-I ARFs suggesting that highly related ARFs are able to discriminate between common effectors/interactors. These observations are consistent with a recent report arguing that individual ARF activities are to some extent non-redundant and may exert unique and non-overlapping functions key to the control of discrete segments of intracellular trafficking pathways (12).
The fluorescence complementation appeared to be specific as it was never observed with control constructs or with certain specific pairings of constructs and efficient in that essentially all transfected cells gave evidence of complementation for those pairs of constructs that interacted. The specificity of the interaction between ARF isoforms and PI4Kβ is unlikely to be fortuitous as the ARF5 and 6 constructs which did not result in complementation with PI4Kβ clearly produced fluorescence complementation when expressed with the NCS-1 construct. In addition, independent assays using RNA interference and assay of regulated exocytosis fully supported the data from the fluorescence complementation approach for the specificity of ARF isoforms interactions with PI4Kβ. For reasons that are unclear, ARF4-YC expressed less well than the other isoform constructs and so we can not conclude whether or not this isoform would be capable of interaction with NCS-1 or PI4Kβ. From the use of fluorescent protein fragment complementation we have been able to visualise spatially localised protein-protein interactions involving the ARF family in live mammalian cells. Certain caveats should be noted however. First, the experimental approach is dependent on over-expression of tagged proteins (we did not however see any obvious effects of expression of the ARF isoform constructs in the HeLa cells). Second, the interactions could conceivably occur in other compartments but then become trapped after complementation at the observed sites such as seen for the non-myristoylated NCS-1 construct. This could also explain the appearance of the ARF1 and NCS-1 complementation at the plasma membrane. It should be noted, however, that despite the existence of soluble pools of the proteins examined a cytoplasmic signal for fluorescence complementation was never observed. Alternatively, the plasma membrane signal could result from the accumulation of complexes following low affinity interactions. This would be difficult to test by other approaches. In fact the fluorescence complementation technique may be useful in the detection of low-affinity/transient interactions that might perhaps evade other modes of investigation. Indeed, pull down assays previously suggested that NCS-1 interacted with ARF1 but not ARF3 (17) whereas we now show interaction of NCS-1 with all ARFs but at distinct intracellular locations. This approach for analysis of protein-protein interactions represents a potentially powerful means for determination of not only the existence of direct protein binding but also to localise the potential sites of this interaction in the physiological setting of the live mammalian cell. Our FRAP experiments suggest, however, that fluorescence fragment complementation would not occur fast enough for it to be used in dynamic studies of protein-protein interactions over short time scales.
The ARF1-5 isoforms, PI4Kβ and NCS-1 are localised to a greater or lesser extent to the Golgi complex with PI4Kβ (35) and NCS-1 (25) being associated with the TGN and all of these proteins have roles in the regulation of membrane traffic from the TGN to the cell surface (3, 17, 36, 37). PI4Kβ appears to be a key enzyme for the synthesis of PI4P in the Golgi complex (38). This lipid is crucial for traffic from the Golgi complex (39) where it is required for ARF-dependent vesicle coat recruitment (40). NCS-1 stimulates regulated and constitutive exocytosis (17, 41); this has been attributed to its ability to activate PI4Kβ (32, 42, 43) and over-expression of PI4Kβ can itself lead to increased evoked exocytosis. ARF1 has been shown to recruit PI4Kβ to the Golgi (5) and to directly activate the enzyme (17). ARFs1-5 could potentially have either specific or overlapping roles at the Golgi complex. Previous work has identified some specific sites of action for the class I compared to the other ARFs in membrane traffic (12) and our results suggest that activation of PI4Kβ may be a specific function of the ARF1/3 isoforms. This conclusion was based not only on the data from fluorescence complementation but independent assay of the effects of over-expression of PI4Kβ and ARF isoforms in stimulating exocytosis. These data thereby implicate the ARF1/3 isoforms in regulation of membrane traffic at the TGN and in the control of evoked exocytosis through activation of PI4Kβ. The application of the fluorescence complementation assay used here for the analysis of other ARF effectors may give further complimentary insights into specific functions of the ARF isoforms.
Materials and Methods
Plasmids
The ECFP-Golgi plasmid encoding ECFP linked to the N-terminus of beta 1,4-galactosyltransferase was constructed by substituting ECFP for EYFP in EYFP-Golgi obtained from Clontech (Palo Alto, California, USA) as described previously (44). Enhanced yellow fluorescent protein (EYFP) fragments encoding amino acids 1-158 and 159-239 were generated by PCR using pEYFP-N1 (Clontech, CA, USA) vector as a template and the following primer pairs to permit sub-cloning back into the pEYFP-N1 backbone after removal of the full length EYFP coding sequence with the enzymes AgeI and NotI:
1-158 sense (AgeI): 5'-GATCACCGGTAATGGTGAGCAAGGGCGAGG-3'
1-158 antisense (NotI): 5'-ATCGGCGGCCGCCATGATATAGACATTGTG-3'
159-239 sense (AgeI): 5'-GATCACCGGTAGCCGACAAGCAGAAGAACG-3'
159-239 antisense (NotI): 5'-ATCGGCGGCCGCTTTACTTGTACAGCTCGTC-3'
The two new vectors, YFP-YN (residues 1-158) and YFP-YC (residues 159-239) were generated such that the reading frame of the multiple cloning site from the parental pEYFP-N1 vector remained unaltered.
The coding sequence of NCS-1 was cloned in frame into YFP-YN by digestion of an existing NCS-1-EYFP plasmid (25) with the enzymes XhoI and BamHI followed by ligation to create NCS-1-YN. A YFP-YN plasmid encoding the myristoylation deficient mutant NCS-1(G2A) (NCS-1(G2A)-YN) was generated from pre-existing EYFP construct (25) in the same manner. All ARF coding sequences were sub-cloned into both full length pEYFP-N1 and the C-terminal YFP fragment vector YFP-YC. Human ARF1 and ARF3 were PCR amplified from pre-existing plasmids encoding ARF1-HA and ARF3-HA (17) and sub-cloned to generate ARF1-EYFP, ARF1-YC, ARF3-EYFP and ARF3-YC. Plasmids containing full length human ARF4 and ARF5 coding sequences were obtained from the rzpd consortium (Berlin, Germany). ARF4 (rzpd clone ID: IRALp962D1132Q2) and ARF5 (rzpd clone ID: IRAUp969E0818D6) coding sequences were amplified by PCR to permit subsequent sub-cloning and generation of ARF4-EYFP, ARF4-YC, ARF5-EYFP and ARF5-YC vectors. PCR primers were as follows:
ARF1 sense (HindIII): 5'-ATCGAAGCTTATGGGGAATATCTTTGCAAAC-3'
ARF1 antisense (SacII): 5'-ATATCCGCGGCTTCTGGTTCCGGAGCTG-3'
ARF3 sense (HindIII): 5'-ATCGAAGCTTATGGGGAATATCTTTGGGAAC-3'
ARF3 antisense (SacII): 5'-CGATCCGCGGCTTCTTGTTTTTGAGCTG-3'
ARF4 sense (HindIII): 5'-ACTGAAGCTTATGGGCCTCACTATCTCCTCC-3'
ARF4 antisense (SacII): 5'-TCAGCCGCGGACGTTTTGAAAGCTCATTTGAC-3'
ARF5 sense (HindIII): 5'-ACTGAAGCTTATGGGCCTCACCGTGTCCGCG-3'
ARF5 antisense (SacII): 5'-TCAGCCGCGGGCGCTTTGACAGCTCGTGGGAC-3'
The coding sequence of human ARF6 was PCR amplified from FirstChoice™ human brain cDNA (Ambion, Austin, TX, USA) using the following primer pair to permit sub-cloning and generation of ARF6-EYFP and ARF6-YC:
ARF6 sense (HindIII): 5'-AGTCAAGCTTATGGGGAAGGTGCTATCC-3'
ARF6 antisense (SacII): 5'-GACTCCGCGGAGATTTGTAGTGAGAGGTTAAC-3'
The full length coding sequence of human PI4Kβ was PCR amplified from a pre-existing plasmid (45) using the following primer pair to permit sub-cloning and generation of the PI4Kβ-YN vector. The full length PI4Kβ sequence was also sub-cloned into a vector encoding a C-terminal enhanced cyan fluorescent protein tag (pECFP-N1, Clontech). The new plasmid, PI4Kβ -ECFP, was used in co-localisation studies with NCS-1-EYFP and ARF1-EYFP expression constructs.
PI4Kβ sense (XhoI): 5'-ATCGCTCGAGATGGGAGATACAGTAGTGGAG-3'
PI4Kβ antisense (SacII): 5'-CGATCCGCGGCATGATGCCGTTGGTGAGGTAC-3'
All PCR reactions were performed using an Omn-E dry-block thermocycler (Hybaid, Middlesex, UK) and Bio-X-act Long DNA polymerase (Bioline, London, UK). All DNA constructs were checked by automated sequencing (DBS Genomics, University of Durham, UK).
Cell culture, transfection and western blotting
For all imaging studies, HeLa cells (0.5 × 106 cells/well) were plated onto glass cover slips and transiently transfected with 2 μg test plasmid using Lipofectamine 2000™ reagent (Invitrogen, The Netherlands) according to the manufacturers instructions. For dual transfections 2 μg of each test plasmid were transfected. Cells were imaged 24 hrs post-transfection. For RNA interference cells transfected with 5 μg per well each of the specified pSuper-ARF silencing plasmids(12) (a kind gift of Dr R Kahn, Emory University School of Medicine, Atlanta, USA) along with 2 μg of PI4K-ECFP plasmid using Lipofectamine 2000 reagent. Five days after transfection cells were fixed and ECFP fluorescence imaged.
For protein over-expression studies, HeLa cells (1 × 106 cells/well) were plated onto sterile 24 well tissue culture trays and transiently transfected with 1 μg test plasmid and 1 μg control empty vector (single transfections) or 1 μg each of both test plasmids (dual transfections) using Lipofectamine 2000™ reagent. Wells were solubilised 24 hrs post-transfection in 100 μl SDS dissociation buffer and samples boiled for 5 min prior to separation on 10% (PI4K), 12% (NCS-1), or 14% (ARFs) SDS-PAGE. Proteins were transferred to nitrocellulose filters for western blotting by transverse electrophoresis. Filters were probed with rabbit polyclonal anti-NCS-1 (1:500, (41)) for detection of over-expressed NCS-1-YN and NCS-1(G2A)-YN fusion proteins and sheep polyclonal anti-GFP (1:1000, a kind gift from Dr Ian Prior, University of Liverpool, UK) for detection of ARFs 1, 3, 4, 5 and 6-YC and PI4K-YN construct expression. Proteins were visualised following the application of appropriate species specific secondary antibodies coupled to HRP and incubation with ECL reagents.
Confocal laser scanning microscopy
For confocal laser scanning microscopy, live transfected HeLa cells were analysed using a Leica TCS-SP-MP microscope (Leica Microsystems, Heidelberg, Germany) using a 22 μm pin hole and a x63 water immersion objective with a 1.2 numerical aperture. For optimal imaging of ECFP, cells were excited at 405 nm and light collected at 450-490 nm. EYFP and reconstituted split YFP transfected cells were excited at 514 nm and light collected at 525-590 nm. Images were exported as TIFF files and compiled in CorelDraw.
Fluorescence recovery after photobleaching
Live HeLa cells plated onto 32 mm glass coverslips (0.5 × 106 cells/coverslip) were transiently co-transfected with 2 μg of either ARF1-EYFP plasmid and 2 μg NCS-1-ECFP or 2 μg of ARF1-YC and 2 μg of NCS-1-YN split-YFP constructs and analysed by confocal microscopy 24 hrs post-transfection. Briefly, EYFP fluorescence of cells was imaged in Flymode (Leica confocal software) at 800 Hz to permit simultaneous bleaching and fluorescence capture at a frame rate of 1 frame/0.5s. A bleach region of interest over the Golgi complex was designated and cells subsequently excited at 4% laser power for 20 frames, bleached in the region of interest at 100% laser intensity for 3 frames and post bleach images acquired for a further 70 frames at 4% laser power to permit analysis of EYFP fluorescence recovery.
Assay of regulated exocytosis in transfected PC12 cells
Analysis of regulated exocytosis from transfected PC12 cells used expression of human growth hormone (hGH) in a reporter assay (46). PC12 cells (0.75 × 106 cells/well) were plated onto Biocoat® Poly-D-Lysine coated 24-well tissue culture trays (BD Biosciences, Oxford, UK) and allowed to adhere for 24 h. Cells were transiently co-transfected using Lipofectamine 2000 reagent (Invitrogen) with 0.75 μg of pXGH5 (encoding human growth hormone) per well and the following test plasmid combinations to give a total of 2 μg transfected plasmid DNA per well. For control transfections 1.25 μg of empty split YFP vector (YFP-YN) was used per well. For ARF1 alone transfections, 0.25 μg of ARF1-YC and 1 μg of YFP-YN were used per well. For PI4Kβ alone transfections, 1 μg of PI4Kβ and 0.25 μg of empty split YFP vector (YFP-YC) were used per well. For PI4Kβ in combination with ARFs 1, 5 and 6, 1 μg of PI4Kβ-YN along with 0.25 μg of either ARF1-YC, ARF5-YC or ARF6-YC were used per well. 48 h post transfection, cells were washed twice in Krebs buffer (20 mM HEPES (pH 7.4), 145 mM NaCl, 10 mM glucose, 5 mM KCl, 3 mM CaCl2, 1.3 mM MgCl2, 1.2 mM NaH2PO4) followed by treatment with or without 300 μM ATP in Krebs for 15 min at room temperature. Samples of secreted growth hormone and cells were assayed using an enzyme linked immunosorbent assay kit (Roche Applied Science).
Acknowledgements
This work was supported by a grant from the Wellcome Trust to RDB. MWS and NJD were supported by Wellcome Trust Prize Studentships. We thank Professors Alan Morgan and Alexei Tepikin for their comments on this paper.
The abbreviations used are
- ARF
ADP-ribosylation factor
- FRAP
fluorescence recovery after photobleaching
- FRET
fluorescence resonance energy transfer
- NCS-1
neuronal calcium sensor-1
- PI4Kβ
phosphatidylinositol-4 kinase-IIIβ
- TGN
trans-Golgi network
References
- 1.Kahn RA, Volpicelli-Daley L, Bowzard B, Shrivastava-Ranjan P, Li Y, Zhou C, et al. Arf family GTPases: roles in membrane traffic and microtubule dynamics. Biochem Soc Trans. 2005;33:1269–72. doi: 10.1042/BST0331269. [DOI] [PubMed] [Google Scholar]
- 2.Donaldson JG, Honda A. Localization and function of Arf family GTPases. Biochem Soc Trans. 2005;33:639–42. doi: 10.1042/BST0330639. [DOI] [PubMed] [Google Scholar]
- 3.Donaldson JG, Honda A, Weigert R. Multiple activities for Arf1 at the Golgi complex. Biochim. Biophys. Acta. 2005;1744:364–373. doi: 10.1016/j.bbamcr.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Nie Z, Hirsch DS, Randazzo PA. Arf and its many interactors. Curr. Biol. 2003;15:396–404. doi: 10.1016/s0955-0674(03)00071-1. [DOI] [PubMed] [Google Scholar]
- 5.Godi A, Pertile P, Meyers R, Marra P, DiTullio G, Lurisci C, et al. ARF mediates recruitment of PtdIns-4-OH kinase-B and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat Cell Biol. 1999;1:280–287. doi: 10.1038/12993. [DOI] [PubMed] [Google Scholar]
- 6.Peters PJ, Hsu VW, Ooi CE, Finazzi D, Teal SB, Oorschot V, et al. Overexpression of wild-type and mutant ARF1 and ARF6: distinct perturbations of nonoverlapping membrane compartments. J Cell Biol. 1995;128:1003–17. doi: 10.1083/jcb.128.6.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paleotti O, Macia E, Luton F, Klein S, Partisani M, Chardin P, et al. The small G-protein Arf6GTP recruits the AP-2 adaptor complex to membranes. J Biol Chem. 2005;280:21661–6. doi: 10.1074/jbc.M503099200. [DOI] [PubMed] [Google Scholar]
- 8.Donaldson JG. Multiple roles for Arf6: sorting, structuring, and signaling at the plasma membrane. J Biol Chem. 2003;278:41573–6. doi: 10.1074/jbc.R300026200. [DOI] [PubMed] [Google Scholar]
- 9.Liang JO, Sung TC, Morris AJ, Frohman MA, Kornfeld S. Different domains of mammalian ADP-ribosylation factor 1 mediate interaction with selected target proteins. J Biol Chem. 1997;272:33001–8. doi: 10.1074/jbc.272.52.33001. [DOI] [PubMed] [Google Scholar]
- 10.Honda A, Nogami M, Yokozeki T, Yamazaki M, Nakamura H, Watanabe H, et al. Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell. 1999;99:521–32. doi: 10.1016/s0092-8674(00)81540-8. [DOI] [PubMed] [Google Scholar]
- 11.Perez-Mansilla B, Ha VL, Justin N, Wilkins AJ, Carpenter CL, Thomas GM. The differential regulation of phosphatidylinositol 4-phosphate 5-kinases and phospholipase D1 by ADP-ribosylation factors 1 and 6. Biochim Biophys Acta. 2006;1761:1429–42. doi: 10.1016/j.bbalip.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Volpicelli-Daley LA, Li Y, Zhang C-J, Kahn RA. Isoform-selective effects of the depletion of ADP-ribosylation factors 1-5 on membrane traffic. Mol Biol Cell. 2005;16:4495–4508. doi: 10.1091/mbc.E04-12-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shin OH, Ross AH, Mihai I, Exton JH. Identification of arfophilin, a target protein for GTP-bound class II ADP-ribosylation factors. J Biol Chem. 1999;274:36609–15. doi: 10.1074/jbc.274.51.36609. [DOI] [PubMed] [Google Scholar]
- 14.Shin OH, Couvillon AD, Exton JH. Arfophilin is a common target of both class II and class III ADP-ribosylation factors. Biochemistry. 2001;40:10846–52. doi: 10.1021/bi0107391. [DOI] [PubMed] [Google Scholar]
- 15.Deretic D, Williams AH, Ransom N, Morel V, Hargrave PA, Arendt A. Rhodopsin C terminus, the site of mutations causing retinal disease, regulates trafficking by binding to ADP-ribosylation factor 4 (ARF4) Proc Natl Acad Sci U S A. 2005;102:3301–6. doi: 10.1073/pnas.0500095102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burgoyne RD, Weiss JL. The neuronal calcium sensor family of Ca2+-binding proteins. Biochem. J. 2001;353:1–12. [PMC free article] [PubMed] [Google Scholar]
- 17.Haynes LP, Thomas GMH, Burgoyne RD. Interaction of neuronal calcium sensor-1 and ARF1 allows bidirectional control of PI(4) kinase and TGN-plasma membrane traffic. J.Biol. Chem. 2005;280:6047–6054. doi: 10.1074/jbc.M413090200. [DOI] [PubMed] [Google Scholar]
- 18.Nyfeler B, Michnick SW, Hauri HP. Capturing protein interactions in the secretory pathway of living cells. Proc. Natl. Acad. Sci. USA. 2005;102:6350–6355. doi: 10.1073/pnas.0501976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu C-D, Kerppola TK. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat Biotechnol. 2003;21:539–545. doi: 10.1038/nbt816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu CD, Chinenov Y, Kerppola TK. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell. 2002;9:789–98. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 21.Johnson AE. Fluorescence approaches for determining protein conformations, interactions and mechanisms at membranes. Traffic. 2005;6:1078–92. doi: 10.1111/j.1600-0854.2005.00340.x. [DOI] [PubMed] [Google Scholar]
- 22.Karpova TS, Baumann CT, He L, Wu X, Grammer A, Lipsky P, et al. Fluorescence resonance energy transfer from cyan to yellow fluorescent protein detected by acceptor photobleaching using confocal microscopy and a single laser. J Microsc. 2003;209:56–70. doi: 10.1046/j.1365-2818.2003.01100.x. [DOI] [PubMed] [Google Scholar]
- 23.Valentin G, Verheggen C, Piolot T, Neel H, Coppey-Moisan M, Bertrand E. Photoconversion of YFP into a CFP-like species during acceptor photobleaching FRET experiments. Nat Methods. 2005;2:801. doi: 10.1038/nmeth1105-801. [DOI] [PubMed] [Google Scholar]
- 24.Niu TK, Pfeifer AC, Lippincott-Schwartz J, Jackson CL. Dynamics of GBF1, a Brefeldin A-sensitive Arf1 exchange factor at the Golgi. Mol Biol Cell. 2005;16:1213–22. doi: 10.1091/mbc.E04-07-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Callaghan DW, Ivings L, Weiss JL, Ashby MC, Tepikin AV, Burgoyne RD. Differential use of myristoyl groups on neuronal calcium sensor proteins as a determinant of spatio-temporal aspects of Ca 2+-signal transduction. J. Biol.Chem. 2002;277:14227–14237. doi: 10.1074/jbc.M111750200. [DOI] [PubMed] [Google Scholar]
- 26.O'Callaghan DW, Hasdemir B, Leighton M, Burgoyne RD. Residues within the myristoylation motif determine intracellular targeting of the neuronal Ca2+ sensor protein KChIP1 to post-ER transport vesicles and traffic of Kv4 K+ channels. J. Cell Sci. 2003;116:4833–4845. doi: 10.1242/jcs.00803. [DOI] [PubMed] [Google Scholar]
- 27.Aikawa Y, Martin TFJ. ARF6 regulates a plasma membrane pool of phosphatidylinositol(4,5)bisphosphate required for regulated exocytosis. J Cell Biol. 2003;162:647–659. doi: 10.1083/jcb.200212142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peters PJ, Gao M, Gaschet J, Ambach A, van Donselaar E, Traverse JF, et al. Characterization of coated vesicles that participate in endocytic recycling. Traffic. 2001;2:885–95. doi: 10.1034/j.1600-0854.2001.21204.x. [DOI] [PubMed] [Google Scholar]
- 29.Hosaka M, Toda K, Takatsu H, Torii S, Murakami K, Nakayama K. Structure and intracellular localization of mouse ADP-ribosylation factors type 1 to type 6 (ARF1-ARF6) J Biochem (Tokyo) 1996;120:813–9. doi: 10.1093/oxfordjournals.jbchem.a021484. [DOI] [PubMed] [Google Scholar]
- 30.Presley JF, T.H. W, Pfeifer AC, Siggia ED, Phair RD, Lippincott-Schwartz J. Dissection of COPI and Arf1 dynamics in vivo and role in Golgi membrane transport. Nature. 2002;417:187–193. doi: 10.1038/417187a. [DOI] [PubMed] [Google Scholar]
- 31.Zhao X, Varnai P, Tuymetovna G, Balla A, Toth ZE, Oker-Blom C, et al. Interaction of neuronal calcium sensor-1 (NCS-1) with phosphatidylinositol 4-kinase beta stimulates lipid kinase activity and affects membrane trafficking in COS-7 cells. J. Biol. Chem. 2001;276:40183–40189. doi: 10.1074/jbc.M104048200. [DOI] [PubMed] [Google Scholar]
- 32.de Barry J, Janoshazi A, Dupont JL, Procksch O, Chasserot-Golaz S, Jeromin A, et al. Functional implication of neuronal calcium sensor-1 and PI4 kinase-β interaction in regulated exocytosis of PC12 cells. J Biol Chem. 2006;281:18098–18111. doi: 10.1074/jbc.M509842200. [DOI] [PubMed] [Google Scholar]
- 33.Hendricks KB, Wang BQ, Schnieders EA, Thorner J. Yeast homologue of neuronal frequenin is a regulator of phosphatidylinositol-4-OH kinase. Nature Cell Biology. 1999;1:234–241. doi: 10.1038/12058. [DOI] [PubMed] [Google Scholar]
- 34.Zhang CJ, Rosenwald AG, Willingham MC, Skuntz S, Clark J, Kahn RA. Expression of a dominant allele of human ARF1 inhibits membrane traffic in vivo. J. Cell Biol. 1994;124:289–300. doi: 10.1083/jcb.124.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong K, Meyers R, Cantley LC. Subcellular locations of phosphatidylinositol 4-kinase isoforms. J. Biol.Chem. 1997;272:13236–13241. doi: 10.1074/jbc.272.20.13236. [DOI] [PubMed] [Google Scholar]
- 36.Hausser A, Storz P, Martens S, Link G, Toker A, Pfizenmaier K. Protein kinase D regulates vesicular transport by phosphorylating and activating phosphatidylinositol-4 kinase IIIβ at the Golgi complex. Nature Cell Biol. 2005;7:880–886. doi: 10.1038/ncb1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weisz OA, Gibson GA, Leung S-M, Roder J, Jeromin A. Overexpression of frequenin, a modulator of phosphatidylinositol 4-kinase, inhibits biosynthetic delivery of an apical protein in polarized Madin-Darby Canine Kidney cells. J. Biol. Chem. 2000;275:24341–24347. doi: 10.1074/jbc.M000671200. [DOI] [PubMed] [Google Scholar]
- 38.Balla A, Balla T. Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol. 2006;16:351–361. doi: 10.1016/j.tcb.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 39.De Matteis MA, Di Campli A, Godi A. The role of phosphoinositides at the Golgi complex. Biochim. Biophys. Acta. 2005;1744:396–405. doi: 10.1016/j.bbamcr.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 40.Wang YJ, Wang J, Sun HQ, Martinez M, Sun YX, Macia E, et al. Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the Golgi. Cell. 2003;114:299–310. doi: 10.1016/s0092-8674(03)00603-2. [DOI] [PubMed] [Google Scholar]
- 41.McFerran BW, Graham ME, Burgoyne RD. NCS-1, the mammalian homologue of frequenin is expressed in chromaffin and PC12 cells and regulates neurosecretion from dense-core granules. J. Biol. Chem. 1998;273:22768–22772. doi: 10.1074/jbc.273.35.22768. [DOI] [PubMed] [Google Scholar]
- 42.Koizumi S, Rosa P, Willars GB, Challiss RAJ, Taverna E, Francolini M, et al. Mechanisms underlying the neuronal calcium sensor-1 evoked enhancement of exocytosis in PC12 cells. J. Biol.Chem. 2002;277:30315–30324. doi: 10.1074/jbc.M201132200. [DOI] [PubMed] [Google Scholar]
- 43.Rajebhosale M, Greenwood S, Vidugiriene J, Jeromin A, Hilfiker S. Phosphatidylinositol 4-OH kinase is a downstream target of neuronal calcium sensor 1 in enhancing exocytosis in neuroendocrine cells. J.Biol. Chem. 2003;278:6075–6084. doi: 10.1074/jbc.M204702200. [DOI] [PubMed] [Google Scholar]
- 44.Hasdemir B, Fitzgerald DJ, Prior IA, Tepikin AV, Burgoyne RD. Traffic of Kv4 K+ channels mediated by KChIP1 is via a novel post-ER vesicular pathway. J Cell Biol. 2005;171:459–469. doi: 10.1083/jcb.200506005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyers R, Cantley LC. Cloning and characterisation of a wortmannin-sensitive human phosphatidylinositol 4-kinase. J.Biol. Chem. 1997;272:4384–4380. doi: 10.1074/jbc.272.7.4384. [DOI] [PubMed] [Google Scholar]
- 46.Graham ME, Fisher RJ, Burgoyne RD. Measurement of exocytosis by amperometry in adrenal chromaffin cells: effects of clostridial neurotoxins and activation of protein kinase C on fusion pore kinetics. Biochimie. 2000;82:469–479. doi: 10.1016/s0300-9084(00)00196-6. [DOI] [PubMed] [Google Scholar]