

Abstract
HOX11, a divergent homeodomain-containing transcription factor, was isolated from the breakpoint of the nonrandom t(10;14)(q24;q11) chromosome translocation found in human T cell acute lymphoblastic leukemias. The translocation places the HOX11 coding sequence under the transcriptional control of TCR α/δ regulatory elements, resulting in ectopic expression of a normal HOX11 protein in thymocytes. To investigate the oncogenic potential of HOX11, we targeted its expression in lymphocytes of transgenic mice by placing the human cellular DNA under the transcriptional control of Ig heavy chain or LCK regulatory sequences. Only IgHμ-HOX11 mice expressing low levels of HOX11 were viable. During their second year of life, all HOX11 transgenic mice became terminally ill with more than 75% developing large cell lymphomas in the spleen, which frequently disseminated to thymus, lymph nodes, and other nonhematopoietic tissues. Lymphoma cells were predominantly clonal IgM+IgD+ mature B cells. Repopulation of severe combined immunodeficient mice with cells from hyperplastic spleens indicated that the HOX11 tumor phenotype was transplantable. Before tumor development, expression of the transgene did not result in perturbations in lymphopoiesis; however, lymphoid hyperplasia involving the splenic marginal zones was present in 20% of spleens. Our studies provide direct evidence that expression of HOX11 in lymphocytes leads to malignant transformation. These mice are a useful model system to study mechanisms involved in transformation from B-lineage hyperplasia to malignant lymphoma and for testing novel approaches to therapy. They represent a novel animal model for non-Hodgkin’s lymphoma of peripheral mature B cell origin.
Translocations in T cell acute lymphoblastic leukemias (T-ALL) activate a variety of transcription factors through aberrant recombinations with T-cell receptor (TCR) loci (1–3). We first reported a novel translocation, t(10;14)(q24;q11), as a nonrandomly acquired change in human T-lineage malignancies (4). The translocations arise as a result of failed attempts at TCR gene rearrangement that deregulate expression of the novel human homeodomain-containing gene, HOX11, in 10q24 (5–10). Aberrant expression of HOX11 in T lymphocytes was hypothesized to contribute to the development of T-ALL through abnormal regulation of HOX11 transcriptional target genes (11).
The homeobox gene family encode helix–turn–helix transcription factors that are spatially and temporally expressed during embryonic development and regulate pattern formation and development in vertebrates (12). Homeobox genes also are expressed in normal hematopoietic and leukemic cells, suggesting they may play roles in both regulating normal hematopoiesis and contributing to the malignant transformation of hematopoietic cells (13). HOX11 is a member of a class of noncluster homeobox genes, which shares greater than 50% amino acid conservation within the homeodomain (5, 14, 15). There are at least three members in this subclass, all of which contain a threonine residue in place of the more common isoleucine or valine in helix 3 of the homeodomain (14, 16). During murine embryonic development HOX11 is expressed in the surface ectoderm and central mesenchyme of branchial arches 1, 2, 3, and 4 and then within motor neurons of the cranial nerves innervating these structures (17–19). HOX11 also is expressed in the developing oral, pharyngeal, salivary, auditory, and splenic structures and transiently in the fetal thymus at days 13.5 and 14.5 and from day E13.5 to 4 weeks of age in the spleen (20). In humans HOX11 is expressed in the liver (10) and in CD34+ human bone marrow cells (21). It remains unclear whether HOX11 is expressed in normal T cells (20, 22, 23). The role of HOX11 in embryogenesis and leukemogenesis remains obscure. HOX11-deficient mice are asplenic at birth but show no other morphological or functional defects (17, 24).
Despite the association of HOX11 gene activation with T-ALL, there is little evidence that inappropriate expression of HOX11 leads to lymphoid malignancies. To assess the oncogenic potential of HOX11 and to determine whether its aberrant expression is a key event in the development of lymphoid neoplasia, we generated transgenic mice in which expression of HOX11 was targeted to the lymphoid lineages.
MATERIALS AND METHODS
Generation of HOX11 Transgenic Mice.
Nucleotides 165–2058 of human HOX11 cellular DNA (cDNA) (5) were subcloned into the SmaI site of pGEM7Zf to generate the pGEMHOX11 vector. A 2.5-kb XbaI fragment containing a 600-bp BamHI fragment carrying the Ig heavy chain (IgH) promoter (25) and a 689-bp Xbal–EcoRI fragment encoding the IgH enhancer (26, 27) were subcloned upstream of the HOX11 cDNA in the Xbal site of pGEMHOX11. The IgHμ-HOX11 transgene (Fig. 1A) was released from the plasmid as a 3.18-kb BglII partial, HindIII complete digest, and purified linear DNA was injected into the pronuclei of CD-1 fertilized eggs. Transgenic founders and progeny were identified by either Southern blot analysis (Fig. 1B) or PCR amplification of a 330-bp fragment by using the forward primer, 5′-AACCGCAGATACACAAAGGA-3′ and the reverse primer, 5′-TGGGCCAGGCTCTTCTGGAA-3′ (Fig. 1C). HOX11 transgenic lines were established by back-crossing HOX11 transgenic mice to CD-1 mice. Animals were maintained in microisolators under pathogen-free conditions. All animal manipulations and housing were in accordance with the Sunnybrook Health Science Center Animal Care Committee guidelines.
Figure 1.

Structure and detection of the IgHμ-HOX11 transgene. (A) Diagrammatic representation of the 3.18-kb HOX11 transgene consisting of the human HOX11 cDNA under the transcriptional control of the murine IgH promoter (IgPμ) and enhancer (IgEμ). Restriction sites: B, BglII; X, XbaI; S, SmaI; E, EcoRI; H, HindIII. (B) Southern blot showing mouse genomic DNA from nontransgenic and transgenic mice from HOX11 mouse lines C2 (lanes 1–14) and D11 (lanes 15–18) digested with BamHI or XhoI/NotI, respectively, and hybridized with either the HOX11 cDNA (lanes 1–14) or a 1.4-kb 5′ HOX11 genomic probe (lanes 15–18). The HOX11 transgene (HOX11 Tg) is indicated. (C) PCR analysis used to distinguish control and HOX11 mice from the C5 founder line. The primers amplify both endogenous murine (mHOX11) and a 499-bp HOX11 transgenic (HOX11 Tg) fragment. Lane 1 contained a negative control, and lanes 2–15 contain amplified DNA from transgenic and nontransgenic mice followed by CD-1 nontransgenic DNA, positive transgenic DNA, and a plasmid containing the entire HOX11 cDNA. (D) Reverse transcription–PCR analysis showing expression of the HOX11 transgene in hematopoietic tissues and tumors from C2 (lanes 9, 11, and 12), C5 (lanes 6 and 10), and D11 (lanes 3–5, 7, 8, and 13) HOX11 mice affected with lymphoma or myeloid hyperplasia. K3P (lane 2), used as a positive control, is a t(10;14) positive human T-ALL cell line (5). Lanes 3–13 contain the 593-bp fragment amplified from cDNA generated from RNA isolated from the indicated tissues: thymus (lanes 3, 7, and 8), kidney (lanes 4 and 6), marrow (lane 5), spleen (lanes 9 and 11), tumor (lanes 10, 12, and 13).
Tumor DNA and RNA Analysis.
Total DNA was isolated from tumor cells by either a standard phenol/chloroform procedure (28) or by using DNAzol (BRL/GIBCO). VDJH rearrangements were assessed by using the 5′ VALL degenerate VH-specific primer and the 3′ JH4 primer (29). VDJH products were detected by probing Southern blots with a JH4 internal probe (29). Band intensities were analyzed by using an Alpha Imager (Alpha Innotech, San Leandro, CA). Total cellular RNA was isolated by standard guanidinium thiocyanate procedures (28) or by using TRIzol (BRL/GIBCO) and reverse transcription–PCR done by using a kit from Boehringer Mannheim. The primers for amplification of a 593-bp fragment of the HOX11 transcript from nucleotides 306–899 were 5′-CGGCTCCTACAACGTGAACA-3′ and 5′-GAGGGTCAGCGGGCAGCGGCTGTGCCA-3′.
Spleen Cell Transplantation Assays.
Spleens from control and HOX11 mice were removed and sampled for histology, and single cell suspensions were generated. Donor cells (1 × 107) were injected i.p. or i.v. into severe combined immunodeficient (SCID) mice (three recipients per group). Two to three months postinjection, two recipients from each group were sacrificed, and hematopoietic tissues were removed, sampled for histology, and prepared for FACS analysis and isolation of DNA. The remaining recipients were maintained for long-term follow-up analysis.
Histopathology.
Hematoxylin- and eosin-stained tissues and immunohistochemical analyses of frozen tissues were processed by using standard procedures. Morphological interpretation of the histological preparations of the diverse tissues were made by a pathologist (Ir.D.) and a hematopathologist (M.D.R.).
Flow Cytometric Analysis.
Single cell suspensions from bone marrow, thymuses, spleens, and lymph nodes were prepared as described (30, 31). Cells were stained with primary antibodies in Hank’s Hepes buffered salt solution containing 2% fetal calf serum and 0.1% sodium azide (HFN) on ice for 30 min, washed twice with HFN, and resuspended in HFN containing 1 μg/ml of propidium iodide. Flow cytometric analysis was performed by using a FACScan flow cytometer equipped with cellquest software (Becton Dickinson).
Antibodies.
Fluorescein isothiocyanate (FITC)-labeled mAbs to Mac-1, CD3ɛ, and CD4 and mAbs to phosphatidylethanolamine (PE)-labeled CD3ɛ, CD8, and B220 (CD45R) were purchased from PharMingen. FITC-labeled hamster IgG, rat IgG2c, and PE-labeled rat IgG2c isotype controls were obtained from Cedarlane (Hornby, Canada). Purified Thy1.2 and FITC-labeled polyclonal anti-rat Ig were obtained from PharMingen. FITC-labeled anti-mouse IgM and PE-labeled anti-mouse IgD were obtained from Southern Biotechnology Associates.
In Vitro Clonogenic Progenitor Assays.
For myeloid progenitor assays, bone marrow (3 × 104) or spleen (1 × 106) cells were plated in 1-ml methylcellulose cultures (StemCell Technologies, Vancouver) in Greiner Petri dishes (StemCell Technologies). Dishes were incubated in humidified chambers at 37°C in 5% CO2, and colonies were enumerated on days 10–14. In some experiments colonies were scored as being derived from colony-forming unit (CFU)-macrophage, CFU-granulocyte, CFU-granulocyte-macrophage, or CFU-granulocyte-erythroid-macrophage-megakaryocyte progenitors according to standard criteria. A proportion of scored colonies were individually plucked, and identification of colony types was confirmed by Wright-Giemsa staining of cytospin preparations of colonies.
Interleukin 7-responsive B cell clonogenic progenitors were assayed by plating bone marrow (5 × 104) or spleen (1 × 106) cells in 1-ml methylcellulose cultures (StemCell Technologies) in Greiner dishes in a humidified chamber at 37°C in 5% CO2. Day 7 colonies were scored, and a proportion was plucked and Wright-Giemsa-stained cytospins were analyzed to confirm the presence of lymphocytes.
Peripheral Blood Cell Analysis.
Hematological parameters of peripheral blood were assessed by using an ABX Argos 5 Diff hematology analyzer (Roche, Mississauga, Canada) set to the veterinarian setting.
RESULTS
Generation of HOX11 Transgenic Mice.
The transgenic vector was constructed by placing human HOX11 cDNA under the transcriptional control of the murine IgH variable region promoter fused to the IgH enhancer (Fig. 1A). The transgene (IgHμ-HOX11) was injected into CD-1-fertilized oocytes, and three founders, designated C2, C5, and D11, showing germ-line transmission of the transgene were generated. IgHμ-HOX11 transgenic mice (from here on referred to as HOX11 mice) appeared normal and healthy at birth with no developmental defects. Southern blot analysis of genomic DNA (Fig. 1B) showed that the three lines each contained approximately 2–4 copies of the transgene. Northern blot analysis of total RNA isolated from various tissues from the three transgenic lines revealed low-level expression of HOX11 in kidney, whereas RNase protection assays demonstrated expression of the transgene in bone marrow and spleen (data not shown).
Normal B and T Lymphocyte Development in Young HOX11 Mice.
HOX11 mice were normal with respect to the absolute number of cells isolated from bone marrow, thymus, spleen, and lymph nodes at 3 or 11 weeks of age (data not shown). Flow cytometric analyses of cells isolated from bone marrow, spleen, and lymph nodes revealed normal B lymphopoiesis from the early pro-B and pre-B stages of development (B220+IgM−) to immature (IgM+IgD−) and mature (IgM+IgD+) B cell stages. Staining of thymocytes and splenocytes with CD4, CD8, or CD3 antibodies indicated no statistically significant differences in immature or mature T lymphocyte subpopulations. The absolute number of Mac-1+ myeloid cells in bone marrow, spleens, thymuses, and lymph nodes were also normal. Both lymphoid and myeloid clonogenic progenitor numbers were normal in HOX11 mice as compared with age- and sex-matched control littermates (pre-B CFU: 14,356 ± 4,918 vs. 23,849 ± 7,079, P = 0.87; myeloid CFU: 50,974 ± 7,356 vs. 53,583 ± 19,846, P = 0.9). There were no significant differences in total white blood cell counts, red blood cell counts, platelets, and hemoglobin or in percentages of hematopoietic subpopulations in peripheral blood between control and 1-year-old HOX11 mice (data not shown).
Hyperplasia in the Spleens of Some Young HOX11 Mice.
Histological analysis of tissue sections of primary and secondary hematopoietic organs from 3- or 4-week-old HOX11 mice showed normal bone marrow, thymus, and lymph node architecture. However, mild lymphoid hyperplasia was noted in one of the spleens obtained from a 3-week-old HOX11 mouse (Fig. 2A). In addition, analysis of the same spectrum of tissues from four apparently healthy 6-month-old HOX11 mice revealed lymphoid hyperplasia in three of the four spleens analyzed (Fig. 2B). Infiltration of the surrounding red pulp was noted in one of the three hyperplastic spleens. Marked myeloid hyperplasia in the bone marrow of one mouse was noted (Fig. 2C). Thymuses and lymph nodes from all four mice appeared normal.
Figure 2.
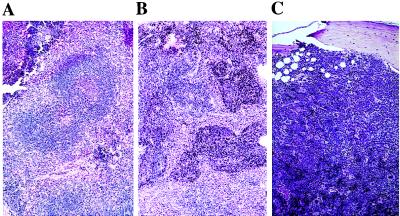
Lymphoid and myeloid hyperplasia in young HOX11 transgenic mouse tissue sections stained with hematoxylin and eosin. (A) Section of a spleen from a 4-week-old mouse showing fusion of germinal centers. (B) Section of a spleen from a 6-month-old transgenic mouse showing lymphoid hyperplasia with characteristic invasion of red pulp areas. (C) Myeloid hyperplasia in the bone marrow of a 6-month-old HOX11 transgenic mouse. Note the increased cellularity and loss of fatty spaces, but all hematopoietic cell types are present. (Magnification: ×40.)
HOX11 Mice Developed Lymphoma in Their Second Year of Life.
To assess the oncogenic potential of HOX11, we grouped 20–25 HOX11 mice from each founder line into three cohorts, together with a control cohort of 30 age- and sex-matched littermates and followed the mice for 20 months (Fig. 3). At the end of this period all but two of the control mice were alive and healthy whereas 100% of the HOX11 mice had died. Death was between the ages of 10 and 20 months (average 14–15 months) for all three lines.
Figure 3.

Survival of HOX11 transgenic mice and control littermates. Mice were monitored for 20 months for signs of terminal illness (at which time mice were humanely killed) or death. Data are expressed in terms of the percentage of live mice in each cohort at monthly intervals between 9 and 20 months of age.
Histological examination of a full spectrum of tissues, including bone marrow, spleen, thymus, lymph nodes, kidney, liver, lungs, gut, pancreas, and reproductive organs from 17 HOX11 mice and hematopoietic tissues from an additional 15 mice indicated 88% of mice had a large cell lymphoma with the spleen being the principal organ involved (Table 1, Fig. 4). The normal splenic architecture was obliterated with extensive invasion of the red pulp by lymphomatous infiltrates (Fig. 4A). The lymphoma cells were a relatively uniform, monotonous population of large cells with loose, speckled, open chromatin and prominent nucleoli (Fig. 4B). Mature lymphocytes and plasma cells also were seen in regions with lymphoma involvement. Twenty-five percent of thymuses isolated from HOX11 mice were enlarged with disruption of the general architecture and extensive replacement of the normal cortex and medulla with lymphoma cells (Fig. 4C). The appearance of the lymphoma cells of the thymus were similar to those described in the spleen. Other organs involved by lymphoma were lymph nodes, liver, lung, kidney, and pancreas (Fig. 4 D–F). Bone marrow involvement was a rare finding seen in only one of 32 marrow samples analyzed. Thirty percent of mice presented with myeloid hyperplasia in the bone marrow with 6% showing no evidence of lymphoma in other organs analyzed. Three percent of mice had myeloid hyperplasia of the spleen with no evidence of lymphoma. Reverse transcription–PCR analysis showed HOX11 transgene expression in hematopoietic tissues and organs infiltrated with lymphoma cells (Fig. 1D).
Table 1.
Summary of histopathological studies of 32 terminally ill HOX11 transgenic mice
| Pathology | Percent |
|---|---|
| Lymphoma in spleen | 78 |
| Thymic lymphoma | 28 |
| Lymphoma in lymph nodes | 32 |
| Bone marrow hyperplasia* | 30 |
| Hyperplasia in spleen† | 9 |
| Disseminated lymphoma | |
| Kidney | 19 |
| Lung | 13 |
| Liver | 19 |
| Pancreas | 6 |
Results are presented as percentages of total mice analyzed.
100% were myeloid hyperplasia.
6% of total spleens showed myeloid hyperplasia, and 3% showed lymphoid hyperplasia.
Figure 4.
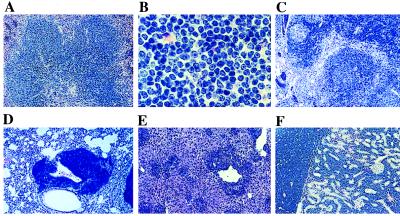
Tissue sections from HOX11 transgenic mice stained with hematoxylin and eosin showing lymphoma. (A) HOX11 transgenic spleen showing extensive invasion and almost complete replacement of red pulp region by lymphoma cells. (B) High-power magnification of lymphoma cells showing large lymphocytes with loose, open chromatin and prominent nucleoli. (C) HOX11 transgenic thymus showing extensive invasion with lymphoma cells and disruption of the normal thymic architecture. Invasion of lung (D), liver (E), and kidney (F) by lymphoma cells is shown. (Magnifications: A and C–F, ×40; B, ×200.)
Lymphoma Cells from HOX11 Mice Exhibit a Mature IgM+IgD+ B Cell Immunophenotype.
Frozen sections from age- and sex-matched control and HOX11 thymuses and spleens were stained with B220 or Thy-1 antibodies. Control thymuses consisted almost exclusively of Thy-1 positive cells with only minor numbers of B cells (Fig. 5 A and B). In sharp contrast, thymuses infiltrated with lymphoma contained a high proportion of strongly positive B220+ cells (Fig. 5 C and D). Analyses of HOX11 spleens confirmed the B cell origin of the lymphoma cells.
Figure 5.
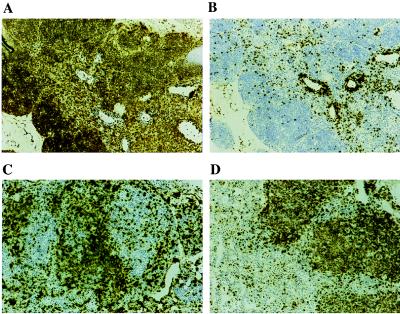
Immunohistochemical analysis of frozen sections of a control thymus (A and B) and thymic lymphomas from HOX11 transgenic mice (C and D) stained with anti-mouse B220 (A and C) or Thy-1 (B and D). Most of the cells of the thymus from an age- and sex-matched control mouse stain with Thy-1 (A) with only a minor component reacting with the B220 antibody (B). In sharp contrast, a high proportion of the cells from the HOX11 transgenic thymus infiltrated with lymphoma cells do not stain with Thy-1 (C) and large regions staining with the B220 antibody are seen (D). (Magnifications: ×40.)
Flow cytometric analyses of age- and sex-matched control and transgenic mice revealed normal thymocyte subpopulations in HOX11 thymuses not affected with lymphoma. However, thymuses infiltrated with lymphoma cells were profoundly abnormal, often showing a complete absence of the double positive thymocyte subpopulation and an accompanying increase in B220+ cells (Fig. 6A). Lymphoma cells expressed both IgM and IgD, indicating they were mature B cells. Tumor cell heterogeneity with respect to expression of B-lineage markers was seen with IgM+IgD+, IgM+IgD−, and B220+IgM− subpopulations being detected. In addition, the absolute number of B220+ B cells in the spleens and lymph nodes of HOX11 mice was increased although not significantly (t test, P = 0.14 and P = 0.09, respectively) (Table 2, Fig. 6 B and C). The lack of a significant increase in B cells in the spleen might be explained by both tumor heterogeneity with respect to expression of B-lineage markers and the loss of expression of B-lineage antigens associated with the differentiation of tumor cells to plasma cells and more mature lymphocytes. Early stage pro- and pre-B cells and immature B cell subpopulations in the bone marrow of HOX11 mice with lymphoma were normal (Table 2, Fig. 6D). In addition, the total number of interleukin 7 responsive B cell clonogenic progenitors per femur, the size of the colonies, and the microscopic appearance of lymphocytes within the colonies were normal (1,975 ± 866 vs. 2,092 ± 2,077 t test, P = 0.92). These findings are consistent with our conclusion that the target cell for transformation by HOX11 is not an early stage lymphocyte originating in the marrow but rather a mature, peripheral B cell.
Figure 6.

Flow cytometric analysis of cells isolated from bone marrow, spleen, lymph nodes, and thymus of HOX11 transgenic diagnosed with lymphoma and control littermates. (A) Size comparisons of cells isolated from thymuses of control (broken line) and HOX11 transgenic mice with lymphoma (solid line). Also shown are two-color dot plots of thymocytes stained with CD4 and CD8, B220 and IgM, or IgM and IgD. (B) Two-color analyses of spleen cells stained with B220/IgM, IgM/IgD, or CD4/CD8. (C) Size comparisons of cells isolated from lymph nodes of control (broken line) and HOX11 transgenic mice with lymphoma (solid line). Also shown are two-color analyses of cells stained with either IgM and IgD or CD4 and CD8. (D) Two-color dot plots of bone marrow cells stained for expression of B220 and IgM or IgM and IgD.
Table 2.
Absolute cell numbers of hemopoietic subpopulations in control and terminally ill HOX11 transgenic mice
| Control, n = 5 | HOX11, n = 8 | |
|---|---|---|
| Bone marrow | ||
| Total cell number/femur | 2.14 ± 0.3 × 107 | 2.63 ± 1.6 × 107 |
| B220 | 1.72 ± 1.1 × 106 | 1.86 ± 1.4 × 106 |
| Mac-1 | 1.09 ± 0.2 × 107 | 1.34 ± 0.6 × 107* |
| Thymus | ||
| Total cell number | 2.66 ± 1.9 × 107 | 2.60 ± 1.0 × 107 |
| B220 | 1.77 ± 2.1 × 106 | 4.44 ± 5.2 × 106 |
| CD4−CD8− | 3.83 ± 3.4 × 106 | 8.44 ± 5.8 × 106 |
| CD4+CD8+ | 1.51 ± 1.0 × 107 | 1.14 ± 1.1 × 107 |
| CD4+CD8− | 4.97 ± 4.2 × 106 | 4.46 ± 2.8 × 106 |
| CD4−CD8+ | 2.71 ± 1.8 × 106 | 1.74 ± 0.8 × 106 |
| Spleen | ||
| Total cell number | 3.68 ± 0.6 × 108 | 5.62 ± 3.0 × 108 |
| B220 | 1.77 ± 0.8 × 108 | 1.88 ± 0.9 × 108 |
| Mac-1 | 1.82 ± 0.3 × 107 | 8.72 ± 8.5 × 107 |
| CD4+ | 5.98 ± 0.3 × 107 | 5.22 ± 3.0 × 107 |
| CD8+ | 2.55 ± 0.4 × 107 | 2.43 ± 2.19 × 107 |
| Lymph nodes | ||
| Total cell number | 2.31 ± 1.1 × 106 | 5.01 ± 3.1 × 106 |
| B220 | 6.58 ± 3.1 × 105 | 1.46 ± 0.8 × 106 |
| Mac-1 | 6.64 ± 1.9 × 104 | 2.46 ± 1.2 × 105* |
| CD4+ | 1.15 ± 0.9 × 106 | 2.30 ± 2.3 × 106 |
| CD8+ | 4.30 ± 4.1 × 106 | 6.72 ± 6.4 × 105 |
| Peripheral blood | ||
| Total number leukocytes | 3.03 ± 1.4 × 106 | 3.33 ± 0.9 × 106 |
| B220 | 1.18 ± 0.8 × 106 | 1.19 ± 0.7 × 106 |
| Mac-1 | 5.58 ± 1.9 × 105 | 9.17 ± 3.1 × 105 |
| CD4+ | 4.33 ± 0.5 × 105 | 5.41 ± 4.4 × 105 |
| CD8+ | 1.54 ± 0.7 × 105 | 2.40 ± 1.1 × 105 |
Results are expressed as mean ± SD.
Statistical analysis using a Student’s t test showed a significant difference between control and HOX11 transgenic mice (P ≤ 0.05).
The total cellularity of spleens and lymph nodes was increased. This increase correlated with both myeloid hyperplasia and the associated statistically significant increase in Mac-1+ cells (t test, P = 0.02). These mice also showed a minor increase in the frequency of myeloid clonogenic progenitors in both the marrow (46,620 ± 30,181 vs. 49,065 ± 26,731 t test, P = 0.88) and spleen (63,837 ± 46,684 vs. 183,120 ± 200,160 t test, P = 0.22). No significant differences between control and HOX11 mice were noted in the absolute numbers of CD4+ and CD8+ T cells in spleens or lymph nodes. Normal white blood cell counts were noted in HOX11 mice with lymphoma (data not shown).
Analysis of IgH Rearrangements.
We used a VDJ-specific PCR assay to assess the status of the IgH gene rearrangements in cells isolated from spleens and thymuses of mice diagnosed with lymphoid hyperplasia, myeloid hyperplasia, and lymphoma. As shown in Fig. 7, VDJ-PCR of age-matched control spleen and thymus DNA amplified the expected 1.6-, 1.3-, 1.0-, and 0.4-kb fragments representing the products of VDJ1, VDJ2, VDJ3, and VDJ4 joinings, respectively. PCR analysis of DNA isolated from hematopoietic tissues from HOX11 mice affected with either myeloid or lymphoid hyperplasia also showed the four amplified fragments with no preferential amplification of one VDJ rearranged product. However, analysis of spleen and thymus samples, shown by histological analysis to contain lymphoma cells, indicated a 2- and 3-fold expansion, respectively, of a VDJ4 product. Coincident with the VDJ4 expansion was the reduction in the relative intensity of the other VDJ amplification products. Analysis of other HOX11 samples revealed 2- to 4-fold expansions of VDJ1, VDJ2, VDJ3, or VDJ4 clones (data not shown). In two mice, the same VDJ product was seen in both the spleen and thymus, suggesting expansion, dissemination, and infiltration of the same clone. In another mouse a 2-fold expansion of a VDJ1 product was seen in the spleen whereas a VDJ4 product was detected in the thymus, suggesting two independent sites of conversion of B cells to malignant lymphoma.
Figure 7.

PCR analysis of IgH rearrangements. PCR products of different VDJ recombination events are indicated on the left. Lanes 1 and 2 contain amplified DNA from the spleen and thymus of a control mouse. Lanes 3 and 4 contain DNA from a mouse with a myeloid hyperplasia of the spleen and the unaffected thymus. Lanes 5 and 6 contain DNA from a spleen and thymus with lymphoid hyperplasia in the spleen. Lanes 7–9 contain amplified DNA from a mouse with lymphoma cells in both the spleen (lane 8) and thymus (lane 9). The liver DNA sample (lane 7) served as a negative control.
Transplantation of HOX11 Transgenic Splenocytes into SCID Recipients.
Spleen cells from one control and four HOX11 transgenic mice designated HOX11-Tg1 to HOX11-Tg4) were injected i.p. or i.v. (three mice per group) into a total of 30 SCID recipients. Histological examination of the HOX11-Tg1 donor spleen revealed a focus of lymphoma in a background of lymphoid hyperplasia. Examination of four SCID spleens repopulated with HOX11-Tg1 splenocytes 14 weeks after injection revealed lymphoid hyperplasia with two spleens showing regions of clonal lymphocyte expansion. VDJ-specific PCR analysis indicated an approximate 2-fold expansion of a VDJ4 clone in two spleens. A 5-fold increase in B220+IgM+ cells was detected in SCID spleens repopulated with HOX11-Tg1 transgenic splenocytes (t test, P = 0.0005) (Fig. 8).
Figure 8.

Repopulation of SCID mice with HOX11 splenocytes. Spleen cells isolated from four HOX11 mice, designated HOX11 Tg-1 to Tg-4 were injected either i.p. or i.v. into SCID mice. Bars show B cell percentages in bone marrow, thymuses, or spleens of SCID recipients 10–14 weeks after injection of the indicated donor cells.
Histological examination of the spleen from HOX11-Tg2 indicated myeloid hyperplasia, whereas donor spleen HOX11-Tg3 showed lymphoid hyperplasia with no evidence of lymphoma. Both lymphoid and myeloid hyperplasia were noted in the spleen of HOX11-Tg4. Histological and flow cytometric analyses 10 or 14 weeks postinjection of SCID spleens repopulated with HOX11-Tg2, HOX11-Tg3, or HOX11-Tg4 splenocytes confirmed the transplantibility of both the myeloid and lymphoid hyperplasia (Fig. 8) but, to date, we have no data suggesting conversion to overt leukemia or lymphoma.
DISCUSSION
Despite the strong association of HOX11 activation with T cell neoplasia (5–10, 22), there has been little direct evidence that expression of HOX11 in T cells actually leads to malignant transformation. A preliminary report describing Lck-HOX11 transgenic mice suggested that ectopic expression of HOX11 in developing thymocytes of some transgenic mice resulted in perturbations in T cell development and that a proportion of these transgenic mice developed T cell lymphoma (32). In addition, two founder H-2K-HOX11 transgenic mice developed a lymphoproliferative disorder (33), but to date, detailed studies have not been published. In another study, two of eight mice reconstituted with bone marrow infected with a HOX11-bearing retrovirus developed T-ALL-like malignancies (34).
To directly assess the oncogenic potential of HOX11, we attempted to generate transgenic mice in which the human HOX11 cDNA was under the transcriptional control of either the murine IgH regulatory sequences or the LCK proximal promoter. Despite several attempts, only IgHμ-HOX11 founders expressing low levels of the transgene were obtained. Although numerous progeny from multiple heterozygous matings were produced, no homozygous HOX11 mice were identified (data not shown). Thus, high-level or T-lineage expression of HOX11 may be lethal.
Low-level HOX11 expression did not cause gross perturbations in hematopoiesis in transgenic mice. The total cellularity of all hematopoietic tissues analyzed and the absolute numbers of both lymphocyte and myeloid subpopulations from young HOX11 mice were within normal ranges. In addition, normal numbers of myeloid and B-lymphoid clonogenic progenitors were seen. However, both lymphoid and myeloid hyperplasia were noted in the spleens and marrow, respectively, of young mice, suggesting that low-level HOX11 expression may enhance the proliferative potential of hematopoietic cells.
A cohort study of HOX11 mice and control littermates for a 20-month observation period clearly demonstrated that HOX11 expression in lymphoid cells was oncogenic. All HOX11 transgenic mice became terminally ill during their second year of life with most mice developing lymphoma. The lymphomas were detected predominantly in spleens, suggesting that the spleen was the primary site of their development. With time, the lymphomas progressed to an aggressive phenotype characterized by dissemination to lymph nodes, thymus, liver, lungs, kidney, and pancreas. Transplantation of tumor containing splenocytes into SCID recipients demonstrated the malignant phenotype of these cells. The long latency period suggests that expression of HOX11 in lymphocytes was insufficient for malignant transformation and that the accumulation of additional genetic mutations are required.
The target cell for transformation in HOX11 mice is most likely a mature IgM+IgD+ B cell. Two- to four-fold clonal B cell expansions were detected in a polyclonal background of spleens and thymuses of several HOX11 mice. Heterogeneity with respect to expression of B-lineage markers was noted with some B cells expressing only IgM and others expressing neither IgM nor IgD. This heterogeneity most likely reflects genetic changes associated with tumor progression and the differentiation of tumor cells into more mature lymphocytes and plasma cells.
Histological examination of hematopoietic tissues from HOX11 mice identified a premalignant phase characterized by lymphoid hyperplasia in the spleens of apparently healthy mice. However, transplantation studies failed to show conversion of these cells to overt lymphoma although long-term follow-up studies are ongoing. A low percentage of both young and mature HOX11 mice developed mild to severe myeloid hyperplasia in bone marrow and/or spleens but we found no evidence for conversion to leukemia. These findings are in agreement with Hawley et al. (34, 35) who showed that expression of HOX11 in myeloid progenitors, although not leukemogenic, favored myeloid proliferation.
Low levels of HOX11 expression in lymphoid cells of HOX11 mice may contribute to lymphomagenesis by allowing cells with genomic abnormalities to bypass a G2/M cell cycle check point. Microinjection of HOX11 into Xenopus oocytes arrested at the G2 phase of the cell cycle promoted progression to the M phase (36). In addition, expression of HOX11 in a T cell line allowed the cells to bypass a normal G2 arrest induced by γ-irradiation (36). Thus HOX11 may play a role in progression from G2 to M in the cell cycle, and inappropriate HOX11 expression may disrupt normal cell cycling, thereby promoting genomic instability and facilitating tumor progression. The fact that normal lymphopoiesis involves physiological, error-prone genetic recombination underscores the importance of this G2 checkpoint.
This study clearly demonstrates the oncogenic potential of ectopic expression of HOX11 in murine lymphocytes. Furthermore, HOX11 mice provide a novel model in which to identify both putative HOX11 cofactors and potential downstream targets and thus help clarify the mechanisms underlying lymphomagenesis by HOX11. In addition, this animal model will provide a valuable tool for assessing therapies for human lymphoma because both the long latency period and the initiation of lymphoma in mature B cells are features of HOX11 transgenic mice that closely resemble the human condition.
Acknowledgments
We thank Dr. Megan Lim for critical review of this manuscript, Dr. Robert G. Hawley, The Toronto Hospital, for providing the murine IgHμ promoter and enhancer sequences used to construct the IgHμ-HOX11 transgenic vector, and Dr. Gillian E. Wu for assistance with the analysis of VDJ-PCR data. In addition, we gratefully acknowledge the excellent technical assistance of the pathology laboratory technologists of SD Laboratories, Sunnybrook Health Sciences Centre for preparation of tissue sections. Finally, we thank Denise Vanner and Josephine Correia for their assistance with animal husbandry and Kin Chan for preparation of figures. This work was supported by grants from the Medical Research Council of Canada, the National Cancer Institute of Canada, and the Hospital for Sick Children Foundation.
ABBREVIATIONS
- T-ALL
T cell acute lymphoblastic leukemias
- cDNA
cellular DNA
- SCID
severe combined immunodeficient
- IGH
Ig heavy chain
- CFU
colony-forming unit
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Nowell P C. Cancer Genet Cytogenet. 1997;94:13–19. doi: 10.1016/s0165-4608(96)00227-0. [DOI] [PubMed] [Google Scholar]
- 2.Korsmeyer S J. Annu Rev Immunol. 1992;10:785–807. doi: 10.1146/annurev.iy.10.040192.004033. [DOI] [PubMed] [Google Scholar]
- 3.Rabbitts T H. Nature (London) 1994;372:143–149. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- 4.Dubé I D, Raimondi S C, Pi D, Kalousek D K. Blood. 1986;67:1181–1184. [PubMed] [Google Scholar]
- 5.Dubé I D, Kamel-Reid S, Yuan C C, Lu M, Wu X, Corpus G, Raimondi S C, Crist W M, Carroll A J, Minowada J, Baker J B. Blood. 1991;78:2996–3003. [PubMed] [Google Scholar]
- 6.Lu M, Dubé I, Raimondi S, Carroll A, Zhao Y, Minden M, Sutherland P. Genes Chromosomes Cancer. 1990;2:217–222. doi: 10.1002/gcc.2870020309. [DOI] [PubMed] [Google Scholar]
- 7.Zutter M, Hockett R D, Roberts C W, McGuire E A, Bloomstone J, Morton C C, Deaven L L, Crist W M, Carroll A J, Korsmeyer S J. Proc Natl Acad Sci USA. 1990;87:3161–3165. doi: 10.1073/pnas.87.8.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kennedy M A, Gonzalez-Sarmiento R, Kees U R, Lampert F, Dear N, Boehm T, Rabbitts T H. Proc Natl Acad Sci USA. 1991;88:8900–8904. doi: 10.1073/pnas.88.20.8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kagan J, Joe Y S, Freireich E J. Cancer Res. 1994;54:226–230. [PubMed] [Google Scholar]
- 10.Hatano M, Roberts C W, Minden M, Crist W M, Korsmeyer S J. Science. 1991;253:79–82. doi: 10.1126/science.1676542. [DOI] [PubMed] [Google Scholar]
- 11.Lichty B D, Ackland-Snow J, Noble L, Kamel-Reid S, Dubé I D. Leukemia Lymphoma. 1995;16:209–215. doi: 10.3109/10428199509049759. [DOI] [PubMed] [Google Scholar]
- 12.Krumlauf R. Cell. 1994;78:191–201. doi: 10.1016/0092-8674(94)90290-9. [DOI] [PubMed] [Google Scholar]
- 13.Lawrence H J, Sauvageau G, Humphries R K, Largman C. Stem Cells. 1996;14:281–291. doi: 10.1002/stem.140281. [DOI] [PubMed] [Google Scholar]
- 14.Dear T N, Sanchez-Garcia I, Rabbitts T H. Proc Natl Acad Sci USA. 1993;90:4431–4435. doi: 10.1073/pnas.90.10.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wen X Y, Tang S, Breitman M L. Genomics. 1994;24:388–390. doi: 10.1006/geno.1994.1634. [DOI] [PubMed] [Google Scholar]
- 16.Tang S, Breitman M L. Nucleic Acids Res. 1995;23:1928–1935. doi: 10.1093/nar/23.11.1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dear T N, Colledge W H, Carlton M B, Lavenir I, Larson T, Smith A J, Warren A J, Evans M J, Sofroniew M V, Rabbitts T H. Development (Cambridge, UK) 1995;121:2909–2915. doi: 10.1242/dev.121.9.2909. [DOI] [PubMed] [Google Scholar]
- 18.Raju K, Tang S, Dubé I D, Kamel-Reid S, Bryce D M, Breitman M L. Mech Dev. 1993;44:51–64. doi: 10.1016/0925-4773(93)90016-q. [DOI] [PubMed] [Google Scholar]
- 19.Roberts V J, van Dijk M A, Murre C. Mech Dev. 1995;51:193–198. doi: 10.1016/0925-4773(95)00364-9. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto H, Hatano M, Iitsuka Y, Mahyar N S, Yamamoto M, Tokuhisa T. Mol Immunol. 1995;32:1177–1182. doi: 10.1016/0161-5890(95)00100-x. [DOI] [PubMed] [Google Scholar]
- 21.Moretti P, Simmons P, Thomas P, Haylock D, Rathjen P, Vadas M, D’Andrea R. Gene. 1994;144:213–219. doi: 10.1016/0378-1119(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 22.Salvati P D, Ranford P R, Ford J, Kees U R. Oncogene. 1995;11:1333–1338. [PubMed] [Google Scholar]
- 23.Lu M, Zhang N, Ho A D. Oncogene. 1992;7:1325–1330. [PubMed] [Google Scholar]
- 24.Roberts C W, Shutter J R, Korsmeyer S J. Nature (London) 1994;368:747–749. doi: 10.1038/368747a0. [DOI] [PubMed] [Google Scholar]
- 25.Gillies S D, Morrison S L, Oi V T, Tonegawa S. Cell. 1983;33:717–728. doi: 10.1016/0092-8674(83)90014-4. [DOI] [PubMed] [Google Scholar]
- 26.Clarke C, Berenson J, Goverman J, Boyer P D, Crews S, Siu G, Calame K. Nucleic Acids Res. 1982;10:7731–7749. doi: 10.1093/nar/10.23.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hawley R G, Covarrubias L, Hawley T, Mintx B. Proc Natl Acad Sci USA. 1987;84:2406–2410. doi: 10.1073/pnas.84.8.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 29.Pennycook J L M H, Marshal A J, Wu G E. In: Immunology Methods Manual. Lefkovits I, editor. New York: Academic; 1997. pp. 239–254. [Google Scholar]
- 30.Hough M R, Chappel M S, Sauvageau G, Takei F, Kay R. J Immunol. 1995;156:479–488. [PubMed] [Google Scholar]
- 31.Sauvageau G, Thorsteinsdottir U, Hough M R, Hugo P, Lawrence H J, Largman C, Humphries R K. Immunity. 1997;6:13–22. doi: 10.1016/s1074-7613(00)80238-1. [DOI] [PubMed] [Google Scholar]
- 32.Hatano, M., Roberts, C. W. M., Kawabe, T., Shutter, J. & Korsmeyer, S. J. (1992) Blood 80, Suppl., 355a (Abstr.).
- 33.Cheng S H, Mak T W. Dev Growth Differ. 1993;35:655–663. doi: 10.1111/j.1440-169X.1993.00655.x. [DOI] [PubMed] [Google Scholar]
- 34.Hawley R G, Fong A Z, Reis M D, Zhang N, Lu M, Hawley T S. Cancer Res. 1997;57:337–345. [PubMed] [Google Scholar]
- 35.Hawley R G, Fong A Z, Lu M, Hawley T S. Oncogene. 1994;9:1–12. [PubMed] [Google Scholar]
- 36.Kawabe T, Muslin A J, Korsmeyer S J. Nature (London) 1997;385:454–458. doi: 10.1038/385454a0. [DOI] [PubMed] [Google Scholar]