

Abstract
Aims
Deletion of the transcription factor Cited2 causes penetrant and phenotypically heterogenous cardiovascular and laterality defects and adrenal agenesis. Heterozygous human CITED2 mutation is associated with congenital heart disease, suggesting haploinsufficiency. Cited2 functions partly via a Nodal→Pitx2c pathway controlling left–right patterning. In this present study we investigated the primary site of Cited2 function and mechanisms of haploinsufficiency.
Methods and results
A Cited2 conditional allele enabled its deletion in particular cell lineages in mouse development. A lacZ reporter cassette allowed indication of deletion. Congenic Cited2 heterozygous mice were used to investigate haploinsufficiency. Embryos were examined by magnetic resonance imaging, by sectioning and by quantitative real-time polymerase chain reaction (qRT-PCR). Epiblast-specific deletion of Cited2 using Sox2Cre recapitulated penetrant and phenotypically heterogenous cardiovascular and laterality defects. Neural crest-specific deletion using Wnt1Cre affected cranial ganglia but not cardiac development. Mesodermal deletion with Mesp1Cre resulted in low penetrance of septal defect. Mesodermal deletion with T-Cre resulted in adrenal agenesis, but infrequent cardiac septal and laterality defects. β-Galatactosidase staining and qRT-PCR demonstrated the efficiency and location of Cited2 deletion. Murine Cited2 heterozygosity is itself associated with cardiac malformation, with three of 45 embryos showing ventricular septal defect. Cited2 gene expression in E13.5 hearts was reduced 2.13-fold in Cited2+/− compared with wild-type (P = 2.62 × 10−6). The Cited2 target gene Pitx2c was reduced 1.5-fold in Cited2+/− (P = 0.038) hearts compared with wild-type, and reduced 4.9-fold in Cited2−/− hearts (P = 0.00031). Pitx2c levels were reduced two-fold (P = 0.009) in Cited2+/− embryos, in comparison with wild-type. Cited2 and Pitx2c expression were strongly correlated in wild-type and Cited2+/− hearts (Pearson rank correlation = 0.68, P = 0.0009). Cited2 expression was reduced 7474-fold in Sox2Cre deleted hearts compared with controls (P = 0.00017) and Pitx2c was reduced 3.1-fold (P = 0.013). Deletion of Cited2 with Mesp1Cre resulted in a 130-fold reduction in cardiac Cited2 expression compared with control (P = 0.0002), but Pitx2c expression was not affected.
Conclusion
These results indicate that phenotypically heterogenous and penetrant cardiac malformations in Cited2 deficiency arise from a primary requirement in epiblast derivatives for left–right patterning, with a secondary cell-autonomous role in the mesoderm. Cardiac malformation associated with Cited2 haploinsufficiency may occur by reducing expression of key Cited2 targets such as Pitx2c.
Keywords: Cited2, Cardiac development, Phenotypic heterogeneity, Haploinsufficiency, Left–right patterning, Pitx2c
1. Introduction
The transcription factor CITED2 binds the histone acetyltransferase CREBBP/EP300 with high affinity,1 acting as a co-activator for transcription factors, such as TFAP2, LHX2, PPARA, and SMAD2/3.2–5 It also acts as a repressor of hypoxia-activated transcription.1 Genetic evidence indicates that Cited2 is essential for cardiac, adrenal, neural, and placental development, and also for embryonic left–right patterning.6–8 The cardiac malformations in mice lacking Cited2 are highly penetrant and phenotypically heterogeneic, and include atrial, atrioventricular, and ventricular septal defects (ASD, AVSD, VSD), outflow tract defects [double outlet right ventricle (DORV), common arterial trunk (CAT), tetralogy of Fallot (TOF), transposition of great arteries (TGA)], and interrupted and right-sided aortic arch.6,9 Mutation in Cited2 also results in adrenal agenesis, fusion of cranial ganglia, abnormal cardiac neural crest migration, and exencephaly.6
In addition, Cited2−/− mice show left–right patterning defects characterized by right atrial and pulmonary isomerism, and abnormal ventricular topology at a lower penetrance.7,8 Cardiovascular left–right patterning is created, in part, by the left-determining Nodal→Pitx2c pathway (reviewed by Hamada et al.10 and Ramsdell11). Genetic evidence shows that Cited2 is necessary for expression of Pitx2c, Nodal, and Lefty2 in the left-lateral plate mesoderm and of Lefty1 in the prospective floor plate, and is present at the promoter of the endogenous Pitx2c gene during development.7,8 Thus, Cited2 is necessary for Nodal-activated gene transcription.7 The mechanism for the cardiovascular phenotypic heterogeneity and the partial penetrance of left–right patterning defect, which is observed even on a congenic Cited2-deficient background,7 is not clear. Importantly, loss-of-function mutations in CITED2 have been found in patients with phenotypically variable congenital heart disease.12 These mutations are heterozygous, suggesting haploinsufficiency, but the mechanism for this is unclear.
Cited2 is expressed before gastrulation in the visceral endoderm.13 Following gastrulation it is present in the newly forming mesoderm, developing blood islands, cardiac crescent, presomitic and splanchnic mesoderm, cranial neuroectoderm, and migrating neural crest before becoming ubiquitously expressed by embryonic day (E)8.5.13 In the heart, it is highly expressed in the AV endocardial cushions.8 One possible mechanism for cardiac phenotypic heterogeneity in Cited2 deficiency is that not only is it required for the earlier process of left–right patterning but also independently for the later processes of atrioventricular septation, outflow tract, and aortic arch development.8 Abnormal patterning of aortic arches and CAT observed in Cited2 deficiency could, for instance, arise from defective neural crest development.6 An alternative possibility is that an abnormality in the earlier developmental process of left–right patterning may secondarily affect the subsequent processes of atrioventricular septal, outflow tract, or aortic arch development.7 These different mechanisms were investigated with a conditional knockout approach. We also investigated the mechanism of haploinsufficiency using quantitative analysis of Cited2 and its target gene Pitx2c. We show that the phenotypic heterogeneity in Cited2 deficiency is because of a primary requirement in epiblast derivatives with a secondary cell-autonomous role in the mesoderm. Also, Cited2 haploinsufficiency may cause cardiac malformation by reducing expression of key Cited2 targets such as Pitx2c.
2. Methods
2.1. Molecular biology
Standard molecular biology procedures were used for plasmid constructions.14
2.2. Generation of Cited2flox/flox mice
The targeting vector strategy and generation of correctly targeted embryonic stem cells with a conditional Cited2 allele (Cited2flox or Cited2f) is shown in Supplementary material online, Figure S1. Methods are also provided in detail in the Supplementary data. The allele was designed such that, after successful Cre recombination, a lacZ expression cassette came under the control of the endogenous Cited2 promoter. This allowed indication of efficient recombination by β-galactosidase staining.
2.3. Conditional deletion of Cited2
Cited2+/− mice (Cited2tm1Bha) on a C57BL/6J background7 were crossed with Sox2Cre (Tg(Sox2-cre)1Amc),15Wnt1Cre (Tg(Wnt1-cre)11Rth)16 (both gifts from Andrew McMahon, Harvard University), Mesp1Cre (Mesp1tm2(cre)Ysa)17 or TCre18 mice to generate males with Cited2+/−;Cre genotypes. These males were crossed to Cited2flox/flox females on a mixed 129Sv × C57BL/6J background to generate embryos with Cited2−/flox;Cre and Cited2+/flox;Cre (control) genotypes. This strategy prevented global recombination owing to Cre expression in the maternal germline.19Cited2−/flox;Cre embryos lack Cited2 expression in cells expressing Cre-recombinase. Cited2+/flox;Cre embryos continue to express Cited2, providing littermate controls. Embryos were harvested at the indicated time points after the detection of a vaginal plug (E0.5), and genotyped using allele-specific polymerase chain reaction (PCR) (primer details are available on request). All mouse experiments conform with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23, revised 1996).
2.4. Analysis of embryos
Magnetic resonance imaging (MRI) was performed on a horizontal 9.4 T/21 cm VNMRS Direct Drive™ MR system (Varian Inc., Palo Alto, CA, USA) essentially as described previously20 on embryos at E15.5. Whole mount in situ immunostaining was performed on E9.5 embryos using anti-neurofilament antibody (clone 2H3, Developmental Studies Hybridoma Bank, University of Iowa, IA, USA).21 X-Gal and nuclear fast red (Sigma, St. Louis, MO, USA) staining were performed according to standard procedures.14,16
2.5. Quantitative real time-polymerase chain reaction
Embryo hearts (E13.5) were dissected out initially in DEPC treated phosphate-buffered saline (PBS), and then finely dissected in RNAlater (Ambion, Austin, TX, USA) to remove mediastinal tissue. Embryos at E8.5 were dissected free from the yolk sac in cold DEPC–PBS and flash-frozen in liquid nitrogen. RNA was isolated using a Nucleospin RNA purification kit (Clontech, Palo Alto, CA, USA), and an Archive cDNA kit (Ambion) used to synthesize complementary DNA. Quantitative RT-PCR (qRT-PCR) reactions were carried out using a Bio-Rad I-Cycler (Bio-Rad, CA, USA) and pre-optimized TaqMan® primer-probe sets from Applied Biosytems (Mus musculus assays Mm00516121_m1 (for NM_010828 Cited2), Mm00440826_m1 (for NM_001042502 Pitx2c), and eukaryotic 18S rRNA). Expression levels were normalized to 18S rRNA using the R0 method of analysis.22,23 Multiplex reactions were performed with 18S rRNA and the TaqMan primer-probe set for the gene of interest. Data were normalized to the level obtained for 18S RNA in each case. All reactions were performed in triplicates and, as a quality control, only samples for which the threshold cycle (Ct) replicate values were within 1 Ct value of each other were used for subsequent analysis.24 Mean values for each reaction triplicate were analysed.
2.6. Statistical analyses
We used the chi-squared test for deviation from expected Mendelian ratios (http://www.kursus.kvl.dk/shares/vetgen/_Popgen/genetik/applets/ki.htm), and calculated probability of a type I error using the CHIDIST function in Microsoft Excel (Redmond, WA, USA). For analysis of the qRT-PCR data, we used a two-tailed, two-sample heteroscedastic t-test assuming unequal variance.
3. Results
3.1. Deletion of Cited2 in the epiblast
To determine the role of Cited2 in the epiblast it was deleted using Sox2Cre which induces recombination in all epiblast cells by E6.5.15. Genotyping of mice at weaning showed a complete absence of Cited2−/flox;Sox2Cre embryos, but genotyping at E15.5 showed that these embryos were present in the expected Mendelian ratio (Supplementary material online, Table S1). This indicated that Cited2−/flox;Sox2Cre embryos die either in late gestation or perinatally. Two of 10 Cited2−/flox;Sox2Cre embryos at E15.5 had visible exencephaly (not shown). Analysis of Cited2−/flox;Sox2Cre embryos at E15.5 using MRI showed that they frequently had a classical cardiac laterality defect, i.e. right-atrial isomerism, associated with right-pulmonary isomerism (six of 10 embryos) (Figure 1; see also see Supplementary material online, Table S2), indicating a defect in embryonic left–right patterning. VSDs were present in all embryos (see Supplementary material online, Table S2). The embryos invariably had adrenal agenesis (see Supplementary material online, Figure S2). No defects were seen in the 11 littermate control Cited2+/flox;Sox2Cre or in eight Cited2−/flox embryos.
Figure 1.
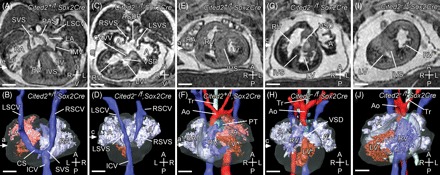
Deletion of Cited2 in the epiblast: cardiac morphology. Magnetic resonance imaging of E15.5 embryos. (A–D) Transverse sections and three-dimensional (3D) reconstructions (dorsal views) of Cited2+/flox;Sox2Cre (control) and Cited2−/flox;Sox2Cre hearts. (A, B) Control normal heart showing a pectinated (P) right atrium (RA), with a systemic venous sinus (SVS), into which drains the right superior caval vein (RSCV), the left superior caval vein (LSCV) via the coronary sinus (CS) and the inferior caval vein (ICV). The left atrium (LA) is characterized by the primary atrial septum (PAS). Other structures seen are right and left ventricles (RV, LV), interventricular septum (IVS), and the mitral and tricuspid valves (MV, TV not labelled). (C, D) Cited2−/flox;Sox2Cre heart showing a large primum atrial septal defect (ASDP), resulting in a common atrium (A). This is pectinated on each side and has bilateral systemic venous sinuses (LSVS, RSVS), into which drains the bilateral superior and inferior caval veins. The coronary sinus is absent. These appearances indicate right atrial isomerism. A common atrioventricular valve (CAVV) over-rides a ventricular septal defect (VSD). Note also that the heart is malpositioned to the right. (E–J) Coronal sections and 3D reconstructions (ventral views) of Cited2+/flox;Sox2Cre (control) and Cited2−/flox;Sox2Cre hearts. (E, F) Control normal heart where the right ventricle is dextral to the left, and gives rise to the main pulmonary trunk (PT). The left ventricle gives rise to the aorta (Ao), which arches to the left of the trachea (Tr) over the left main bronchus. (G, H) Cited2−/flox;Sox2Cre heart with normal ventricular topology, cardiac dextroposition, and a right-sided aortic arch arising from the right ventricle. (I, J) Cited2−/flox;Sox2Cre heart with abnormal ventricular topology: the right ventricle is sinistral (and anterior) to the left ventricle. Scale bars = 500 µm; axes: D, dorsal; V, ventral; R, right; L, left; A, anterior; P, posterior.
3.2. Deletion of Cited2 in the neural crest
To determine the requirement for Cited2 in the neural crest it was deleted using Wnt1Cre, which drives expression of Cre-recombinase in all neural crest-derived tissues, including the cardiac neural crest, from E9.16,25,26 There was a significant reduction in the numbers of Cited2−/flox;Wnt1Cre mice in comparison with Cited2+/flox;Wnt1Cre mice at weaning [21 of 61 vs. 40 of 61, respectively (χ2 = 5.9; P < 0.015). There was no observed reduction when genotyping was performed between E9.5 and E18.5. Examination of embryos at E15.5–E18.5 (n = 16) using MRI revealed none with heart or great vessel defects, but one embryo had exencephaly (Figure 2B). Exencephaly was not observed in Cited2+/flox;Wnt1Cre embryos (n = 16). Examination of cranial ganglia by anti-neurofilament staining in Cited2−/flox;Wnt1Cre embryos showed that 11 of 14 sets had fusions of ganglia IX and X (Figure 2E and F) and hypomorphic ganglia V and VII/VIII. In contrast, two of 28 sets of ganglia from Cited2+/flox;Wnt1Cre embryos had fusions of ganglia IX and X (χ2 = 22.28; P < 0.001). In both Cited2−/flox;Wnt1Cre and in Cited2+/flox;Wnt1Cre embryos, we observed extensive lacZ staining in the cranial mesenchyme, and in the first and second pharyngeal arches (Figure 2G and H). Staining was also observed in the cardiac neural crest (the third and fourth/sixth pharyngeal arches). The staining pattern corresponded to that described with Wnt1Cre-driven recombination in R26R mice that express lacZ conditional to Cre-mediated recombination.16
Figure 2.
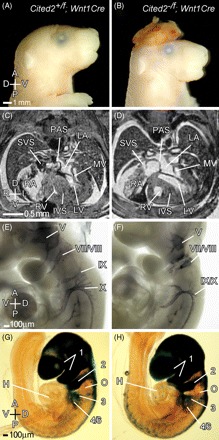
Deletion of Cited2 in the neural crest. Control (Cited2+/flox;Wnt1Cre, left panels) and mutant (Cited2−/flox;Wnt1Cre, right panels) embryos were analysed at different stages of development. (A, B) Control and mutant E17.5 embryos, respectively. The mutant embryo has exencephaly. (C, D) Magnetic resonance imaging (MRI) transverse sections of E17.5 control and mutant embryos. No cardiac or aortic arch abnormalities were identified in 14 mutant embryos examined by MRI. (E, F) Anti-neurofilament antibody staining of control and mutant E9.5 embryos showing cranial ganglia. The fifth (trigeminal ganglion) is smaller, and the ninth and tenth (glossopharyngeal and vagal) ganglia are fused in the mutant. (G, H) LacZ staining of control and mutant embryos. Extensive staining is observed in the cranial mesenchyme, and in the first and second pharyngeal arches. Staining is also observed in the cardiac neural crest (the third and fourth/sixth pharyngeal arches). No difference was observed in either case. The otic vesicle (O) is indicated. Axes and abbreviations are as shown in Figure 1.
3.3. Deletion of Cited2 in the mesoderm
The role of Cited2 in the cranial, cardiac, and extraembryonic mesoderm was examined by deletion using crosses with Mesp1Cre mice, which deletes in these tissues by E7.5.17,27 As this Cre allele is a ‘knockin’ and disrupts the endogenous gene, we analysed genotypic ratios of offspring born by crossing these lines with Cited2+/− mice to determine if there was a genetic interaction. The number of Cited2+/−;Mesp1Cre+/KI mice at weaning was similar to Cited2+/+;Mesp1Cre+/KI mice indicating no significant genetic interaction (Supplementary material online, Table S3). Isolated septal defects (but no outflow or aortic arch defects) were seen in three of 18 Cited2−/flox;Mesp1Cre embryos and no defects were seen in the seven Cited2+/flox;Mesp1Cre or nine Cited2−/flox littermate control embryos studied (Supplementary material online, Figure S3). The deletion of Cited2 using a TCre driver was then investigated. This would be expected to delete extensively in the lateral-plate, intermediate, and paraxial mesoderm and the hindgut endoderm, with earliest deletion being detected by E7.518. Deletion of Cited2 using TCre resulted in frequent adrenal agenesis (10 of 14 embryos), and infrequent cardiac septal and left–right patterning defect (three of 14 and one of 14, respectively, Figure 3). No defects were identified in the 11 Cited2+/flox;TCre littermate control embryos studied.
Figure 3.
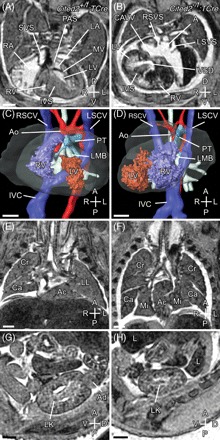
Deletion of Cited2 using TCre. Control (Cited2+/flox;TCre, left panels) and mutant (Cited2−/flox;TCre, right panels) embryos were analysed at E15.5 using magnetic resonance imaging. (A) Transverse section of a control embryo showing normal cardiac anatomy. (B) Corresponding transverse section from the single mutant embryo (of 14 analysed) that showed a laterality defect. Right atrial isomerism is indicated by bilateral systemic venous sinuses and a common atrium created by complete absence of the primary atrial septum. The heart is dextroposed, with abnormal ventricular topology [dextral left ventricle (LV)] and a ventricular septal defect. (C) Three-dimensional (3D) reconstruction of the control embryo heart showing normal topology of the ventricles. The aorta (Ao) arises from the LV, and pulmonary trunk (PT) from the right ventricle (RV). Both Ao and PT arch over the left main bronchus (LMB). (D) 3D reconstruction of the mutant embryo heart showing abnormal ventricular topology. Both Ao and PT arise from the RV and arch over the LMB. (E) Sagittal section through the control embryo showing normal pulmonary topology. The three lobes of the right lung are seen. The left lung (LL) has a single lobe. (F) Sagittal section through the mutant embryo showing right pulmonary isomerism. Both lungs have three lobes. A fused accessory (Ac) lobe is also seen. (G, H) Sagittal sections through the left kidney (LK) of control and mutant embryos, respectively. The adrenal gland (Ad) is seen in the control but not in the mutant embryo. Scale bars = 500 µm. Axes and abbreviations are as shown in Figure 1.
3.4. Analysis of Cited2 recombination
3.4.1. LacZ staining
The above results indicate that Cited2 deficiency induced by Wnt1Cre, Mesp1Cre, or TCre drivers cannot explain either the phenotypic heterogeneity or penetrance of cardiac malformation and left–right patterning defects observed in the epiblast-specific deletion of Cited2. To explore the mechanism and evaluate the efficiency of recombination we examined the expression of lacZ in these different Cited2+/flox;Cre embryos at E9.5 in comparison with Cited2+/flox;Sox2Cre (Figure 4 and Supplementary material online, Table S4). Deletion of exon 2 by Cre-recombinase brings the lacZ cassette under control of the endogenous Cited2 promoter, so analysis of lacZ expression allowed the confirmation of Cited2flox recombination in the embryonic lineages where both the Cre driver and Cited2 are expressed. In the heart, we observed staining resembling the Sox2Cre pattern using Mesp1Cre, consistent with effective recombination in the cardiogenic mesoderm.17,27 Reduced lacZ staining was observed in the outer curvature of the left ventricle suggesting that Cited2 is not highly expressed here (Figure 4). With TCre we observed complete recombination in the common atrial chamber, but partial recombination in the atrioventricular canal, primitive ventricle, bulbus cordis, and outflow tract. The lateral plate mesoderm is thought to play a key role in establishing left–right patterning (reviewed in Hamada et al.10). We therefore examined the expression of lacZ in structures derived from the lateral plate mesoderm: these include the mesenchyme of the body wall (somatopleure) and of the viscera (splanchnopleure).28 In Cited2+/flox;Sox2Cre embryos, strong lacZ expression was observed in the splanchnopleure, and weaker expression in the somatopleure. With TCre and Mesp1Cre, lacZ expression was detected chiefly in the posterior splanchnopleure surrounding the midgut. No lacZ staining was detected in the somatopleure.
Figure 4.

LacZ expression in Cited2+/flox;Cre embryos. The left panels show pictures of embryos (E9.5) with the indicated genotypes after staining for lacZ. Subsequent panels show transverse embryo sections counterstained with nuclear fast red. ACV, anterior cardinal vein; AVC, atrioventricular canal; BA, branchial arch; BC, bulbus cordis; CAC, common atrial chamber; CCV, common cardinal vein; DA, dorsal aorta; FG, foregut; HD, hepatic diverticulum; HG, hindgut; HGD, hindgut diverticulum; HM, head mesenchyme; LB, lung bud; LM, lateral mesenchyme from splanchnopleure; MG, midgut; NC, nephric cord (intermediate mesenchyme); NT, neural tube; OFT, outflow tract; PC, peritoneal cavity; PPC, pleuroperitoneal canal; PV, primitive ventricle; ST, septum transversum; S, somite. Scale bars = 500 µm. Axes are as shown in Figure 1.
We also examined lacZ expression at earlier developmental stages (Supplementary material online, Figure S4). At earlier stages, in Cited2+/flox;Sox2Cre embryos at E7.5, lacZ expression was present in all epiblast-derived tissues, including the extraembryonic mesoderm. In Cited2+/flox;TCre embryos, we observed lacZ staining in the extraembryonic mesoderm, amnion, and blood islands, and also to some extent in the nascent embryonic mesoderm. This became more extensive in the primitive streak and migrating embryonic mesoderm at slightly later stages. Examination of head-fold stage Cited2+/flox;Mesp1Cre embryos showed extensive recombination in the cardiac crescent (Supplementary material online, Figure S4).
3.4.2. Quantitative real-time polymerase chain reaction
To verify efficient deletion of Cited2 in the Sox2Cre and Mesp1Cre conditional knockouts at the mRNA level, and to determine the effect on the Cited2 target gene Pitx2c, qRT-PCR was performed. Cited2 expression was reduced 7474-fold (P = 0.00017) in Cited2−/flox;Sox2Cre hearts compared with littermate controls (Figure 5A). There was also a 3.1-fold reduction in Pitx2c expression (P = 0.013; Figure 5B). Cited2 expression was reduced 130-fold in Cited2−/flox;Mesp1Cre hearts compared with littermate controls (Figure 5C). However, no reduction in Pitx2c expression was seen in these hearts (Figure 5D).
Figure 5.
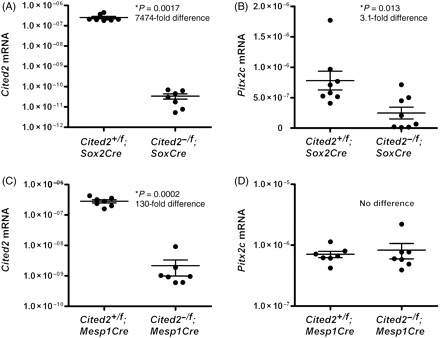
Cited2 and Pitx2c mRNA levels in Cited2 conditionally deleted hearts at E13.5. Sample mean and SEM are indicated. (A) Cited2 mRNA levels were reduced 7474-fold in Cited2−/flox;Sox2Cre hearts compared with controls. (B) A significant 3.1-fold decrease in Pitx2c mRNA levels was found in Cited2−/flox;Sox2Cre hearts compared with controls. (C) In Cited2−/flox;Mesp1Cre hearts, Cited2 levels were reduced 130-fold compared with controls. (D) No significant difference in Pitx2c expression was found between Cited2−/flox;Mesp1Cre hearts and controls.
3.5. Cited2 haploinsufficiency in the mouse
The mutations found in human congenital heart disease patients12 suggested that Cited2 haploinsufficiency may occur. We tested for this in the mouse by examining the genotypic ratios of Cited2+/− intercrosses where we obtained 500 wild-type and 782 Cited2+/− pups (expected 855). The 8.5% reduction in Cited2+/− pups was statistically significant (χ2 = 18.5; P < 0.0001). A similar reduction was not observed however when the genotypic ratios of E13.5 or E15.5 embryos were examined (Supplementary material online, Table S5).
To determine a mechanism for reduced survival, we examined Cited2+/− embryos at E15.5 by MRI (Figure 6). This was followed by histological sectioning of embryos that had either oedema or a cardiac abnormality visible on MRI. We found that three of 45 Cited2+/− embryos had a clearly identifiable VSD, and one presented with DORV with hypoplastic pulmonary trunk (Figure 6). These defects were not seen in the wild-type littermate controls. Thus, heterozygosity for a Cited2 null allele is associated, at low penetrance, with cardiac malformation in the mouse, implying haploinsufficiency.
Figure 6.
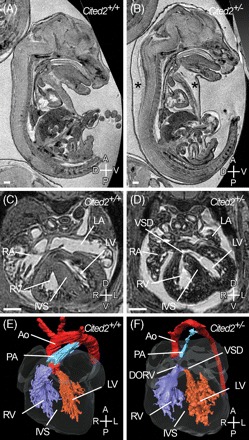
Oedema and cardiac malformations in Cited2+/− embryos. Magnetic resonance imaging of wild-type and littermate Cited2+/− embryos at E15.5. Sagittal sections of wild-type (A) and Cited2+/− (B) embryos. Severe oedema (asterisks) is seen in the Cited2+/− embryo. (C) Transverse section of wild-type and Cited2+/− embryos. The Cited2+/− embryo has a ventricular septal defect (VSD) (D). (E, F) Three-dimensional reconstruction of the heart in the wild-type and Cited2+/− embryos shown in the previous panel. The aorta (Ao) and pulmonary artery (PA) arise from the left ventricle and right ventricle (RV), respectively, in the wild-type embryo, but in the Cited2+/− embryo they arise from the RV creating a double-outlet right ventricle (DORV). VSD is indicated. Scale bars, 500 µm. Axes are as shown in Figure 1.
3.6. Quantitative real-time-polymerase chain reaction analysis of Pitx2c and Cited2 expression
To determine if haploinsufficiency in Cited2-deficient hearts is linked to abnormality in Pitx2c expression, we examined gene expression using qRT-PCR. Embryos and hearts on a C57BL/6J background, generated by backcrossing for 14 generations, were used to minimize the effects of genetic background. Hearts at E13.5 were used as this is a key time-point in cardiac septation, and embryos at E8.5 were selected as Pitx2c is expressed asymmetrically in the left lateral plate mesoderm at this stage. In E13.5 hearts, Cited2 was reduced 2.13-fold in Cited2+/− compared with wild-type (P = 2.62 × 10−6) (Figure 7A). We found that the Cited2 mRNA in Cited2−/− hearts was either undetectable or, if detectable, had values ranging between 1.45 × 10−12 and 3.47 × 10−11 relative to 18S (Figure 7C), likely to represent the limits of the assay. The Cited2 target gene Pitx2c was reduced 1.5-fold in Cited2+/− (P = 0.038) hearts and 4.7-fold (P = 0.00031) in Cited2−/− hearts compared with wild-type controls (Figure 7B and D). Cited2 and Pitx2c mRNA were strongly correlated in wild-type and Cited2+/− hearts (Pearson rank correlation = 0.68, P = 0.0009; Figure 7F). In the E8.5 embryos, we found a two-fold reduction in Pitx2c levels in Cited2+/− compared with wild-type (P = 0.009; Figure 7E).
Figure 7.
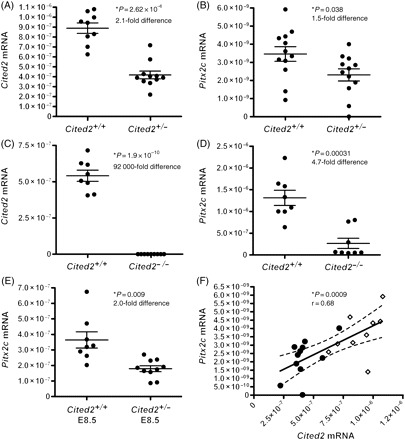
Cited2 and Pitx2c mRNA levels in E13.5 embryo hearts and E8.5 embryos. Sample mean and SEM are indicated. (A) A 2.1-fold reduction in Cited2 mRNA levels was seen in Cited2+/− hearts compared with wild-type. (B) Pitx2c levels were reduced 1.5-fold in Cited2+/− hearts compared with wild-type. (C) A 92 000-fold reduction in Cited2 mRNA is seen in the Cited2−/− hearts compared with wild-type. (D) Pitx2c mRNA levels were reduced 4.7-fold in the Cited2−/− hearts compared with wild-type. (E) Pitx2c levels in wild-type and Cited2+/− E8.5 embryos. A two-fold reduction in Pitx2c mRNA is seen in the Cited2+/− embryos. (F) Pearson correlation between Cited2 and Pitx2c E13.5 heart mRNA levels with line-of-best-fit and 95% confidence intervals (dotted lines) shown. Filled circles represent data from Cited2+/− hearts and diamonds those from wild-type. Cited2 and Pitx2c mRNA levels were strongly correlated in wild-type and Cited2+/− hearts.
4. Discussion
In these experiments, we used a conditional knockout approach to understand how Cited2 deficiency results in cardiac phenotypic heterogeneity. To define the role of Cited2 in the epiblast, we deleted it with Sox2Cre.15 This resulted in 100% penetrance of cardiovascular malformations and adrenal agenesis. The most common cardiac malformation was a VSD, which was present in all embryos, and was associated with DORV in some embryos. In addition we observed left–right patterning, atrial septal, outflow tract, and aortic arch defects. These observations are consistent with, and fully recapitulate, those previously reported with the global deletion of Cited2.6–9,29 They indicate that Cited2 is essential in the epiblast or its derivatives for normal embryonic left–right patterning, and for cardiac and adrenal development. The complete penetrance of the defects, and the widespread expression of lacZ in embryos containing a Cited2flox allele and the Cre transgene also indicate that efficient recombination occurs in response to Cre expression. This was further validated by the large reduction seen in Cited2 mRNA levels in the hearts of Cited2−/flox;Sox2Cre embryos. We observed reduced lacZ staining in the outer curvature of the left ventricle following conditional deletion of Cited2 with Sox2Cre (as well as with Mesp1Cre and TCre). This is consistent with the published data,8 and indicates that Cited2 is expressed at a lower level in this region of the heart.
To define the role of Cited2 in the neural crest, we deleted it with Wnt1Cre.16,30 This resulted in cranial ganglia fusions and exencephaly, but did not affect cardiac septal, outflow tract, aortic arch or adrenal development, or left–right patterning. These results indicate that although Cited2 does have a cell autonomous role in the neural crest, as evidenced by fusion of the cranial ganglia, it is not required here for outflow tract septation or aortic arch patterning.
To determine the requirement of Cited2 in the cranial, cardiac, and extraembryonic mesoderm it was deleted with Mesp1Cre, which results in extensive recombination in these tissues by E7.5.17,27 Subsequently, by E9.5, recombination is observed extensively in myocardium and endocardium including the mesenchyme of the cono-truncal and atrioventricular cushions, in the epicardium, and in the pharyngeal arch mesoderm and vascular endothelium.17,27,31 Deletion of Cited2 using Mesp1Cre resulted in low penetrance of septal defect, and no outflow tract, aortic arch, or laterality defects. These results indicate that Cited2 function in the mesoderm does not explain the phenotypically heterogeneic cardiac malformations observed in the epiblast-specific deletion. Efficient recombination at the Cited2 locus in response to Cre expression as demonstrated by lacZ staining and qRT-PCR indicated that lack of a phenotypically heterogeneic phenotype is unlikely to be owing to inefficient recombination.
As Cited2 is expressed in the lateral-plate mesoderm,8 and as its deficiency also leads to loss of Nodal-activated transcripts such as a Nodal, Lefty2, and Pitx2c in the left-lateral plate mesoderm,7,8 we investigated its role here by deletion using TCre.18 This resulted in infrequent septal and laterality defect. Deletion of Cited2 induced by TCre was predominantly in the posterior rather than the anterior extent of the lateral plate mesoderm, making this one possible mechanism for the infrequency of left–right patterning defect observed with a TCre-induced deletion. The lacZ staining pattern seen in Cited2+/flox;TCre embryos was comparable with that seen with TCre × R26R lacZ embryos.18 Moreover, in the heart the staining pattern was highly reminiscent of that observed for Isl132 suggesting that TCre may also drive recombination in second heart field-derived regions of the heart. These results indicate that the penetrant and phenotypically heterogeneic cardiac malformations in Cited2 deficiency arise from a primary requirement in epiblast derivatives for left–right patterning with a secondary cell-autonomous role in the mesoderm for septation. A limitation of our studies, however, is that the role of Cited2 in the endoderm—which has a major role in cardiac induction33—could not be established. We are currently limited here by the lack of a suitable Cre driver that expresses in the endoderm at the appropriate time-point.
Our data also show that Cited2 haploinsufficiency occurs in the mouse and is associated with cardiac malformation. Although 8.5% of Cited2 heterozygous pups are lost by weaning, consistent with the numbers of cardiac malformations observed, no deviation from expected numbers was seen during development. The mechanistic link between cardiac malformations and Cited2 haploinsufficiency is unclear, and to address this, we quantitatively examined the expression of Cited2 and its target gene Pitx2c in embryonic hearts and E8.5 embryos. Using qRT-PCR we showed that Cited2 expression is significantly reduced in Cited2 heterozygous embryonic hearts. We also showed that this was accompanied by a significant reduction in Pitx2c expression, and that the two were strongly correlated. We also observed a significant reduction in Pitx2c levels in E8.5 Cited2+/− embryos. As the dose of Pitx2c is crucial for normal left–right patterning and cardiac development,34,35 we suggest that Pitx2c deficiency observed in Cited2 heterozygous hearts and embryos could explain, at least in part, the observed cardiac malformations.
In conclusion, these results indicate that phenotypically heterogeneic and penetrant cardiac malformations in Cited2 deficiency arise from a primary requirement in epiblast derivatives for left–right patterning with a secondary cell-autonomous role in the mesoderm for septation. Cardiac malformation associated with Cited2 haploinsufficiency may occur by reducing the expression of key Cited2 targets, such as Pitx2c from E8.5. Importantly, cardiovascular phenotypic heterogeneity is observed, for instance, with mutation in NKX2-5, 36–38CFC139,40 or ZIC3.41–43 Consistent with our proposed model, these genes, like Cited2, are also known to control or modulate left–right patterning pathways.42,44–47 Our results support the emerging idea7,11 that genes controlling early left–right patterning are candidates for diverse forms of human congenital heart disease, even in the absence of a classical laterality defect such as isomerism.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
The Wellcome Trust (Grant 054528), the British Heart Foundation (Grant PG/06/075/21127), and the Center for Cancer Research, National Cancer Institute. S.T.M. is a Wellcome Trust Prize Student (Grant 076257/Z/04/Z) and S.B. a Wellcome Trust Senior Research Fellow.
Supplementary Material
Acknowledgements
We thank Stuart Peirson for advice on quantitative real-time polymerase chain reaction and Hannah Barnes for technical help with magnetic resonance imaging.
Conflict of interest: none declared.
References
- 1.Bhattacharya S, Michels CL, Leung MK, Arany ZP, Kung AL, Livingston DM. Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev. 1999;13:64–75. doi: 10.1101/gad.13.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braganca J, Eloranta JJ, Bamforth SD, Ibbitt JC, Hurst HC, Bhattacharya S. Physical and functional interactions among AP-2 transcription factors, p300/CREB-binding protein, and CITED2. J Biol Chem. 2003;278:16021–16029. doi: 10.1074/jbc.M208144200. [DOI] [PubMed] [Google Scholar]
- 3.Glenn DJ, Maurer RA. MRG1 binds to the LIM domain of Lhx2 and may function as a coactivator to stimulate glycoprotein hormone alpha-subunit gene expression. J Biol Chem. 1999;274:36159–36167. doi: 10.1074/jbc.274.51.36159. [DOI] [PubMed] [Google Scholar]
- 4.Tien ES, Davis JW, Vanden Heuvel JP. Identification of the CREB-binding protein/p300-interacting protein CITED2 as a peroxisome proliferator-activated receptor alpha coregulator. J Biol Chem. 2004;279:24053–24063. doi: 10.1074/jbc.M401489200. [DOI] [PubMed] [Google Scholar]
- 5.Chou YT, Wang H, Chen Y, Danielpour D, Yang YC. Cited2 modulates TGF-beta-mediated upregulation of MMP9. Oncogene. 2006;25:5547–5560. doi: 10.1038/sj.onc.1209552. [DOI] [PubMed] [Google Scholar]
- 6.Bamforth SD, Braganca J, Eloranta JJ, Murdoch JN, Marques FI, Kranc KR, et al. Cardiac malformations, adrenal agenesis, neural crest defects exencephaly in mice lacking Cited2 a new Tfap2 co-activator. Nat Genet. 2001;29:469–474. doi: 10.1038/ng768. [DOI] [PubMed] [Google Scholar]
- 7.Bamforth SD, Braganca J, Farthing CR, Schneider JE, Broadbent C, Michell AC, et al. Cited2 controls left-right patterning and heart development through a Nodal→Pitx2c pathway. Nat Genet. 2004;36:1189–1196. doi: 10.1038/ng1446. [DOI] [PubMed] [Google Scholar]
- 8.Weninger WJ, Floro KL, Bennett MB, Withington SL, Preis JI, Barbera JP, et al. Cited2 is required both for heart morphogenesis and establishment of the left-right axis in mouse development. Development. 2005;132:1337–1348. doi: 10.1242/dev.01696. [DOI] [PubMed] [Google Scholar]
- 9.Yin Z, Haynie J, Yang X, Han B, Kiatchoosakun S, Restivo J, et al. The essential role of Cited2, a negative regulator for HIF-1{alpha}, in heart development and neurulation. Proc Natl Acad Sci USA. 2002;99:10488–10493. doi: 10.1073/pnas.162371799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamada H, Meno C, Watanabe D, Saijoh Y. Establishment of vertebrate left-right asymmetry. Nat Rev Genet. 2002;3:103–113. doi: 10.1038/nrg732. [DOI] [PubMed] [Google Scholar]
- 11.Ramsdell AF. Left-right asymmetry and congenital cardiac defects: getting to the heart of the matter in vertebrate left-right axis determination. Dev Biol. 2005;288:1–20. doi: 10.1016/j.ydbio.2005.07.038. [DOI] [PubMed] [Google Scholar]
- 12.Sperling S, Grimm CH, Dunkel I, Mebus S, Sperling HP, Ebner A, et al. Identification and functional analysis of CITED2 mutations in patients with congenital heart defects. Hum Mutat. 2005;26:575–582. doi: 10.1002/humu.20262. [DOI] [PubMed] [Google Scholar]
- 13.Dunwoodie SL, Rodriguez TA, Beddington RSP. Msg1 and Mrg1, founding members of a gene family, show distinct patterns of gene expression during mouse embryogenesis. Mech Dev. 1998;72:27–40. doi: 10.1016/s0925-4773(98)00011-2. [DOI] [PubMed] [Google Scholar]
- 14.Ausubel F, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, et al. Short Protocols in Molecular Biology. Hoboken: John Wiley & Sons, Inc.; 1995. [Google Scholar]
- 15.Hayashi S, Lewis P, Pevny L, McMahon AP. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech Dev. 2002;119(Suppl. 1):S97–S101. doi: 10.1016/s0925-4773(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 16.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- 17.Saga Y, Miyagawa-Tomita S, Takagi A, Kitajima S, Miyazaki J, Inoue T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development. 1999;126:3437–3447. doi: 10.1242/dev.126.15.3437. [DOI] [PubMed] [Google Scholar]
- 18.Perantoni AO, Timofeeva O, Naillat F, Richman C, Pajni-Underwood S, Wilson C, et al. Inactivation of FGF8 in early mesoderm reveals an essential role in kidney development. Development. 2005;132:3859–3871. doi: 10.1242/dev.01945. [DOI] [PubMed] [Google Scholar]
- 19.Hayashi S, Tenzen T, McMahon AP. Maternal inheritance of Cre activity in a Sox2Cre deleter strain. Genesis. 2003;37:51–53. doi: 10.1002/gene.10225. [DOI] [PubMed] [Google Scholar]
- 20.Schneider JE, Bose J, Bamforth SD, Gruber AD, Broadbent C, Clarke K, et al. Identification of cardiac malformations in mice lacking Ptdsr using a novel high-throughput magnetic resonance imaging technique. BMC Dev Biol. 2004;4:16–27. doi: 10.1186/1471-213X-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilkinson DG. Whole mount in situ hybridization of vertebrate embryos. In: Wilkinson DG, editor. In situ Hybridization. Oxford: IRL Press; 1992. pp. 75–83. [Google Scholar]
- 22.Peirson SN. Quantitative analysis of ocular gene expression. In: Dorak TM, editor. Real-time PCR. New York: Taylor & Francis Group; 2006. pp. 107–126. [Google Scholar]
- 23.Peirson SN, Butler JN, Foster RG. Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res. 2003;31:e73–e79. doi: 10.1093/nar/gng073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams PS. Data analysis and reporting. In: Dorak TM, editor. Real-time PCR. New York: Taylor & Francis Group; 2006. pp. 41–62. [Google Scholar]
- 25.Tallquist MD, Soriano P. Cell autonomous requirement for PDGFRalpha in populations of cranial and cardiac neural crest cells. Development. 2003;130:507–518. doi: 10.1242/dev.00241. [DOI] [PubMed] [Google Scholar]
- 26.Chai Y, Jiang X, Ito Y, Bringas P, Jr, Han J, Rowitch DH, et al. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 2000;127:1671–1679. doi: 10.1242/dev.127.8.1671. [DOI] [PubMed] [Google Scholar]
- 27.Saga Y, Kitajima S, Miyagawa-Tomita S. Mesp1 expression is the earliest sign of cardiovascular development. Trends Cardiovasc Med. 2000;10:345–352. doi: 10.1016/s1050-1738(01)00069-x. [DOI] [PubMed] [Google Scholar]
- 28.Kaufman M, Bard JBL. The Anatomical Basis of Mouse Development. San Diego: Academic Press; 1999. [Google Scholar]
- 29.Weninger WJ, Mohun T. Phenotyping transgenic embryos: a rapid 3-D screening method based on episcopic fluorescence image capturing. Nat Genet. 2002;30:59–65. doi: 10.1038/ng785. [DOI] [PubMed] [Google Scholar]
- 30.Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Z, Cerrato F, Xu H, Vitelli F, Morishima M, Vincentz J, et al. Tbx1 expression in pharyngeal epithelia is necessary for pharyngeal arch artery development. Development. 2005;132:5307–5315. doi: 10.1242/dev.02086. [DOI] [PubMed] [Google Scholar]
- 32.Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, et al. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell. 2003;5:877–889. doi: 10.1016/s1534-5807(03)00363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moorman AF, Christoffels VM. Cardiac chamber formation: development, genes, and evolution. Physiol Rev. 2003;83:1223–1267. doi: 10.1152/physrev.00006.2003. [DOI] [PubMed] [Google Scholar]
- 34.Liu C, Liu W, Lu MF, Brown NA, Martin JF. Regulation of left-right asymmetry by thresholds of Pitx2c activity. Development. 2001;128:2039–2048. doi: 10.1242/dev.128.11.2039. [DOI] [PubMed] [Google Scholar]
- 35.Gage PJ, Suh H, Camper SA. Dosage requirement of Pitx2 for development of multiple organs. Development. 1999;126:4643–4651. doi: 10.1242/dev.126.20.4643. [DOI] [PubMed] [Google Scholar]
- 36.McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E. NKX2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol. 2003;42:1650–1655. doi: 10.1016/j.jacc.2003.05.004. [DOI] [PubMed] [Google Scholar]
- 37.Goldmuntz E, Geiger E, Benson DW. NKX2.5 mutations in patients with tetralogy of fallot. Circulation. 2001;104:2565–2568. doi: 10.1161/hc4601.098427. [DOI] [PubMed] [Google Scholar]
- 38.Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, et al. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest. 1999;104:1567–1573. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bamford RN, Roessler E, Burdine RD, Saplakoglu U, dela Cruz J, Splitt M, et al. Loss-of-function mutations in the EGF-CFC gene CFC1 are associated with human left-right laterality defects. Nat Genet. 2000;26:365–369. doi: 10.1038/81695. [DOI] [PubMed] [Google Scholar]
- 40.Goldmuntz E, Bamford R, Karkera JD, dela Cruz J, Roessler E, Muenke M. CFC1 mutations in patients with transposition of the great arteries and double-outlet right ventricle. Am J Hum Genet. 2002;70:776–780. doi: 10.1086/339079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Megarbane A, Salem N, Stephan E, Ashoush R, Lenoir D, Delague V, et al. X-linked transposition of the great arteries and incomplete penetrance among males with a nonsense mutation in ZIC3. Eur J Hum Genet. 2000;8:704–708. doi: 10.1038/sj.ejhg.5200526. [DOI] [PubMed] [Google Scholar]
- 42.Gebbia M, Ferrero GB, Pilia G, Bassi MT, Aylsworth A, Penman-Splitt M, et al. X-linked situs abnormalities result from mutations in ZIC3. Nat Genet. 1997;17:305–308. doi: 10.1038/ng1197-305. [DOI] [PubMed] [Google Scholar]
- 43.Ware SM, Peng J, Zhu L, Fernbach S, Colicos S, Casey B, et al. Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am J Hum Genet. 2004;74:93–105. doi: 10.1086/380998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shiratori H, Sakuma R, Watanabe M, Hashiguchi H, Mochida K, Sakai Y, et al. Two-step regulation of left-right asymmetric expression of Pitx2: initiation by nodal signaling and maintenance by Nkx2. Mol Cell. 2001;7:137–149. doi: 10.1016/s1097-2765(01)00162-9. [DOI] [PubMed] [Google Scholar]
- 45.Yan YT, Gritsman K, Ding J, Burdine RD, Corrales JD, Price SM, et al. Conserved requirement for EGF-CFC genes in vertebrate left-right axis formation. Genes Dev. 1999;13:2527–2537. doi: 10.1101/gad.13.19.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaio U, Schweickert A, Fischer A, Garratt AN, Muller T, Ozcelik C, et al. A role of the cryptic gene in the correct establishment of the left-right axis. Curr Biol. 1999;9:1339–1342. doi: 10.1016/s0960-9822(00)80059-7. [DOI] [PubMed] [Google Scholar]
- 47.Purandare SM, Ware SM, Kwan KM, Gebbia M, Bassi MT, Deng JM, et al. A complex syndrome of left-right axis, central nervous system and axial skeleton defects in Zic3 mutant mice. Development. 2002;129:2293–2302. doi: 10.1242/dev.129.9.2293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.