

Abstract
Zebrafish carrying heterozygous mutations for 17 different ribosomal protein (rp) genes are prone to developing malignant peripheral nerve sheath tumors (MPNSTs), a tumor type that is seldom seen in laboratory strains of zebrafish. Interestingly, the same rare tumor type arises in zebrafish that are homozygous for a loss-of-function point mutation in the tumor suppressor gene p53. For these reasons, and because p53 is widely known to be mutated in the majority of human cancers, we investigated the status of p53 in the rp+/− MPNSTs. Using monoclonal antibodies that we raised to zebrafish p53, we found that cells derived from rp+/− MPNSTs are significantly impaired in their ability to produce p53 protein even in the presence of a proteasome inhibitor and γ-irradiation. Although the coding regions of the p53 gene remain wild type, the gene is transcribed, and overall protein production rates appear normal in rp+/− MPNST cells, p53 protein does not get synthesized. This defect is observed in all MPNSTs we examined that were derived from our 17 zebrafish lines with rp gene mutations. To date, studies of p53 in malignancies have focused predominantly on either p53 gene mutations or the aberrant posttranslational regulation of the p53 protein. Our results show that the appropriate amount of numerous ribosomal proteins is required for p53 protein production in vivo and that disruption of this regulation most likely contributes to tumorigenesis.
Keywords: MPNST, protein synthesis
Among several hundred insertional mutants isolated in a screen for zebrafish genes essential for early development were mutations in 28 different ribosomal protein (rp) genes (1–4). Although the homozygous rp mutations are embryonic lethals, fish with heterozygous mutations develop into adults normally, albeit with a slight growth defect (5), as is seen in other animal models of heterozygous rp gene mutations (6, 7). However, heterozygous mutation of 17 of these 28 rp genes resulted in the development of malignant peripheral nerve sheath tumors (MPNSTs) as early as 8 months of age (1). The rps that when mutated give rise to MPNSTs include S3a, S5, S7, S8, S11, S15a, S18, S28, S29, L7, L13, L14, L19, L23a, L35, L36, and L36a, whereas those that do not include SA, S12, S15, acidic LP1, L3, L6, L9, L11, L12, L24, and L28 (5). The MPNST is a very rare tumor type only observed in wild-type zebrafish in our colony with a frequency of ≈1:2,000 fish (A.A., unpublished data). Surprisingly, a homozygous loss-of-function point mutation in the zebrafish p53 gene (p53M214K/M214K) results in tumors that appear similar (8). This observation led us to consider the possibility that heterozygosity at certain rp loci results in the aberrant regulation of the p53 pathway.
The tumor suppressor p53 is one of the most intensely studied proteins in biology because it has been shown that between 50% and 70% of all human tumors contain mutations in the p53 gene (9). Furthermore, it is thought that most of the remaining tumors that do not harbor p53 gene mutations contain other gene amplifications or deletions that result in the down-regulation of the p53 protein. When regulated under normal conditions, p53 is usually kept at low levels in cells by binding to the MDM2 protein, which rapidly ubiquitinates p53 and targets it for degradation by the proteasome (10, 11). DNA damage is one of several signals that results in the disruption of the association of MDM2 with p53, an event that triggers p53 stabilization and transcriptional activation of p53 target genes that instigate cell cycle arrest and/or apoptosis (12, 13). p53 can be similarly stabilized experimentally via inhibition of the proteasome with drugs such as MG132 (14). Deregulation of p53 protein by constitutive ubiquitination has been shown to occur in tumor cells that preserve wild-type copies of the p53 gene yet contain gene amplifications of mdm2 or deletions of the MDM2 inhibitor Arf (15, 16). Thus, in addition to the vast number of p53 gene mutations found associated with tumors, it is safe to say that many, if not all, cancerous cells contain some impairment of the p53 pathway. Notably, zebrafish maintain the core components of the p53 pathway. As mentioned above, p53M214K/M214K zebrafish develop tumors (8); although there is no known arf, an mdm2 ortholog has been identified. There also is genetic confirmation that the epistasis between mdm2 and p53 evident in mammals is conserved: mdm2 knockdown in zebrafish embryos causes widespread apoptosis, and this phenotype is rescued by simultaneous knockdown of p53 (17).
Although there has been no direct study to show that in humans the heterozygous loss of a ribosomal protein leads to cancer, there are several lines of intriguing evidence to suggest such a link may exist. One is the disease Diamond-Blackfan anemia (DBA). It has been shown that >25% of all patients with this disease carry heterozygous mutations in ribosomal protein genes rpS19, rpS24, or rpS17 (18–20). It also is known that a significant number of DBA patients develop leukemia, lymphomas, or solid tumors later in life (21, 22). Additionally, the ribosomal protein gene rpS14 has been identified as one of the nonbiallelic deletions in 5q syndrome, a subtype of myelodysplastic syndrome (MDS) known to predispose individuals to both acute and chronic myeloid leukemia (23). Very recent evidence suggests that the knockdown of the rpS14 gene in hematopoetic progenitor cells recapitulates the phenotype of MDS and that exogenous expression of rpS14 rescues the phenotype in MDS patient bone marrow samples (24). However, for both DBA and MDS, the role of the ribosomal protein gene mutations in the manifestation of disease and the progression to cancer remains unknown.
There also have been a number of cellular studies that link the rps to p53 regulation. Several previous reports have shown that rpL11, rpL5, rpL23, and rpS7 are able to bind to the p53 inhibitor MDM2 in cells, resulting in p53 stabilization (25–29). Furthermore, there is evidence that rpL26 is able to bind to p53 mRNA and influence its rate of translation (30). rpL22 also has been implicated as being able to affect the biosynthesis of p53 (30, 31).
In this article, we investigate how heterozygosity of numerous rp genes might contribute to the formation of tumors in zebrafish. This analysis reveals that there is a widespread link between rp gene dosage and the regulation of p53 protein synthesis. Specifically, we show that, irrespective of the affected rp gene, heterozygous rp gene mutant zebrafish tumor cells maintain wild-type copies of the p53 gene and mRNA transcript, yet do not translate this transcript into p53 protein. This article provides in vivo evidence that alterations in rp gene dosage impair p53 protein synthesis and promote tumorigenesis.
Results
We have previously shown that rp+/− mutant zebrafish develop MPNSTs. However, the molecular basis for this increased tumorigenicity was unclear (1). Subsequent studies showed that p53M214K/M214K zebrafish developed histologically similar tumors (8), raising a possible relationship between rp heterozygosity and p53 regulation. To further explore this notion, we first used expression array profiling to determine whether the rp+/− mutant tumors were indeed the same tumor type as those arising in the p53M214K/M214K zebrafish. mRNAs used for profiling were isolated from tumor cells dissected from p53M214K/M214K (three tumors) and rp+/− mutants (six tumors from four different lines representing rps that participate in both large and small ribosomal subunits). Based on gene expression analysis, the MPNSTs from rp+/− and p53M214K/M214K mutant fish coclustered rather than segregating by genotype. To provide a context for the clustering, we also analyzed mRNAs from seminoma tumors (a commonly occurring tumor type in zebrafish) and compared these data with samples from other publicly available datasets representing a variety of cell types and developmental stages [supporting information (SI) Fig. S1]. We found that one of the rp+/− samples (hi258.3) clustered most closely with a heart sample likely because a subset of the MPNSTs are highly vascularized and contain a high number of blood cells. With the exception of this single outlier, the MPNST samples are more closely related to one another than they are to any other available sample, including the seminoma tumor samples. This finding is consistent with the notion that the rp and p53 mutant tumors share common origins.
Because p53 gene mutations occur so frequently in human tumors, we first sequenced the coding exons of the p53 gene in genomic DNA isolated from rp+/− MPNSTs. In 18 of 18 samples we examined from several different lines of rp+/− mutants, the p53 gene was found to be wild type. As an added control, we also examined the sequence of p53 mRNA transcripts isolated from rp+/− MPNST cells. We cloned and sequenced the coding and noncoding regions of the p53 cDNA from six different MPNSTs (originating from different mutant lines). In every case, the p53 cDNA sequence in the MPNSTs was found to be identical to the published sequence of zebrafish p53 (data not shown). This result would suggest that the transcription of the p53 gene in rp+/− MPNST cells does not introduce genetic mutations, and the p53 mRNA transcript in these cells remains wild type. Because there has been no prior investigation of spontaneous p53 mutations in zebrafish tumors, we also sequenced the p53 gene in 12 seminoma samples from non-rp mutant backgrounds (data not shown). We found that three of these tumors had missense mutations (V124L, H161Y, and L286S) that were not germ-line mutations, as judged by comparative sequencing of tail DNA (which remained wild type). Importantly, the amino acid H161 is analogous to H193 in human p53, which also is found mutated in a number of different cancers (IARC TP53 Mutation Database, www-p53.iarc.fr). These data suggest that p53 gene mutations do occur spontaneously in zebrafish and may contribute to tumorigenesis. However, we did not observe p53 mutations in rp+/− MPNSTs.
Given the absence of p53 gene mutations in the rp+/− MPNSTs, we next sought to determine whether the p53 protein in these cells was deregulated. To this end, we raised monoclonal antibodies to the full-length zebrafish p53 protein. Several of these antibodies recognize zebrafish p53 in ELISA, Western blot, immunoprecipitation, and whole-mount immunohistochemistry assays (data not shown). Three select αp53 antibodies, LLzp53-9E10, LLzp53-12E6, and LLzp53-16H12, were shown to immunoprecipitate HA-tagged zebrafish p53 expressed in transfected 293 cells (Fig. 1A). (From this point on, LLzp53-16H12 is used for Western blots and LLzp53-9E10 is used for immunoprecipitations.) Using the αp53 antibody for Western blotting, we determined that p53 levels are undetectable in zebrafish embryos 24 h after fertilization (Fig. 1B). This finding is entirely consistent with the known instability of human and murine p53 protein in the absence of DNA damage. Accordingly, after treatment of the embryos with γ-irradiation, the αp53 antibody detected two bands of 49 kDa, the predicted size of full-length zebrafish p53, and 44 kDa. To verify that these species were zebrafish p53, we injected embryos with either a translation-blocking morpholino directed to the start of the p53 transcript (p53MO) or a similar morpholino with four base mismatches (p53misMO) at the one-cell stage. Both protein bands were lost exclusively when the p53MO was injected, confirming both the identity of these protein species as p53 and the specificity of the αp53 antibody (Fig. 1B). Based on significant mobility differences, the 44 kDa does not appear to be the previously reported truncated zebrafish p53, Δ113 (32). Thus, its identity is currently unclear. Notably, beyond embryogenesis, we only detect the presumptive full-length 49-kDa p53 species.
Fig. 1.
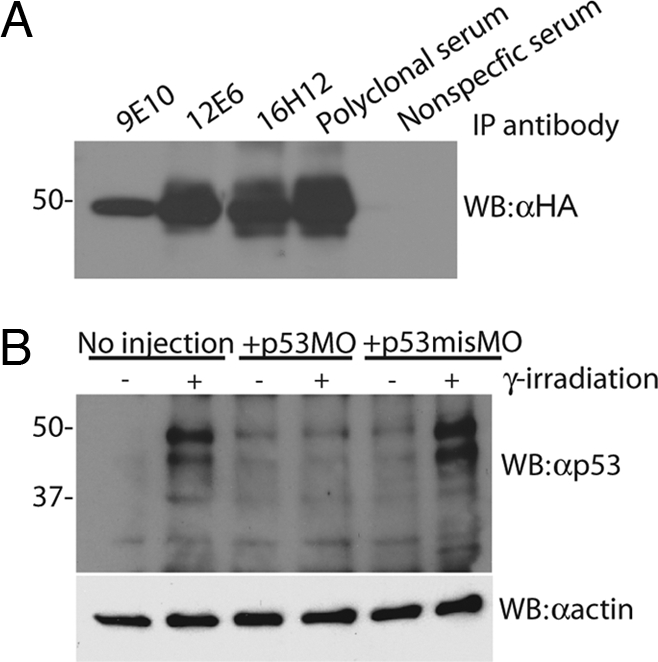
Monoclonal antibodies specifically recognize zebrafish p53. (A) 293 cells were transiently transfected with HA-tagged zebrafish p53 and immunoprecipitated with monoclonal antibodies raised against zebrafish p53 (LLzp53-9E10, LLzp53-12E6, and LLzp53-16H12), followed by Western blotting with αHA. (B) Zebrafish embryos that were either uninjected or injected with p53MO or p53misMO, γ-irradiated with 20 Gy, and Western blotted with αp53.
Having verified the specificity of our antibodies in zebrafish embryos, we next examined p53 response in zebrafish tumors. Initially, we examined the levels of p53 in control tumors, specifically seminomas from non-rp mutant backgrounds. For these experiments, the tumor cells were plated and then left untreated or treated either with γ-irradiation, the proteasome inhibitor MG132, or both. In the absence of treatment, p53 protein is present at low or undetectable levels in most of the seminoma samples (Fig. 2 A–C). Either γ-irradiation or MG132 increased zebrafish p53 levels, and treatment with both led to further p53 stabilization (Fig. 2 A–C), exactly as is observed in mammalian cells. Notably, in a small subset of the seminomas, we observed similar, high levels of p53 protein in either the absence or presence of treatment with γ-irradiation and MG132 (Fig. 2B). This finding is entirely consistent with our finding that a subset of seminomas carries p53 mutations and the fact that mutant p53 protein is often stabilized in human tumors. In agreement with this notion, we also see high levels of p53 protein expressed in p53 mutant tumor cells (p53M214K/M214K MPNST cells) in the absence of DNA damage and proteasome inhibition (Fig. 2 D–F).
Fig. 2.

rp+/− MPNST cells from many mutant rp lines do not express p53 protein. (A–F) Plated seminoma cells from non-rp mutant backgrounds, p53M214K/M214K MPNST cells, or rp+/− MPNST cells were exposed to 20 Gy γ-irradiation and/or overnight incubation with 20 μM MG132 and Western blotted with αp53.
Having established that both proteosome inhibition and DNA damage are capable of inducing p53 protein in zebrafish seminoma cells, we next examined the rp+/− MPNST cells. Remarkably, although these cells retain wild-type p53, we detected little or no p53 protein in plated rp+/− MPNST cells even after treatment with γ-irradiation and MG132 (Fig. 2). Importantly, we examined one or more MPNSTs from 17 different tumor-prone rp mutant zebrafish lines, and in every single case we observed a similar absence of p53 in the presence of DNA damage and proteosome inhibition (7 of 17 lines are shown in Fig. 2; 10 lines are data not shown). This finding shows that loss of p53 is a universal feature of tumors that result from rp haploinsufficiency, which is in stark contrast to the high expression of p53 in p53M214K/M214K MPNST cells, ruling out the possibility that peripheral nerve sheath cells per se are unable to synthesize p53 protein (Fig. 2 D–F).
To determine whether the loss of p53 protein in the rp+/− MPNSTs was due to a loss of transcription of the p53 gene, we measured the p53 mRNA in tumor cells by Northern blot analysis. Tumor cells from seminomas, p53M214K/M214K MPNSTs, and rp+/− MPNSTs were plated and subjected to γ-irradiation and MG132 as described above. Before cell lysis, the samples were divided, and a fraction of each sample was then used for either northern or Western blot analysis. p53 mRNA is transcribed by seminoma cells in the absence of DNA damage and proteasome inhibition (Fig. 3A). This level of p53 mRNA significantly increases upon treatment of the cells with γ-irradiation and MG132 most likely due to the reported p53-dependent induction of p53 mRNA in response to γ-irradiation (8). p53 mRNA also is present in MPNST cells from rp+/− and p53M214K/M214K mutants (Fig. 3A). However, in contrast to seminoma cells, neither of these tumor samples exhibit an increase of p53 mRNA upon γ-irradiation, suggesting that, although p53 is transcribed in both MPNST samples, both reveal a loss of p53 protein function.
Fig. 3.

rp+/− MPNST cells transcribe sufficient p53 mRNA to detect p53 protein, but fail to induce p53 transcription upon DNA damage. (A) Plated seminoma cells from non-rp mutant backgrounds, p53M214K/M214K MPNST cells, or rp+/− MPNST cells were exposed to 20 Gy γ-irradiation and/or overnight incubation with 20 μM MG132. Cells were then split, with one part lysed for Western blot analysis with αp53 and αactin and the other part for Northern blot analysis with p53 and actin probes. (B) The whole-cell protein lysate from seminoma cells treated with 20 Gy γ-irradiation and overnight incubation with 20 μM MG132 was serially diluted for Western blot analysis with αp53 compared with the lysate from undiluted rp+/− MPNST cells. Total RNA from the same seminoma cells was diluted as for the protein lysates, and the level of p53 mRNA was measured by Northern blot analysis compared with undiluted total RNA from the rp+/− MPNST cells. Phosphoimaging quantification of the p53 transcript levels in the Northern blot is shown. Asterisks denote lanes of approximately equivalent levels of p53 mRNA.
p53 mRNA is consistently found at lower levels in rp+/− MPNST cells compared with seminoma cells, as seen in Fig. 3A. Therefore, to determine that the amount of p53 mRNA expressed in the rp+/− MPNSTs was sufficiently high to be translated into enough protein to be within the detection limits of the αp53 antibody, we compared the p53 mRNA and p53 protein levels in an rp+/− MPNST sample to a serial dilution of both total protein and RNA in seminoma cells exposed to both DNA damage and proteasome inhibition. The amount of p53 mRNA was similar between the MPNST sample and the 4-fold dilution of the seminoma sample. At this dilution of the seminoma sample, p53 protein was still detectable by the αp53 antibody (Fig. 3B). In contrast, the equivalent amount of p53 mRNA in rp+/− MPNSTs did not result in any detectable p53 protein despite the application of DNA damage and proteasome inhibition. This finding suggests that, although an adequate level of p53 mRNA exists in rp+/− MPNST cells to produce detectable protein, the process does not occur.
These data strongly suggest that the p53 mRNA in the rp+/− MPNSTs is not being translated into protein. Wild-type p53 mRNA is sufficiently present in the cells. Moreover, the nature of our assay suggests that alterations in the expression and/or activity of upstream p53 regulators, like Arf and MDM2, cannot account for the observed p53 protein loss. Specifically, if elevated ubiquitination were responsible for the lack of p53 protein either by mdm2 gene amplification or through deletion of Arf (or some other negative regulator of MDM2 because Arf has not been found in zebrafish), either proteosome inhibition or DNA damage would relieve p53 from this regulation and allow its accumulation. However, even under conditions of proteasome inhibition and DNA damage, p53 is not induced in the rp+/− MPNSTs. Furthermore, the fact that this phenomenon occurs in MPNSTs from 17 different tumor prone rp+/− mutant lines strongly suggests that there is a common mechanism by which decreased rp dosage causes p53 protein loss.
The fact that the loss of p53 protein observed in the rp+/− MPNST cells is linked to mutations of genes associated with ribosome integrity immediately suggests that the mechanism of this loss could be due to a global translation defect in the rp+/− MPNST cells. However, we found that there was no difference in the overall translation capacity between rp+/− and p53M214K/M214K MPNST cells. Pulse-labeled cells from both rp+/− and p53M214K/M214K MPNSTs synthesize the same amount of total protein as demonstrated by autoradiography (Fig. 4A) or TCA precipitation (Fig. 4B) of the labeled proteins after the pulse. In addition to being unable to detect a difference in the amount of total protein being synthesized in rp+/− compared with p53M214K/M214K MPNST cells, we also found no difference in the amount of a specific protein synthesized in the cells as shown by immunoprecipitation of actin in pulse-labeled cells (Fig. 4C). Although total and actin protein synthesis did not vary between the tumor samples, the levels of p53 protein synthesis were dramatically different. Immunoprecipitation with αp53 antibodies demonstrated that there was no synthesis of p53 in the rp+/− MPNST cells even when cells were subjected to DNA damage and proteasome inhibition (Fig. 4D). In contrast, both p53M214K/M214K MPNST cells and seminoma cells demonstrated robust synthesis of p53 protein in the presence and absence of γ-irradiation and MG132. These data would suggest that the loss of p53 protein synthesis resulting from rp+/− gene mutations is specific to the p53 transcript and not due to an overall loss of translation capacity.
Fig. 4.

rp+/− MPNSTs do not exhibit an overall decrease of translation capacity, but specifically fail to synthesize p53 protein. (A and B) rp+/− MPNST cells and p53M214K/M214K MPNST cells were counted, and an equal number of cells were plated and labeled for a 30-min pulse with [35S]cysteine/methionine. Cells were then lysed for visualization of newly synthesized labeled proteins by autoradiography (A) or for quantification of labeled proteins by TCA precipitation and scintillation counting (B). Both methods reveal equal labeling of newly synthesized proteins in both rp+/− MPNST cells and p53M214K/M214K MPNST cells. (C) Plated rp+/− MPNST cells, p53M214K/M214K MPNST cells, and seminoma pulse-labeled with [35S]cysteine/methionine and immunoprecipitated with antibodies against actin reveal no difference in the amount of actin protein synthesized during the pulse. (D) Plated p53M214K/M214K MPNST and seminoma cells pulse-labeled with [35S]cysteine/methionine and immunoprecipitated with αp53 antibodies display newly synthesized p53 protein in the presence or absence of γ-irradiation and MG132, in contrast to rp+/− MPNST cells, which do not synthesize p53 protein in either condition.
Discussion
The fact that zebrafish carrying mutations in many different rp genes are prone to developing the same rare MPNST type as zebrafish carrying a loss-of-function p53 point mutation compelled us to consider the possibility that the heterozygous loss of these rp genes leads to aberrations in the p53 pathway. We found that MPNST cells derived from rp+/− mutant zebrafish, in contrast to other zebrafish cells, lack p53 protein even under conditions where it should be stabilized, including γ-irradiation and proteasome inhibition. We only observed loss of p53 protein in tumor cells in the rp+/− mutant zebrafish; this phenomenon is not seen in all cells throughout the body (data not shown). Thus, we cannot rule out the possibility that p53 loss is an indirect effect of selection by the tumor cells. However, the tumor cells maintain wild-type copies of the p53 gene and transcribe wild-type p53 mRNA. Frequently, mammalian tumors that retain wild-type p53 have defects in upstream p53 regulators (typically amplifications of mdm2 or deletions of arf) that prevent p53 stabilization by constitutive ubiquitination. However, this mechanism cannot account for the loss of p53 in rp+/− mutant MPNST cells because p53 stabilization is not restored with proteasome inhibition. Given these observations and the role of rps in translation, we hypothesize that there is a defect in translation of the p53 mRNA. Notably, we do not detect a global defect of protein synthesis in these rp+/− mutant tumors, suggesting that there is a selective impairment of p53 translation. Our data do not rule out the possibility that there are other specific transcripts whose translation is also selectively abrogated, but they clearly establish a link between rp gene dosage and p53 regulation.
As mentioned previously, a number of connections have been drawn between the rps and p53 regulation. First, specific rps L5, L11, L23, and S7 (25–29) have been shown to bind to MDM2 and induce p53 stabilization. This regulation is extremely intriguing, but it cannot account for the observed loss of p53 in our rp mutant zebrafish tumors because, if it were the operating mechanism, one would expect to see p53 protein expression rescued by MG132, which we do not.
As a second link between rps and p53 regulation, Takagi et al. (30) have shown that specific rps, especially L26, bind to the 5′ and/or 3′ UTRs of human p53 mRNA and can selectively promote p53 translation in response to DNA damage. It is formally possible that the loss of p53 in our rp mutant tumors somehow results from the disruption of this mechanism. For example, although there is no homology between the UTR sequences of zebrafish and human p53 mRNA, it is conceivable that different rps could regulate p53 mRNAs in different species. Alternatively, perhaps a precise combination of rps is required to bind p53 mRNA for normal regulation, and this combination is somehow perturbed by the heterozygous loss of certain rps. However, our data point to a much more widespread connection between rp dosage, tumorigenicity, and p53 regulation. First, we find that, among the 27 rp+/− mutant lines that are represented in our zebrafish mutant collection, two thirds of these are highly tumor prone. Although we have been unable to establish a clear relationship between those rps that when mutated give rise to tumors and those that do not, it is possibly due to the effect of gene dosage in the precise tissue type (because it has been suggested that different tissues have different requirements for specific rp dosages) (33). Second, we detected universal loss of p53 in MPNSTs derived from all 17 of the rp mutant lines we have examined. Based on these observations, we favor the notion that there is a general mechanism that links alterations in dosage of the rps, and perhaps of functional ribosomes, to the regulation of p53. Intriguing, we have found that the young tumor prone rp+/− zebrafish display a slight growth defect (5). This defect is probably due to a subtle translation deficiency, which is clearly overcome in the MPNST cells. It seems possible that, in order for the MPNST cells to overcome this deficiency, they need to selectively discard the translation of mRNAs that do not confer a growth advantage, such as those coding for tumor suppressors like p53.
We favor the notion that rp haploinsufficiency selectively impairs p53 translation. However, our data do not establish whether this defect reflects a direct effect on the translation machinery or is operating through a more indirect mechanism. For example, rps might directly or indirectly interact with RNAi machinery. It is well established that mircoRNAs (miRNAs) are capable of down-regulating the expression of specific proteins, and it has been proposed that translational silencing of mRNAs by miRNAs is possible with or without a detectable decrease in the levels of the target transcripts (34–36). It is entirely possible that the p53 transcript in rp+/− MPNST cells is being specifically regulated by miRNAs not active in seminomas or p53M214K/M214K MPNSTs. It is not clear how stoichiometry of rps could result in the expression of p53 specific miRNAs in the rp+/− MPNST cells, but one possibility is that the scarcity of a single rp impairs ribosome biogenesis and results in excess rRNA that is processed into miRNAs. Interestingly, a number of short RNAs cloned have been found to contain rRNA sequences (Phillip Sharp, personal communication), although their function remains unknown. It would be interesting to try to map cis-acting elements in either the 3′ or 5′ UTRs of the zebrafish p53 gene and then determine whether they show a relationship to target sequences of any detectable miRNAs. Identification of such elements also would allow investigation of other possible mechanisms, such as the possibility of direct regulation by the binding of individual ribosomal proteins like that described for human L26 (30).
Our finding that the rp+/− MPNST cells do not synthesize p53 protein provides a new mechanism for p53 regulation and perhaps a new mechanism for tumorigenesis. Adding to the quantity of data linking p53 gene mutations and faulty posttranslational regulation of p53 protein to tumor formation, we provide evidence for a loss of p53 protein synthesis in tumors. It will be of great interest to determine whether this mechanism is evident in human tumors and if it explains why tumors arise in some cells that maintain wild-type copies of the p53 gene and no detectable gene amplifications of mdm2 or deletions of Arf.
Materials and Methods
Zebrafish.
The rp+/− and p53M214K/M214K zebrafish (Danio rerio) have been described previously (2–4, 8).
Microarray Analysis.
Total RNA from zebrafish tumors was isolated with TRIzol. (Invitrogen), and cRNA was hybridized to zebrafish microarrays (Affymetrix). Summarization and normalization were done with robust multiarray analysis with sequence-based (GC) background adjustment (gcRMA, www.bioconductor.org). The samples included were the 11 tumor samples from this study (GSE11493), a heart sample (GSM112796), a retina sample (GSM113859), a 16-cell stage embryo sample (GSM95612), a 10-somite embryo sample (GSM209542), a 5-day postfertilization (dpf) sample (Niss.wt1; A5005 wt1.CEL, unpublished data), and a 90-dpf sample (Emb.zon; Tue-90d-1.CEL, unpublished data). Hierarchical clustering of samples was done by using pvclust (37). Full datasets are publicly available at www.ncbi.nlm.nih.gov/projects/geo/query/acc.cgi?acc = GSE11493.
Genomic Sequencing.
Genomic DNA was isolated from tumor cells and tail tissue and used as the template in PCR using primers designed to amplify the coding exons of the p53 gene. After gel isolation, the PCR products were sequenced with nested primers. All primer sequences are available upon request. Chromatograms were carefully observed for double peaks, indicating one wild-type copy and one mutant copy.
Antibody Development.
Murine monoclonal antibodies were raised against full-length zebrafish p53 protein purified from bacteria transformed with zebrafish p53 gene cloned into the pQE-30 vector (Qiagen) as described previously (38). Positive cell lines were identified by ELISA, single cell cloned, and tested for immunoreactivity against zebrafish p53 by Western blot, immunoprecipitation, and whole-mount immunohistochemistry.
Transfections/Immunoprecipitations.
Briefly, 293 cells were transfected with 1 μg of HA-tagged zebrafish p53 cloned into pCDNA3.1 (Invitrogen) using Lipofectamine (Invitrogen) according to the manufacturer's protocol. Immunoprecipitations were conducted with 1,000 μg of total protein and either an equal volume of the antibody supernatant or 5 μl of nonimmunized mouse serum or serum extracted from the mice immunized with zebrafish p53 protein.
Western Blotting.
Tumors were dissected from zebrafish, finely diced, treated with dispase, plated on coatings of 0.2% gelatin in DME + 10% FCS, and incubated at 28°C + 5% CO2. MG132 (Calbiochem) incubations were done for either 6 h (for Western blots with corresponding Northern blots) or 18 h (for all other samples) at a concentration of 20 μM. Cells were treated with 20 Gy γ-irradiation 4 h before lysis. Then 40 μg of total protein was run on a 10% SDS/PAGE gel for Western blotting of the tumor cells. Embryos were injected with p53MO and p53misMO morpholinos at the 1–2 cell stage as previously described (17), treated with 20 Gy γ-irradiation at 1-dpf, and lysed 4 h later. The protein from two embryos per lane was run on a 10% SDS/PAGE gel. Western blotting was performed with supernatant from αp53 hybridoma cell lines at a 1:1 dilution in TBST blocking solution (+5% powdered milk) or a 1:1,000 dilution of α-actin (Santa Cruz Biotechnology). For Western blotting of immunoprecipitations from transfected 293 cells, a 1:1 dilution of anti-influenza hemagglutinin antibody (αHA) 12CA5 supernatant was used.
Northern Blotting.
RNA was isolated with TRIzol (Invitrogen), and 5–10 μg of RNA was electrophoresed through a formaldehyde denaturing agarose gel, blotted, and probed as described previously (39) using radiolabeled probes against the coding sequence of zebrafish p53 or actin. Blots were exposed to a phosphoimaging screen (Molecular Dynamics) overnight and analyzed using ImageQuant software.
Pulse Labeling/Immunoprecipitations.
Tumors were dissected and treated as for Western blotting. Then 2.5 h after γ-irradiation, the media were changed to DME without methionine or cysteine (Sigma); 45 min later, 200 μCi of [35S]methionine/cystenine (PerkinElmer) was added to each well for 45 min. Cells were lysed and 1,200 μg of protein was incubated with an equal volume of αp53 antibody supernatant at 4°C overnight and precipitated with Protein A/G Plus agarose beads (Calbiochem) for 1 h at 4°C. Samples were run on a 10% SDS/PAGE gel that was fixed and incubated in Autofluor (National Diagnostics) for 2 h before being dried and exposed to autoradiography film (Kodak). For assessment of protein synthesis, cells were plated and labeled with 500 μCi [35S]methionine/cystenine for 30 min, and then an equal number of cells (≈2 × 106) from each sample was removed, lysed in equal volumes of lysis buffer, and loaded on a 10% SDS/PAGE gel that was treated for autoradiography as described above. Immunoprecipitations of actin were done with 1 μg of actin antibody (Santa Cruz Biotechnology) and lysate from an equal number of cells (≈4 × 107) labeled with 500 μCi [35S]methionine/cystenine for 30 min. TCA precipitations were done by labeling an equal number of cells (≈1 × 106) with 100 μCi [35S]methionine/cystenine for 30 min, samples were lysed, and protein was precipitated with 0.25 volumes of TCA, filtered by vacuum, and 35S cpm calculated with a scintillation counter.
Supplementary Material
Acknowledgments.
We thank Dr. Sarah Farrington, Kate Anderson, Tim Angelini, and Sam Farrington for care of the zebrafish facility; Manlin Luo (BioMicro Center, Massachusetts Institute of Technology) for processing of microarrays; Robert Lindsay for assistance with tumor sample sequencing; and Dr. Thomas Look (Dana-Farber Cancer Institute, Boston, MA) for the kind gift of p53M214K/M214K mutant zebrafish. This work was supported by National Institutes of Health/National Cancer Institute Grant CA106416 (to J.A.L.), National Institutes of Health/National Cancer Institute funds (to N.H.), and Jane Coffin Childs Memorial Foundation and Genentech Postdoctoral Fellowships (to A.W.M.). J.A.L. is a Ludwig Scholar.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0805036105/DCSupplemental.
References
- 1.Amsterdam A, et al. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2004;2:E139. doi: 10.1371/journal.pbio.0020139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Golling G, et al. Insertional mutagenesis in zebrafish rapidly identifies genes essential for early vertebrate development. Nat Genet. 2002;31:135–140. doi: 10.1038/ng896. [DOI] [PubMed] [Google Scholar]
- 3.Amsterdam A, et al. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci USA. 2004;101:12792–12797. doi: 10.1073/pnas.0403929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amsterdam A, et al. A large-scale insertional mutagenesis screen in zebrafish. Genes Dev. 1999;13:2713–2724. doi: 10.1101/gad.13.20.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai K. Cambridge, MA: Massachusetts Institute of Technology; 2006. A cancer screen in zebrafish identifies many ribosomal proteins as haploinsufficient tumor suppressor. PhD dissertation. [Google Scholar]
- 6.Oliver ER, Saunders TL, Tarle SA, Glaser T. Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute. Development (Cambridge, UK) 2004;131:3907–3920. doi: 10.1242/dev.01268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marygold SJ, et al. The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol. 2007;8:R216. doi: 10.1186/gb-2007-8-10-r216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berghmans S, et al. tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc Natl Acad Sci USA. 2005;102:407–412. doi: 10.1073/pnas.0406252102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lane DP. Exploiting the p53 pathway for the diagnosis and therapy of human cancer. Cold Spring Harb Symp Quant Biol. 2005;70:489–497. doi: 10.1101/sqb.2005.70.049. [DOI] [PubMed] [Google Scholar]
- 10.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 11.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 12.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 13.Lu X, Lane DP. Differential induction of transcriptionally active p53 following UV or ionizing radiation: Defects in chromosome instability syndromes? Cell. 1993;75:765–778. doi: 10.1016/0092-8674(93)90496-d. [DOI] [PubMed] [Google Scholar]
- 14.Maki CG, Huibregtse JM, Howley PM. In vivo ubiquitination and proteasome-mediated degradation of p53(1) Cancer Res. 1996;56:2649–2654. [PubMed] [Google Scholar]
- 15.Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–83. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- 17.Langheinrich U, Hennen E, Stott G, Vacun G. Zebrafish as a model organism for the identification and characterization of drugs and genes affecting p53 signaling. Curr Biol. 2002;12:2023–2028. doi: 10.1016/s0960-9822(02)01319-2. [DOI] [PubMed] [Google Scholar]
- 18.Draptchinskaia N, et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet. 1999;21:169–175. doi: 10.1038/5951. [DOI] [PubMed] [Google Scholar]
- 19.Gazda HT, et al. Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2006;79:1110–1118. doi: 10.1086/510020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cmejla R, Cmejlova J, Handrkova H, Petrak J, Pospisilova D. Ribosomal protein S17 gene (RPS17) is mutated in Diamond-Blackfan anemia. Hum Mutat. 2007;28:1178–1182. doi: 10.1002/humu.20608. [DOI] [PubMed] [Google Scholar]
- 21.Willig TN, Gazda H, Sieff CA. Diamond-Blackfan anemia. Curr Opin Hematol. 2000;7:85–94. doi: 10.1097/00062752-200003000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Lipton JM, et al. Osteogenic sarcoma associated with Diamond-Blackfan anemia: A report from the Diamond-Blackfan Anemia Registry. J Pediatr Hematol Oncol. 2001;23:39–44. doi: 10.1097/00043426-200101000-00009. [DOI] [PubMed] [Google Scholar]
- 23.Boultwood J, Lewis S, Wainscoat JS. The 5q-syndrome. Blood. 1994;84:3253–3260. [PubMed] [Google Scholar]
- 24.Ebert BL, et al. Identification of RPS14 as a 5q-syndrome gene by RNA interference screen. Nature. 2008;451:335–339. doi: 10.1038/nature06494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3:577–587. doi: 10.1016/s1535-6108(03)00134-x. [DOI] [PubMed] [Google Scholar]
- 26.Marechal V, Elenbaas B, Piette J, Nicolas JC, Levine AJ. The ribosomal L5 protein is associated with mdm-2 and mdm-2-p53 complexes. Mol Cell Biol. 1994;14:7414–7420. doi: 10.1128/mcb.14.11.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai MS, Lu H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem. 2004;279:44475–44482. doi: 10.1074/jbc.M403722200. [DOI] [PubMed] [Google Scholar]
- 28.Chen D, et al. Ribosomal protein S7 as a novel modulator of p53-MDM2 interaction: Binding to MDM2, stabilization of p53 protein, and activation of p53 function. Oncogene. 2007;26:5029–5037. doi: 10.1038/sj.onc.1210327. [DOI] [PubMed] [Google Scholar]
- 29.Jin A, Itahana K, O'Keefe K, Zhang Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol Cell Biol. 2004;24:7669–7680. doi: 10.1128/MCB.24.17.7669-7680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell. 2005;123:49–63. doi: 10.1016/j.cell.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 31.Anderson SJ, et al. Ablation of ribosomal protein L22 selectively impairs alphabeta T cell development by activation of a p53-dependent checkpoint. Immunity. 2007;26:759–772. doi: 10.1016/j.immuni.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 32.Chen J, et al. Loss of function of def selectively up-regulates Delta113p53 expression to arrest expansion growth of digestive organs in zebrafish. Genes Dev. 2005;19:2900–2911. doi: 10.1101/gad.1366405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bortoluzzi S, d'Alessi F, Romualdi C, Danieli GA. Differential expression of genes coding for ribosomal proteins in different human tissues. Bioinformatics. 2001;17:1152–1157. doi: 10.1093/bioinformatics/17.12.1152. [DOI] [PubMed] [Google Scholar]
- 34.O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 35.Poy MN, et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432:226–230. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- 36.Jackson RJ, Standart N. How do microRNAs regulate gene expression? Sci STKE. 2007;2007:re1. doi: 10.1126/stke.3672007re1. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki R, Shimodaira H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22:1540–1542. doi: 10.1093/bioinformatics/btl117. [DOI] [PubMed] [Google Scholar]
- 38.Trimarchi JM, Fairchild B, Wen J, Lees JA. The E2F6 transcription factor is a component of the mammalian Bmi1-containing polycomb complex. Proc Natl Acad Sci USA. 2001;98:1519–1524. doi: 10.1073/pnas.041597698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaiano N, et al. Insertional mutagenesis and rapid cloning of essential genes in zebrafish. Nature. 1996;383:829–832. doi: 10.1038/383829a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.