

Abstract
Gene therapy for β-thalassemia requires stable transfer of a β-globin gene into hematopoietic stem cells (HSCs) and high and regulated hemoglobin expression in the erythroblastic progeny. We developed an erythroid-specific lentiviral vector driving the expression of the human β-globin gene from a minimal promoter/enhancer element containing two hypersensitive sites from the β-globin locus control region. Transplantation of transduced HSCs into thalassemic mice leads to stable and long-term correction of anemia with all red blood cells expressing the transgene. A frequency of 30–50% of transduced HSCs, harboring an average vector copy number per cell of 1, was sufficient to fully correct the thalassemic phenotype. In the mouse model of Cooley's anemia transplantation of transduced cells rescues lethality, leading to either a normal or a thalassemia intermedia phenotype. We show that genetically corrected erythroblasts undergo in vivo selection with preferential survival of progenitors harboring proviral integrations in genome sites more favorable to high levels of vector-derived expression. These data provide a rationale for a gene therapy approach to β-thalassemia based on partially myeloablative transplantation protocols.
Keywords: gene therapy, hematopoietic stem cells, lentiviral vector
Thalassemias are a heterogeneous group of inherited anemias that collectively represent the most common monogenic disorders. The β-thalassemias are characterized by reduced or absent production of hemoglobin β-chains. The most severe form, β-thalassemia major or Cooley's anemia, is characterized by a profound anemia that, if not treated, leads to death in the first year of life. The only available cure is allogeneic bone marrow (BM) transplantation which is, however, available for less than 30% of patients (1). Autologous transplantation of genetically corrected hematopoietic stem cells (HSCs) is considered an attractive therapeutic alternative for patients lacking a compatible donor. The development of lentiviral vectors (LVs) and the optimization of HSC transduction conditions has led to the recent application of LVs expressing the human β-globin gene in preclinical murine models and in human thalassemic cells (2–4). LVs expressing β-globin contain different combinations of DNase I-hypersensitive sites from the β-globin locus control region (LCR) and β-globin promoter/enhancers (5). Inclusion of insulator elements has been reported to increase globin synthesis (4) but impairs viral production because of genetic instability (6). Large-scale production of β-globin LVs, transduction of human HSCs at safe, low vector copy number (VCN), and engraftment of therapeutic levels of genetically corrected HSCs are currently major challenges to a successful clinical translation.
The clinical history of thalassemia and over 20 years of experience with BM transplantation allow for predictions that even a mild correction of the globin chain imbalance in a fraction of maturing erythroblasts would be sufficient to reduce the morbidity caused by ineffective erythropoiesis, to improve the clinical management of the disease, and to increase the patients' life expectancy (1). Studies of long-term transplanted thalassemic patients revealed that persistent mixed chimerism, with a proportion of donor cells of 20–30%, results in resolution of thalassemia (7). The rise in Hb in the presence of a limited number of donor normal cells is suggestive of a selective growth and survival advantage of erythroid precursors and RBCs. Studies on β-thalassemic mice transplanted with normal BM cells demonstrated that 10–20% chimerism is sufficient to significantly increase Hb and to diminish extramedullary hematopoiesis (EMH). Amplification of normal red blood cells (RBCs) in the peripheral blood of mice engrafted with low numbers of HSCs suggested an in vivo selection during erythropoiesis (8).
In the context of gene therapy for thalassemia, the minimal dose of transduced HSCs required to correct the phenotype and the potential selective advantage of the genetically corrected erythroblasts need to be determined. Survival advantage of genetically corrected erythroblasts has never been studied, and it is not predictable from BM transplantation experiments because the amount of β-globin produced by vector-transduced cells is unlikely to match that synthesized by normal cells. This issue is relevant to develop gene therapy protocols based on mildly myeloablative conditioning regimens. In this study, we have developed and produced at high titer a LV that expresses β-globin (GLOBE), and demonstrated that transplantation of transduced HSCs corrects the pathological features of the most severe mouse models of thalassemia intermedia and major (th3 mutant). A frequency of 30–50% of transduced hematopoietic cells harboring an average VCN of 1 was sufficient to fully correct the thalassemic phenotype in th3/+ mice. Importantly, molecular analysis of proviral integrations in BM subpopulations provides evidence for in vivo selection of genetically corrected erythroblasts differentiating from transduced HSCs.
Results
A LV for Expression of Human β-Globin from Minimal βLCR.
We have constructed a minimal LCR-β-globin transcription unit containing a 2.7-kb fragment encompassing LCR elements HS2 and HS3, which have been reported to have classical enhancer and dominant chromatin-opening functions, respectively (9). This minimal LCR HS2/HS3 combination was linked to a fully functional mini-β-globin gene with 265 bp of 5′ and 300 bp of 3′ flanking sequences (10). The minimized LCR-β-globin transcription unit was inserted into a self-inactivating, HIV-derived LV in reverse orientation with respect to the 5′LTR-driven genomic transcript, generating the GLOBE LV (Fig. 1A). We routinely produced viral stocks at a titer up to 2 × 109 transducing units per ml (350 fold concentration), which is 1/2 the titer of LV expressing GFP.
Fig. 1.

GLOBE LV-derived human β-globin expression in RBCs of th3/+ transplanted mice. (A) Schematic representation of GLOBE LV in its proviral form. LTRs deleted of 400 bp in the HIV U3 region (ΔLTR), rev-responsive element (RRE), splicing donor (SD) and splicing acceptor (SA) sites, human β-globin gene, β-globin promoter (βp), and DNase I-hypersensitive sites HS2 and HS3 from β-globin LCR are shown. (B) FACS analysis of peripheral blood samples 6 months after BM transplantation from mice transplanted with mock-transduced (th3/+) and transduced (th3/+ LV) cells. PE, phycoerythrin.
Transplantation of Transduced HSCs in th3/+ Mice Corrects Anemia and Restores Normal Hematological Parameters.
To test the therapeutic potential of the GLOBE vector, we transplanted transduced BM cells into lethally irradiated β-thalassemic mice and evaluated the correction of the pathological features that characterize this disease model. We used heterozygous C57BL6/Hbbth3 knockout mice (th3/+), which show a pathophysiology comparable to that of patients with severe β-thalassemia intermedia (11). BM cells were isolated from th3/+ male donors, transduced with the GLOBE vector, and transplanted into eight lethally irradiated th3/+ female recipients. As controls, eight and nine mice were transplanted with mock-transduced th3/+ and normal cells, respectively (referred as th3/+ and normal control). Recipient mice were killed nine months after BM transplantation, and the peripheral blood and the hematopoietic organs were harvested and subjected to multiple analysis. The percentages of RBCs expressing human β-globin, detected by FACS [please see supporting information (SI) Materials and Methods], in th3/+ mice transplanted with transduced HSCs ranged from 64% to 99% (Fig. 1B and Table 1). Vector-derived β-globin ranged from 14% to 37% of endogenous murine α-chain [HPLC analysis (SI Materials and Methods) and Table 1] and was sufficient to fully correct anisocytosis and poikilocytosis in peripheral blood (Fig. 2A iii and iv). In contrast, th3/+ control mice remained severely anemic and showed abnormal RBC morphology (Fig. 2Aii). Hematocrit (Hct), RBC count, Hb level, and reticulocyte count were significantly improved compared with th3/+ control mice (Fig. 2B and Table S1). Molecular analysis of VCN, performed by quantitative PCR on DNA extracted from BM cells, indicated that in most of the mice the degree of correction of the thalassemic phenotype was commensurate to VCN (Table S1). Transduction of long-term repopulating HSCs, persistence of vector-derived globin expression, and long-term correction of the thalassemic phenotype were demonstrated in secondary transplanted mice (Fig. S1, data not shown).
Table 1.
Human β-globin expression, Hb levels, and VCN in th3/+ mice transplanted with transduced BM
| Mouse no. | Peripheral blood |
BM |
CFU-S |
|||
|---|---|---|---|---|---|---|
| % huβ+ RBCs | % huβ/mα* | Hb, g/dl | VCN | % vector+ | VCN | |
| 14 | 64 | ND | 9.69 | 0.23 | 35 | 1.06 ± 0.10 |
| 19 | 99 | 19 | 12.48 | 0.91 | 57 | 1.11 ± 0.12 |
| 21 | 95 | 19 | 14.01 | 0.74 | ND | ND |
| 4 | 99 | 23 | 12.30 | 0.55 | 45 | 1.18 ± 0.08 |
| 6 | 86 | 14 | 9.40 | 0.27 | ND | ND |
| 5 | 99 | 37 | 13.86 | 0.83 | ND | ND |
| 2 | 95 | 30 | 13.20 | 2.20 | 100 | 3.77 ± 0.51 |
| 55 | 96 | ND | 11.65 | 0.55 | 63 | 1.17 ± 0.10 |
CFU-S, spleen colony-forming units; ND, not determined.
*Ratio between the areas of HPLC peaks corresponding to human β - and murine α-globin chains (huβ/mα).
Fig. 2.

Correction of hematological parameters in mice transplanted with transduced BM. (A) May–Grunwald/Giemsa staining of blood smears derived from mice transplanted with mock-transduced normal (i), th3/+ (ii), and transduced th3/+ (iii and iv) HSCs. Abnormal RBC morphology and reticulocytosis, which are typical in thalassemic mice (ii), were completely corrected in mice transplanted with transduced HSCs (iii and iv). (Original magnification ×40.) (B) Hb concentration, RBC count, Hct, and percentages of reticulocytes in peripheral blood of mice transplanted with mock-transduced th3/+ (white column; n = 8), normal (WT; black column; n = 9), and transduced th3/+ (th3/+ LV; gray column; n = 8) cells. Values represent the mean and SEM for each group of animals. Hematological parameters were significantly different in th3/+ mice transplanted with transduced HSCs compared with control th3/+ mice (Hb concentration, P < 0.001; RBC count, P < 0.001; Hct, P < 0.01; reticulocyte count, P < 0.001). No statistically significant difference was found compared with normal controls.
Restoration of Effective Erythropoiesis and Correction of Organ Pathology.
To determine the impact of sustained β-globin gene expression on hematopoiesis, we studied the erythroid (Ter119+) BM and splenic populations and the degree of splenomegaly and EMH. In thalassemic mice, we observed a massive expansion of the Ter119+ erythroid compartment (60.0 ± 2.9% of total BM cells compared with 37.1 ± 1.8% in normal control mice, Table S2), which was significantly reduced in mice transplanted with GLOBE-transduced cells (43.5 ± 2.9%, P = 0.003 in mock-transduced vs. transduced th3/+ group; P > 0.05 in WT vs. transduced th3/+ group, Table S2). The level of splenic EMH in th3/+ mice, represented by a high proportion of Ter119+ cells (47.1 ± 8.0% of total splenocytes), was also reduced in treated mice (11.5 ± 2.8%, Table S2). The regression of EMH was confirmed by histological examination of spleen and liver from long-term chimeras. In treated mice, the proportion between spleen white and red pulp, which was completely lost in th3/+ control mice because of erythroid precursors expansion, was reconverted to normal. The amount of iron deposits was also comparable to those of normal mice (Fig. S2A and Table S2). Livers from untreated th3/+ mice showed several foci of intrasinusoidal EMH and iron accumulation because of increased gastrointestinal iron uptake, whereas livers from treated mice were similar to those of normal animals (Fig. S2B and Table S2).
In vivo Selection of Genetically Corrected Erythroid Progenitors.
To study the clonality of engrafted cells, genomic DNA extracted from BM samples of primary transplanted mice was subjected to linker-mediated PCR (LM-PCR), and proviral integration sites were sequenced and mapped on the mouse genome. As control we used a group of th3/+ mice transplanted with HSCs transduced with a GFP-expressing LV (LV-GFP). Proviral localization, targeted genes, and their biological function are reported in Tables S3 and S4. We found a finite number of integrations (2–15 per animal) in both groups of transplanted mice, indicating oligoclonal contribution of transduced HSCs to the hematopoiesis. Southern blot analysis confirmed this estimate (data not shown). None of the 61 GLOBE integrations and only 1 of the 28 LV-GFP integrations mapped in the vicinity (±30 kb) of cancer-associated retroviral common integration sites [Retrovirus Tagged Cancer Gene Database (RTCGD), http://rtcgd.abcc.ncifcrf.gov]. These results are different from those obtained in human CD34+ cells (12), although both analyses were performed on a number of samples too small to draw any significant conclusion. The analysis of VCN indicated that the proportion of vector-harboring cells on the total BM cells ranged from 0.23 to >1 (Table 1). BM cells harvested from primary hosts (mice 14, 19, 4, 2, and 55) were transplanted in secondary recipients to perform clonal analysis on day 12 for spleen colony-forming units (CFU-S). Quantitative PCR analysis showed that in most of the mice transduced, on average, CFU-S harbored a single integrant per genome. Frequency of transduction in CFU-S correlated with that observed in BM cells of primary recipients (Table 1). The proportion of RBCs producing Hb was invariably high, ranging from 64% to 99%, independently from the number of transduced HSCs in the marrow. In particular, mice engrafted with a low number of genetically modified HSCs (mice 14, 6, 4, and 55) had a VCN of 0.23, 0.27, 0.55, and 0.55 which correlated with 64%, 86%, 99%, and 96% of RBCs expressing human β-globin (Table 1). This observation suggests an in vivo selection of transduced erythroblastic progenitors or a survival advantage of genetically corrected RBCs.
We investigated this hypothesis by sorting the Ter119+ fraction from the nonerythroid (Ter119−) subpopulation of BM cells from transplanted mice and determined VCN by using quantitative PCR. In most of the mice, the proportion of transduced cells in the Ter119+ fraction was 2- to 10-fold higher than that observed in the Ter119− compartment (VCN, 1.73 ± 0.40 vs. 0.70 ± 0.20; P > 0.005, n = 8), indicating an in vivo selective advantage of highly transduced erythroid progenitors (Fig. 3A). In one mouse, no. 14, although the proportion of BM Ter119+ transduced cells was 43% (VCN 0.43), a higher proportion of RBC expressing β-globin (64%) was found, suggesting also a survival advantage of transduced mature erythroid cells in the periphery. In marked contrast, in a control group of th3/+ mice transplanted with th3/+ BM cells transduced by a LV-GFP, no significant difference was found in the VCN of Ter119+ vs. Ter119− cells (0.81 ± 0.34 vs. 0.78 ± 0.32; P = 0.44, n = 5; Fig. 3B). We confirmed the evidence of in vivo selection also in secondary transplanted mice where the level of transduced Ter119+ cells remained significantly higher than that of the Ter119− cells (2.72 ± 0.53 vs. 1.32 ± 0.35; P = 0.05, n = 5; Fig. 3A). In a control group of secondary th3/+ mice transplanted with HSCs transduced by LV-GFP, no significant difference was found in VCN between Ter119+ and Ter119− cells (1.18 ± 0.16 vs. 1.26 ± 0.25; P = 0.5, n = 3; Fig. 3B).
Fig. 3.
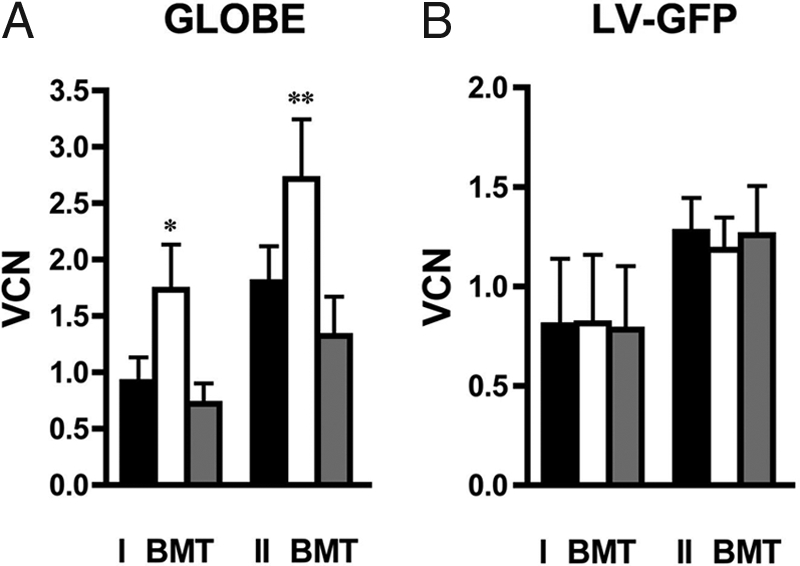
In vivo selection of transduced erythroid progenitors. (A) VCN in total BM (black bar), Ter119+ (white bar), and Ter119− (gray bar) cells from primary (I BMT) and secondary (II BMT) recipients transplanted with HSCs transduced by GLOBE. (B) VCN analysis in control th3/+ recipients of HSCs transduced with LV-GFP. *, P > 0.005; **, P = 0.05).
Analysis of the proviral integration sites revealed that in 50% of transplanted mice, Ter119+ and Ter119− cells shared common integrations (2–5) (Table S3, mice 2, 4, and 19). The remaining 50% of the mice, which were reconstituted with a higher number of transduced HSCs, showed few shared integrants (4 of 15, 2 of 11, and 4 of 16 in mice 14, 21, and 55, respectively) with unique predominant events in the Ter119+ and Ter119− fractions (Table S3). This observation indicates that in these cases the erythroblastic compartment was mainly sustained by the expansion of progenitors harboring specific proviral integrations. Interestingly, the integrations mapping to intergenic regions were significantly more abundant in the Ter119+ than in the Ter119− fraction (52.9% vs. 23.9%, P = 0.01), suggesting that proviral expression in such locations is less prone to interference by chromatin environment and associated transcriptional activity (Table 2). The increased proportion of intergenic sites remained statistically significant in ex vivo GLOBE-transduced Ter119+ cells compared with GLOBE- and LV-GFP-transduced pretransplant cells (P = 0.04 and P = 0.001, respectively, Table 2). In the total unfractionated BM cell population, the proportion of intergenic (38.7%) and intragenic (53.2%) integration sites was contributed by the Ter119+ and Ter119− subpopulations. Importantly, no evidence of skewing after transplantation was found since in the Ter119− fraction, the proportion of intergenic integrations was comparable to that observed in lineage-negative (Lin−) BM progenitors transduced by GLOBE (27.6% vs. 23.9%), and to that reported (13) for Lin− BM cells transduced with a LV-GFP (28.1%) (Table 2). Compared with randomly cloned control sequences (Table 2 and Table S4), the GLOBE vector showed a preference for intragenic integration in nonerythroid cells, similar to our recent reported results for LV in human CD34+ cells (14).
Table 2.
In vivo and in vitro vector integration sites distribution
| Intergenic, % | Intragenic, % | Perigenic, % | |
|---|---|---|---|
| GLOBE total BM (n = 61) | 38.7 | 53.2 | 8.1 |
| GLOBE Ter119+ BM (n = 34) | 52.9 | 38.2 | 8.8 |
| GLOBE Ter119− BM (n = 46) | 23.9 | 65.2 | 10.9 |
| GLOBE in vitro (n = 57) | 27.6 | 56.9 | 15.5 |
| LV-GFP in vitro* (n = 202) | 28.1 | 57.9 | 14.0 |
| Control sequences (n = 61) | 49.2 | 39.3 | 11.5 |
Distribution of GLOBE integrations sites unambiguously mapped in total BM, Ter119+, and Ter119− sorted cells of th3/+ transplanted mice, and GLOBE and LV GFP integrations in cultured lineage-negative BM cells. Integrations were distributed as inside (intragenic), outside (intergenic), or at a distance of <30 kb upstream or downstream (perigenic) from known genes (University of California Santa Cruz annotation). Control sequences were obtained from a randomly cloned library of MseI/NarI-restricted, LM-PCR-amplified murine BM DNA.
*LV-GFP integrations in lineage-negative BM cells from a previously published collection (10).
Correction of Thalassemia Major in the Murine Model of Cooley's Anemia.
The therapeutic potential of the GLOBE LV was also tested in a murine model of Cooley's anemia (15). We analyzed the efficacy of th3/th3 fetal liver cells (FLCs), transduced with the GLOBE LV, by rescuing the survival and ameliorating the pathophysiology of transplanted co-isogenic C57BL/6-CD45.1 mice (n = 16). As controls, recipient mice were lethally irradiated (n = 8) or irradiated and transplanted with normal (n = 8), th3/+ (n = 8), and th3/th3 FLCs (n = 9), respectively. Recipients of mock-transduced th3/th3 FLCs (referred to as th3/th3 control) died 30–80 days after transplantation (T50 = 50 days), because of the profound state of anemia that occurred coincidentally with the loss of normal recipient RBCs. All of the mice transplanted with GLOBE transduced th3/th3 FLCs showed long-term survival (P < 0.0001 th3/th3 GLOBE vs. th3/th3; Fig. 4A). Further analysis determined that 4 of 16 transplanted mice showed a percentage of chimeric Hb tetramers (Hbhu/mu) of >90% of the total Hb (Hbhu/mu + Hbmu), as determined by using cellulose acetate electrophoresis (Fig. 4B). Six months after FLC transplantation, all RBCs expressed β-globin (Fig. S3) with amelioration of anisocytosis and poikilocytosis (Fig. S4A). Hct, RBC count, and Hb level were markedly improved compared with those of th3/th3 controls (Hct: 39.49 ± 3.04% vs. 12.64 ± 2.22%; RBC: 6.40 ± 0.48 × 106/μl vs. 2.02 ± 0.22 × 106/μl; Hb: 10.20 ± 1.08 g/dl vs. 3.52 ± 0.50 g/dl; Table S5), and in 2 mice (21 and 22), Hb concentration reached the value obtained in mice transplanted with normal cells (11.9 and 12.2 g/dl vs. 13.4 ± 1.1 g/dl). Erythroid expansion, measured as percentage of Ter119+ cells, was decreased in BM and spleen of treated mice in comparison with th3/th3 controls (BM: 61.50 ± 4.52% vs. 80.10 ± 2.75%; spleen: 63.50 ± 3.97% vs. 90.40 ± 1.75%, Table S5). The extent in reduction of iron deposits and hepatic EMH was commensurate to the degree of correction of hematological parameters and erythroid hyperplasia (Table S6 and Fig. S4 B and C). Finally, molecular analysis showed that BM cells of long-term, full-donor chimeras harbored an average VCN of 4 (Table S5).
Fig. 4.

Rescue from death and Hb production in th3/th3 hematopoietic chimeras. (A) Lethally irradiated recipients were transplanted with mock-transduced (th3/th3; n = 9) and transduced th3/th3 FLCs (th3/th3 GLOBE; n = 16). Control mice received mock-transduced th3/+ (n = 8), normal (WT) FLCs (n = 8), or were lethally irradiated (irradiation controls, n = 8). Survival was monitored and Kaplan–Meier analysis was performed. Mice transplanted with mock-transduced th3/th3 FLCs died (T50 = 50 days) significantly later than the irradiation controls (T50 = 12 days; P < 0.0001, log-rank test). All mice transplanted with transduced th3/th3 FLCs survived long-term (P < 0.0001 for th3/th3 vs. th3/th3 GLOBE, log-rank test). (B) Analysis of murine (Hbmu) and chimeric (Hbhu/mu) Hb tetramers by cellulose acetate. In mice transplanted with transduced FLCs (th3/th3 GLOBE), the percentage of Hbhu/mu tetramers reached more than 90% of total Hb, as determined by densitometric analysis of Ponceau S-stained bands.
Discussion
The high levels of expression of the β-globin-like gene family are mediated at the transcriptional level by the LCR working in concert with gene promoter/enhancer elements. Each of the four transcriptionally active LCR hypersensitive sites possesses a functional core of 2–300 bp, which contains a high density of erythroid-specific and ubiquitous transcription factor binding elements (9). Mechanistically, the LCR mediates activation of gene expression by means of DNA/chromatin looping. This explains the need to retain minimum distances between the core regions and between the LCR and downstream globin promoters (16). Genetic correction of β-thalassemia requires the use of control sequences from the LCR to drive therapeutic levels of transcription. The size constraints of the LV system force trimming and compaction of genomic fragments spanning the active cores of the LCR elements, which significantly reduce the efficiency of this regulatory region by preventing insertion site position effects. Nevertheless, LV harboring LCR HS2/HS3/HS4-mini-β-globin gene combinations have encouragingly resulted in therapeutic levels of β-globin expression at an average VCN of 1–2 per cell (2, 4, 17). More recently, it has been shown that the addition of βLCR HS1 may further improve expression levels (17).
In this work, we developed a LV for β-globin expression carrying only βLCR HS2 and HS3 sequences, which were reported to possess the most crucial LCR classical enhancer and dominant chromatin-opening functions, respectively (9). The size and position of these genomic fragments was chosen to preserve the distance between their active cores that give rise to full functionality (18). In addition, GLOBE features a larger internal deletion of intron 2, reducing its size to 257 bp but maintaining the 3′ enhancer end and full splicing capability (10, 19, 20), and lacks the 3′ β-globin enhancer that was dispensable in this transcription unit design (M.N.A., unpublished results). The absence of HS4 does not appear to affect the therapeutic potential of the GLOBE vector. Furthermore, the introduction of this β-globin transcriptional cassette into the LV backbone did not adversely affect vector production and stability, as reported for other large LCR-β-globin vectors (4, 12, 15). The high viral titer of GLOBE and its molecular stability will facilitate large-scale production and utilization at a clinical level.
The therapeutic potential of GLOBE was evaluated in one of the most severe murine models of β-thalassemia intermedia, the th3/+ mutant. Transplantation of LV-transduced HSCs into primary and secondary thalassemic recipients led to complete and persistent correction of anemia and hematological parameters, rescue of ineffective erythropoiesis, and correction of secondary organ associated pathology. Molecular analysis indicated that the engraftment of a finite number of transduced HSCs was sufficient to fully correct thalassemia, with an average VCN of less than 1. These data are comparable with those recently obtained by using a vector expressing β-globin under the control of larger LCR elements (17). A high VCN of GLOBE into engrafted HSCs was not necessary to correct thalassemia, in contrast to some previous studies (3, 21–23), indicating a good safety profile for the GLOBE vector. We observed a finite number of dominant transduced HSCs (2–15 per mouse), as expected by their frequency in the transplanted BM, the engraftment defect associated with the in vitro stimulation, and the fluctuation in the expansion of single HSCs, which gives rise to committed progenitors (24, 25). Clonal dominance driven by vector insertion is highly unlikely given the features of the GLOBE integration sites. Indeed, oligoclonality is a common finding in experiments of transplantation of retrovirus-transduced hematopoietic cells, as reported by others for different systems (2, 26, 27).
Correction of thalassemia was also achieved in the murine model of Cooley's anemia. Transplantation of transduced β0 FLCs in lethally irradiated recipients resulted in all RBCs expressing chimeric human–murine Hb, rescue of all of the transplanted mice from lethality, reversion to a normal phenotype in half of the treated animals, and conversion to a thalassemia intermedia phenotype in the remaining ones. Long-term full chimeras had a VCN between 3 and 5. These values are not unexpected, considering that a consistent increase in Hb levels (from 3.5 to 13.4 g/dl) is necessary to correct this phenotype, and that human β-globin associates poorly with the murine α-chain (28), forming tetramers that might not be as functional as the murine ones. Results obtained in this model should therefore be considered indicative but not fully predictive for future clinical translation.
A crucial aspect of gene therapy for hematopoietic diseases is the proportion of engrafted, genetically corrected HSCs that is needed to achieve a therapeutic benefit. Results emerging from gene therapy trials of two different forms of severe combined immunodeficiency indicated that selective advantage conferred by transgenes (adenosine deaminase or γc-receptor) is key to a successful outcome in the presence of a small number of transduced hematopoietic progenitors (0.1–10%) (29–31). BM transplantation in thalassemia demonstrates that it is not necessary to completely ablate the patient's BM to obtain clinical control of the disease. Transplanted patients with long-term, persistent mixed chimerism ranging from 20% to 50% are transfusion independent (32), allowing one to predict that partial replacement of thalassemic HSCs with genetically corrected cells would be sufficient, with no need for a full myeloablative regimen. In transplanted β-thalassemic mice, low-level chimerism (10–20%) resulted in a large majority of donor-derived RBCs, a significant increase in Hb level, and diminished EMH (8). The proportion of donor erythroid progenitors/precursors (burst-forming units-erythroid or pro-erythroblasts) in BM of transplanted patients or mice paralleled the proportion observed for donor leukocytes (8, 32), demonstrating that the amplification of the normal erythroid component does not occur up to the pro-erythroblast stage. However, data from BM transplantation cannot be directly translated to gene therapy because the amount of β-globin produced by vector-transduced cells is unlikely to be comparable to that synthesized by normal cells. Our findings provide evidence that a significant, therapeutic amplification of a genetically corrected erythroid component occurs with HSC chimerism as low as 30–50%. In most of the mice, the transduction level in the Ter119+ BM population was on average 4-fold higher than that observed in the nonerythroid compartment, indicating a selective advantage of highly transduced and possibly highly expressing erythroid precursors. Because the most immature, pro-erythroblastic precursors account for <6% of the Ter119+ component of a steady-state BM, it is very likely that selection occurs at the level of mature basophilic, polychromatophilic, and orthochromic erythroblasts. Molecular and biochemical analysis on CD71+/Ter119+ BM subpopulations is needed to determine the stage at which selection is established. The normal proportion of the overall Ter119+ component in the BM of genetically corrected mice indicates a survival rather than a proliferative advantage. In at least one mouse, we obtained evidence for a survival advantage of transduced erythroid cells, also in the periphery. Correction of ineffective erythropoiesis and increased red cell survival are most likely the basis of the observed selective advantage of genetically corrected cells. This selective advantage was most likely overlooked in previous studies, which did not separately analyze BM subpopulations in genetically corrected mice (2, 3).
Interestingly, the analysis of the proviral integration sites in the Ter119+ and Ter119− subpopulations revealed that integrations in intergenic regions were significantly more abundant in the former fraction. We speculate that the erythroblastic compartment was mainly sustained by the expansion of progenitors harboring proviral integrations in genome sites more favorable to high levels of vector-derived expression. A high level of transgene expression from intergenic integrations was previously reported in CFU-S transduced with a globin-expressing vector (33). These findings are unexpected based on previous data on the genomic location of latent and stably expressed HIV proviruses (34), suggesting that the genomic requirements for expression of globin LCR-driven promoters in a LV context may differ from those of the HIV LTR.
In conclusion, this study demonstrates that correction of β-thalassemia is achieved by in vivo selection of genetically corrected erythroblastic progenitors, differentiating from a relatively limited number of transduced HSCs. These findings have relevant implications for the design of future clinical trials because they indicate that full myeloablation may not be necessary for gene therapy of thalassemia.
Materials and Methods
DNA Cloning and Analysis.
To generate the pRRL.sin-18.GLOBE LV, a human β-globin minigene and a 2.7-kb βLCR HS2-HS3 element (18) were cloned into pRRL.sin-18.CMV.GFP plasmid (35), by replacing the CMV-GFP-WPRE fragment. β-Globin minigene, deleted from a 600-bp RsaI to SspI sequence in intron II, extends from a ClaI-linkered SnaBI site at −265 bp from the transcriptional start site to a Acc65I-linkered BspHI site ≈300 bp after the poly(A)-addition site. Viral vector stocks were prepared as described in ref. 36. Viral titers were determined by transduction of HEL cells with serial dilution of the vector stocks followed by quantitative PCR after 3 weeks of culture to allow dilution of unintegrated vector below detection level. Genomic DNA was isolated by using the Qiagen QIAmp DNA mini Kit. VCN was measured with quantitative PCR, by using primers and probe specific for the rev-responsive element (RRE) region: forward primer 5′-TGAAAGCGAAAGGGAAACCA-3′, reverse primer 5′-CCGTGCGCGCTTCAG- 3′ and probe 5′-VIC-AGC TCT CTC GAC GCA GGA CTC GGC-MGB-3′ (VIC, a reporter from Applied Biosystems; MGB, minor groove binder).
Analysis of GLOBE Integration Sites.
Integration sites were amplified by LM-PCR, cloned, and sequenced as described in ref. 14. Sequences were mapped within the murine genome by employing the BLAT genome browser (University of California Santa Cruz Human Genome Project Working Draft, October 2005). A genuine integration contained both LTR- and linker-specific sequences and a genomic sequence featuring a unique best hit with 95% identity to the murine genome. The list of mapped integrations is provided in Tables S3 and S4.
Transplantation of Hematopoietic Cells.
C57BL/6 mice were purchased from Charles River. C57BL6/Hbbth3 mice (th3/+) were kindly provided by M. Sadelain (Memorial Sloan–Kettering Cancer Center, New York). C57BL/6-CD45.1 (B/6.SJL-CD45a-Pep3b) were purchased from The Jackson Laboratory. Murine BM cells were harvested from control C57BL/6 and C57BL6/Hbbth3 mice and infected for 18 h with viral stocks at a multiplicity of infection of 20 as described in ref. 36. Transduced cells were injected (2 × 106 cells per mouse) i.v. in 8-week-old C57BL/6-CD45.1 and C57BL6/Hbbth3 mice given 1,100 cGy of total body irradiation. CFU-S were isolated at day 12 from the spleen of recipient mice injected with 1 × 105 BM cells from primary recipients. FLCs were harvested, infected as previously described (15), and injected (3–4 × 106 cells per mouse) into lethally irradiated C57BL/6-CD45.1 mice. All procedures were approved by the Animal care and Use Committee of the Fondazione San Raffaele del Monte Tabor and of the University of Washington and were communicated to the Ministry of Health and local authorities according to Italian law.
RBC Morphology and Hematological Analysis.
Blood samples were collected and Hb concentration, RBC counts, and Hct were measured on the Sysmex KX-21 automated blood cell analyzer (Sysmex). Blood smears were stained with May–Grunwald/Giemsa. Reticulocyte counts were determined by flow cytometry after staining with thiazole orange dye (Aldrich).
Histopathological Analyses.
Liver and spleen samples were fixed in 4% paraformaldehyde solution, paraffin-embedded, sectioned, stained with hematoxylin and eosin (H&E), Giemsa, and Perls staining, and examined by light microscopy.
Statistical Analyses.
We used the two-tailed Student's t test to determine whether hematological parameters and erythroid expansion differed between groups. The Kaplan–Meier analysis was performed to construct survival curves that were compared by using the log-rank test. All of the statistical analyses were performed by using GraphPad Prism Version 0.4 (GraphPad).
Supplementary Material
Acknowledgments.
We thank Francesca Tiboni for technical assistance and Fulvio Mavilio for helpful discussion and critical comments. This work was supported by grants from the Italian Telethon Foundation, Fondazione Istituto Mediterraneo di Ematologia, the European Commission (VI FP, CONSERT, BMH4-CT96-1279), the National Institutes of Health, and the United Kingdom Thalassaemia Society.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0711666105/DCSupplemental.
References
- 1.Lucarelli G, Andreani M, Angelucci E. The cure of thalassemia by bone marrow transplantation. Blood Rev. 2002;16:81–85. doi: 10.1054/blre.2002.0192. [DOI] [PubMed] [Google Scholar]
- 2.May C, et al. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature. 2000;406:82–86. doi: 10.1038/35017565. [DOI] [PubMed] [Google Scholar]
- 3.Imren S, et al. Permanent and panerythroid correction of murine β thalassemia by multiple lentiviral integration in hematopoietic stem cells. Proc Natl Acad Sci USA. 2002;99:14380–14385. doi: 10.1073/pnas.212507099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Puthenveetil G, et al. Successful correction of the human beta-thalassemia major phenotype using a lentiviral vector. Blood. 2004;104:3445–3453. doi: 10.1182/blood-2004-04-1427. [DOI] [PubMed] [Google Scholar]
- 5.Sadelain M. Recent advances in globin gene transfer for the treatment of beta-thalassemia and sickle cell anemia. Curr Opin Hematol. 2006;13:142–148. doi: 10.1097/01.moh.0000219658.57915.d4. [DOI] [PubMed] [Google Scholar]
- 6.Hino S, Fan J, Taguwa S, Akasaka K, Matsuoka M. Sea urchin insulator protects lentiviral vector from silencing by maintaining active chromatin structure. Gene Ther. 2004;11:819–828. doi: 10.1038/sj.gt.3302227. [DOI] [PubMed] [Google Scholar]
- 7.Gaziev J, Lucarelli G. Stem cell transplantation for hemoglobinopathies. Curr Opin Pediatr. 2003;15:24–31. doi: 10.1097/00008480-200302000-00005. [DOI] [PubMed] [Google Scholar]
- 8.Persons DA, et al. Functional requirements for phenotypic correction of murine beta-thalassemia: Implications for human gene therapy. Blood. 2001;97:3275–3282. doi: 10.1182/blood.v97.10.3275. [DOI] [PubMed] [Google Scholar]
- 9.Mahajan MC, Karmakar S, Weissman SM. Control of beta globin genes. J Cell Biochem. 2007;102:801–810. doi: 10.1002/jcb.21507. [DOI] [PubMed] [Google Scholar]
- 10.Antoniou M, Geraghty F, Hurst J, Grosveld F. Efficient 3′-end formation of human beta-globin mRNA in vivo requires sequences within the last intron but occurs independently of the splicing reaction. Nucleic Acids Res. 1998;26:721–729. doi: 10.1093/nar/26.3.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang B, et al. A mouse model for β0-thalassemia. Proc Natl Acad Sci USA. 1995;92:11608–11612. doi: 10.1073/pnas.92.25.11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imren S, et al. High-level beta-globin expression and preferred intragenic integration after lentiviral transduction of human cord blood stem cells. J Clin Invest. 2004;114:953–962. doi: 10.1172/JCI21838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montini E, et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol. 2006;24:687–696. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- 14.Cattoglio C, et al. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood. 2007;110:1770–1778. doi: 10.1182/blood-2007-01-068759. [DOI] [PubMed] [Google Scholar]
- 15.Rivella S, May C, Chadburn A, Riviere I, Sadelain M. A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene transfer. Blood. 2003;101:2932–2939. doi: 10.1182/blood-2002-10-3305. [DOI] [PubMed] [Google Scholar]
- 16.Palstra RJ, de Laat W, Grosveld F. beta-Globin regulation and long-range interactions. Adv Genet. 2008;61:107–142. doi: 10.1016/S0065-2660(07)00004-1. [DOI] [PubMed] [Google Scholar]
- 17.Lisowski L, Sadelain M. Locus control region elements HS1 and HS4 enhance the therapeutic efficacy of globin gene transfer in beta-thalassemic mice. Blood. 2007;110:4175–4178. doi: 10.1182/blood-2007-08-108647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collis P, Antoniou M, Grosveld F. Definition of the minimal requirements within the human beta-globin gene and the dominant control region for high level expression. EMBO J. 1990;9:233–240. doi: 10.1002/j.1460-2075.1990.tb08100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Behringer RR, Hammer RE, Brinster RL, Palmiter RD, Townes TM. Two 3′ sequences direct adult erythroid-specific expression of human β-globin genes in transgenic mice. Proc Natl Acad Sci USA. 1987;84:7056–7060. doi: 10.1073/pnas.84.20.7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antoniou M, deBoer E, Habets G, Grosveld F. The human beta-globin gene contains multiple regulatory regions: Identification of one promoter and two downstream enhancers. EMBO J. 1988;7:377–384. doi: 10.1002/j.1460-2075.1988.tb02824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levasseur DN, Ryan TM, Pawlik KM, Townes TM. Correction of a mouse model of sickle cell disease: Lentiviral/antisickling beta-globin gene transduction of unmobilized, purified hematopoietic stem cells. Blood. 2003;102:4312–4319. doi: 10.1182/blood-2003-04-1251. [DOI] [PubMed] [Google Scholar]
- 22.Pawliuk R, et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. 2001;294:2368–2371. doi: 10.1126/science.1065806. [DOI] [PubMed] [Google Scholar]
- 23.Persons DA, et al. Successful treatment of murine beta-thalassemia using in vivo selection of genetically modified, drug-resistant hematopoietic stem cells. Blood. 2003;102:506–513. doi: 10.1182/blood-2003-03-0677. [DOI] [PubMed] [Google Scholar]
- 24.Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci USA. 1999;96:3120–3125. doi: 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peters SO, Kittler EL, Ramshaw HS, Quesenberry PJ. Ex vivo expansion of murine marrow cells with interleukin-3 (IL-3), IL-6, IL-11, and stem cell factor leads to impaired engraftment in irradiated hosts. Blood. 1996;87:30–37. [PubMed] [Google Scholar]
- 26.Mohamedali A, et al. Self-inactivating lentiviral vectors resist proviral methylation but do not confer position-independent expression in hematopoietic stem cells. Mol Ther. 2004;10:249–259. doi: 10.1016/j.ymthe.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 27.Richard E, et al. Gene therapy of a mouse model of protoporphyria with a self-inactivating erythroid-specific lentiviral vector without preselection. Mol Ther. 2001;4:331–338. doi: 10.1006/mthe.2001.0467. [DOI] [PubMed] [Google Scholar]
- 28.Fabry ME, et al. A second generation transgenic mouse model expressing both hemoglobin S (HbS) and HbS-Antilles results in increased phenotypic severity. Blood. 1995;86:2419–2428. [PubMed] [Google Scholar]
- 29.Aiuti A, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–2413. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 30.Cavazzana-Calvo M, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 31.Gaspar HB, et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet. 2004;364:2181–2187. doi: 10.1016/S0140-6736(04)17590-9. [DOI] [PubMed] [Google Scholar]
- 32.Andreani M, et al. Long-term survival of ex-thalassemic patients with persistent mixed chimerism after bone marrow transplantation. Bone Marrow Transplant. 2000;25:401–404. doi: 10.1038/sj.bmt.1702151. [DOI] [PubMed] [Google Scholar]
- 33.Hanawa H, et al. Extended beta-globin locus control region elements promote consistent therapeutic expression of a gamma-globin lentiviral vector in murine beta-thalassemia. Blood. 2004;104:2281–2290. doi: 10.1182/blood-2004-03-0863. [DOI] [PubMed] [Google Scholar]
- 34.Lewinski MK, et al. Genome-wide analysis of chromosomal features repressing human immunodeficiency virus transcription. J Virol. 2005;79:6610–6619. doi: 10.1128/JVI.79.11.6610-6619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Follenzi A, Sabatino G, Lombardo A, Boccaccio C, Naldini L. Efficient gene delivery and targeted expression to hepatocytes in vivo by improved lentiviral vectors. Hum Gene Ther. 2002;13:243–260. doi: 10.1089/10430340252769770. [DOI] [PubMed] [Google Scholar]
- 36.Lotti F, et al. Transcriptional targeting of lentiviral vectors by long terminal repeat enhancer replacement. J Virol. 2002;76:3996–4007. doi: 10.1128/JVI.76.8.3996-4007.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.