

Abstract
Peroxynitrite (ONOO−) is a potent oxidant implicated in a number of pathophysiological processes. The activity of ONOO− is related to its accessibility to biological targets before its spontaneous decomposition (t1/2 ≈ 1 s at pH 7.4, 37°C). Using model phospholipid vesicular systems and manganese porphyrins as reporter molecules, we demonstrated that ONOO− freely crosses phospholipid membranes. The calculated permeability coefficient for ONOO− is ≈8.0 × 10−4 cm⋅s−1, which compares well with that of water and is ≈400 times greater than that of superoxide. We suggest that ONOO− is a significant biological effector molecule not only because of its reactivity but also because of its high diffusibility.
Peroxynitrite (ONOO−) has emerged as an important member of the family of reactive oxygen and nitrogen species (1–5) since the recognition of its rapid formation from nitric oxide (NO−·) and superoxide anion (O2−·) (6) The production of ONOO− in vivo has been demonstrated in the macrophage immune response (7, 8) and under conditions of oxidative stress such as ischemia/reperfusion (9–11). The reactions of ONOO− with biological substrates are known to include the nitration of tyrosine residues in proteins (12) and the oxidation of redox metal centers (13, 14), DNA (15, 16), lipids (17), sulfhydryls (18), and methionine (19). In light of this reactivity, ONOO− has been implicated in a number of pathological conditions including neurological disorders (20–23) such as Alzheimer disease and amyotrophic lateral sclerosis, in atherosclerosis (24, 25), and a variety of conditions precipitated by endothelial injury (26). Furthermore, since nitration of tyrosine has been shown to block tyrosine phosphorylation, a key event in signal transduction cascades, the role of ONOO− as a signal molecule has been under investigation (27, 28). It also has been demonstrated that ONOO− nitrates and inactivates manganese superoxide dismutase in chronic rejection of human renal allografts, which was proposed to be a general mechanism for the amplification of ONOO− oxidative damage (29). Given the short lifetime of ONOO− (t1/2 ≈ 1 s at pH 7.4, 37°C) (30), a diffusion distance of ≈100 μm has been estimated in physiological buffers (31). However, cells are compartmentalized into membrane-protected organelles (32). Thus, an important determinant of toxicity or signaling effectiveness of ONOO− will lie in its invasiveness and ability to access biological targets. Here, we demonstrate that ONOO− can diffuse freely across phospholipid membrane bilayers to react with target substrates. Thus, the significance of ONOO− as a biological effector molecule will derive not only from its reactivity but also its diffusibility.
MATERIALS AND METHODS
Materials.
5,10,15,20-Tetrakis(N-methyl-4′-pyridyl)porphinatomanganese(III) chloride [Mn(III)TMPyP(Cl)] and Fe(III)TMPyP(Cl) were purchased from Midcentury (Posen, TX). Peroxymonosulfate (HSO5− or OXONE), hypochlorite (OCl−), and 1-O-octyl-β-d-glucopyranoside were obtained from Aldrich. Sephadex G25 was obtained from Pharmacia. Anhydrous monosodium phosphate, anhydrous disodium phosphate, Sepharose 4B, dimyristol l-α-phosphatidylcholine (DMPC), and Liposome Kit Positive (lyophilized powder containing 63 μmol of l-α-phosphatidylcholine, 18 μmol of stearyl amine, and 9 μmol of cholesterol) were purchased from Sigma. (α,β,α,β-Meso-tetrakis[O-((3β-hydroxy-5-cholenyl)amido)phenyl]-porphyrinato)manganese(III) chloride [Mn(III)ChP(Cl)] was synthesized according to literature methods (33). The sodium salt of ONOO− was prepared from the reaction of acidic H2O2 with sodium nitrite following a published procedure (34). All solvents were analytical grade. Water used in all the experiments was distilled and deionized (Milli-Q, Millipore).
Vesicles Preparation.
Small unilamellar vesicles (SUV) were prepared by using the method of ultrasonication of DMPC thin films containing Mn(III)ChP (35). The porphyrin was replaced by retinoic acid (40 μM) or tocopherol (80 μM) in the lipid thin films for SUV used in the Fe(III)TMPyP protection experiments. Buffer solutions were added to the dry lipid thin films and sonicated using a probe tip sonicator (Branson model 250) for ≈5 min at 20–22°C. SUV generated were allowed to equilibrate for 1/2 h before centrifugation to remove particulate matter. The final concentration of DMPC in the vesicular solutions was 1.67 mM. The Mn(III)ChP concentration was varied between 2 and 8 μM.
Large unilamellar vesicles (LUV) were prepared using the detergent removal method (36) with modifications. In brief, a mixture of l-α-phosphatidylcholine (63 μmol), stearyl amine (18 μmol), and cholesterol (9 μmol) was dissolved in a phosphate buffer (50 mM, pH 7.4) solution containing Mn(III)TMPyP (200 μM) and 1-O-octyl-β-d-glucopyranoside (900 μmol). This solution was applied to a Sephadex G-25 column (1 × 20 cm) presaturated with an eluant consisting of Mn(III)TMPyP (200 μM) in phosphate buffer (50 mM, pH 7.4). The exclusion of the detergent on the column resulted in the formation of LUV containing Mn(III)TMPyP (200 μM) in the entrapped internal volume. The vesicles were eluted from the column by using the same solution used to presaturate the column. The vesicles were purified further on a Sepharose 4B column (2 × 10 cm) using phosphate buffer (50 mM, pH 7.4). Neutral LUV without the positively charged stearyl amine were prepared similarly. Transmission electron micrographs of the LUV with entrapped Mn(III)TMPyP and SUV were obtained after fixing with 0.5% OsO4 and staining with 2% uranyl acetate. Typical SUV diameters were 35 nm, and the LUV diameters were 150 nm.
Stopped-Flow Spectrophotometry.
Time-resolved UV-vis spectra were recorded on a HI-TECH SF-61 DX2 stopped-flow spectrophotometer (HI-TECH, Salisbury, U.K.) by using the photo-diode array fast scan mode. The spectral resolution was ≈1 nm. The stoichiometric oxidation of Mn(III)TMPyP by ONOO− was achieved by stopped-flow single mixing to give final concentrations of 5 μM Mn(III)TMPyP and 6.7 μM ONOO− in phosphate buffer (25 mM, pH 7.4). The reaction was followed by UV-vis (0–0.5 s, 100 scans, 0.005-s integration time). The kinetic profiles of the Mn(III)TMPyP oxidation by ONOO− in solution or inside the LUV were collected by using the stopped-flow photomultiplier mode. All measurements were performed at ambient temperatures (22–24°C). The reactions were conducted in either a phosphate buffer (25 mM, pH 7.4) or a Tris buffer (25 mM, pH 8.2). Typically, the concentration of Mn(III)TMPyP was held constant (2 μM) while the ONOO− concentration was varied (50–250 μM) to provide pseudo first-order conditions. For experiments involving Mn(III)TMPyP entrapped in LUV, the concentration of Mn(III)TMPyP in the bulk solution was used (typically 2–2.5 μM). The decay of Mn(III)TMPyP and the appearance of oxoMn(IV) complexes were monitored at 462 nm and 428 nm, respectively. The kinetic profiles could be fitted nicely into a single exponential to give the pseudo first-order rate constants [k (s−1)] under different ONOO− concentrations. The oxidants H2O2, perchlorate (OCl−), and peroxymonosulfate (HSO5−) were used for the control experiments.
The reactions of the membrane-bound Mn(III)ChP with ONOO− also were performed in the stopped-flow photomultiplier mode by following changes at 474 nm [Mn(III)ChP] and 424 nm [oxoMn(IV) species]. The initial rate of oxoMn(IV) formation was linearly dependent on the ONOO− concentration. This allowed an estimation of the second-order rate constant (k) for the oxidation reaction after taking into account the spontaneous decay rates of ONOO− and the oxoMn(IV) species.
Protection of Membrane Components by Fe(III)TMPyP.
Retinoic acid (40 μM) and tocopherol (80 μM) were incorporated into SUV preparations containing DMPC (1.67 mM). The reactions of ONOO− with retinoic acid were followed at 340 nm, and those with tocopherol were followed at 300 nm. The membrane-bound substrates were mixed with 250 μM ONOO−, which resulted in a complete loss of chromophore in both cases indicating oxidation. For the protection experiments, the concentration of Fe(III)TMPyP was varied between 2 and 10 μM. Under these conditions, the ONOO− concentration was exhausted quickly (t½ = 0.16 − 0.035).
RESULTS AND DISCUSSION
The membrane permeability of ONOO− has been examined in a phospholipid vesicular model system by exploiting the diagnostic reactions of manganese porphyrins we have described recently (37, 38). Manganese porphyrins known to occupy different regions of the membrane system have been used to assess the accessibility of ONOO− to these sites. Thus, a membrane-spanning steroidal porphyrin [Mn(III)ChP] (33, 39) and a water-soluble [Mn(III)TMPyP] porphyrin were deployed within the hydrophobic bilayer and inside the aqueous interior compartment, respectively, of unilamellar vesicles (Fig. 1). Oxidation of Mn(III) to the corresponding oxo Mn(IV) species was followed by observing the signature changes in the UV-vis spectra on addition of ONOO− to the medium.
Figure 1.

A schematic representation of the reporter manganese porphyrins in the model vesicular systems. Mn(III)TMPyP, when entrapped inside the vesicles, is prevented from sampling the bulk volume during the experiments. Mn(III)ChP spans the membrane with the position of the macrocycle at the center of the membrane and porphyrin plane parallel to the membrane surface (33). The reaction of the manganese(III) porphyrin with peroxynitrite to afford oxomanganese(IV) and NO2 is depicted below.
The solution reaction of Mn(III)TMPyP with ONOO− was found to be fast and stoichiometric by rapid mixing stopped-flow spectrophotometry (Fig. 2). The depletion of Mn(III)TMPyP to generate the oxoMn(IV) species followed clear stoichiometric behavior with isosbestic points at 389, 448, 536, and 603 nm. Kinetic measurements determined that the intrinsic rate of Mn(III)TMPyP oxidation by ONOO− was 1.8 × 106 M−1⋅s−1 at pH 7.4. Notably, the rates for the ONOO− oxidation of metalloporphyrins and heme peroxidases like myeloperoxidase are the fastest reactions reported for ONOO− (14). Control experiments showed that H2O2 was incapable of oxidizing Mn(III)TMPyP to oxoMn(IV) species under these conditions.
Figure 2.

Time resolved UV-vis profile of the stoichiometric oxidation of Mn(III)TMPyP by ONOO− to generate the oxoMn(IV) species. Stopped-flow mixing was used to give final concentrations of Mn(III)TMPyP (5 μM) and ONOO− (6.7 μM) in 25 mM phosphate (pH 7.4) buffer. The reaction was followed by UV-vis (0–0.5 s, 100 scans, 0.005-s integration time) by using a HI-TECH SF-61 DX2 rapid-mixing stopped-flow spectrophotometer (1 in 10 scans shown for clarity). The rate constant for the oxidation of Mn(III)TMPyP by ONOO− obtained under pseudo first-order conditions at pH 7.4 was kintrinsic = 1.8 × 106 M−1⋅s−1 (see Fig. 3D).
The rate of oxidation of Mn(III)ChP by ONOO−, in the bilayer region, was much slower (k = 3 × 104 M−1⋅s−1) but demonstrated the ability of ONOO− to enter and react within the hydrophobic region of the membrane (Fig. 1). Likewise, retinoic acid and tocopherol were oxidized readily by ONOO− under these conditions. Mn(III)TMPyP redox coupled with antioxidants, which catalytically reduces ONOO−, has been shown to afford a dose-dependent protection of these lipid components (40). Fe(III)TMPyP, which has been reported to isomerize ONOO− to nitrate (41), also effectively prevented the oxidation of these lipid components by ONOO−. Liposomal assemblies of both manganese and iron porphyrin amphiphiles have been found to be effective as well but only when the molecular design of the membrane anchor allowed the charged porphyrin moiety to occupy the aqueous region of the membrane interface (42).
Significantly, the reaction of ONOO− with Mn(III)TMPyP entrapped within LUV resulted in the efficient production of the oxoMn(IV) species (Fig. 3A). Similar results were obtained with the oxidant OCl−. By contrast, HSO5− (43) failed to react under these conditions (Fig. 3 B and C), demonstrating that the LUV were impermeable to this less chaotropic and less basic anion. The high membrane permeability of ONOO− and OCl− may be attributed to the relatively high pKa of their conjugate acids, 6.8 (18, 19) and 7.4 (44), respectively. The order described by the Eisenman sequence I for anions crossing artificial membranes is: SCN− > I− > NO3− > Br− > Cl− > F− > CH3OSO3− ≈ CH3SO3− ≈ SO42− (45). Whereas HSO5− behaves like the membrane-impermeable CH3SO3−, ONOO− resembles NO3− in its permeability. Curiously, however, the oxidation rate of the entrapped Mn(III)TMPyP by ONOO− showed little dependence on pH. The second-order rate constants at pH 7.4 (25 mM, phosphate) and at pH 8.2 (25 mM Tris) were kobsd = 0.8 × 106 M−1⋅s−1 (R = 0.996) and kobsd = 1.2 × 106 M−1⋅s−1 (R = 0.996), respectively. We also determined that the presence of the positively charged stearyl amine did not assist in ONOO− transport across membranes because its removal to generate neutral vesicles did not change the observed rates of the oxidation reactions. We suggest that the hydrophobic vesicular barrier may modulate the pKa of ONOO− (46), thus facilitating the transport of the neutral HOONO species across membranes.
Figure 3.

ONOO− is shown to cross membranes freely. Difference spectra of the UV-vis absorbances before and after addition of oxidants to LUV—encapsulated Mn(III)TMPyP. (A) ONOO− (1 mM) addition resulted in an instantaneous oxidation of Mn(III) (462 nm) to the oxoMn species (428 nm), indicating that ONOO− diffused rapidly across the lipid bilayer. Rapid oxidation was seen between pH 7.4 and 8.8, suggesting that the ONOO− anion as well as HOONO can cross membranes (pKa ONOO− 6.8). Similar results were obtained with OCl−. (B) Addition of HSO5− (1 mM) to the Mn(III)TMPyP/LUV resulted in no oxidation, verifying that the LUV prevented intermixing of the entrapped porphyrin and HSO5− in this construct. (C) Oxidation by HSO5− was observed only after the LUV had been sonicated to relocate Mn(III)TMPyP into the bulk solution. (D) Comparison of the pseudo first-order rates for the solution reaction compared with the reaction in LUV. All of the experiments were conducted using rapid mixing stopped-flow methods as described in Materials and Methods. Reaction rates obtained from the slope of the pseudo first-order plots were: kintrinsic = 1.8 × 106 M−1⋅s−1 (R = 0.999); and kobsd = 0.8 × 106 M−1⋅s−1 (R = 0.996).
Comparison of rates of reaction between Mn(III)TMPyP and ONOO− in solution and in the Mn(III)TMPyP/LUV system (Fig. 3D) showed that membrane entrapment retarded the rate of manganese oxidation by only 2-fold. Accordingly, the barrier offered to ONOO− transport by the membrane is small. This rate difference allows an estimate of the permeability coefficient (P) for ONOO− crossing a membrane bilayer using Fick’s Law. The permeability coefficient is defined as P = J/Δc, where J (mol⋅cm−2⋅s−1) is the flux of ONOO− across unit surface area of the membrane and Δc (mol/L) is the difference in the concentration ([ONOO−]out − [ONOO−]in). The ratio of these concentrations is directly related to kobsd and kintrinsic, the observed and intrinsic rate constants for the reaction of ONOO− with Mn(III)TMPyP we have measured (Eq. 1).
 |
1 |
In the steady state, the membrane crossing rate of ONOO− was taken to be equal to the rate of the oxidation of the entrapped porphyrin inside the LUV, reflecting the barrier offered by the lipid bilayer to the diffusion of ONOO− (Eq. 2). Thus, P is expressed as in Eq. 3, where A is the surface area of vesicles, and Vin is the volume entrapped in LUV.
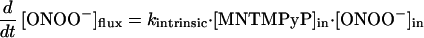 |
2 |
 |
3 |
Calculated values for A = 4.0 × 106 cm2/L and Vin = 10 ml/L derive from the observed radius of the vesicles and afforded a value for P = 8.0 × 10−4 cm⋅s−1 at pH 7.4. Simulations using this permeability coefficient agreed well with the experimental data. A simple, sequential reaction model was assumed, taking into consideration the ONOO− diffusion across phospholipid membrane and subsequent reaction with the entrapped Mn(III)-TMPyP.
We also have considered the recently reported reaction between ONOO− and CO2 to generate an unstable intermediate, presumably ONOOCO2− (k ≈ 1 × 104 M−1⋅s−1) (47, 48). Because the physiological concentration of HCO3− is relatively high (≈25 mM) and is in equilibrium with CO2, this reaction has been suggested to be an important pathway for ONOO− and a determinant of toxicity. However, the rate of diffusion for ONOO− crossing lipid bilayers determined here is at least 30 times faster than the ONOO− reaction with the physiological concentration of CO2 (see Table 1). Preliminary experiments showed that the oxidation of manganese by ONOO− in the Mn(III)TMPyP/LUV system remained unaffected by 25 mM HCO3−, indicating further that the transmembrane diffusion of ONOO− is faster than its reaction with CO2. Therefore, we conclude that the transmembrane diffusion of ONOO− is considerably faster than all documented reaction rates of ONOO− with its biological targets.
Table 1.
Permeability across lipid bilayers
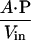
Significantly, the permeability coefficient P obtained for ONOO− in this way is close to that reported for water (49) and is ≈400 times faster than that of superoxide (50, 51) (Table 1). Accordingly, the availability of superoxide across biological membranes is limited both by its relatively slow diffusion and the high concentrations of superoxide dismutase within the cell (25, 52, 53). The high membrane permeability observed here for ONOO− may have important consequences regarding its biochemistry and pathophysiology. Thus, in marked contrast to superoxide ion (54), ONOO− can be expected to have free access to cell interiors across membranes and over distances of cellular dimensions, necessary properties for a biological effector molecule.
Acknowledgments
We thank Dr. M. K. Stern, Monsanto Chemical Co., for stimulating discussions. Support of this research by the National Institutes of Health (GM36928) is acknowledged gratefully.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: ONOO−, peroxynitrite; Mn(III)ChP(Cl), (α,β,α,β-meso-tetrakis[O-((3β-hydroxy-5-cholenyl)amido) phenyl]-porphyrinato)manganese(III) chloride; Mn(III)TMPyP, 5,10,15,20-tetrakis(N-methyl-4′-pyridyl)porphinatomanganese(III) chloride; HSO5−, peroxymonosulfate; SUV, small unilamellar vesicles; LUV, large unilamellar vesicles.
References
- 1.Beckman J S, Koppenol W H. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 2.Beckman J S. Chem Res Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- 3.Rubbo H, Darley-Usmar V, Freeman B A. Chem Res Toxicol. 1996;9:809–820. doi: 10.1021/tx960037q. [DOI] [PubMed] [Google Scholar]
- 4.Radi R. Chem Res Toxicol. 1996;9:828–835. doi: 10.1021/tx950176s. [DOI] [PubMed] [Google Scholar]
- 5.Khan A U, Wilson T. Chem Biol. 1995;2:437–445. doi: 10.1016/1074-5521(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 6.Huie R E, Padmaja S. Free Radical Res Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 7.Ischiropoulos H, Zhu L, Beckman J S. Arch Biochem Biophys. 1992;298:446–451. doi: 10.1016/0003-9861(92)90433-w. [DOI] [PubMed] [Google Scholar]
- 8.Evans T J, Buttery L D K, Carpenter A, Springall D R, Polak J M, Cohen J. Proc Natl Acad Sci USA. 1996;93:9553–9558. doi: 10.1073/pnas.93.18.9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beckman J S, Beckman T W, Chen J, Marshall P A, Freeman B A. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carreras M C, Pargament G A, Catz S, Poderosso J J, Boveris A. FEBS Lett. 1994;341:65–68. doi: 10.1016/0014-5793(94)80241-6. [DOI] [PubMed] [Google Scholar]
- 11.Huang Z, Huang P L, Panahian N, Dalkara T, Fishman M C, Moskowitz M A. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 12.Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin J C, Smith C D, Beckman J S. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- 13.Castro L, Rodriguez M, Radi R. J Biol Chem. 1994;269:29409–29415. [PubMed] [Google Scholar]
- 14.Floris R, Piersma R, Yang G, Jones P, Wever R. Eur J Biochem. 1993;215:767–775. doi: 10.1111/j.1432-1033.1993.tb18091.x. [DOI] [PubMed] [Google Scholar]
- 15.King P A, Anderson V E, Edwards J O, Gustafson G, Plumb R C, Suggs J W. J Am Chem Soc. 1992;114:5430–5432. [Google Scholar]
- 16.King P A, Jamison E, Strahs D, Anderson V E, Brenowitz M. Nucleic Acids Res. 1993;21:2473–2478. doi: 10.1093/nar/21.10.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Radi R, Beckman J S, Bush K M, Freeman B A. Arch Biochem Biophys. 1991;288:481–487. doi: 10.1016/0003-9861(91)90224-7. [DOI] [PubMed] [Google Scholar]
- 18.Radi R, Beckman J S, Bush K M, Freeman B A. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 19.Pryor W A, Jin X, Squadrito G L. Proc Natl Acad Sci USA. 1994;91:11173–11177. doi: 10.1073/pnas.91.23.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipton S A, Chol T-B, Pan Z-H, Lei S Z, Chen H-S, Sucher N J, Loscalzo J, Singel D J, Stamler J S. Nature (London) 1993;364:626–631. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 21.Beckman J S, Carson M, Smith C D, Koppenol W H. Nature (London) 1993;364:584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- 22.Good P F, Werner P, Hsu A, Olanow C W, Perl D P. Am J Pathol. 1996;149:21–28. [PMC free article] [PubMed] [Google Scholar]
- 23.Wiedau-Pazos M, Goto J J, Rabizadeh S, Gralla E B, Roe J A, Lee M K, Valentine J S, Bredesen D E. Science. 1996;271:515–518. doi: 10.1126/science.271.5248.515. [DOI] [PubMed] [Google Scholar]
- 24.Hogg N, Darley-Usmar V M, Graham A, Moncada S. Biochem Soc Trans. 1993;21:358–362. doi: 10.1042/bst0210358. [DOI] [PubMed] [Google Scholar]
- 25.White C R, Brock T A, Chang L, Crapo J, Briscoe P, Ku D, Bradley W A, Gianturco S H, Gore J, Freeman B A, Tarpey M M. Proc Natl Acad Sci USA. 1994;91:1044–1048. doi: 10.1073/pnas.91.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haddad I Y, Pataki G, Hu P, Beckman J S, Matalon S. J Clin Invest. 1994;94:2407–2413. doi: 10.1172/JCI117607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kong S-K, Yim M B, Stadtman E R, Chock P B. Proc Natl Acad Sci USA. 1996;93:3377–3382. doi: 10.1073/pnas.93.8.3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berlett B S, Friguet B, Yim M B, Chock P B, Stadtman E R. Proc Natl Acad Sci USA. 1996;93:1776–1780. doi: 10.1073/pnas.93.5.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacMillan-Crow L A, Crow J P, Kerby J D, Beckman J S, Thompson J A. Proc Natl Acad Sci USA. 1996;93:11853–11858. doi: 10.1073/pnas.93.21.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfeiffer S, Gorren A C F, Schmidt K, Werner E R, Hansert B, Bohle D S, Mayer B. J Biol Chem. 1997;272:3465–3470. doi: 10.1074/jbc.272.6.3465. [DOI] [PubMed] [Google Scholar]
- 31.Beckman J S. In: Nitric Oxide: Principles and Actions. Lancaster J, editor. San Diego: Academic; 1996. pp. 43–44. [Google Scholar]
- 32.Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson J D. Molecular Biology of the Cell. New York: Garland; 1989. [Google Scholar]
- 33.Groves J T, Neumann R. J Am Chem Soc. 1989;111:2900–2909. [Google Scholar]
- 34.Papée H M, Petriconi G L. Nature (London) 1964;204:142–144. [Google Scholar]
- 35.Huang C. Biochemistry. 1969;8:344–352. doi: 10.1021/bi00829a048. [DOI] [PubMed] [Google Scholar]
- 36.Mimms L T, Zampighi G, Nozaki Y, Tanford C, Reynolds J A. Biochemistry. 1981;20:833–840. doi: 10.1021/bi00507a028. [DOI] [PubMed] [Google Scholar]
- 37.Groves J T, Lee J, Marla S S. J Am Chem Soc. 1997;119:6269–6273. [Google Scholar]
- 38.Groves J T, Marla S S. J Am Chem Soc. 1995;117:9578–9579. [Google Scholar]
- 39.Groves J T, Neumann R. J Am Chem Soc. 1987;109:5045–5047. [Google Scholar]
- 40.Lee J, Hunt J A, Groves J T. Bioorg Med Chem Lett. 1997;7:2913–2918. [Google Scholar]
- 41.Stern M K, Jensen M P, Kramer K. J Am Chem Soc. 1996;118:8735–8736. [Google Scholar]
- 42.Hunt J A, Lee J, Groves J T. Chem Biol. 1997;4:845–858. doi: 10.1016/s1074-5521(97)90117-4. [DOI] [PubMed] [Google Scholar]
- 43.Bernadou J, Fabiano A-S, Robert A, Meunier B. J Am Chem Soc. 1994;116:9375–9376. [Google Scholar]
- 44.Sanderson R T. In: Simple Inorganic Substances. Sanderson R T, editor. Malabar, FL: Robert E. Krieger Publishing; 1989. p. 298. [Google Scholar]
- 45.Wright E M, Diamond J M. Physiol Rev. 1977;57:109–156. doi: 10.1152/physrev.1977.57.1.109. [DOI] [PubMed] [Google Scholar]
- 46.Pryor W A, Jin X, Squadrito G L. J Am Chem Soc. 1996;118:3125–3128. [Google Scholar]
- 47.Lymar S V, Hurst J K. J Am Chem Soc. 1995;117:8867. [Google Scholar]
- 48.Lymar S V, Jiang Q, Hurst J K. Biochemistry. 1996;35:7855–7861. doi: 10.1021/bi960331h. [DOI] [PubMed] [Google Scholar]
- 49.Finkelstein A, Cass A. Nature (London) 1967;216:717–718. doi: 10.1038/216717a0. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi M-A, Asada K. Arch Biochem Biophys. 1983;226:558–566. doi: 10.1016/0003-9861(83)90325-9. [DOI] [PubMed] [Google Scholar]
- 51.Mao G D, Poznansky M J. FEBS Lett. 1992;305:233–236. doi: 10.1016/0014-5793(92)80675-7. [DOI] [PubMed] [Google Scholar]
- 52.Halliwell B, Gutteridge J M C. Methods Enzymol. 1990;186:1–85. doi: 10.1016/0076-6879(90)86093-b. [DOI] [PubMed] [Google Scholar]
- 53.Chevion M. Free Radical Biol Med. 1988;5:27–37. doi: 10.1016/0891-5849(88)90059-7. [DOI] [PubMed] [Google Scholar]
- 54.Crow J P, Beckman J S. In: Advances in Pharmacology. Ignarro L, Murad F, editors. Vol. 35. San Diego: Academic; 1995. pp. 17–43. [Google Scholar]